

Abstract
We investigated the separate and combined effects of hyperglycemia and hyperinsulinemia on markers of endothelial function, proinflammatory and proatherothrombotic responses in overweight/obese nondiabetic humans. Twenty-two individuals (13 F/9 M, BMI 30.1 ± 4.1 kg/m2) were studied during four randomized, single-blind protocols. The pancreatic clamp technique was combined with 4-h glucose clamps consisting of either 1) euinsulinemia-euglycemia, 2) euinsulinemia-hyperglycemia, 3) hyperinsulinemia-hyperglycemia, or 4) hyperinsulinemia-euglycemia. Insulin levels were higher (998 ± 66 vs. 194 ± 22 pmol/l) during hyperinsulinemia compared with euinsulinemia. Glucose levels were 11.1 mmol/l during hyperinsulinemia compared with 5.1 ± 0.1 mmol/l during euglycemia. VCAM, ICAM, P-selectin, E-selectin, IL-6, adiponectin, and PAI-1 responses were all increased (P < 0.01-0.0001), and endothelial function was decreased (P < 0.0005) during euinsulinemia-hyperglycemia compared with other protocols. Hyperinsulinemia in the presence of hyperglycemia prevented the increase in proinflammatory and proatherothrombotic markers while also normalizing vascular endothelial function. We conclude that 4 h of moderate hyperglycemia can result in increases of proinflammatory markers (ICAM, VCAM, IL-6, E-selectin), platelet activation (P-selectin), reduced fibrinolytic balance (increased PAI-1), and disordered endothelial function in a group of obese and overweight individuals. Hyperinsulinemia prevents the actions of moderate hyperglycemia to reduce endothelial function and increase proinflammatory and proatherothrombotic markers.
Keywords: hyperglycemia, endothelial function, hyperinsulinemia, inflammation
the prevalence of obesity has increased dramatically worldwide over the last 30 years (14). Obesity is now an independent risk factor for type 2 diabetes, cancer, and cardiovascular disease (12, 29, 36, 38, 50). Recent data from Schmidt et al. (51) report that obese individuals have an increased risk of venothrombotic disease in addition to myocardial infarction and/or stroke. Previous studies have demonstrated that obesity is a proinflammatory state (30, 47, 55). However, the acute specific effects of insulin and hyperglycemia, either alone or in combination, on endothelial function and proinflammatory and proatherothrombotic markers in obese and overweight individuals remain largely unknown.
There are accumulating data that hyperglycemia can induce atherothrombotic mechanisms, reduce fibrinolytic balance, and impair endothelial function in both nondiabetic and diabetic individuals (9, 15–17, 44). The in vivo vascular biological effects of insulin are controversial, with studies reporting either deleterious or beneficial effects on endothelial function and atherothrombotic balance (24, 28, 32, 52). Complicating interpretation of insulin's effects per se is the presence of insulin resistance. Thus, in insulin-resistant, nondiabetic individuals, the β-cells can compensate for insulin resistance and maintain normoglycemia at the expense of hyperinsulinemia. Insulin resistance per se can increase systemic blood pressure and triglycerides and lower HDL. All are powerful risk factors for macrovascular disease. Thus, in cross-sectional population studies (9, 28), it is difficult to determine the effects of insulin resistance as opposed to hyperinsulinemia on the pathogenesis of cardiovascular disease (4, 40, 54). To determine insulin's specific acute effects on markers of vascular function and proinflammatory and proatherothrombotic responses, several carefully controlled physiological studies have been performed. These studies have also provided conflicting results. Dandona, et al. (22) have demonstrated significant anti-inflammatory and antiatherogenic properties of insulin during euglycemic conditions. Similarly, Baron, et al. (7) have also reported beneficial effects on endothelial function with insulin. Contrary to these findings, Arcaro et al. (5) in humans and Pandolfi et al. (48) in rats have reported that insulin decreases endothelial function and fibrinolytic balance. Of additional clinical relevance is the question of insulin's vascular biological effects during hyperglycemia. To date, only two studies appear to have addressed this topic. Stegenga et al. (53), using the pancreatic clamp technique in six young lean males, reported that both hyperglycemia and hyperinsulinemia are prothrombotic. On the other hand, Williams et al. (59) demonstrated that even modest increases in insulin could reduce the deleterious effects of hyperglycemia on endothelial function. However, no data are available regarding the independent and combined acute effects of hyperinsulinemia and hyperglycemia on integrated markers of endothelial function and proinflammatory and proatherothrombotic responses in overweight and/or obese individuals. Thus, this present study used pancreatic and glucose clamp techniques to determine the specific acute effects of moderate hyperglycemia (11.1 mmol/l) and physiological hyperinsulinemia (∼900 pmol/l) either separately or combined in overweight and obese humans.
Research Design and Methods
Subjects.
Twenty-two adult volunteers (13 F/9 M), age 41 ± 3 yr, BMI 30.1 ± 1 k/gm2, HbA1c 5.5 ± 0.1%, fat 29 ± 2%, were studied. None of the subjects smoked, received anticoagulants, clopidogrel, statins, angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, or oral insulin sensitizers (metformin, thiazolidinediones). Subjects over age 40 yr were screened for silent ischemia with an electrocardiogram. Each subject had normal blood count, plasma electrolytes, and liver and renal function and no evidence of either impaired fasting glucose or overt diabetes mellitus. Participants were randomized to undergo one of four protocols (Fig. 1), which consisted of an initial pancreatic clamp (8) followed by a single-step 4-h glucose clamp. The intention was for subjects to complete all four protocols. However, withdrawals occurred for the following reasons: 1) time schedule conflicts, 2) intolerance to nitroglycerin, or 3) inability to maintain iv access. Additional subjects were recruited to replace the individuals that withdrew and complete the remaining experiments in the randomized four-protocol block. Thus, seven participants completed all four protocols; four participants completed three protocols; six participants completed two protocols; and five participants completed one study protocol. All gave written informed consent. Studies were approved by the Vanderbilt University Human Subjects Institutional Review Board.
Fig. 1.

Experimental protocols.
Experimental design.
All individuals refrained from caffeine, exercise, and alcohol 24 h prior to the study. Participants were instructed to not use aspirin, NSAIDs, or COX 2 inhibitors for 3 days prior to a study. Subjects were admitted to the General Clinical Research Center the night prior to the study at 5:00 PM. After an overnight 10-h fast, two intravenous cannulae were inserted under 1% Lidocaine local anesthesia. One cannula was placed in a retrograde fashion into a vein on the back of the hand of the nondominant arm. This hand was placed in a heated box (55–60°C) during the study so that arterialized blood could be obtained (1). The other cannula was placed in the ipsilateral arm for infusions.
