

Abstract
Oxidative stress-induced mitochondrial dysfunction and mitochondrial DNA (mtDNA) damage in peripheral neurons is considered to be important in the development of diabetic neuropathy. Mitochondrial transcription factor A (TFAM) wraps mtDNA and promotes mtDNA replication and transcription. We studied whether overexpression of TFAM reverses experimental peripheral diabetic neuropathy using TFAM transgenic mice (TFAM Tg) that express human TFAM (hTFAM). Levels of mouse mtDNA and the total TFAM (mouse TFAM + hTFAM) in the dorsal root ganglion (DRG) increased by approximately twofold in the TFAM Tg mice compared with control (WT) mice. WT and TFAM Tg mice were made diabetic by the administration of streptozotocin. Neuropathy end points were motor and sensory nerve conduction velocities, mechanical allodynia, thermal nociception, and intraepidermal nerve fiber density (IENFD). In the DRG neurons, mtDNA copy number and damage to mtDNA were quantified by qPCR, and TFAM levels were measured by Western blot. Mice with 16-wk duration of diabetes developed motor and sensory nerve conduction deficits, behavioral deficits, and intraepidermal nerve fiber loss. All of these changes were mostly prevented in diabetic TFAM Tg mice and were independent of changes in blood parameters. Mice with 16 wk of diabetes had a 40% decrease in mtDNA copy number compared with nondiabetic mice (P < 0.01). Importantly, the mtDNA copy number in diabetic TFAM Tg mice reached the same level as that of WT nondiabetic mice. In comparison, there was upregulation of mtDNA and TFAM in 6-wk diabetic mice, suggesting that TFAM activation could be a therapeutic strategy to treat peripheral neuropathy.
Keywords: mitochondria, mitochondrial transcription factor A, diabetes, neuropathy
diabetes-induced oxidative damage in neurons, axons, and Schwann cells has been proposed as a unifying mechanism for diabetic neuropathy (3, 13, 18, 22, 43, 51, 54, 55, 60). One mechanism for generation of oxidative stress is that an increased metabolic influx into mitochondria increases respiration and results in a high proton gradient with high redox equivalents, leading to increased production of reactive oxygen species (ROS) (13, 18, 22, 43, 51, 54, 55, 60). There is accumulating evidence that neuronal mitochondria are impaired in diabetic neuropathy (56). The inner membrane potential of mitochondria in sensory neuronal cultures from diabetic animals is depolarized, dorsal root ganglia (DRG) neurons from diabetic rodents exhibit reduced respiratory chain activity, and mitochondrial DNA (mtDNA) levels are decreased in chronic diabetic neuropathy (45, 56). In contrast, factors that promote mitochondrial regeneration would rescue impaired mitochondria and may be protective in diabetic neuropathy.
Mitochondrial biogenesis requires the participation of two genetic systems, nuclear DNA (nDNA) encoding the majority of proteins that are transported to mitochondria and the mitochondrial genomic system (19, 20, 25, 26, 28, 33). mtDNA contains two promoters, the light- (LSP) and heavy-strand promoters (HSP), from which transcripts are produced and then processed to yield the individual mRNAs encoding 13 subunits of the oxidative phosphorylation system, ribosomal, and transfer RNAs (4, 6, 14, 26). Mitochondrial transcription factor A (TFAM) is an nDNA-encoded protein that binds upstream of the LSP and HSP of mtDNA and promotes replication and transcription of mtDNA (8, 27, 53). TFAM is essential for maintenance of mtDNA and for cell survival. Homozygous knockout of TFAM is embryonic lethal (29). In contrast, tissue-specific knockout mice survive and have been used to study the role of TFAM in disease models (52). Mice with TFAM knockout in pancreatic β-cells develop diabetes from the age of 5 wk and display mtDNA depletion, deficient oxidative phosphorylation, and abnormal-appearing mitochondria in islets at the ages of 7–9 wk (50). Overexpression of TFAM in mice protects against delayed neuronal death due to forebrain transient ischemia (23), improves working memory (21), and protects mitochondria against β-amyloid-induced oxidative damage (59). Therefore, there is a rationale to test the effect of TFAM overexpression in diabetic neuropathy.
TFAM plays a dual role. First, TFAM maintains mtDNA copy number by promoting mtDNA replication, which is essential for preservation of mitochondrial function (8). mtDNA copy number correlates with mitochondrial gene expression levels as well as with mitochondrial respiratory activity (15). In contrast, age-related depletion of mtDNA in human islets contributes to decreased mitochondrial function and risk of type 2 diabetes (38). The second role of TFAM is structural. TFAM wraps mtDNA entirely to form a nucleoid structure similar to histones in the nucleosome (1, 39, 58) that may also protect mtDNA against ROS (9). Recent results show that it is important to maintain the ratio of TFAM to mtDNA within a narrow range, since small TFAM variations may affect transcription and mtDNA replication (16). A small increase in TFAM levels in vivo (∼2-fold) leads to a proportional increase in mtDNA.
The pathogenic event proposed in diabetic neuropathy is that chronic diabetes causes an increase the generation of ROS, resulting in increased mtDNA mutations, and loss of mtDNA. These changes in turn cause impaired mitochondrial respiratory function (11, 17), loss of cellular energy, reduced ATP, and neuronal degeneration. We tested whether overexpression of TFAM would upregulate the mtDNA levels and protect against peripheral neuropathy.
MATERIALS AND METHODS
Generation of TFAM transgenic mice.
TFAM transgenic (Tg) mice were generated as described previously (24). In brief, a modified chicken β-actin promoter with cytomegalovirus enhancer-driven human TFAM (hTFAM) cDNA construct was microinjected into the pronuclei of fertilized eggs of C57BL/6 mice. Founder lines were identified by the presence of the hTFAM cDNA in the tail DNA (24). Human TFAM-positive Tg mice were mated to C57BL/6 wild-type (WT) mice to generate WT and heterozygous Tg mice. WT and TFAM Tg mice were used at 10 to 13 wk of age.
Animals.
All animal procedures were approved by the Institutional Animal Care and Use Committee of the University of Maryland and the Veterans Affairs Maryland Health Care System. Male C57Bl/6J mice were fed standard chow (no. 7001; Harlan-Teklad, Madison, WI) with a 12:12-h light-dark cycle and free access to food and water. At 3 mo of age, diabetes was induced via serial intraperitoneal injections of streptozotocin (STZ) over 6 days, following our published procedure (11). Mice having blood glucose levels of 300 mg/dl (16.7 mM) or greater were considered to be diabetic. Age-matched nondiabetic control male mice were injected with vehicle instead of STZ. WT and TFAM Tg mice were tested at 6 and 16 wk after the induction of diabetes. There were four groups of mice at each time point: WT nondiabetic (n = 30), WT diabetic (n = 60), TFAM Tg nondiabetic (n = 30), and TFAM Tg diabetic (n = 60).
Evaluation of nerve conduction velocity, von Frey sensory testing, thermal latency, and intraepidermal nerve fiber density.
For phenotyping neuropathy in animals, the guidelines provided by the diabetic neuropathy study group of the European Association for the Study of Diabetes (Neurodiab) were followed (7). For nerve conduction studies, mice were anesthetized with either ketamine and xylazine or isoflurane. Thermal support was provided, and tail and sciatic-peroneal nerve conduction studies were performed as described previously (11, 57). Tail and limb temperatures were maintained at 32–33°C. Tail orthodromic sensory responses were obtained using low-intensity, long-duration supramaximal stimulation, and averaging of the responses until the baseline and the recording were stable.
Mechanical allodynia was assessed using Somedic von Frey monofilaments, using the Semmes-Weinstein series (Somedic Sales) as described in detail (11). Ordinal numbers >4 were applied gently on the fat part of both plantar heels until the hair started to bend and maintained for ∼2 s. The threshold was defined as the minimal bending force of the thinnest filament sensed by the mouse in an ascending and descending series of applications. A withdrawal response is considered valid only if the hindpaw is completely removed from the platform. Hargreaves' test was used to test thermal nociception, which assesses small nerve fiber function. Mice were left in a multiple animal enclosure cage (Harvard Apparatus) to acclimatize for 30 min. The temperature of the glass floor was maintained at 30°C. Light from a halogen bulb lamp was delivered to the plantar surface of the mouse hindpaw through the base of the glass panel to induce the heat stimuli. The time taken for the mouse to lift or lick its hindpaw was recorded automatically by the device. The intensity of the radiant heat was adjusted to reach a basal latency of 8–10 s. A cutoff time of 20 s was used to avoid tissue damage. Three measurements were performed with intervals of 1–2 min.
