

Key Points
MUC1-C oncoprotein contributes toward maintenance of redox balance in CTCL.
Targeting the MUC1-C oncoprotein in CTCL cells induces ROS-mediated death highlighting its potential as an effective strategy.
Abstract
Cutaneous T-cell lymphoma (CTCL) is an aggressive neoplasm with limited treatments for patients with advanced disease. The mucin 1 C-terminal subunit (MUC1-C) oncoprotein plays a critical role in regulating cell proliferation, apoptosis, and protection from cytotoxic injury mediated by reactive oxygen species (ROS). Although CTCL cells exhibit resistance to ROS-induced apoptosis, the expression and functional significance of MUC1 in CTCL have not been previously investigated. Present studies demonstrate that MUC1-C is overexpressed in CTCL cell lines and primary CTCL cells but is absent in resting T cells from healthy donors and B-cell lymphoma cells. We have developed a cell-penetrating peptide that disrupts homodimerization of the MUC1-C subunit necessary for its nuclear translocation and downstream signaling. We show that treatment of CTCL cells with the MUC1-C inhibitor is associated with downregulation of the p53-inducible regulator of glycolysis and apoptosis and decreases in reduced NAD phosphate and glutathione levels. In concert with these results, targeting MUC1-C in CTCL cells increased ROS and, in turn, induced ROS-mediated late apoptosis/necrosis. Targeting MUC1-C in CTCL tumor xenograft models demonstrated significant decreases in disease burden. These findings indicate that MUC1-C maintains redox balance in CTCL cells and is thereby a novel target for the treatment of patients with CTCL.
Introduction
Primary cutaneous T-cell lymphomas (CTCL) comprise a heterogeneous group of non-Hodgkin lymphomas arising from skin tropic T cells. The most frequent entities are mycosis fungoides (MF) and Sézary syndrome (SS). MF typically presents with skin patches and/or plaques, which can progress to skin tumors, and SS includes diffuse erythema of the skin and involvement of the lymph nodes, peripheral blood, and in advanced cases, visceral organs. Large-cell transformation increases the likelihood of systemic dissemination and is associated with a worse prognosis because of limited treatment options and poor insight into the pathogenesis of disease.1 Hence there is a great interest in developing novel targeted therapies that can selectively kill the malignant population without impacting the T-cell repertoire. Notably, maintenance of redox balance appears to be a critical factor in protecting CTCL cells from apoptosis in comparison with normal T cells.2,3 Mucin 1 (MUC1) is a heterodimeric protein that regulates critical pathways of oncogenesis including those governing cell proliferation, self-renewal, tissue invasion, and apoptosis. Of note, MUC1 protects against reactive oxygen species (ROS)–mediated cell death because of hypoxic or other stress-induced injury. MUC1 is aberrantly expressed in epithelial tumors and selected hematologic malignancies including multiple myeloma (MM) and acute myeloid leukemia (AML).4-9 Knockdown experiments of MUC1 in adult T-cell lymphoma and leukemia revealed its role in tumor progression.10 The extracellular MUC1 N-terminal subunit (MUC1-N) contains glycosylated tandem repeats that are a characteristic of mucin family members (supplemental Figure 1, modified after permission obtained from D.K.; see the Blood Web site).11 MUC1-N forms a complex with the transmembrane MUC1 C-terminal subunit (MUC1-C) at the cell surface. MUC1-C consists of a 58-amino-acid extracellular domain that associates with galectin-3 and a 72-amino-acid cytoplasmic domain that interacts with diverse effectors that have been linked to transformation.11,12 The MUC1-C cytoplasmic domain contains a CQC motif that is necessary for its homodimerization and thereby its oncogenic function.13 Based on these findings, cell-penetrating peptide drugs were developed to block the MUC1-C CQC motif and inhibit MUC1-C homodimerization.14 The peptide inhibitors contain the MUC1-C CQCRRKN amino acid sequence linked at the N terminus to 9 arginine residues for cell permeability.14,15 Notably, treatment of MM and AML cells with MUC1-C inhibitors has been associated with increases in ROS and thereby cell death in vitro and in xenograft models.15-17
Here, we demonstrate that MUC1 is overexpressed in CTCL cell lines in comparison with B-cell lymphoma cell lines and normal T cells. Investigation of primary CTCL cells confirmed high levels of MUC1 expression in the malignant T-cell population, in contrast to T cells from normal individuals. Exposure of CTCL cells to the MUC1-C inhibitor, GO-203, was associated with downregulation of the TP53-induced glycolysis and apoptosis regulator (TIGAR), depletion of reduced NAD phosphate (NADPH) and glutathione (GSH), and a subsequent increase in ROS levels, promoting oxidative stress-induced late apoptosis/necrosis. GO-203-induced killing of CTCL cells was observed in cell lines and primary samples. In a murine xenograft model, treatment with GO-203 resulted in reduction of tumor volume of established CTCL lesions, highlighting its potential to be a novel target.