Pancreatic clamp.
The somatostatin analog octreotide was infused at 30 ng·kg−1·min−1 to inhibit endogenous insulin, glucagon, and growth hormone secretion. Basal replacement amounts of insulin (1.8 pmol·kg−1·min−1), human growth hormone (3 ng·kg−1·min−1), and a variable basal amount of glucagon were infused throughout each experiment (until time 240 min). Glucagon was adjusted as needed to maintain euglycemia of ∼5 mmol/l. Arterialized venous blood was sampled every 10–15 min to monitor plasma glucose.
Glucose clamp studies.
After stable euglycemia was established during the pancreatic clamp, 4-h single-step glucose clamps at differing glycemia and hyperinsulinemia (euinsulinemia-euglycemia n = 14; euinsulinemia-hyperglycemia n = 15; hyperglycemia-hyperinsulinemia n = 14; hyperinsulinemia-euglycemia n = 14) were performed (Fig. 1). At time −120 min, a constant (1.8 pmol·kg−1·min−1) infusion of insulin (Human Regular Insulin; Eli Lilly, Indianapolis, IN) was started via a precalibrated infusion pump (Harvard Apparatus, South Natick, MA). At time 0 min, the insulin infusion was either maintained at 1.8 pmol·kg−1·min−1 or increased to 9 pmol·kg−1·min−1 and continued until 240 min. Ends of pancreatic clamp glucagon and growth hormone infusion rates were maintained unchanged during the 240 min glucose clamp procedures. Glucose targets of (5 mmol/l, 90 mg/dl), or (11.1 mmol/l, 200 mg/dl), depending on protocol, were achieved using a modification of the glucose clamp technique (23). During the clamp period, plasma glucose was measured every 5 min, and a 20% dextrose infusion was adjusted so that plasma glucose levels were held constant. Potassium chloride (20 mmol/l) was infused during hyperinsulinemic clamp studies to reduce insulin-induced hypokalemia.
Analytic methods.
The collection and processing of blood samples have been described elsewhere (18). Plasma glucose concentrations were measured in triplicate every 5 min using the glucose oxidase method with a glucose analyzer (Beckman, Fullerton, CA). Blood for insulin, catecholamines (epinephrine and norepeinephrine), nonesterified fatty acids (NEFA), glucagon, cortisol, growth hormone, and C-peptide was drawn every 30 min during the experimental period. Insulin was measured as previously described (58) with an interassay coefficient of variation (CV) of 9%. Catecholamines were determined by HPLC (13) with an interassay CV of 12% for epinephrine and 8% for norepinephrine. Two modifications to the procedure for catecholamine determination were made: 1) a five-point rather than a one-point standard calibration curve was used; and 2) the initial and final samples of plasma with known amounts of epinephrine and norepinephrine were spiked so that accurate identification of the relevant respective catecholamine peaks could be made. NEFA were measured using the WAKO kit adapted for use on a Packard instrument (Meriden, CT) (35). Glucagon was measured according to a modification of the method of Aguilar-Parada, et al. (2), with an interassay CV of 12%. Cortisol was assayed using the Clinical Assays Gamma Coat Radioimmunoassay (RIA) kit with an interassay CV of 6%. Growth hormone was determined by RIA (37) with a CV of 8.6%. C-peptide was measured by a specific double-antibody RIA (33).
Blood for vascular cell adhesion molecule-1 (VCAM-1), intercellular adhesion molecule-1 (ICAM-1), E-selectin, P-selectin, IL-6, and adiponectin was drawn every 60 min and every 30 min for PAI-1 and tissue plasminogen activator (tPA) during the experimental period. All vascular adhesion molecules and adiponectin were assayed using LINCO Research Kits (St. Charles, Missouri) with an interassay CV of 8.5% (VCAM), CV of 9.7% (ICAM), CV of 13.4%, (sE-selectin), CV of 15.9% (adiponectin), CV of 9.02% (IL-6), respectively (43). Plasminogen activator inhibitor 1 (PAI-1) and tPA were determined by TintElize Platinum Kit with interassay CV of 3.3% (10). P-selectin was measured using the Meso Scale Discovery assay kit (Gathersburg, MD) with a CV of 9.9%.
Endothelial function.
Measurements of endothelial function were conducted at baseline (before the pancreatic clamp) and during the final 30 min of each glucose clamp. Endothelial measurements of the dominant brachial artery were measured using 2-D Doppler ultrasound during reactive hyperemia and exogenous nitroglycerin administration. Before any study intervention while the patient was in a resting state, three measurements of the brachial artery over a 1-min period were recorded. The average of the three measurements provided the baseline artery diameter. Baseline images of the brachial artery were obtained during systole by scanning the artery in longitudinal section 5–10 cm above the antecubital fossa of the dominant arm with the focal zone set to the depth of the mid vessel. Intravenous cannulas for the clamp portion of the study were placed on the nondominant arm. Boundaries for diameter measurements were identified manually with electronic calipers. Flow-mediated dilation was obtained by inflating the sphygmomanometric cuff around the proximal forearm to a pressure of 40 mmHg greater than the patient's systolic blood pressure for 4 min. Brachial artery diameter measurements were taken at time points 30, 60, 90, 120 s before and and after cuff deflation. The peak change in vessel diameter during these time points was calculated as the maximum percent change from the baseline. Then, after a 15-min rest period, subjects received 0.4-mg sublingual nitroglycerin [as an exogenous nitric oxide (NO) donor]. Additional scans were performed as above, with vessel diameter measurements obtained at 1, 2, 3, and 4 min (26). Maximal increases in percent brachial artery diameter (vasodilation) were compared from baseline to end of study within each glucose clamp protocol and overall within the four groups.
Statistical analysis.
Data are expressed as means ± SE and were analyzed using standard, parametric, one- and two-way analysis of variance (ANOVA) and with repeated measures where appropriate (GraphPad Software, San Diego, CA). Once overall group mean difference was identified by ANOVA, Tukey's post hoc analysis was used to delineate statistical significance within each group. Data were also analyzed using paired and unpaired two-tailed t-tests to determine if there were changes from baseline for an individual parameter within each protocol. In all cases, a P value of <0.05 was accepted as statistically significant.
RESULTS
Glucose and insulin.