Staining for intraepidermal nerve fiber density (IENFD) was performed as described previously (11, 30, 31). The mean IENFD were measured using standardized measurement protocols and compared with controls (11). IENFD was determined by the number of complete baseline crossings of nerve fibers at the dermoepidermal junction divided by the calculated length of the epidermal surface.
Western blot analysis.
DRG neurons were homogenized in lysis buffer (50 mM Tris·HCl, pH 7.4, 1% SDS, 1% Triton X-100, and 150 mM NaCl). Proteins (25 μg) were extracted and SDS-PAGE gels prepared as described previously (12). Nitrocellulose membranes were probed with anti-mouse-specific TFAM antibody (cat. no. 28-597; Prosci), anti-human-specific TFAM antibody (cat. no. 7495; Cell Signaling Technology), or antibody recognizing both mouse and human TFAM (cat. no. SAB1401383; Sigma-Aldrich). Following application of species-specific secondary antibodies, the signal was detected using the Super Signal chemiluminescence kit (Pierce), and the signal intensity was evaluated using an Alpha Innotech imaging system.
Mitochondiral oxygen consumption.
Oxygen consumption was measured using a thermostatically controlled Clark-type O2 electrode (Hansatech Instruments, Norfolk, UK), as described previously (49). State 3 respiration was measured with malate and glutamate substrates in the presence of ADP (10, 11). Approximately 2 min later state 3 was terminated, and state 4 respiration (resting) was initiated with the addition of the ATP synthase inhibitor oligomycin (1.25 μg/ml). The maximal rate of uncoupled respiration was subsequently measured by titration with 54 nM FCCP.
Quantitative real-time PCR measurement of mtDNA and other genes.
Lumbar DRG were harvested from diabetic (after 16 wk of diabetes) and nondiabetic mice. RNA and DNA were isolated using a standard Invitrogen protocol. The ratio of mtDNA to nDNA was determined using a quantitative PCR method (10, 11). The cycle threshold (CT) values of mtDNA and nDNA were used to determine the relative ratio of mtDNA to nDNA in DRG neurons. Relative ratio of NADH dehydrogenase subunit 1 (ND1; encoded on the mitochondrial genome) over lipoprotein lipase (LPL; encoded on the nuclear genome) was determined. Primers and PCR conditions were used as described previously (11). For other real-time PCR experiments, the Applied Biosystems primer assay IDs are as follows: TFAM, Mm00447485_m1; manganese superoxide dismutase (MN SOD/SOD2), Mm00449726_m1; glutathione peroxidase (GPX1), Mm00656767-g1. Individual genes were run against GAPDH or β-actin as controls.
Mitochondrial DNA copy number and mitochondrial DNA damage.
Genomic DNA was isolated from the L5 DRG neurons. Mitochondrial DNA copy number was determined by PCR of two mtDNA targets, a 197-bp ND1 gene and a 199-bp Cyt B gene, and the CT values were compared with a standard plasmid carrying ND1 and CytB mtDNA fragments (10). Nuclear DNA copy number was determined by PCR for a nuclear DNA 175-bp B2M gene target and the CT values were compared with a plasmid carrying a B2M nuclear gene fragment. The ratio of mtDNA to nDNA was calculated by 2 × (ND1 copies/20 ng DNA)/(B2M copies/20 ng DNA) and by 2 × (Cyt B copies/20 ng DNA)/(B2M copies/20 ng DNA). Damage to mtDNA was measured by long-range (LR) qPCR of an 8.9-kb mtDNA target (10). The amount of PCR product was quantified, and the amount was inversely related to mtDNA damage.
Adult mouse neuron culture and measurement of oxidative stress and cellular injury.
DRG were collected from adult WT and TFAM Tg C57Bl/6J mice (11). DRG were placed in Leibovitz's L-15 media and centrifuged to pellet, and 0.5 ml of papain (2 mg/ml in Hanks' balanced solution) and 0.5 ml of collagenase (2.5% in sterile water; Worthington) were added and incubated for 30 min at 37°C. After 30 min, 2 ml of FBS (Atlanta Biological) was added to inhibit the enzymes, and cells were centrifuged, processed, and plated as described (11). The final concentration of media components was as follows: selenium (5.2 μg/ml), hydrocortisone (7.6 μg/ml), transferrin (10 μg/ml), estradiol (5.4 μg/ml), FUDR, and penicillin-streptomycin. The cells were cultured in a total of 5.5 mM glucose for 24 h and then exposed to either 5.5 or 25 mM glucose in serum-free media. As a measure of oxidative stress, 2′,7′-dichlorofluorescein (DCF) fluorescence was measured as described previously (5). Briefly, 15 min prior to the end of the 5-h glucose treatment period, cultures were rinsed with HBSS and further incubated with 2-dichlorofluorescin diacetate (2.5 μmol/l) in HBSS for 15 min at 37°C. Cells were rinsed once with phosphate-buffered saline (PBS) and then fixed for 30 min with 4% paraformaldehyde in PBS and mounted on slides with Biomedia Gel Mount. For each slide, images from 10 random fields of 25–100 cells per field were collected in bright-field and fluorescent channels. DCF pixel intensity was determined, mean pixel intensity for each field of neurons was calculated, and background intensity was subtracted.
Statistics.
Comparison of dependent variables was performed on transformed data using factorial analysis of variance (ANOVA) with an ad hoc Tukey test used to determine the significance among the groups. Individual comparisons were made using Student's t-test, assuming unequal variances (42). Levels of significance were determined from two-tailed t-tests. An observer blinded to the experimental condition made measurements. Bar graphs illustrate means ± SE.
RESULTS
Characterization of hTFAM transgenic mouse.
hTFAM cDNA was used to generate the Tg mice and was mated with WT C57BL6 mice to generate heterozygous hTFAM-Tg mice. The Tg mice were identified from tail DNA by PCR using primers specific to hTFAM (Fig. 1A). The promoter used to drive the expression of hTFAM was a modified chicken β-actin promoter with cytomegalovirus enhancer. To ensure that the Tg mice expressed hTFAM in DRG neurons and did not affect the expression of endogenous mouse TFAM (mTFAM), RT-PCR and Western blot analysis were performed using species-specific primers and antibodies (24). The results are shown in Fig. 1, B and C. We found in DRG neurons that hTFAM mRNA and protein were expressed only in the TFAM Tg mice and not in the WT mice. Furthermore, in DRG neurons, the mTFAM mRNA and protein levels were similar in both WT and Tg mice, which demonstrated that the expression of hTFAM did not affect the expression of endogenous mTFAM gene. Quantification of the intensity of the TFAM bands suggests that that the total levels of TFAM (mTFAM + hTFAM) increased 80–100% in the Tg mice compared with WT mice (Fig. 1).
Fig. 1.