Methods
Cell culture
H9, HuT-78, and HuT-102 CTCL cell lines were obtained from American Tissue Culture Collection (Manassas, VA). CTCL lines, Myla, and SeAx were obtained from Dr Robert Gniadecki (University of Copenhagen, Denmark). B-cell lymphoma cell lines (SUDHL-8, SUDHL-2, SUDHL-5, RCK-8, Toledo, K422, Val, OCI-LY3, OCI-LY7, OCI-LY8, Daudi, Ramos, and JVM-2) were provided by Dr Margaret A. Shipp (Dana-Farber Cancer institute, Boston, MA). All cell lines were grown as previously described.18
RT-PCR
Total cellular RNA was extracted in Trizol as described.16 MUC1-C specific primers were used as previously described.19 RNA-specific primers for human glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were used for the control. The RNA was reverse transcribed and amplified using SuperScript One-Step reverse transcription–polymerase chain reaction (RT-PCR) with Platinum Taq (Invitrogen). Amplified fragments were analyzed by electrophoresis in 2% agarose gels.
Immunoblot analysis
Cell lysates were prepared as described.16 Soluble proteins were analyzed by immunoblotting with anti-MUC1-C (Neomarkers), anti-TIGAR (Abcam), and anti-GAPDH (Cell-Signaling Technology).
Detection of MUC1 expression by flow cytometry
Cells were analyzed for MUC1 expression by multichannel flow cytometric analysis.16,17 Briefly, cells were incubated with monoclonal antibody (mAb) DF3 (anti-MUC1-N) or a control mouse immunoglobulin (Ig) G1 for 30 minutes, followed by secondary labeling of the cells with phycoerythrin-conjugated goat anti-mouse IgG for an additional 30 minutes. Stained cells were analyzed by flow cytometry using FACScan and CellQuest Pro software (BD Biosciences).
Detection of MUC1 expression in primary CTCL samples and normal healthy donor controls by flow cytometry and immunohistochemistry
These studies were performed in accordance with the Declaration of Helsinki. Blood samples were obtained from 6 patients with leukemic CTCL (L-CTCL) seen at the Cutaneous Lymphoma Program at the Dana-Farber/Brigham and Women’s Cancer Center. Collection was approved by the Dana-Farber Harvard Cancer Institute Institutional Review Board (Partners Research Management).20 As controls, peripheral blood samples were obtained from 6 normal healthy donors as per a Beth Israel Deaconess Medical Center institutionally approved protocol. Peripheral blood mononuclear cells (PBMCs) were isolated from these samples by Ficoll centrifugation. Cells were incubated with mAb DF3 (anti-MUC1-N) for 30 minutes, followed by secondary labeling of the cells with allophycocyanin-conjugated goat anti-mouse IgG for an additional 30 minutes. The cells were then incubated with Pacific Blue–conjugated anti-CD4 and malignant clone specific phycoerythrin-conjugated anti-T-cell receptor (TCR) Vβ mAb’s as previously described.19 For each L-CTCL sample with a unique TCR Vβ identifiable malignant clone, PBMCs from normal healthy donors were analyzed as controls by flow cytometry using FACScan and CellQuest Pro software (BD Biosciences). In addition, formalin-fixed and paraffin-embedded sections of cutaneous lesions from 3 MF patients were analyzed for MUC1-C overexpression by immunohistochemical staining with the anti-MUC1-C antibody (1:200 dilution). Histology and immunohistochemistry was confirmed by a trained pathologist (P.B.) in accordance with the World Health Organization European Organization of Research and Treatment of Cancer Classification. Tumors were scored for the percentage of cells positive for MUC1-C using an Olympus BX41microscope, total magnification ×400 (Olympus America Inc., New York, NY). The malignant cells were deemed as negative (no visible staining) or positive in a majority (>50%) of the tumor cells. Blocks containing samples of tonsils were used as negative controls.