Plasma glucose was maintained at equivalent levels during euglycemic clamps (5.1 ± 0.1 vs. 5.0 ± 0.1 mmol/l) and were equivalent during all hyperglycemia clamp protocols (11.2 ± 0.1 vs. 11.1 ± 0.2 mmol/l) (Fig. 2). Insulin levels during euinsulinemia (194 ± 22 vs. 210 ± 18 pmol/l) and hyperinsulinemia (998 ± 66 vs. 988 ± 114 pmol/l) groups were similar during the respective clamp studies (Fig. 2). The glucose infusion rates (μmol·kg−1·min−1) required to maintain euglycemia were 11.8 ± 2.3 during the euinsulinemic-euglycemic control study and 41.3 ± 3.3 during the hyperinsulinemic-euglycemic clamps. Glucose infusion rates during the euinsulinemic-hyperglycemic and hyperinsulinemic-hyperglycemic clamps were 28.9 ± 4.6 and 70.5 ± 6.1 μmol·kg−1·min−1, respectively.
Fig. 2.
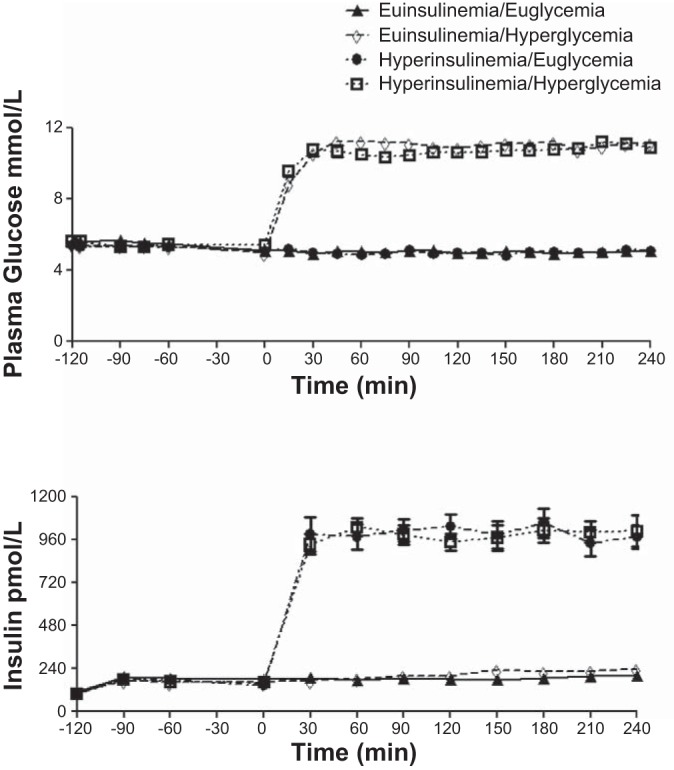
Glucose and insulin values in 23(14 F/9 M) adult healthy volunteers during euglycemic (5.0 mmol/l) and hyperglycemic (11.1 mmol/l) glucose clamps in the presence of octreotide and insulin infused at either 1.8 or 9 pmol·kg−1·min−1.
Neuroendocrine counterregulatory hormones.
Baseline (time 0) epinephrine, norepinephrine, glucagon, growth hormone, and cortisol levels were similar at the start of all glucose clamp studies. Norepinephrine increased similarly during all protocols. Other neuroendocrine counterregulatory hormones remained similar to baseline in all studies (Table 1).
Table 1.
Comparison of baseline to end of clamp growth hormone, glucagon, cortisol, epinephrine, norepinephrine, free fatty acids, and triglycerides
| Euinsulinemia/Euglycemia |
Euinsulinemia/Hyperglycemia |
Hyperinsulinemia/Hyperglycemia |
Hyperinsulinemia/Euglycemia |
|||||
|---|---|---|---|---|---|---|---|---|
| Basal | Final 60 min of clamp | Basal | Final 60 min of clamp | Basal | Final 60 min of clamp | Basal | Final 60 min of clamp | |
| Growth Hormone (ng/ml) | 2.2 ± 0.2 | 2.7 ± 0.2 | 2.3 ± 0.2 | 2.7 ± 0.2 | 2.6 ± 0.3 | 2.2 ± 0.2 | 2.1 ± 0.1 | 2.2 ± 0.1 |
| Glucagon (ng/ml) | 54 ± 4 | 47 ± 3 | 56 ± 6 | 46 ± 5 | 65 ± 6 | 49 ± 5 | 51 ± 5 | 43 ± 3 |
| Cortisol (μg/ml) | 12 ± 1.5 | 11.8 ± 2 | 9.4 ± 2 | 13.7 ± 3 | 9.9 ± 1 | 9.9 ± 2 | 13 ± 2 | 9.9 ± 1 |
| Epinephrine (pg/ml) | 29 ± 3 | 45 ± 8 | 21 ± 3 | 29 ± 7 | 26 ± 3 | 29 ± 10 | 26 ± 4 | 38 ± 12 |
| Norepinephrine (pg/ml) | 154 ± 15 | 226 ± 22* | 160 ± 19 | 246 ± 29* | 132 ± 13 | 215 ± 33* | 146 ± 39 | 239 ± 40* |
| Free fatty acid (μmol/l) | 220 ± 44 | 196 ± 44† | 192 ± 53 | 88 ± 28* | 194 ± 31 | 65 ± 16* | 167 ± 25 | 52 ± 10* |
| Triglycerides (mg/dl) | 129 ± 20 | 71 ± 15* | 137 ± 26 | 105 ± 28* | 135 ± 25 | 109 ± 25* | 122 ± 29 | 85 ± 21* |
Results are means ± SE.
P < 0.05-0.0001 vs. baseline;
P < 0.008 vs. other protocols.
Intermediary metabolism.
Blood NEFA and triglyceride levels were similar at the start of all protocols (time 0) and fell by a similar and a significant amount during hyperglycemic and hyperinsulinemic clamps (P < 0.008; Table 1).
Atherogenic vascular adhesion molecules.
Baseline values of VCAM, ICAM, and E-selectin were similar at baseline (time 0) during all four protocols (Table 2). VCAM, ICAM, and E-selectin responses increased (P < 0.0001) during euinsulinemia-hyperglycemia but fell (P < 0.0001) compared with baseline during the other protocols (Table 2 and Fig. 3). The responses of VCAM, ICAM, and E-selectin during euinsulinemia-hyperglycemia were significantly increased (P < 0.0001) compared with the other three protocols when compared as either time course or as baseline to end of clamp responses (Fig. 3). VCAM, ICAM and E-selectin fell from baseline (P < 0.0001) during all hyperinsulinemic protocols.
Table 2.