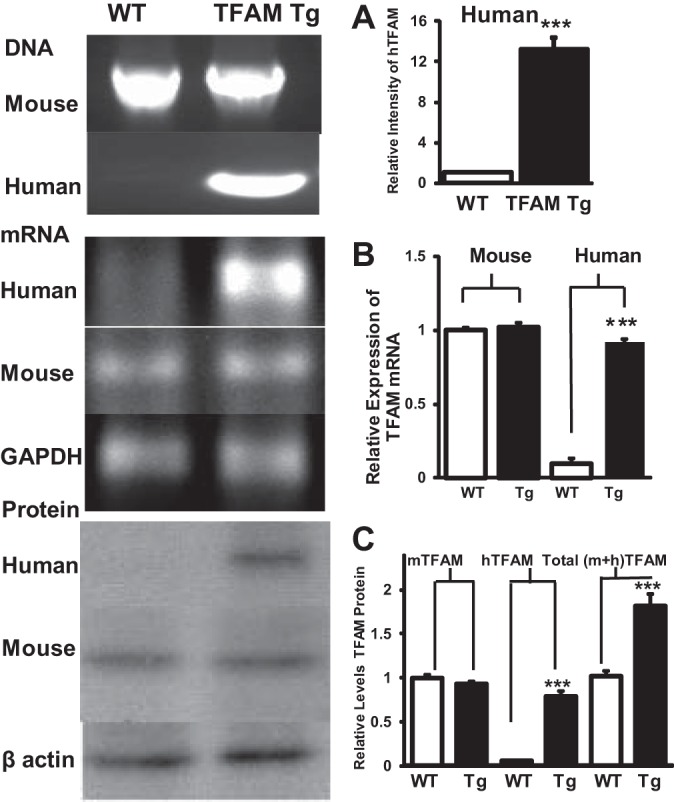
Presence and expression of mouse and human mitochondrial transcription factor A (TFAM) gene, mRNA, and protein in wild-type (WT) and TFAM transgenic (Tg) mice. A: PCR reaction using human TFAM (hTFAM)-specific primers detected the presence of the transgene band only in the Tg mice tail DNA and was absent in the tail DNA from WT mice. B: RT-PCR reaction, using species-specific primers, in DRG neuronal RNA from Tg and WT mice showed the presence of hTFAM only in the Tg. In contrast, RT-PCR reaction using mouse primers showed the presence of mouse TFAM in both WT and Tg mice. Right: quantification of the CT values were normalized to control gene (GAPDH) and expressed as relative gene expression. Results are means ± SE. C: Western blot analysis in the protein extracts from doral root ganglia (DRG) neurons of Tg and WT mice showed the presence of hTFAM only in the Tg when hTFAM antibody was used. In contrast, mouse TFAM (mTFAM) was detected in both WT and Tg mice (see materials and methods). Quantification of the band intensity of the Western blots showed that the total TFAM [mouse TFAM plus human (m + h TFAM) TFAM] was increased ∼2-fold in Tg DRG protein extracts compared with WT. Data are normalized to controls and expressed as means ± SE. ***P < 0.001 by t-test between WT and Tg mice.
Expression of mtDNA and mitochondrial respiration.
We tested whether the expression of hTFAM is functional in the Tg mouse, and does it regulate the levels of mouse mtDNA? The result is shown in in Fig. 2. We performed Southern blot analyses to assess mtDNA levels in total DNA extracts from DRG neurons from WT and Tg animals (Fig. 2A, left), and found a clear increase in mtDNA levels in DRG neurons from the Tg animals. The overall increase in mtDNA in Tg mice compared with the WT mice was ∼90% (Fig. 2A, middle), and this increase in mtDNA paralleled the increase in hTFAM protein levels. The calculated ratio between TFAM protein and mtDNA remained the same in both WT and Tg mice (Fig. 2A, right). Mitochondria were isolated from lumbar sensory DRG neurons of 6-mo-old WT and Tg mice, and mitochondrial state 3 and state 4 respiration was measured. The results are shown in Fig. 2B. ADP-stimulated state 3 and oligomycin-sensitive state 4 respiration rates were ∼20% higher in Tg mice compared with the WT mice. The increase was not statistically significant. There was no significant difference in respiratory control ratio between the groups.
Fig. 2.

mtDNA and mitochondrial function in DRG neurons of WT and TFAM Tg mice. A, left: Southern blot analysis of mtDNA in total DNA extracts from the DRG neurons of WT and TFAM Tg mice. Top bands show signals from the mtDNA fragment, and bottom bands show signals from the nuclear DNA fragment containing the 18S rRNA gene. Middle: quantification of mtDNA levels (n = 4). Data were obtained by densitometry quantification of the Southern blots, such as those shown at left. Right: ratio of mtDNA to total TFAM protein in WT and Tg mice. Values were calculated from the densitometry intensity of the bands measured by Southern blot for mtDNA and Western intensity for TFAM protein. TFAM values in Tg were calculated as a ratio to WT values. B: mitochondrial respiration in isolated mitochondria from DRG neurons (n = 5). Data are compared with controls and expressed as means ± SE. **P < 0.01 by t-test between WT and Tg mice. RCR, respiratory control ratio.
Characteristics of WT and TFAM Tg control and diabetic mice.
Diabetes was induced in 3-mo-old WT and TFAM Tg mice by the administration of STZ. The body weight, blood glucose levels, and blood lipid levels are shown in animal groups at 6 and 16 wk (Table 1). Both WT diabetic and TFAM Tg diabetic mice lost a significant amount of weight. Fasting blood glucose was significantly elevated, and insulin was decreased in both WT diabetic and TFAM Tg diabetic mice. There was no significant difference between the diabetic groups in these values, even though TFAM transgene was expressed in all tissues. Total cholesterol was significantly increased in TFAM Tg diabetic compared with WT diabetic mice (232 ± 64.34 vs. 128.3 ± 29.6). More importantly, HDL cholesterol, the critical cholesterol scavenger and transporter, was significantly higher in TFAM Tg diabetic mice compared with WT diabetic mice(110.5 ± 6.83 vs. 73.9 ± 7.56).
Table 1.
| Nondiabetic vs. Diabetic Animals | WT | WT Diab | TFAM Tg | TFAM Tg Diab | Significance (P Value) |
|||
|---|---|---|---|---|---|---|---|---|
| 1 vs. 2 | 3 vs. 4 | 1 vs. 3 | 2 vs. 4 | |||||
| 6 Wk | ||||||||
| Weight, g | 29.7 ± 0.7 | 24.4 ± 1.1 | 31.1 ± 0.5 | 23.8 ± 1.1 | <0.001 | <0.001 | NS | NS |
| Blood glucose, mg/dl | 159 ± 8 | 479 ± 53 | 142 ± 15 | 459 ± 45 | <0.001 | <0.001 | NS | NS |
| Insulin, μU/ml | 4.50 ± 0.3 | 1.87 ± 0.36 | 6.15 ± 0.12 | 1.52 ± 0.02 | <0.001 | <0.01 | NS | NS |
| Total cholesterol, mg/dl | 85.3 ± 5.9 | 120.3 ± 19 | 86.8 ± 1.7 | 225 ± 52.5 | NS | <0.01 | NS | <0.01 |
| HDL, mg/dl | 59 ± 1.9 | 64.4 ± 6.33 | 58.8 ± 1.67 | 107.5 ± 7.0 | <0.05 | <0.01 | NS | <0.01 |
| Triglycerides, mg/dl | 58 ± 9.2 | 133.2 ± 20 | 59.33 ± 6 | 144.6 ± 7.1 | <0.01 | <0.01 | NS | NS |
| 16 Wk | ||||||||
| Weight, g | 31.7 ± 0.9 | 22.4 ± 1 | 33.1 ± 0.9 | 21.8 ± 1.6 | <0.001 | <0.001 | NS | NS |
| Blood glucose, mg/dl | 163 ± 12 | 467 ± 48 | 146 ± 18 | 454 ± 52 | <0.001 | <0.001 | NS | NS |
| Insulin, μU/ml | 5.49 ± 0.3 | 1.78 ± 0.3 | 5.85 ± 0.23 | 1.63 ± 0.08 | <0.001 | <0.001 | NS | NS |
| Total cholesterol, mg/dl | 88.6 ± 6.98 | 128.3 ± 30 | 89.68 ± 3.7 | 232 ± 64.3 | <0.05 | <0.01 | NS | <0.05 |
| HDL, mg/dl | 58.97 ± 3.7 | 73.9 ± 7.56 | 54.82 ± 3.5 | 110.5 ± 6.8 | <0.05 | <0.01 | NS | <0.01 |
| Triglycerides, mg/dl | 48 ± 8.7 | 126.2 ± 16 | 54.33 ± 9.5 | 102.5 ± 9.3 | <0.01 | <0.01 | NS | NS |
Values are means ± SE. WT, wild type; Diab, diabetic; TFAM, mitochondrial transcription factor A; Tg, transgenic; NS, not significant. For 6-wk nondiabetic vs. diabetic animals, n = 6 WT, 12 WT Diab, 6 TFAM Tg, and 9 TFAM Tg Diab; for 16-wk nondiabetic vs. diabetic animals, n = 5 WT, 10 WT Diab, 8 TFAM Tg, and 8 TFAM Tg Diab.
Mitochondrial degeneration in DRG neurons of WT and TFAM Tg diabetic mice.