Measurement of ROS levels
Briefly, cells were incubated with 5 μM carboxy-H2DCFDA (2′,7′-dichlorodihydrofluorescein diacetate) (Molecular Probes) for 30 minutes at 37°C to assess hydrogen peroxide–mediated oxidation to the fluorescent compound DCF (2′,7′-dichlorofluorescein) as measured by excitation at 480 nm and emission at 590 nm. Each experiment was done in triplicate and repeated at least twice.
Determination of NADPH and GSH levels
Intracellular NADPH concentrations were measured using the Enzychrom NADP/NADPH Assay Kit (BioAssay Systems). Intracellular GSH concentrations were measured using the Bioxytech GSH-400 kit (OXIS International) as previously described.16,21 Each experiment was done in triplicate and repeated at least twice.
Cytotoxicity assays
For all in vitro assays, cells were counted and resuspended at an approximate concentration of 2.5 × 105 per well in a 96-well plate (Becton Dickinson Labware) and incubated at 37°C in a 5% CO2 humidified incubator for up to 96 hours. MUC1-C inhibitor, GO-203, and inactive analog CP-2 were added at concentrations from 1 to 10 μmol/L daily to cultures of HuT-78, Myla, and H9 cells.16 Following incubation for 48, 72, and 96 hours at 37°C in a 5% CO2 humidified incubator, 100 μL from each well was transferred to a 96-well opaque-walled plate. CellTiter-Glo reagent (Promega Corporation) was used according to the manufacturer’s instructions. The plates were allowed to incubate at room temperature for 10 minutes before recording luminescence with a Synergy HT Multi-Detection Microplate Reader (Biotek Instruments Inc.). Each experiment was done in triplicate and repeated at least twice.
Analysis of cell death
Cells were incubated with propidium iodide (PI)/annexin V–fluorescein isothiocyanate (BD BioSciences) for 15 minutes at room temperature and then analyzed by flow cytometry. Each experiment was done in triplicate and repeated at least twice.
FACS sorting of primary CTCL cells and cytotoxicity assay
The MUC1 and TCR Vβ double-positive population was fluorescence-activated cell sorter (FACS) sorted from one of the primary L-CTCL sample (details mentioned previously) on a Mo-Flo Legacy cell sorter (Beckman Coulter, Brea, CA). Cells were counted and resuspended at an approximate concentration of 1.0 × 105 per well in a 96-well plate (Becton Dickinson Labware) and incubated at 37°C in a 5% CO2 humidified incubator for up to 72 hours. MUC1-C inhibitor, GO-203, and inactive analog CP-2 were added at concentrations of 3.5 μmol/L daily to cultures. Following incubation for 48 and 72 hours at 37°C in a 5% CO2 humidified incubator, 100 μL from each well was transferred to a 96-well opaque-walled plate. CellTiter-Glo reagent (Promega Corporation) was used according to the manufacturer’s instructions, and luminescence was recorded as mentioned previously. The experiment was done in triplicate.
GO-203 treatment of murine xenograft models
Five- to 7-week-old female NSG mice were injected with 2.5 million HuT-78 or Myla cells in the flank via a subcutaneous route. Three-dimensional ultrasound imaging data sets were collected for each xenograft using a Vevo 2100 ultrasound microimaging system (VisualSonics Inc., Toronto, ON, Canada) designed for small-animal imaging. For analysis of ultrasound data, images were imported into Amira 5.2 (Visage Imaging, San Diego, CA) for volumetric analysis. Mice were imaged once a week for 3 weeks starting 4 days after inoculation of cells. After the xenograft tumors reached an average of 5 to 7 mm in diameter on imaging, mice were randomized to the 2 treatment groups of 6 animals each: (1) a control group treated with normal saline alone and (2) a GO-203-treated group that received the drug at 14 mg/kg by intradermal administration each day for 21 days. All the mice were monitored twice a week and managed as per Institutional Animal Care and Use Committee regulations mentioned previously. In the HuT-78 xenograft murine model, all mice were euthanized after 21 days of treatment, and tumor tissue was harvested for immunohistochemical analysis. Formalin-fixed and paraffin-embedded 5-μm sections of tumor samples were analyzed for apoptotic cells by terminal uridine deoxynucleotidyltransferase-mediated dUTP nick end labeling (TUNEL) staining using the tumor TACS in situ apoptosis detection kit (R&D Systems Inc.) as per the manufacturer’s instructions. Tumors were scored by the percentage of cells positive for necrosis and for TUNEL (as a measure of apoptosis). Tumors were also analyzed for MUC1-C immunohistochemical staining with the anti-MUC1-C antibody. In the Myla xenograft murine model, mice were followed for survival analysis.