Baseline and values during the final hour of inflammatory atherothrombotic and fibrinolytic balance during glucose clamps
| Euinsulinemia/Euglycemia |
Euinsulinemia/Hyperglycemia |
Hyperinsulinemia/Hyperglycemia |
Hyperinsulinemia/Euglycemia |
|||||
|---|---|---|---|---|---|---|---|---|
| Basal | Final 60 min of clamp | Basal | Final 60 min of clamp | Basal | Final 60 min of clamp | Basal | Final 60 min of clamp | |
| VCAM (ng/ml) | 844 ± 76 | 745 ± 70* | 737 ± 39 | 795 ± 48† | 752 ± 45 | 641 ± 45* | 763 ± 34 | 637 ± 26* |
| ICAM (ng/ml) | 89 ± 11 | 76 ± 8* | 76 ± 10 | 101 ± 9† | 81 ± 9 | 68 ± 6* | 87 ± 7 | 73 ± 6* |
| P-selectin (pg/ml) | 67 ± 8 | 53 ± 9* | 74 ± 7 | 110 ± 11† | 79 ± 14 | 58 ± 9* | 76 ± 12 | 55 ± 6* |
| E-selectin (ng/ml) | 23 ± 3 | 19 ± 3* | 19 ± 2 | 21 ± 2† | 22 ± 3 | 18 ± 3* | 21 ± 2 | 18 ± 2* |
| PAI-1 (ng/ml) | 16 ± 3 | 8 ± 1* | 15 ± 3 | 17 ± 3 | 24 ± 6 | 10 ± 2* | 13 ± 2 | 8 ± 1* |
| IL-6 (ng/ml) | 23 ± 6 | 22 ± 6 | 16 ± 5 | 19 ± 6† | 20 ± 9 | 18 ± 7 | 12 ± 4 | 12 ± 4 |
| Adiponectin (ng/ml) | 11,130 ± 557 | 10,576 ± 618 | 10,635 ± 685 | 11,176 ± 655† | 10,890 ± 659 | 9,876 ± 687* | 11,907 ± 693 | 10,660 ± 741* |
| tPA (ng/ml) | 5.4 ± 0.3 | 5.6 ± 1.1 | 5.8 ± 1.2 | 5.9 ± 1.2 | 5.3 ± 1.3 | 5.2 ± 1.3 | 5.1 ± 0.8 | 4.6 ± 0.6 |
Results are means ± SE.
VCAM, vascular cell adhesion molecule; ICAM, intercellular adhesion molecule; PAI-1, plasminogen activator inhibitor 1; tPA, tissue plasminogen activator.
P < 0.02-0.0001 reduced vs. baseline;
P < 0.01-0.0001 increased vs. baseline.
Fig. 3.

Timeline responses from baseline of adiponectin, ICAM, P-selectin, IL-6, E-selectin, PAI-1, and VCAM during euglycemic (5.0 mmol/l) and hyperglycemic (11.1 mmol/l) clamps in the presence of octreotide and insulin at 1.8 or 9 pmol·kg−1·min−1 in overnight-fasted obese/overweight humans. *Response during euinsulinemic hyperglycemia is significantly increased (P < 0.01-0.0001) vs. responses in the other groups. †Response is significantly decreased (P < 0.02-0.0001) vs. baseline. #Response is significantly increased (P < 0.02-0.0001) vs. baseline.
Platelet activation and fibrinolytic balance.
Baseline P-selectin, tPA, and PAI-1 were similar at the start of the four glucose clamp protocols (Table 2). P-selectin and PAI-1 responses and levels were higher (P < 0.02-.0001) during euinsulinemica-hyperglycemia and were significantly increased (P < 0.0001) compared with the other three protocols (Table 2 and Fig. 3). PAI-1 and P-selectin levels fell from baseline (P < 0.02-0.0001) during the hyperinsulinemic protocols. There was no difference in tPA during any protocol (Table 2).
Adiponectin and IL-6.
Adiponectin responses increased (P < 0.0003) during euinsulinemia-hyperglycemia compared with falls during the other three protocols (Table 2 and Fig. 3).
IL-6 increased significantly (P < 0.02) from baseline during euinsulinemia-hyperglycemia (Table 2), and responses were increased (P < 0.02) compared with all other protocols (Table 2 and Fig. 3).
Endothelial function.
Brachial artery diameter was similar at the start of all endogenously NO-mediated vasodilation studies (4.4 ± 0.1 and 4.5 ± 0.1 mm) at baseline and end of clamp measurements. The increase in flow-mediated dilation during endogenous NO stimulation was significantly blunted during euinsulinemia-hyperglycemia compared with all other protocols (P = 0.0005; Fig. 4). Similar to the above proinflammatory and proatherothrombotic biomarkers, this effect was reversed during hyperinsulinemic hyperglycemia and euglycemic protocols (Fig. 4).
Fig. 4.

Responses at baseline and end of clamp flow-mediated (endogenous) and nitroglycerin (exogenous) -mediated dilation during euglycemic (5.0 mmol/l) and hyperglycemic clamps (11.1 mmol/l) in the presence of octreotide and insulin infused at 1.8 or 9 pmol·kg−1·min−1 in overnight-fasted obese/overweight humans. *Response is significantly (P < 0.01) different from baseline. ≠Response of mean maximal %change is significantly (P < 0.01-0.001) different from euinsulinemia-euglycemia.
There were similar increases in vasodilation among all protocols during exogenous NO donation with nitroglycerin administration.
There was no correlation between changes in baseline flow-mediated dilation during endogenous NO stimulation and insulin action (glucose infusion rates) as measured during the euglycemic hyperinsulinemic clamps (r = 0.15, P = 0.61). The CV of the baseline FMD measurements was 4.5 ± 0.7% with a range of 1 to 10% per individual.
DISCUSSION
In the present study, we have demonstrated, by incorporating the pancreatic clamp technique to allow determination of the independent effects of hyperglycemia and hyperinsulinemia, that 4 h of moderate hyperglycemia (11.1 mmol/l) produces acute impairment of endothelial function and increases proatherothrombotic biomarkers in overweight and/or obese individuals. Our study extends previous reports demonstrating that hyperglycemia can simultaneously impair multiple components of vascular function (9, 15–17, 44). The proatherogenic adhesion molecules VCAM-1, E-selectin, and ICAM-1 were increased acutely by modest hyperglycemia. P-selectin, a marker of platelet activation and endothelial dysfunction (27), also increased during hyperglycemia. Furthermore, hyperglycemia impaired fibrinolytic balance by limiting the usual diurnal fall in PAI-1. This created a condition of relatively increased levels of PAI-1 with unchanged systemic values of tPA. Hyperglycemia also blunted endogenous NO-mediated endothelial function, which can be a predictor of future cardiovascular events (6, 46, 49). Last, the significant increases of IL-6 and adiponectin demonstrate that acute hyperglycemia also results in activation of powerful systemic cytokines. The present study has also demonstrated that acute high physiological levels of insulin are able to prevent the proinflammatory and proatherothrombotic effects of moderate hyperglycemia.