We compared the ratio of mtDNA to nDNA in DRG neurons from WT and TFAM Tg mice before and after 6 or 16 wk of STZ-induced diabetes. We used real-time PCR to obtain a relative ratio of ND1 (a gene coded by mtDNA) over LPL (a unigene encoded by nDNA). The ratio is an indicator for relative mtDNA levels. The results (Fig. 3A) showed a significant decrease (40%, P < 0.05) in ND1 to LPL in DRG neurons after 16 wk of diabetes compared with nondiabetic WT mice. In TFAM Tg diabetic mice, there was a decrease (∼30%) in mtDNA in in DRG neurons after 16 wk of diabetes compared with nondiabetic TFAM Tg mice. The decrease was not significant when compared with WT nondiabetic mice. Thus, compared with WT nondiabetic mice, after 16 wk of diabetes, there was no decrease in mtDNA in TFAM Tg diabetic mice. Chronic diabetes decreases mtDNA levels in DRG neurons, and the overexpression of TFAM Tg was able to retain the levels of mtDNA within the WT nondiabetic range.
Fig. 3.
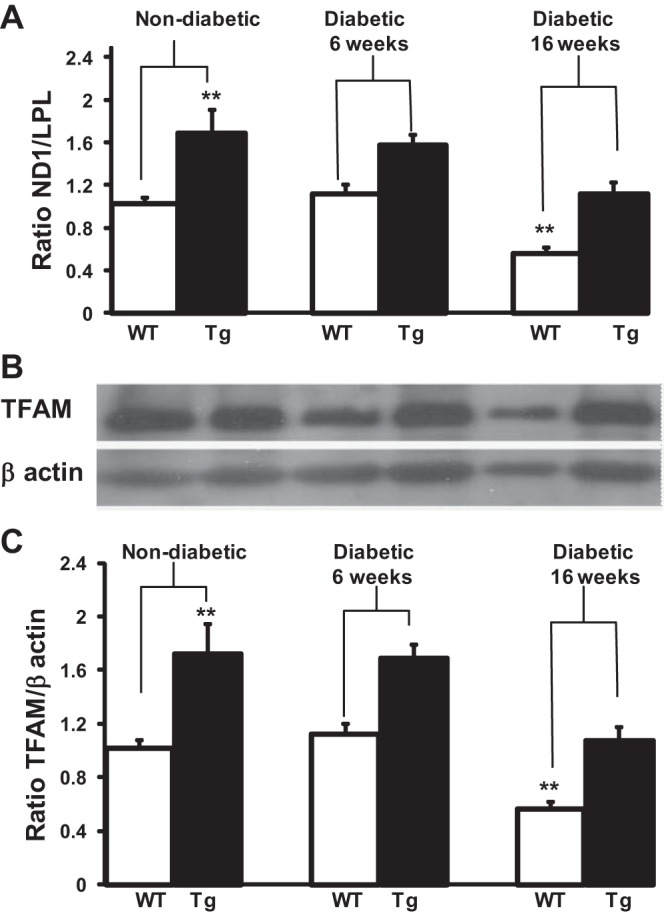
mtDNA content in DRG neurons of WT and TFAM Tg nondiabetic and 16-wk diabetic mice. A: total DNA was isolated from WT and TFAM Tg DRG neurons of nondiabetic control and diabetic mice at 6 and 16 wk after induction of diabetes. Quantitative real-time PCR was done to obtain a relative ratio of NADH dehydrogenase subunit 1 (ND1; a gene coded on the mtDNA) over lipoprotein lipase (LPL; a unigene coded by nDNA). Ratio of ND1 to LPL was determined as an indicator for relative mtDNA content. Data are normalized to nondiabetic WT and expressed as means ± SE. B: total protein was extracted from WT and TFAM Tg DRG neurons of nondiabetic control and diabetic mice at 6 and 16 wk after induction of diabetes. Western blot analysis was with a TFAM antibody that recognizes both hTFAM and mTFAM. Blots were reprobed with β-actin to normalize for loading and transfer. C: ratio of TFAM to β-actin was determined from the intensity of the bands to determine the levels of TFAM. Data are expressed as means ± SE. One-way ANOVA analysis of multiple comparisons with ad hoc Tukey test was used to determine the significance among the groups. **P < 0.01 between WT and Tg mice. Both mtDNA and TFAM protein levels were reduced (P < 0.01) in WT 16-wk diabetic DRG compared with both WT and TFAM Tg diabetic DRG at baseline and 6 wk.
Western blot analysis was done in the protein extracts of DRG neurons to quantify TFAM levels. The results are shown in Fig. 3B. In nondiabetic TFAM Tg mice, TFAM levels showed an approximately twofold increase compared with WT nondiabetic mice, similar to the results shown in Fig. 1. In WT 16-wk diabetic DRG neurons, TFAM levels decreased 40%, and in TFAM Tg diabetic DRG neurons there was a similar decrease. Although quantification of the intensity of the band showed that the decrease in TFAM protein levels in diabetic TFAM Tg mice was significant when compared with nondiabetic TFAM Tg mice, the decrease in TFAM protein in diabetic TFAM Tg mice was not significant when compared with nondiabetic WT mice. On the other hand, the decrease in TFAM levels in diabetic WT mice was significant when compared with nondiabetic WT or TFAM Tg. Considering that the changes in mtDNA (Fig. 3A) and TFAM protein levels (Fig. 3B) are similar (Fig. 3, A and B), this reinforces the idea that the interaction between TFAM and mtDNA may be part of a mechanism of regulation of mtDNA expression.
In addition to determining the ratio of mtDNA to nDNA, we also quantified the copy number of mtDNA. The results (Table 2) showed that there was a significant, approximately twofold increase in mtDNA copy number in DRG neurons from TFAM Tg mice compared with WT mice (TFAM Tg: 2,206 ± 320 vs. WT: 1,145 ± 175, P < 0.01). Sixteen weeks of diabetes caused a significant decrease in the mtDNA copy number in WT mice (nondiabetic: 1,145 ± 175 vs. diabetic: 630 ± 75, P < 0.01) and in TFAM Tg mice (nondiabetic: 2,206 ± 320 vs. diabetic: 1,070 ± 190, P < 0.01). The mitochondrial DNA copy number in TFAM Tg diabetic mice was not significant when compared with WT nondiabetic mice (TFAM Tg diabetic: 1,070 ± 190 vs. WT nondiabetic: 1,145 ± 175). We also determined the damage to mtDNA by LR-QPCR of an 8.9-kb mtDNA target. This was based upon the principle that DNA damage slows down or blocks the progression of DNA polymerase along a template. The results showed that there was higher number of long-range mtDNA PCR products in TFAM Tg diabetic mice compared with WT diabetic (TFAM Tg diabetic: 965 ± 103 vs. WT diabetic: 480 ± 53). The calculated ratio of mtDNA damage to ND1 copy number showed no significant difference between WT diabetic and TFAM diabetic (WT diabetic: 64 ± 12 vs. TFAM Tg diabetic: 68 ± 18), suggesting that there is only a decrease in mtDNA copy number in chronic diabetes.
Table 2.
Mitochondrial DNA copy number and DNA damage in WT and TFAM Tg mice
| Tissue | B2M Copies/10 ng DNA | ND1 Copies/10 ng DNA | Ratio (mtDNA/nDNA) |
|---|---|---|---|
| WT | 10,600 ± 1,215 | 12,237K ± 130K | 1,145 ± 175 |
| TFAM Tg | 10.250 ± 1,350 | 22,350K ± 210K | 2,206 ± 320** |
| WT Diab | 11,350 ± 1,250 | 7,160K ± 69.7K | 630 ± 75 |
| TFAM Tg Diab | 12,720 ± 1,200 | 13,890K ± 190K | 1,070 ± 190** |
| LR DNA amplified | LR DNA amplified/106 ND1 copies | ||
| WT Diab | 480 ± 93 | 64 ± 12 | |
| TFAM Tg Diaby | 965 ± 103 | 68 ± 18 | |
Values are means ± SE.
P < 0.01. ND1, NADH dehydrogenase subunit 1; mtDNA, mitochondrial DNA; nDNA, nuclear DNA. K = 1,000.
Nerve conduction and sensory testing evidence of neuropathy in TFAM Tg mice.