Statistical analysis
The Student t test was used to assess statistical significance. Kaplan-Meier survival functions were calculated respectively for each group in the xenograft murine models. The log-rank test was used to compare the median survival times among the treatment groups.
Results
Expression of MUC1 by CTCL cell lines
SS (HuT-78, H9, and SeAx) and MF (Myla and HuT-102) cell lines were analyzed for MUC1 messenger RNA expression by RT-PCR. The results demonstrated prominent expression of MUC1 in the CTCL cells and minimal levels in normal T cells from healthy donors and in B-cell lymphoma cells (Figure 1A). These findings were confirmed by immunoblot analysis, which showed high levels of the MUC1-C cytoplasmic protein in CTCL cell lines in comparison with T cells from normal healthy donors and B-cell lymphoma cell lines including DLBCL (Val, Toledo, RCK-8, OCI-LY7, OCI-LY3, SUDHL2, and SUDHL5), Burkitt lymphoma (Ramos and Daudi) and mantle cell lymphoma (JVM-2) (Figure 1B). Flow cytometric analysis of the cells with another antibody, DF3 (anti-MUC1-N) targeting the extracellular N-terminal subunit further demonstrated that MUC1 is expressed in 11% to 62% of the Sézary cells and 13% to 43% of the mycosis cells as opposed to 0% to 2% of the B-cell lymphoma cells (Figure 1C-D). These findings demonstrate that MUC1 expression is upregulated in CTCL cells, in contrast to normal resting T cells and B-cell lymphoma cells.
Figure 1.
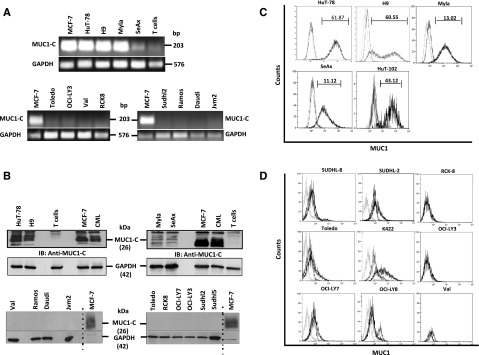
Selective expression of MUC1 in CTCL cells. (A) Sézary (HuT-78, H9, and SeAx) and mycosis (Myla, HuT-102) cell lines were analyzed for MUC1-C messenger RNA expression by RT-PCR (Michigan Cancer Foundation-7 breast adenocarcinoma cell line [MCF-7] used as positive control, GAPDH as loading control) in comparison with normal T cells from healthy individual and diffuse large B-cell lymphoma (DLBCL; Toledo, OCI-LY3, Val, RCK-8, and SUDHL-2), Burkitt lymphoma (Ramos and Daudi), and mantle cell lymphoma (JVM-2) cell lines. (B) Sézary, mycosis, and normal T cells from healthy individuals; additional DLBCL (OCI-LY7 and SUDHL-5), Burkitt lymphoma, and mantle cell lymphoma cells were also analyzed for the oncogenic subunit MUC1-C by immunoblotting (Chronic myeloid leukemia [CML] and MCF-7 used as positive controls). (C-D) Sézary and mycosis cell lines were further analyzed for MUC1 expression by flow cytometry (C) in comparison with additional DLBCL cell lines (SUDHL-8, K422, OCI-LY3, and OCI-LY8) (D). The difference in the mean fluorescent intensity between unstained, isotype control, and MUC1-stained cells are highlighted in each histogram in the top.