The effects of insulin on in vivo integrated vascular biological function are controversial. Large cross-sectional studies in nondiabetics or individuals with impaired glucose tolerance have linked hyperinsulinemia with an increased risk of cardiovascular events (9, 28). Complicating interpretation of these studies is that hyperinsulinemia may be resultant on an underlying putative mechanism such as insulin resistance. Thus, it is unclear what role insulin per se may be playing in the occurrence of these cardiovascular events. Somewhat surprisingly, there have been relatively few in vivo human studies performed investigating the integrated effects of insulin on inflammation and atherothrombotic balance. The majority, but not all, report beneficial effects of insulin during experimental euglycemic studies, even under significant clinical conditions such as myocardial infarction (22). However, data addressing the integrated vascular biological effect of hyperinsulinemia during conditions of hyperglycemia are very scarce. Only two studies appear to have addressed this topic, with somewhat differing results (53, 59). Williams et al. (59) reported that modest increases of insulin could moderate the deleterious effects of hyperglycemia on endothelial function, whereas Stegenga et al. (53) reported that hyperinsulinemia impairs fibrinolysis in six healthy, young males. Recently, Ceriello et al. (15) have also demonstrated that hyperglycemia of 15 mmol/l produces proinflammatory changes and endothelial dysfunction in a group of lean, young type 1 diabetic individuals, thus reinforcing that hyperglycemia per se in the presence of only basal insulin levels can result in changes of a wide range of vascular biomarkers. However, no study has addressed the vascular biological effects of insulin during hyperglycemia in overweight/obese individuals. Our results clearly demonstrate that hyperinsulinemia can prevent the wide spectrum of deleterious effects caused by hyperglycemia on proatherothrombotic balance and endothelial function. Hyperinsulinemia in the presence of hyperglycemia normalized the responses of adhesion molecules (ICAM, VCAM, E-selectin), platelet activation (P-selectin), fibrinolytic balance (PAI-1), inflammation (IL-6, adiponectin), and endothelial function created by hyperglycemia. In fact, the responses of the above vascular biological biomarkers during hypersinulinemic hyperglycemia were no different compared with the time control of euglycemic euinsulinemia or the hyperinsulinemic-euglycemic control. These latter two control protocols demonstrate that 1) acute hyperinsulinemia has no deleterious effects on atherothrombotic and fibrinolytic balance under euglycemic conditions; 2) acute hyperinsulinemia has widespread anti-inflammatory effects by reducing proatherothrombotic markers and improving endothelial function; and 3) it is acute hyperglycemia rather than hyperinsulinemia that has detrimental vascular biological effects.
This study also provides some mechanistic insights regarding the protective effects of insulin on vascular physiology. Hyperglycemia and hyperinsulinemia appear to be exerting opposite physiological effects by direct actions on vascular smooth muscle. Our endothelial function studies clearly demonstrate that hyperglycemia is acting via an endogenous NO mechanism to impair endothelial function, which can be reversed by hyperinsulinemia and prevented by an exogenous NO donor. IL-6 was significantly increased by hyperglycemia, but this elevation was prevented by hyperinsulinemia. Hyperglycemia as well as insulin resistance are known to activate NF-κB, which stimulates production of the proinflammatory cytokine IL-6, whereas insulin has been demonstrated to inhibit NF-κB activity (21). Adiponectin also increased during isolated hyperglycemia. Adiponectin is known to have insulin-sensitizing and antiatherogenic properties and thus may be acting in a “counterregulatory” manner to oppose the proinflammatory properties of hyperglycemia. Hyperglycemia also had a profound effect on suppressing free fatty acids (FFA). In fact, FFAs were similarly suppressed during the two hyperinsulinemic protocols compared with isolated hyperglycemia. This demonstrates that in this study increases in FFA cannot be implicated as a mechanism for the deleterious effects of hyperglycemia per se on the vascular endothelium. This point is worth noting as there are several studies demonstrating that high levels of FFAs (and triglycerides) can also result in adverse vascular biological effects (20, 56). Catecholamines (epinephrine, norepinephrine), cortisol, glucagon, and growth hormone were similar among all protocols. All of the above endocrine hormones have been demonstrated to have significant effects on the vascular endothelium and fibrinolytic balance (3, 19, 25, 34, 41, 57). The use of the pancreatic clamp allowed equivalent replacement of growth hormone during all protocols. This element of the experimental design is distinct to this present study. Other studies using the pancreatic clamp have not replaced growth hormone, and hyperglycemia is also known to suppress the hormone. Both would result in a state of growth hormone deficiency. This may have some relevance, as growth hormone deficiency has recently been reported to increase PAI-1 levels (39).
The PAI-1 gene is known to have glucose and insulin response elements (11). Although there is general agreement that hyperglycemia can increase PAI-1 levels, the effects of insulin on the molecule are more contentious (3). Our present results clearly demonstrate that insulin can reduce PAI-1 levels. Perhaps explaining our results is that previous studies have not controlled for the above metabolic (NEFA) and endocrine (catecholamines, cortisol, growth hormone) factors that are known to influence PAI-1 levels (31, 45).
This present study also provides new information regarding the independent effects of hyperglycemia and hyperinsulinemia on proinflammatory and proatherothrombotic biomarkers in obese and overweight humans. These individuals have an accelerated incidence of coronary artery disease, type 2 diabetes, and cancer, all of which have important inflammatory components. End of clamp or time course responses demonstrate that isolated hyperglycemia on a background of euinsulinemia increased a broad spectrum of proinflammatory and proatherothrombotic biomarkers (including platelet aggregation and reducing fibrinolytic balance). Thus, hyperglycemia can potentially amplify an existing proinflammatory and procoagulant state in obese and/or overweight individuals. Hyperinsulinemia, on the other hand, reversed the proinflammatory and proatherothrombotic effects of hyperglycemia. Furthermore, hyperinsulinema suppressed proinflammatory and proatherothrombotic biomarkers on a background of euglycemia.
Our study utilized the pancreatic clamp as this is the only methodology that allows breaking normal physiological insulin and glucose feedback loops in nondiabeteic humans. Octreotide was infused in all studies to allow us to take control of the endocrine pancreas and growth hormone secretion. Octreotide has been reported to have anti-inflammatory effects and thus may have suppressed some or all of our biomarker responses (42). In addition, there was a mild increase in basal insulin levels (90–162 pmol/l), which in the presence of euglycemia would also have suppressed FFA, triglycerides, and potentially atherothrombotic biomarkers. Furthermore, circadian rhythms would have resulted in decreases in PAI-1 during the duration of our studies. However, this would not have affected our qualitative study conclusions as all results were compared with the time and pancreatic clamp control of euinsulinemic euglycemia. We selected a moderate hyperglycemic stimulus of 11.1 mmol/l (200 mg/dl) and cannot comment on whether a higher glycemic target for a longer duration would have produced a greater proinflammatory and/or proatherothrombotic biomarker response.