After 16 wk of diabetes, sciatic and tail motor conduction velocities were reduced and tail sensory latencies prolonged in WT diabetic compared with WT nondiabetic mice (Table 3). In contrast, there was no significant difference in conduction velocities and latencies between TFAM Tg diabetic mice compared with nondiabetic TFAM Tg mice (Table 3). The results for von Frey sensory testing for mechanical allodynia and Hargreaves' testing for thermal nociception after 16 wk of diabetes is shown in Fig. 4. The threshold for mechanical allodynia was lower in WT diabetic animals compared with TFAM Tg diabetic mice, consistent with more severe neuropathy in WT diabetic compared with TFAM Tg diabetic mice. The threshold for mechanical allodynia was similar in TFAM Tg diabetic mice compared with WT nondiabetic mice. Similarly diabetic mice had an increase in the thermal nociception latency after 16 wk of diabetes (Fig. 4). In the TFAM Tg mouse, the thermal nociception latency was similar to WT nondiabetic mice.
Table 3.
| Significance (P Value) |
||||||||
|---|---|---|---|---|---|---|---|---|
| Nondiabetic vs. Diabetic Animals | WT | WT Diab | TFAM Tg | TFAM Tg Diab | 1 vs. 2 | 3 vs. 4 | 1 vs. 3 | 2 vs. 4 |
| 6 Wk | ||||||||
| Sciatic NCV, m/s | 48.5 ± 1.7 | 42.4 ± 2.3 | 48.4 ± 2.5 | 49.7 ± 2 | <0.05 | NS | NS | <0.01 |
| Tail SL, m/s | 1.04 ± 0.01 | 1.11 ± 0.02 | 0.98 ± 0.02 | 0.98 ± 0.03 | <0.01 | NS | NS | <0.01 |
| Tail ML, ms | 2.13 ± 0.03 | 2.35 ± 0.03 | 2.20 ± 0.04 | 2.25 ± 0.03 | <0.05 | NS | NS | <0.05 |
| 16 Wk | ||||||||
| Sciatic NCV, m/s | 49.9 ± 3.2 | 36.6 ± 3.2 | 49.74 ± 4.52 | 47.45 ± 1.97 | <0.01 | NS | NS | <0.01 |
| Tail SL, m/s | 1.06 ± 0.02 | 1.19 ± 0.04 | 1.12 ± 0.03 | 1.05 ± 0.02 | <0.01 | NS | NS | <0.01 |
| Tail ML, ms | 2.16 ± 0.04 | 2.55 ± 0.04 | 2.19 ± 0.03 | 2.65 ± 0.04 | <0.01 | NS | NS | <0.01 |
Values are means ± SE. NCV, nerve conduction velocity; SCV, sensory conduction velocity; ML, motor latency. For 6-wk nondiaebtic vs. diabetic animals, n = 6 WT, 12 WT Diab, 6 TFAM Tg, and 9 TFAM Tg Diab; for 16-wk nondiabetic vs. diabetic animals, n = 5 WT, 10 WT Diab, 8 TFAM Tg, and 8 TFAM Tg Diab.
Fig. 4.

Von Frey mechanical allodynia and thermal nociception in WT and TFAM Tg nondiabetic and 16-wk diabetic mice. Mechanical allodynia was performed as described in materials and methods. The threshold for mechanical allodynia was decreased in WT diabetic mice and was significantly different when compared with all other groups, including TFAM Tg diabetic mice. The threshold for mechanical allodynia was normalized in TFAM Tg diabetic mice compared with WT diabetic mice. Thermal nociception was impaired, with a prolonged latency in WT diabetic (WT Diab) mice compared with all other groups, including TFAM Tg diabetic (Tg Diab) mice. Thermal nociception was normalized in TFAM Tg Diab mice. One-way ANOVA analysis of multiple comparisons with ad hoc Tukey test was used to determine the significance among the groups. ++P < 0.01.
Intraepidermal nerve fiber innervation in WT diabetic and TFAM Tg diabetic mice.
As shown in Fig. 5A, protein gene product (PGP) 9.5-immunoreactive nerve fibers were abundant in both the epidermis and dermis of WT and TFAM Tg nondiabetic control mice. Sixteen-week diabetic WT mice showed a significant decrease in the IENFD compared with WT nondiabetic mice and TFAM Tg diabetic mice (Fig. 5A). The mean IENFD was significantly decreased in diabetic WT mice compared with TFAM Tg diabetic mice (P < 0.001; Fig. 5B).
Fig. 5.

Intraepidermal nerve fiber density (IENFD) in WT and TFAM Tg nondiabetic and 16-wk diabetic mice. A: photomicrographs immunostained with anti-protein gene product 9.5 antibody in 50-μm cryocut paw skin sections. Arrows indicate intraepidermal nerve fibers (scale bar, 60 μm). There is a marked reduction in epidermal fibers (arrows) in WT diabetic mice compared with WT nondiabetic mice. B: graph showing that the IENFD (no. of intraepidermal nerve fibers crossing the dermoepidermal junction per length of epidermis) is significantly reduced in WT diabetic mice compared with WT nondiabetic mice. In contrast, IENFD is preserved in TFAM Tg diabetic mice. Data indicate means ± SE.
TFAM Tg expression prevents oxidative stress in DRG neurons.
To test whether TFAM Tg expression prevents oxidative injury in neurons, adult mouse DRG neurons were prepared from 3-mo-old TFAM Tg and WT mice. Figure 6A represents an average of four separate experiments and shows that TFAM Tg expression prevents high-glucose-induced oxidative stress over 5 h using H2DCF, a marker of oxidative stress. In contrast, WT DRG neurons were highly susceptible to high glucose-induced oxidative stress. The bright-field and fluorescence images of the neurons are shown in Fig. 6B.
Fig. 6.

Effect of exposure to high glucose on oxidative stress in cultured adult mouse DRG neurons. A: DRG cultures were prepared from 16-wk-old WT and TFAM Tg mice. Neurons were cultured in serum-free medium with 5.5 mM glucose. On the day of the experiment, the culture was treated with either 5.5 or 25 mM glucose in serum-free medium for 5 h. Cultures were further incubated with 2.5 μmol/l 2-dichlorofluorescin diacetate, processed, fixed, and then mounted on slides. 2′,7′-dichlorofluorescein (DCF) pixel intensity was calculated as described in materials and methods. Data indicate means ± SE. There was no significant difference in DCF between WT and TFAM Tg DRG neurons cultured in 5.5 mM glucose. B: representative merged images of bright-field and DCF fluorescence with low (5.5 mM) and high (25 mM) glucose.
We determined whether the protection against oxidative stress by high glucose in TFAM Tg DRG neurons was due to increased expression of antioxidant enzymes. Total RNA was isolated from 3-mo-old WT and TFAM Tg mice, and the expression levels were measured by RT-PCR of scavenging enzymes as mitochondrial superoxide dismutase (SOD2) and GPX. The results are shown in Fig. 7. No significant differences in the expression of SOD2 mRNA and GPX mRNA were observed in diabetic DRG neurons, suggesting that there is no increase in the capacity of the antioxidant pathway in TFAM Tg mice.
Fig. 7.
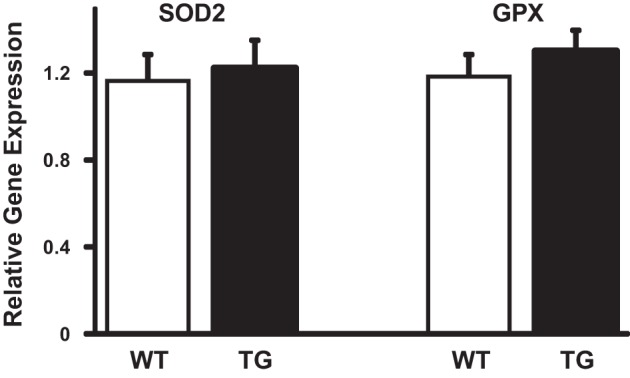
RT-PCR quantitation of expression of manganese superoxide dismutase (MnSOD) and glutiathone peroxidase (GPX) mRNA in WT and TFAM Tg mice. qRT-PCR reaction was performed with gene-specific primers in RNA isolated from DRG neurons of Tg and WT mice. Quantification of the CT values is normalized to GAPDH and expressed as relative gene expression. Values are means ± SE; t-test comparison between WT and Tg mice showed no significant difference in the expression of MnSOD and GPX mRNA.