MUC1 expression in primary CTCL cells as compared with T cells derived from normal healthy donors
Based on the findings with CTCL cell lines, PBMCs from 6 L-CTCL patients with previously identified CD4 dominant TCR Vβ malignant clones were analyzed for MUC1 expression by flow cytometry and compared with PBMCs from 6 healthy donors as controls.20 Analysis of the unselected CD4+ population demonstrated high levels of MUC1 expression in L-CTCL samples (average 61.5%) as compared with normal healthy donor controls (average 5.34%) that was statistically significant (Figure 2A). The dominant malignant clonal population was further isolated using the previously identified specific TCR Vβ, which constituted a mean of 40.65% as compared with 1.7% of the CD4+ cells derived from the CTCL patients and normal controls, respectively (Figure 2B). Consistent with the prior findings, a majority of the cells in the CTCL patients that expressed the TCR Vβ malignant clone also exhibited MUC1 expression (average 85.43%) in comparison with the normal healthy donor controls (average 3.66%) (Figure 2C). A representative FACS plot of a CTCL patient (in which TCR Vβ1 is rearranged in the malignant cells) and corresponding healthy donor is shown in Figure 2D. These findings indicate that upregulation of MUC1 expression is selective for the malignant clonal population in CTCL patients as compared with normal T cells. Further, we performed immunohistochemical staining of skin specimens from 3 MF patients with the anti-MUC1-C antibody. All 3 cases displayed positivity for MUC1-C expression in tumor cells as characterized by irregular nuclei and/or pleomorphic nuclei. The anti-MUC1-C stain was mainly nuclear and cytoplasmic. In the skin biopsies, epidermis demonstrated positive cytoplasmic staining to varying degrees as expected. Representative photomicrographs of MUC1 immunohistochemical stained skin biopsy from a patient with MF are shown in supplemental Figure 2A-C. These results confirm high MUC1 expression in lymphoma cells in CTCL patients with peripheral blood and skin involvement.
Figure 2.
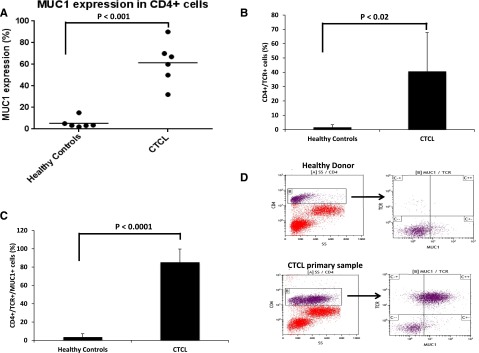
Selective expression of MUC1 in CTCL patients. (A-D) PBMCs from 6 patients with L-CTCL with previously identified unique TCR Vβ rearranged malignant clone and 6 corresponding normal donors were isolated and analyzed for MUC1 and CD4 expression by flow cytometry. Each dot represents an individual patient or control, and results are presented as percentage of cells expressing MUC1 in the CD4+ population (A); TCR Vβ expression was analyzed in the CD4+ subset of cells (B), and MUC1 expression was further analyzed in the CD4+, TCR Vβ+ subset of cells (C). Representative FACS plots of a patient with L-CTCL and healthy donor are shown (D).
Inhibition of MUC1-C increases ROS in CTCL cells
Treatment of HuT-78 Sézary cells with the MUC1-C inhibitor, GO-203, was associated with increases in hydrogen peroxide levels in contrast to that obtained with CP-2 (Figure 3A). Similar results were obtained in GO-203-treated H9 Sézary cells (Figure 3B). The induction of ROS by GO-203 was reversed by concurrent exposure of HuT-78 and H9 cells to the antioxidant NAC that promotes the scavenging of ROS (Figure 3C-D). A dose-dependent effect with GO-203 on increases in ROS and reversal by NAC were observed in both cell lines (supplemental Figure 3A-B).
Figure 3.
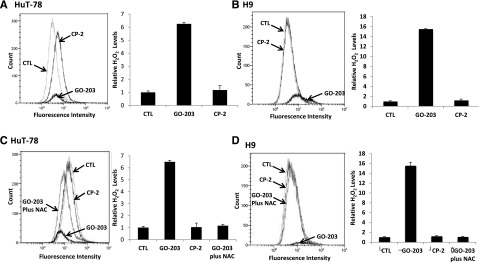
GO-203 increases hydrogen peroxide (H2O2) levels in CTCL cells, which is abrogated by addition of N acetyl cysteine (NAC). (A-D) HuT-78 and H9 cells were left untreated and treated with 5 μM GO-203 or CP-2 each day for 3 days. (C and D) The GO-203-treated cells were also incubated in the presence of 5 mM NAC for the last 2 days. Cells were incubated with c-H2DCFDA for 30 minutes. Fluorescence of oxidized DCF was measured by flow cytometry (left). The results are expressed as the relative hydrogen peroxide level (mean ± standard deviation [SD] of 3 determinations) compared with that obtained for control cells (right).