In summary, the present study has demonstrated, using pancreatic and glucose clamps to control glucose and insulin levels, that hyperglycemia results in significant increases in atherogenic adhesion molecules (ICAM, VCAM, E-selectin), platelet activation (P-selectin), inflammatory cytokines (IL-6), reduced fibrinolytic balance (increased PAI-1), and impaired endothelial flow-mediated dilation. High physiological levels of insulin (albeit higher than levels obtained during insulin replacement therapy in diabetic individuals) prevented the deleterious, proinflammatory, procoagulant, and prothrombotic effects of hyperglycemia. Hyperinsulinemia also reversed the effects of hyperglycemia to impair endogenous NO mediated endothelial function.
In conclusion, this study has identified the specific effects of acute moderate hyperglycemia and hyperinsulinemia on in vivo vascular biomarkers in obese and overweight humans. Plasma glucose levels of 11.1 mmol/l produced a wide spectrum of adverse vascular pathophysiological responses. Concurrent hyperinsulinemia prevented these effects and protected endothelial function and atherothrombotic and fibrinolytic balance against hyperglycemia. Thus, 1) hyperinsulinemia was able to prevent an amplification of the proinflammatory and procoagulant state present in obese and overweight individuals, and 2) an important acute vascular biological action of insulin may be to protect the vasculature against the harmful effects of hyperglycemia.
GRANTS
This work was supported by the following NIH grants: P50 HL-081009 NIH/NHLBI, RO1 DK-069803 NIH/NIDDK, PO1 HL-056693 NIH/NHLBI, Vanderbilt Diabetes Research and Training grant (DRTC) P60 DK-020593 NIH/NIDDK, Vanderbilt General Clinical Research Center TL1 TR-000447 NIH/NCRR. We also thank Takeda Pharmaceuticals for a fellowship award to Nino G. Joy.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: J.M.P., N.G.J., and D.B.T. performed experiments; J.M.P., N.G.J., and D.B.T. analyzed data; J.M.P. and S.N.D. interpreted results of experiments; J.M.P., N.G.J., and D.B.T. prepared figures; J.M.P. drafted manuscript; J.M.P., N.G.J., D.B.T., and S.N.D. approved final version of manuscript; D.B.T. and S.N.D. edited and revised manuscript; S.N.D. conception and design of research.
ACKNOWLEDGMENTS
We thank Wanda Snead, Eric Allen, the Vanderbilt Hormone Assay Core laboratory, Joe Covington, Jesse Gilliam, Antoinette Richardson, Cheryl Williams and Lisa Younk for their excellent technical assistance. We also thank the nursing staff of the Vanderbilt Clinical Research Center for their excellent care. We also acknowledge the expert technical advice and help of Hegen Chen PhD in the analysis of this data.
REFERENCES
- 1.Abumrad NN, Rabin D, Diamond MC, Lacy WW. Use of a heated superficial hand vein as an alternative site for measurement of amino acid concentration and for the study of glucose and alanine kinetics in man. Metabolism 30: 936–940, 1981. [DOI] [PubMed] [Google Scholar]
- 2.Aguilar-Parada E, Eisentraut AM, Unger RH. Pancreatic glucagon secretion in normal and diabetic subjects. Am J Med Sci 257: 415–419, 1969. [DOI] [PubMed] [Google Scholar]
- 3.Alessi MC, Poggi M, Juhan-Vague I. Plasminogen activator inhibitor-1, adipose tissue and insulin resistance. Curr Opin Lipidol 18: 240–245, 2007. [DOI] [PubMed] [Google Scholar]
- 4.Anderson EA, Hoffman RP, Balon TW, Sinkey CA, Mark AL. Hyperinsulinemia produces both sympathetic neural activation, and vasodilation in normal humans. J Clin Invest 87: 22446–2252, 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arcaro G, Cretti A, Balzano S, Lechi A, Muggeo M, Bonaro E, Bonadonna RC. Insulin causes endothelial dysfunction in humans: sites and mechanisms. Circulation 105: 576–582, 2002. [DOI] [PubMed] [Google Scholar]
- 6.Avogaro A, Vigili de Kreutzenberg S, Fadini G. Endothelial dysfunction: causes and consequences in patients with diabetes mellitus. Diabetes Res Clin Prac 82S: S94–S101, 2008. [DOI] [PubMed] [Google Scholar]
- 7.Baron AD, Brechtel G. Insulin differentially regulates systemic and skeletal muscle vascular resistance. Am J Physiol Endocrinol Metab 265: E61–E67, 1993. [DOI] [PubMed] [Google Scholar]
- 8.Basu R, Schwenk WF, Rizza RA. Both fasting glucose production and disappearance are abnormal in people with “mild” and “severe” type 2 diabetes. Am J Physiol Endocrinol Metab 287: E55–E62, 2004. [DOI] [PubMed] [Google Scholar]
- 9.Beckman JA, Creager MA, Libby P. Diabetes and atherosclerosis: epidemiology, pathophysiology, and management. JAMA 287: 2570–2581, 2002. [DOI] [PubMed] [Google Scholar]
- 10.Brown NJ, Agirbasli MA, Williams GH, Litchfield WR, Vaughan DE. Effect of activation and inhibition of the renin-angiotensin system on plasma PAI-1. Hypertension 32: 965–971, 1998. [DOI] [PubMed] [Google Scholar]
- 11.Byrne CD, Wareham NJ, Martensz ND, Humphries SE, Metcalfe JC, Grainger DJ. Increase PIA-1 activity and PAI-1 antigen occuring with and oral fat load: associations with PAI-1 genotype and plasma TGF-b levels. Atherosclerosis 140: 45–53, 1998. [DOI] [PubMed] [Google Scholar]
- 12.Calle EE, Rodriquez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of US adults. N Engl J Med 348: 1625–1638, 2003. [DOI] [PubMed] [Google Scholar]
- 13.Causon R, Caruthers M, Rodnight R. Assay of plasma catecholamines by liquid chromatography with electrochemical detection. Anal Biochem 116: 223–226, 1982. [DOI] [PubMed] [Google Scholar]
- 14.Centers for Disease Control, and Prevention. National Diabetes Fact Sheet: General Information and National Estimates on Obesity in the US. Atlanta, GA: US Dept of Health and Human Services, Centers for Disease Control and Prevention; www.cdc.gov/obesity/data/adult.html 2014. [Google Scholar]