DISCUSSION
TFAM is located primarily in mitochondria, and its level regulates mtDNA copy number (8, 36, 36, 41). Using a different TFAM gene construct, TFAM is shown to increase mtDNA levels (29, 35). Using the same TFAM Tg mouse as that used in our study, increased mtDNA levels have been observed in cardiac and brain tissues by other investigators (24, 27). Our results have extended the observation to include similar regulation between TFAM levels and mtDNA number in peripheral DRG neurons, namely that TFAM increases mtDNA in DRG neurons. In all of these studies, the TFAM Tg has been shown to increase only the mtDNA; the mitochondrial mass remains the same, and there is no increase in oxidative phosphorylation or increase in ATP levels (24, 27). This contrasts with overexpression of the upstream regulator of TFAM, transcriptional coactivator PPARγ coactivator-1α (PGC-1α). PGC-1α has the capacity to not only increase mtDNA levels but also increase mitochondrial number both in cultured cells and in vivo in tissues (32, 40). Similarly, overexpression of the master regulator sirtuin 1 (SIRT1) also increases not only mtDNA levels but also mitochondrial number (41). The plausible reason is that both PGC-1α and SIRT1, via PGC-1α, also increase the expression of nuclear DNA-encoded genes, including the nuclear respiratory factors 1 and 2 (46–48). These factors in turn stimulate expression of other nDNA-encoded mitochondrial oxidative phosphorylation subunits in addition to the nDNA-encoded TFAM and increase mitochondrial mass.
In contrast, TFAM acts only in mitochondria and increases mtDNA replication and transcription. Overactivation of PGC-1α or SIRT1 is found have deleterious pathological effects. For example, in Tg mouse models, forcing high overexpression of SIRT1 or PGC-1α causes cardiac hypertrophy, whereas moderate overexpression of SIRT1 or PGC-1α is cardioprotective (2, 34, 44). Thus, there appear to be physiological and pathological signals that differentially affect tissue function. Such pathological side effects are not noted in TFAM overexpression model systems. In the Tg mouse model that we used, TFAM levels increased only twofold in DRG neurons. In vivo and in vitro results show that human TFAM protein promotes mtDNA replication but poorly activates transcription from mouse LSP and HSP in vitro despite its high nonspecific DNA binding capacity. Thus, it is possible human TFAM in the mouse increases only the mtDNA copy number without altering respiratory chain capacity or mitochondrial mass so that it is possible to experimentally dissociate mtDNA copy number regulation from regulation of oxidative phosphorylation capacity.
In the hTFAM Tg mouse, the endogenous mouse TFAM protein was expressed in DRG neurons at levels similar to that of WT DRG neurons. The Tg mice expressing human TFAM protein increased total TFAM levels and mtDNA copy number to the same extent. This suggests that the mtDNA copy number per se does not affect TFAM gene expression. The proposed explanation is that the interaction between TFAM and mtDNA is dynamic and that the presence of one increases the stability of the other. This interaction is probably beneficial from a regulatory point of view because small changes in TFAM protein levels or mtDNA levels result in rapid adjustment to maintain a constant optimal ratio between TFAM and mtDNA (16). This is supported by the findings that TFAM interacts with mtDNA to wrap mtDNA to form a nucleoid structure similar to histones in the nucleosome (1, 39, 58). Calculation of the molar ratio of TFAM protein to mtDNA provides an estimate of ∼1,000 molecules of TFAM per mtDNA molecule, or one TFAM molecule per 15–20 bp of mtDNA. Since mitochondria can generate ROS due their respiratory activity, perhaps TFAM functions in multiple roles to promote mtDNA transcription, replication, and wrapping of mtDNA to protect it from attack by ROS (1, 39, 58).
The current study presents novel evidence that the expression of hTFAM protects against diabetic neuropathy. This observation is supported by the preservation of measures of neuropathy, including nerve conduction velocity, mechanical allodynia, thermal nociception, and IENFD in diabetic TFAM Tg mice. TFAM prevents slowing of nerve conduction velocity, reduces mechanical allodynia, and decreases the loss of intraepidermal nerve fibers. The blood results showed that there was no difference in glucose or insulin levels between WT diabetic and TFAM Tg diabetic mice, although hTFAM is likely to be expressed in all tissues because the promoter is not tissue or cell specific and TFAM overexpression does not reduce the severity of diabetes. The protection against peripheral nerve injury is local and independent of glycemic control.
The current study shows that there is a net loss of both mtDNA and TFAM in chronic experimental diabetes. In 6-wk diabetic DRG neurons, there is an attempt to upregulate both mtDNA and TFAM, although the increase was not significant. It could be argued that acute exposure to hyperglycemia increases TFAM levels, mtDNA, and mitochondrial biogenesis to meet the high energy demand in neurons (56). However, where there is chronic hyperglycemia with concurrent generation of ROS, the regulation might change from physiological to pathological and could eventually lead to a decline in mtDNA, TFAM, and mitochondrial function. mtDNA is particularly susceptible to oxidative injury, which is due in part to the following factors: 1) its location within mitochondria where the respiratory complexes I and III are potential sites for the generation of·O2− and 2) the limited repair activity against DNA damage within mitochondria (37). Under normal conditions, the toxic effects of ROS are prevented by scavenging enzymes such as SOD, GPX, and catalase as well as by other nonenzymatic antioxidants. However, when the production of ROS becomes excessive, or if the levels of antioxidant enzymes decrease, then oxidative stress might have a harmful effect on the functional and structural integrity of biological tissue. Our results showed that the DRG neurons from TFAM Tg mice are able to scavenge high glucose-induced ROS much more efficiently than the DRG neurons from WT mice. But this scavenging ability did not appear to be due an increase in the expression of the antioxidant enzymes manganese superoxide dismutase and GPX. Our results on mitochondrial respiration (Fig. 2) showed no significant increase in ADP-stimulated state 3 or resting state 4 respiration. All of these findings suggest that the protective effect of TFAM overexpression is apparent only under chronic conditions of oxidative stress. A direct proof of this would have been to show that the mitochondrial respiratory complex activities are decreased in WT diabetic DRG mitochondria but remained normal in TFAM Tg DRG mitochondria. Paucity of tissue amount required to do such experiments prevented us from this undertaking. However, results have been obtained with isolated mitochondria from other tissues (e.g., heart) using the same hTFAM transgenic mice, the original source of our TFAM Tg mouse. The results show that despite the significant increase in mtDNA copy number in the heart from TFAM Tg mice, mitochondrial respiratory complex I, complex II, complex III, and complex IV demonstrated no significant changes in enzymatic activity compared with WT heart mitochondria (24). In infarcted myocardial (MI) heart mitochondria from WT mice, the enzymatic activities of complex I, complex III, and complex IV were significantly lower than those from WT-sham. Most importantly, there was no such decrease in the enzymatic activities of complex I, complex III, or complex IV in TFAM Tg-MI mitochondria (24). These results, together with our results, suggest that the regulation of mtDNA copy number is dissociated from that of electron transport function, although protection is noted in conditions of increased oxidative stress.
In summary, our results show that TFAM overexpression prevented a decrease in mtDNA copy number in diabetic DRG neurons, helped prevent experimental diabetic neuropathy, and protected DRG neurons from oxidative stress.
GRANTS
This work was supported in part by the Office of Research Development, Department of Veterans Affairs (Biomedical and Laboratory Research Service and Rehabilitation Research and Development; 101RX001030), NIH-RR-024888, the Juvenile Diabetes Research Foundation, the American Diabetes Association, a Veterans Administration Research and Development REAP award (J. Choi, K. Chandrasekaran, and J. W. Russell), the Veterans Affairs Baltimore Research and Education Foundation (J. Choi) and the Mid-Atlantic Nutrition Obesity Research Center, and Grant P30-DK-072488 from the National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
K.C., A.M., T. Inoue, T. Ide, and J.W.R. conception and design of research; K.C., A.M., T. Inoue, J.C., A.R.a., C.C., and J.W.R. performed experiments; K.C., A.M., T. Inoue, J.C., A.R.S., C.C., T. Ide, and J.W.R. analyzed data; K.C., A.M., T. Inoue, J.C., T. Ide, and J.W.R. interpreted results of experiments; K.C. and J.W.R. prepared figures; K.C. and J.W.R. drafted manuscript; K.C., A.M., T. Inoue, J.C., A.R.a., C.C., T. Ide, and J.W.R. edited and revised manuscript; K.C., A.M., T. Inoue, J.C., A.R.S., C.C., T. Ide, and J.W.R. approved final version of manuscript.