MUC1 inhibition is associated with decreased levels of TIGAR
To investigate the mechanism by which MUC1 inhibition results in increased ROS levels in CTCL, we examined the impact of GO-203 on TIGAR expression, a p53-inducible protein that regulates glycolysis and protects against oxidative stress. We demonstrated that exposure of CTCL cells to GO-203 was associated with the marked reduction of levels of TIGAR in HuT-78 and H9 cells (Figure 4A-B). TIGAR redirects glycolytic intermediates to the pentose phosphate pathway, increasing NADPH production.15,20 Accordingly, inhibition of MUC1-C and downregulation of TIGAR in HuT-78 cells was associated with a marked decrease in NADPH levels (Figure 5A). NADPH is necessary for the production of GSH and the scavenging of ROS. In this regard, GO-203 treatment resulted in a reduction in GSH levels in HuT-78 cells (Figure 5B). Similar findings were observed in H9 cells (Figure 5C-D). In summary, inhibition of MUC1-C results in increased levels of ROS in the context of reduction in TIGAR and thereby NADPH and GSH.
Figure 4.
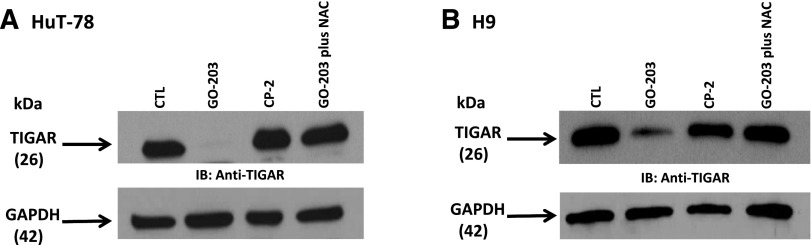
TIGAR expression is decreased by MUC1 inhibition via ROS. (A-B) HuT-78 and H9 cells were left untreated and treated with 5 μM GO-203 or 5 μM CP-2 each day for 3 days. The GO-203-treated cells were also incubated in the presence of 5 mM NAC for the last 2 days. Lysates were immunoblotted with the indicated antibodies.
Figure 5.
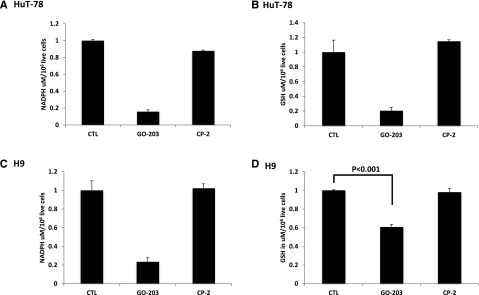
GO-203 treatment decreases NADPH and GSH levels. (A-D) HuT78 and H9 cells were left untreated and treated with 5 μM GO-203 or 5 μM CP-2 each day for 3 days. The HuT78 (A) and H9 (C) cells were analyzed for NADPH levels on day 3. The results are expressed as the NADPH level (mean ± SD of 3 determinations). The HuT-78 (B) and H9 (D) cells were also analyzed for GSH levels (mean ± SD of 3 determinations) on day 3.
We have previously demonstrated that increasing ROS levels negatively impacts expression of TIGAR creating a positive feedback loop that further escalates ROS expression.13,20 To determine whether inhibition of MUC1-C decreases TIGAR expression, in part, by a ROS-mediated mechanism, HuT-78 cells were treated with GO-203 and as a control with NAC. Under these conditions, NAC blocked the GO-203-induced downregulation of TIGAR (Figure 4A). Similar results were obtained in H9 cells treated with GO-203 and NAC indicating that ROS contributes to decreases in TIGAR expression (Figure 4B). These findings indicate that inhibition of MUC1-C increases ROS and thereby decreases TIGAR expression confirming existence of the positive feedback loop in CTCL cell lines.
GO-203 induces apoptosis and cell death in CTCL
CTCL cells were cocultured with escalating concentrations of the MUC1-C inhibitor GO-203 (1 to 10 μM/L) and levels of apoptosis and cell death was quantified at 48, 72, and 96 hours of exposure. The Sézary cell line HuT-78, H9, and MF cell line Myla demonstrated dose- and time-dependent sensitivity to GO-203 relative to CP-2 starting as low as 3 μM at 48 hours. Cell viability was reduced to 40% to 60% at 72 hours in all cell lines when incubated at 3.5 μM GO-203; by contrast, CP-2 had little effect (Figure 6A-C). Flow cytometric analysis of PI/annexin V–fluorescein isothiocyanate staining in the treated CTCL cells (Figure 6D-F) was consistent with late apoptosis/necrosis in 65%, 62%, and 74% of HuT-78, Myla, and H9 cells, respectively. In contrast, exposure to the control peptide, CP-2 alone, resulted in apoptosis/necrosis in 8%, 12%, and 11% of the HuT-78, Myla, and H9 cells, respectively. Representative flow plots are presented in supplemental Figure 4A-B. Similar results were obtained at 48 and 96 hours of treatment across a spectrum of concentrations for other cell lines (supplemental Figure 5A-J). GO-203-induced death of both HuT-78 and H9 cells (supplemental Figure 6A-B) was attenuated by NAC treatment. These findings indicate that the GO-203 CTCL cell death, at least in large part, by a ROS-mediated mechanism.