- 15.Ceriello A, Novials A, Ortega E, Canivell S, LaSala L, Pujadas G, Esposito K, Giugliano D, Genovese S. Glucagon-like peptide reduces endothelial dysfunction, inflammation, and oxidative stress induced by both hyperglycemia and hypoglycemia in type 1 diabetes. Diabetes Care 36: 2346–2350, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ceriello A, Quagliaro L, Piconi L, Assaloni R, Da Ros R, Maier A, Esposito K, Giugliano D. Effect of postprandial hypertriglyceridemia and hyperglycemia on circulating adhesion molecules and oxidative stress generation and the possible role of simvastatin treatment. Diabetes 53: 701–710, 2004. [DOI] [PubMed] [Google Scholar]
- 17.Ceriello A, Taboga C, Tonutti L, Quagliaro L, Piconi L, Bais B, Da Ros R, Motz E. Evidence for an independent and cumulative effect of postprandial hypertriglyceridemia and hyperglycemia on endothelial dysfunction and oxidative stress generation: effects of short- and long-term simvastatin treatment. Circulation 106: 1211–1218, 2002. [DOI] [PubMed] [Google Scholar]
- 18.Cherrington AD, Lacy W, Chiasson JL. Effect of glucagon on glucose production during insulin deficiency in the dog. J Clin Invest 62: 664–667, 1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Colao A, Di Somma C, Rota F, Pivonello R, Savanelli MC, Spiezia S, Lombardi G. Short-term effects of growth hormone (GH) treatment or deprivation on cardiovascular risk parameters and intima-media thickness at carotid arteries in patients with severe GH deficiency. J Clin Endocrinol Metab 90: 2056–2062, 2005. [DOI] [PubMed] [Google Scholar]
- 20.Creager M, Luscher T, Cosentino F, Beckamn J. Diabetes and vascular disease: pathophysiology, clinical consequences, and medical therapy. Part 1. Circulation 108: 1527–1532, 2003. [DOI] [PubMed] [Google Scholar]
- 21.Dandona P, Aljada A, Mohanty P, Ghanim H, Hamouda W, Assian E, Ahmad S. Insulin inhibits intranuclear nuclear factor kb and stimulates IkB in mononuclear cells in obese subjects: evidence for an anti-inflammatory effect? J Clin Endocrinol Metab 86: 3257–3265, 2001. [DOI] [PubMed] [Google Scholar]
- 22.Dandona P, Chaudhuri A, Ghanim H, Mohanty P. Insulin as an anti-inflammatory and antiatherogenic modulator. J Am Coll Cardiol 53: S14–S20, 2009. [DOI] [PubMed] [Google Scholar]
- 23.DeFronzo RA, Tobin K, Andres R. Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am J Physiol Endocrinol Metab Gastrointest Physiol 237: E216–E223, 1979. [DOI] [PubMed] [Google Scholar]
- 24.Després JP, Lamarche B, Mauriège P, Cantin B, Dagenais GR, Moorjani S, Lupien PJ. Hyperinsulinemia as an independent risk factor for ischemic heart disease. N Engl J Med 334: 952–957, 1996. [DOI] [PubMed] [Google Scholar]
- 25.Devin JK, Blevins LS Jr, Verity DK, Chen Q, Bloodworth JR Jr, Covington J, Vaughan DE. Markedly impaired fibrinolytic balance contributes to cardiovascular risk in adults with growth hormone deficiency. J Clin Endocrinol Metab 92: 3633–3639, 2007. [DOI] [PubMed] [Google Scholar]
- 26.Faulx MD, Wright AT, Hoit BD. Detection of endothelial dysfunction with brachial artery ultrasound scanning. Am Heart J 145: 943–951, 2003. [DOI] [PubMed] [Google Scholar]
- 27.Ferroni P, Martini F, Riondino S, La Farina F, Magnapera A, Ciatti F, Guadagni F. Soluble P-selectin as a marker of the in vivo platelet activation. Clin Chim Acta 399: 88–91, 2009. [DOI] [PubMed] [Google Scholar]
- 28.Festa A, D'Agostino R Jr, Mykkänen L, Tracy RP, Zaccaro DJ, Hales CN, Haffner SM. Relative contribution of insulin and its precursors to fibrinogen and PAI-1 in a large population with different states of glucose tolerance. The Insulin Resistance Atherosclerosis Study (IRAS). Arterioscler Thromb Vasc Biol 19: 562–568, 1999. [DOI] [PubMed] [Google Scholar]
- 29.Flegal KM, Graubard BI, Williamson DF, et al. Cause-specific excess deaths associated with underweight, overweight, and obesity. JAMA 298: 2028–2037, 2007. [DOI] [PubMed] [Google Scholar]
- 30.Florez H, Castillo-Florez S, Mendez A, Casanova-Romero P, Larreal-Urdaneta C, Lee D, Goldberg R. C-reactive protein is elevated in obese patients with the metabolic syndrome. Diabetes Res Clin Pract 71: 92–100, 2006. [DOI] [PubMed] [Google Scholar]
- 31.Halleux CM, Declerck PJ, Tran SL, Detry R, Brichard SM. Hormonal control of plasminogen activator inhibitor-1 gene expression and production in human adipose tissue: stimulation by glucocorticoids and inhibition by catecholamines. J Clin Endocrinol Metab 84: 4097–4105, 1999. [DOI] [PubMed] [Google Scholar]
- 32.Haubner F, Lehle K, Munzel D, Schmid C, Birnbaum D, Preuner J. Hyperglycemia increase the levels of vascular cellular adhesion molecule-1 and monocyte-chemoattractnat-protein-1 in the diabetic endothelial cell. Biochem Biophys Res Commun 360: 560–565, 2007. [DOI] [PubMed] [Google Scholar]
- 33.Heding LG. Radioimmunological determination of human C peptide in serum. Diabetologia 11: 541–548, 1975. [DOI] [PubMed] [Google Scholar]
- 34.Henkel E, Gallo S, Siegert G, Koehler C, Hanefeld M. Glucagon as a determinant of fibrinolytic activity in men with different stages of glucose tolerance: impact of glucagon on fibrinolysis. Blood Coagul Fibrinolysis 18: 327–334, 2007. [DOI] [PubMed] [Google Scholar]
- 35.Ho RJ. Radiochemical assay of long chain fatty acids using 63NI as tracer. Anal Biochem 26: 105–113, 1970. [DOI] [PubMed] [Google Scholar]
- 36.Hubert HB, Feinlab M, McNamara PM, Castelli WP. Obesity as an independent risk factor for cardiovascular disease: a 26-year follow-up of participants in the Framingham Heart Study. Circulation 67: 968–977, 1983. [DOI] [PubMed] [Google Scholar]