REFERENCES
- 1.Alam TI, Kanki T, Muta T, Ukaji K, Abe Y, Nakayama H, Takio K, Hamasaki N, Kang D. Human mitochondrial DNA is packaged with TFAM. Nucleic Acids Res 31: 1640–1645, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alcendor RR, Gao S, Zhai P, Zablocki D, Holle E, Yu X, Tian B, Wagner T, Vatner SF, Sadoshima J. Sirt1 regulates aging and resistance to oxidative stress in the heart. Circ Res 100: 1512–1521, 2007. [DOI] [PubMed] [Google Scholar]
- 3.Anjaneyulu M, Berent-Spillson A, Inoue T, Choi J, Cherian K, Russell JW. Transforming growth factor-beta induces cellular injury in experimental diabetic neuropathy. Exp Neurol 211: 469–479, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Attardi G. Animal mitochondrial DNA: an extreme example of genetic economy. Int Rev Cytol 93: 93–145, 1985. [DOI] [PubMed] [Google Scholar]
- 5.Berent-Spillson A, Russell JW. Metabotropic glutamate receptor 3 protects neurons from glucose-induced oxidative injury by increasing intracellular glutathione concentration. J Neurochem 101: 342–354, 2007. [DOI] [PubMed] [Google Scholar]
- 6.Bestwick ML, Shadel GS. Accessorizing the human mitochondrial transcription machinery. Trends Biochem Sci 38: 283–291, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Biessels GJ, Bril V, Calcutt NA, Cameron NE, Cotter MA, Dobrowsky R, Feldman EL, Fernyhough P, Jakobsen J, Malik RA, Mizisin AP, Oates PJ, Obrosova IG, Pop-Busui R, Russell JW, Sima AA, Stevens MJ, Schmidt RE, Tesfaye S, Veves A, Vinik AI, Wright DE, Yagihashi S, Yorek MA, Ziegler D, Zochodne DW. Phenotyping animal models of diabetic neuropathy: a consensus statement of the diabetic neuropathy study group of the EASD (Neurodiab). J Peripher Nerv Syst 19: 77–87, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Campbell CT, Kolesar JE, Kaufman BA. Mitochondrial transcription factor A regulates mitochondrial transcription initiation, DNA packaging, and genome copy number. Biochim Biophys Acta 1819: 921–929, 2012. [DOI] [PubMed] [Google Scholar]
- 9.Chakrabarty S, D'Souza RR, Kabekkodu SP, Gopinath PM, Rossignol R, Satyamoorthy K. Upregulation of TFAM and mitochondria copy number in human lymphoblastoid cells. Mitochondrion 15: 52–58, 2014. [DOI] [PubMed] [Google Scholar]
- 10.Choi J, Chandrasekaran K, Demarest TG, Kristian T, Xu S, Vijaykumar K, Dsouza KG, Qi NR, Yarowsky PJ, Gallipoli R, Koch LG, Fiskum GM, Britton SL, Russell JW. Brain diabetic neurodegeneration segregates with low intrinsic aerobic capacity. Ann Clin Transl Neurol 1: 589–604, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choi J, Chandrasekaran K, Inoue T, Muragundla A, Russell JW. PGC-1alpha regulation of mitochondrial degeneration in experimental diabetic neuropathy. Neurobiol Dis 64: 118–130, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Choi J, Malakowsky CA, Talent JM, Conrad CC, Gracy RW. Identification of oxidized plasma proteins in Alzheimer's disease. Biochem Biophys Res Commun 293: 1566–1570, 2002. [DOI] [PubMed] [Google Scholar]
- 13.Christianson JA, Riekhof JT, Wright DE. Restorative effects of neurotrophin treatment on diabetes-induced cutaneous axon loss in mice. Exp Neurol 179: 188–199, 2003. [DOI] [PubMed] [Google Scholar]
- 14.Clayton DA. Transcription and replication of mitochondrial DNA. Hum Reprod 15: 11–17, 2000. [DOI] [PubMed] [Google Scholar]
- 15.D'Erchia AM, Atlante A, Gadaleta G, Pavesi G, Chiara M, De Virgilio C, Manzari C, Mastropasqua F, Prazzoli GM, Picardi E, Gissi C, Horner D, Reyes A, Sbisa E, Tullo A, Pesole G. Tissue-specific mtDNA abundance from exome data and its correlation with mitochondrial transcription, mass and respiratory activity. Mitochondrion 20: 13–21, 2015. [DOI] [PubMed] [Google Scholar]
- 16.Farge G, Mehmedovic M, Baclayon M, van den Wildenberg SM, Roos WH, Gustafsson CM, Wuite GJ, Falkenberg M. In vitro-reconstituted nucleoids can block mitochondrial DNA replication and transcription. Cell Rep 8: 66–74, 2014. [DOI] [PubMed] [Google Scholar]
- 17.Feldman EL, Russell JW, Sullivan KA, Golovoy D. New insights into the pathogenesis of diabetic neuropathy. Curr Opin Neurol 12: 553–563, 1999. [DOI] [PubMed] [Google Scholar]
- 18.Fernyhough P, Jonathan M. Mechanisms of disease: Mitochondrial dysfunction in sensory neuropathy and other complications in diabetes. Handb Clin Neurol 126: 353–377, 2014. [DOI] [PubMed] [Google Scholar]
- 19.Garesse R, Vallejo CG. Animal mitochondrial biogenesis and function: a regulatory cross-talk between two genomes. Gene 263: 1–16, 2001. [DOI] [PubMed] [Google Scholar]
- 20.Goffart S, Wiesner RJ. Regulation and co-ordination of nuclear gene expression during mitochondrial biogenesis. Exp Physiol 88: 33–40, 2003. [DOI] [PubMed] [Google Scholar]
- 21.Hayashi Y, Yoshida M, Yamato M, Ide T, Wu Z, Ochi-Shindou M, Kanki T, Kang D, Sunagawa K, Tsutsui H, Nakanishi H. Reverse of age-dependent memory impairment and mitochondrial DNA damage in microglia by an overexpression of human mitochondrial transcription factor a in mice. J Neurosci 28: 8624–8634, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ho EC, Lam KS, Chen YS, Yip JC, Arvindakshan M, Yamagishi S, Yagihashi S, Oates PJ, Ellery CA, Chung SS, Chung SK. Aldose reductase-deficient mice are protected from delayed motor nerve conduction velocity, increased c-Jun NH2-terminal kinase activation, depletion of reduced glutathione, increased superoxide accumulation, and DNA damage. Diabetes 55: 1946–1953, 2006. [DOI] [PubMed] [Google Scholar]
- 23.Hokari M, Kuroda S, Kinugawa S, Ide T, Tsutsui H, Iwasaki Y. Overexpression of mitochondrial transcription factor A (TFAM) ameliorates delayed neuronal death due to transient forebrain ischemia in mice. Neuropathology 30: 401–407, 2010. [DOI] [PubMed] [Google Scholar]
- 24.Ikeuchi M, Matsusaka H, Kang D, Matsushima S, Ide T, Kubota T, Fujiwara T, Hamasaki N, Takeshita A, Sunagawa K, Tsutsui H. Overexpression of mitochondrial transcription factor a ameliorates mitochondrial deficiencies and cardiac failure after myocardial infarction. Circulation 112: 683–690, 2005. [DOI] [PubMed] [Google Scholar]
- 25.Jeong-Yu S, Clayton DA. Regulation and function of the mitochondrial genome. J Inherit Metab Dis 19: 443–451, 1996. [DOI] [PubMed] [Google Scholar]
- 26.Jornayvaz FR, Shulman GI. Regulation of mitochondrial biogenesis. Essays Biochem 47: 69–84, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kang D, Kim SH, Hamasaki N. Mitochondrial transcription factor A (TFAM): roles in maintenance of mtDNA and cellular functions. Mitochondrion 7: 39–44, 2007. [DOI] [PubMed] [Google Scholar]
- 28.Kelly DP, Scarpulla RC. Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev 18: 357–368, 2004. [DOI] [PubMed] [Google Scholar]