Figure 6.
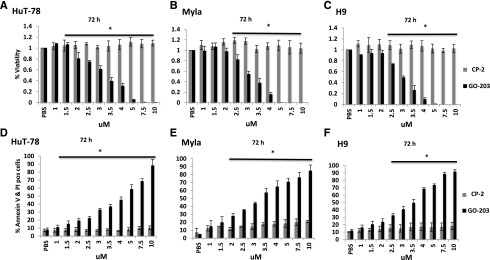
MUC1-C inhibitor GO-203 is cytotoxic in CTCL cells. (A-E) Hut78, Myla, and H9 cells were treated with varying concentrations of GO-203 and the inactive analog CP-2 (1-10 μM) each day for 3 days. Cell Titer Glo was added and viability was measured at 72 hours. The results are expressed as relative percentage viability (mean ± SD of 3 determinations) compared with the control treated with phosphate-buffered saline (PBS; A-C). Cells were incubated with PI and annexin V and analyzed by flow cytometry at 72 hours. The results are expressed as relative percentage of cells with late apoptosis and necrosis (mean ± SD of 3 determinations) compared with the control treated with PBS (D-F). Asterisk indicates a statistically significant P value of <.05.
Targeting of MUC1-C in primary CTCL cells with GO-203
Consistent with these findings, exposure to GO-203 induced cell death in primary CTCL cells. The malignant clonal population was isolated from 3 primary CTCL samples by flow cytometric sorting for the TCR Vβ of interest (Figure 2D). Culture of these cells with different concentrations of GO-203 (3.5-7.5 μM) resulted in decreased viability at 48 hours and 72 hours (10% to 30%) as opposed to CP-2 treated and untreated cells (80% to 100%) (Figure 7A-D and supplemental Figure 7A-B).
Figure 7.
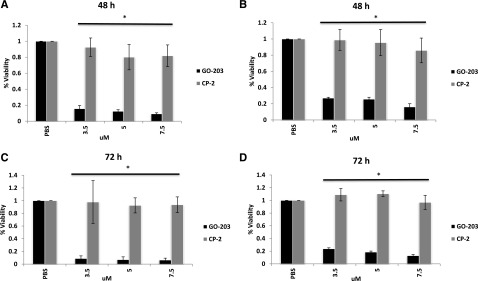
MUC1-C inhibitor GO-203 is cytotoxic in CTCL primary cells at 48 and 72 hours. (A-D) MUC1 and TCR Vβ rearranged double-positive malignant population of our interest was FACS sorted from 2 of the patients with L-CTCL (Figure 2D) and treated with varying concentrations (3.5, 5, and 7.5 μM) of GO-203 and CP-2 daily. Cell viability assessed at 48 (A and B) and 72 hours (C and D) demonstrated increased sensitivity to GO-203 in comparison with CP-2 and untreated cells that were statistically significant. Experiment was performed in triplicate. Asterisk indicates a statistically significant P value of <.05.
Inhibition of MUC1-C induces tumor reduction in xenograft murine models of CTCL
To assess in vivo antitumor activity of GO-203, HuT-78 cells were inoculated subcutaneously in the flank of NSG mice. After the xenograft tumors reached a volume of ∼100 mm3 as determined by 3-dimensional ultrasonography, they were randomized to control (treated with PBS) vs GO-203 treatment daily via intradermal injection for 21 days. At least a 2.5-fold reduction of tumor volume was seen with the administration of GO-203 compared with that obtained with the vehicle (PBS) (supplemental Figure 8A-B). On day 22, all mice from both treatment groups were euthanized for histologic examination of the xenograft tumors and stained for TUNEL analysis. Greater percentage of apoptotic cells were seen in the GO-203-treated mice in comparison with control mice (supplemental Figure 9A-C) in concert with the in vitro findings. Similar abrogation reduction in the tumor volume was obtained in the Myla xenograft model (supplemental Figure 8C). In this model, mice in both control and GO-203-treated groups were followed for survival analysis until they had to be euthanized for disease burden per Institutional Animal Care and Use Committee regulations. Kaplan-Meier survival functions were calculated for each group and demonstrated that the survival distributions for the 2 treatment groups were significantly different in general (supplemental Figure 8D). Log-rank test for comparison of median survival time among the 2 treatment groups (control and GO-203) exhibited a marked increase in survival for the GO-203 group vs the control cohorts that was statistically significant. To whether resistance of the HuT-78 murine xenograft tumors to GO-203 is associated with downregulation of MUC1-C, the tumors were analyzed by immunohistochemistry. Viable tumor cells in GO-203-treated mice demonstrated MUC1-C immunoreactivity indicating that other potential mechanisms contribute to lack of response (supplemental Figure 10A-B).