- 37.Hunter W, Greenwood F. Preparation of [131I]-labeled human growth hormone of high specific activity. Nature 194: 495–496, 1962. [DOI] [PubMed] [Google Scholar]
- 38.Kurukulasuriya LR, Govindarajan G, Sowers J. Stroke prevention in diabetes and obesity. Expert Rev Cardiovasc Ther 4: 487–502, 2006. [DOI] [PubMed] [Google Scholar]
- 39.Kvasnicka J, Marek J, Kvasnicka T, Weiss V, Marková M, Stěpán J, Umlaufová A. Increase of adhesion molecules, fibrinogen, type-1 plasminogen activator inhibitor and orosomucoid in growth hormone (GH) deficient adults and their modulation by recombinant human GH replacement. Clin Endocrinol (Oxf) 52: 543–548, 2000. [DOI] [PubMed] [Google Scholar]
- 40.Laakso M, Edelman V, Brechtel G, Baron AD. Decreased effect of insulin to stimulate skeletal muscle blood flow in obese man. J Clin Invest 85: 1844–1852, 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Laakso M, Edelman V, Brechtel G, Baron AD. Effects of epinephrine in insulin-mediated glucose uptake in whole body and leg muscle in humans: role of blood flow. Am J Physiol Endocrinol Metab 263: E199–E204, 1992. [DOI] [PubMed] [Google Scholar]
- 42.Liu R, Wei N, Guo W, Qiang O, Li X, Ou Y, Huang W, Tang CW. Octreotide alleviates obesity by reducing intestinal glucose absorption and inhibiting low-grade inflammation. Eur J Nutr 52: 1067–1075, 2013. [DOI] [PubMed] [Google Scholar]
- 43.Luminex. Lincoplex Assay. Linco Research. St. Charles, MO. [Google Scholar]
- 44.Meigs JB, Mittleman MA, Nathan DM, Tofler GH, Singer DE, Murphy-Sheehy PM, Lipinska I, D'Agostino RB, Wilson PW. Hyperinsulinemia, hyperglycemia, and impaired hemostasis: the Framingham Offspring Study. JAMA 283: 221–228, 2000. [DOI] [PubMed] [Google Scholar]
- 45.Morange PE, Aubert J, Peiretti F, Lijnen HR, Vague P, Verdier M, Negrel R, Juhan-Vague I, Alessi MC. Glucocorticoids and insulin promote plasminogen activator inhibitor 1 production by human adipose tissue. Diabetes 48: 890–895, 1999. [DOI] [PubMed] [Google Scholar]
- 46.Nacci C, Tarquinio M. Molecular and clinical aspects of endothelial dysfunction in diabetes. Intern Emerg Med 4: 107–116, 2009. [DOI] [PubMed] [Google Scholar]
- 47.Nishimura S, Manabe I, Nagasaki M, Seo K, Yamashita H, Hosoya Y, Ohsugi M, Tobe K, Kadowaki T, Nagai R, Sugiura S. In vivo imaging in mice reveals local cell dynamics and inflammation in obese adipose tissue. J Clin Invest 118: 710–721, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pandolfi A, Giaccari A, Cilli C, Alberta MM, Morviducci L, De Filippis EA, Buongiorno A, Pellegrini G, Capani F, Consoli A. Acute hyperglycemia and acute hyperinsulinemia decrease plasma fibrinolytic activity and increase plasminogen activator inhibitor type 1 in the rat. Diabetologia 38: 71–76, 2001. [DOI] [PubMed] [Google Scholar]
- 49.Pandolfi A, Iacoviello L, Vitacolonna E, Donati MB, Consoli A. Glucose and insulin independently reduce the fibrinolytic potential of human vascular smooth muscle cells in culture. Diabetologia 39: 1425–1431, 1996. [DOI] [PubMed] [Google Scholar]
- 50.Prospective Studies Collaboration. Body-mass index and cause-specific mortality in 900, 000 adults: collaborative analyses of 57 prospective studies. Lancet 373: 1083–1096, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schmidt M, Johannesdottir SA, Lemeshow S, Lash TL, Ulrichsen SP, Botker HE, Sorenson HT. Obesity in young men, and individual and combined risks of type 2 diabetes, cardiovascular morbity and death before 55 years of age: a Danish 33-year follow-up study. Br Med J 3: 1–8, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stegenga ME, van der Crabben SN, Blümer RM, Levi M, Meijers JC, Serlie MJ, Tanck MW, Sauerwein HP, van der Poll T. Hyperglycemia enhances coagulation and reduces neutrophil degranulation, whereas hyperinsulinemia inhibits fibrinolysis during human endotoxemia. Blood 112: 82–89, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stegenga ME, van der Crabben SN, Dessing MC, Pater JM, van den Pangaart PS, de Vos AF, Tanck MW, Roos D, Sauerwein HP, van der Poll T. Effect of acute hyperglycaemia and/or hyperinsulinaemia on proinflammatory gene expression, cytokine production and neutrophil function in humans. Diabet Med 25: 157–164, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stegenga ME, van der Crabben SN, Levi M, de Vos AF, Tanck MW, Sauerwein HP, van der Poll T. Hyperglycemia stimulates coagulation, whereas hyperinsulinemia impairs fibrinolysis in healthy humans. Diabetes 55: 1807–1812, 2006. [DOI] [PubMed] [Google Scholar]
- 55.Trayhurn P, Wood IS. Signaling role of adipose tissue: adipokines and inflammation in obesity. Biochem Soc Trans 33: 1078–1081, 2005. [DOI] [PubMed] [Google Scholar]
- 56.Wang XL, Zhang L, Youker K, Zhang MX, Wang J, LeMaire SA, Coselli JS, Shen YH. Free fatty acids inhibit insulin signaling-stimulated endothelial nitric oxide synthase activation through upregulating PTEN or inhibiting Akt kinase. Diabetes 55: 2301–2310, 2006. [DOI] [PubMed] [Google Scholar]
- 57.Weaver JU, Monson JP, Noonan K, John WG, Edwards A, Evans KA, Cunningham J. The effect of low dose recombinant human growth hormone replacement on regional fat distribution, insulin sensitivity, and cardiovascular risk factors in hypopituitary adults. J Clin Endocrinol Metab 80: 153–159, 1995. [DOI] [PubMed] [Google Scholar]
- 58.Wide L, Porath J. Radioimmunoassay of proteins with the uses of sephadex-coupled antibodies. Biochem Biophys Acta 130: 257–260, 1966. [Google Scholar]
- 59.Williams SB, Goldfine AB, Timimi FK, Ting HH, Roddy M, Simonson DC, Creager MA. Acute hyperglycemia attenuates endothelium-dependent vasodilation in humans in vivo. Circulation 97: 1695–1701, 1998. [DOI] [PubMed] [Google Scholar]