- 29.Larsson NG, Wang J, Wilhelmsson H, Oldfors A, Rustin P, Lewandoski M, Barsh GS, Clayton DA. Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nat Genet 18: 231–236, 1998. [DOI] [PubMed] [Google Scholar]
- 30.Lauria G, Cornblath DR, Johansson O, McArthur JC, Mellgren SI, Nolano M, Rosenberg N, Sommer C. EFNS guidelines on the use of skin biopsy in the diagnosis of peripheral neuropathy. Eur J Neurol 12: 747–758, 2005. [DOI] [PubMed] [Google Scholar]
- 31.Lauria G, Lombardi R, Borgna M, Penza P, Bianchi R, Savino C, Canta A, Nicolini G, Marmiroli P, Cavaletti G. Intraepidermal nerve fiber density in rat foot pad: neuropathologic-neurophysiologic correlation. J Peripher Nerv Syst 10: 202–208, 2005. [DOI] [PubMed] [Google Scholar]
- 32.Lin J, Wu H, Tarr PT, Zhang CY, Wu Z, Boss O, Michael LF, Puigserver P, Isotani E, Olson EN, Lowell BB, Bassel-Duby R, Spiegelman BM. Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature 418: 797–801, 2002. [DOI] [PubMed] [Google Scholar]
- 33.Locker J, Rabinowitz M. An overview of mitochondrial nucleic acids and biogenesis. Methods Enzymol 56: 3–16, 1979. [DOI] [PubMed] [Google Scholar]
- 34.Marin-Garcia J, Goldenthal MJ. Mitochondrial centrality in heart failure. Heart Fail Rev 13: 137–150, 2008. [DOI] [PubMed] [Google Scholar]
- 35.Matsuda T, Kanki T, Tanimura T, Kang D, Matsuura ET. Effects of overexpression of mitochondrial transcription factor A on lifespan and oxidative stress response in Drosophila melanogaster. Biochem Biophys Res Commun 430: 717–721, 2013. [DOI] [PubMed] [Google Scholar]
- 36.Morimoto N, Miyazaki K, Kurata T, Ikeda Y, Matsuura T, Kang D, Ide T, Abe K. Effect of mitochondrial transcription factor a overexpression on motor neurons in amyotrophic lateral sclerosis model mice. J Neurosci Res 90: 1200–1208, 2012. [DOI] [PubMed] [Google Scholar]
- 37.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J 417: 1–13, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nile DL, Brown AE, Kumaheri MA, Blair HR, Heggie A, Miwa S, Cree LM, Payne B, Chinnery PF, Brown L, Gunn DA, Walker M. Age-related mitochondrial DNA depletion and the impact on pancreatic Beta cell function. PLoS One 9: e115433, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ohgaki K, Kanki T, Fukuoh A, Kurisaki H, Aoki Y, Ikeuchi M, Kim SH, Hamasaki N, Kang D. The C-terminal tail of mitochondrial transcription factor a markedly strengthens its general binding to DNA. J Biochem 141: 201–211, 2007. [DOI] [PubMed] [Google Scholar]
- 40.Puigserver P, Spiegelman BM. Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): transcriptional coactivator and metabolic regulator. Endocr Rev 24: 78–90, 2003. [DOI] [PubMed] [Google Scholar]
- 41.Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature 434: 113–118, 2005. [DOI] [PubMed] [Google Scholar]
- 42.Russell JW, Berent-Spillson A, Vincent AM, Freimann CL, Sullivan KA, Feldman EL. Oxidative injury and neuropathy in diabetes and impaired glucose tolerance. Neurobiol Dis 30: 420–429, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Russell JW, Golovoy D, Vincent AM, Mahendru P, Olzmann JA, Mentzer A, Feldman EL. High glucose-induced oxidative stress and mitochondrial dysfunction in neurons. FASEB J 16: 1738–1748, 2002. [DOI] [PubMed] [Google Scholar]
- 44.Russell LK, Mansfield CM, Lehman JJ, Kovacs A, Courtois M, Saffitz JE, Medeiros DM, Valencik ML, McDonald JA, Kelly DP. Cardiac-specific induction of the transcriptional coactivator peroxisome proliferator-activated receptor gamma coactivator-1alpha promotes mitochondrial biogenesis and reversible cardiomyopathy in a developmental stage-dependent manner. Circ Res 94: 525–533, 2004. [DOI] [PubMed] [Google Scholar]
- 45.Ryan MT, Hoogenraad NJ. Mitochondrial-nuclear communications. Annu Rev Biochem 76: 701–722, 2007. [DOI] [PubMed] [Google Scholar]
- 46.Scarpulla RC. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol Rev 88: 611–638, 2008. [DOI] [PubMed] [Google Scholar]
- 47.Scarpulla RC. Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim Biophys Acta 1813: 1269–1278, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Scarpulla RC, Vega RB, Kelly DP. Transcriptional integration of mitochondrial biogenesis. Trends Endocrinol Metab 23: 459–466, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schuh RA, Kristian T, Gupta RK, Flaws JA, Fiskum G. Methoxychlor inhibits brain mitochondrial respiration and increases hydrogen peroxide production and CREB phosphorylation. Toxicol Sci 88: 495–504, 2005. [DOI] [PubMed] [Google Scholar]
- 50.Silva JP, Kohler M, Graff C, Oldfors A, Magnuson MA, Berggren PO, Larsson NG. Impaired insulin secretion and beta-cell loss in tissue-specific knockout mice with mitochondrial diabetes. Nat Genet 26: 336–340, 2000. [DOI] [PubMed] [Google Scholar]
- 51.Song Z, Fu DT, Chan YS, Leung S, Chung SS, Chung SK. Transgenic mice overexpressing aldose reductase in Schwann cells show more severe nerve conduction velocity deficit and oxidative stress under hyperglycemic stress. Mol Cell Neurosci 23: 638–647, 2003. [DOI] [PubMed] [Google Scholar]
- 52.Trifunovic A, Larsson NG. Tissue-specific knockout model for study of mitochondrial DNA mutation disorders. Methods Enzymol 353: 409–421, 2002. [DOI] [PubMed] [Google Scholar]
- 53.Uchiumi T, Kang D. The role of TFAM-associated proteins in mitochondrial RNA metabolism. Biochim Biophys Acta 1820: 565–570, 2012. [DOI] [PubMed] [Google Scholar]
- 54.Vincent AM, Calabek B, Roberts L, Feldman EL. Biology of diabetic neuropathy. Handb Clin Neurol 115: 591–606, 2013. [DOI] [PubMed] [Google Scholar]
- 55.Vincent AM, Callaghan BC, Smith AL, Feldman EL. Diabetic neuropathy: cellular mechanisms as therapeutic targets. Nat Rev Neurol 7: 573–583, 2011. [DOI] [PubMed] [Google Scholar]
- 56.Vincent AM, Edwards JL, McLean LL, Hong Y, Cerri F, Lopez I, Quattrini A, Feldman EL. Mitochondrial biogenesis and fission in axons in cell culture and animal models of diabetic neuropathy. Acta Neuropathol 120: 477–489, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vincent AM, Russell JW, Sullivan KA, Backus C, Hayes JM, McLean LL, Feldman EL. SOD2 protects neurons from injury in cell culture and animal models of diabetic neuropathy. Exp Neurol 208: 216–227, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang YE, Marinov GK, Wold BJ, Chan DC. Genome-wide analysis reveals coating of the mitochondrial genome by TFAM. PLoS One 8: e74513, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xu S, Zhong M, Zhang L, Wang Y, Zhou Z, Hao Y, Zhang W, Yang X, Wei A, Pei L, Yu Z. Overexpression of Tfam protects mitochondria against beta-amyloid-induced oxidative damage in SH-SY5Y cells. FEBS J 276: 3800–3809, 2009. [DOI] [PubMed] [Google Scholar]
- 60.Yagihashi S, Mizukami H, Sugimoto K. Mechanism of diabetic neuropathy: Where are we now and where to go? J Diabetes Investig 2: 18–32, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]