Discussion
Despite combination strategies of conventional agents and incorporation of novel agents such as histone deacetylase inhibitors and antifolates for the treatment of CTCL, durable responses remain elusive and there is a significant ongoing need to define more effective therapeutic targets.22-24 An expanded understanding of the critical pathways that are operative in CTCL is crucial for the development of more effective targeted therapies for this disease. MUC1 is a master regulator of oncogenesis that plays a vital role in promoting cell proliferation, resistance to apoptosis, and maintenance of the malignant phenotype. We have shown that MUC1 modulates cancer metabolism and protection from oxidative stress, which are thought to be important pathways that differentiate CTCL cells from normal activated T cells.2,3,16,21 However, MUC1 expression and its role in the disease pathogenesis of CTCL have yet to be elucidated. Herein, we report that MUC1 is strongly expressed by cell lines derived from SS and MF patients and primary CTCL samples but not in T cells isolated from healthy volunteers. Of note, in L-CTCL patients in whom the TCR Vβ chain of the malignant clonal population was identified, MUC1 expression was found on the CTCL cells but not normal resting T cells. In addition, MUC1 expression was absent in cell lines derived from patients with DLBCL, Burkitt lymphoma, and mantle cell lymphoma suggesting that MUC1 may play a unique role in the evolution of T cell lymphoma.
MUC1 has been shown to be a critical regulator of ROS and the protection of cancer cells from apoptosis and maturation induced by the oxidative stress.16,21 Increased levels of ROS have been shown to induce cell death in activated T cells via CD95L-mediated apoptosis, and CTCL cells have been shown to downregulate ROS promoting cell survival.3 Having demonstrated the presence of MUC1 in CTCL, we sought to define the critical pathways by which MUC1 may modulate ROS in this setting. In previous studies, we have demonstrated that MUC1 regulates TIGAR, a critical mediator of cancer metabolism that protects cells from oxidative stress by inducing production of NADPH and GSH through the pentose phosphate shunt. These molecules act as scavengers preventing the accumulation of ROS. We have developed a cell penetrating peptide, GO-203, that blocked dimerization of MUC1-C necessary for nuclear translocation and interaction with downstream effectors. Exposure of CTCL cells to the MUC1 inhibitor is associated with decreased levels of TIGAR within CTCL cells, decreased levels of NADH and GSH, and an associated rise of ROS. This effect was reversed in part by exposure to NAC suggesting a feedback loop in which ROS levels further modulate TIGAR expression.
Treatment of the CTCL cell lines with GO-203 induced cell death in CTCL cell lines and primary patient–derived samples in a dose-dependent fashion with peak effect seen between 48 and 72 hours of exposure. Cell death was characterized by a pattern of late apoptosis and necrosis as validated by both an adenosine triphosphate–based luminescence assay and PI and annexin V staining. Cell death was abrogated by coculture with NAC suggesting that accumulation of ROS is the primary mechanism by which MUC1 inhibition induces apoptosis in CTCL. Of note, exposure of the CTCL cells to a cell penetrating peptide that does not contain the CQC motif necessary to disrupt MUC1 dimerization did not induce cell death. In a xenograft model, treatment of NSG mice implanted with CTCL with GO-203 resulted in a significant reduction in tumor growth, confirmed by 3-dimensional ultrasound based volumetric analysis. Analysis of tumor harvested after in vivo exposure to GO-203 showed evidence of increased apoptosis compared with control animal. GO-203 was associated with a survival advantage compared with untreated animals.
In summary, the present results indicate that MUC1-C expression is upregulated in CTCL cells and patient samples. MUC1-C inhibition initiates a cascade of (1) downregulation of TIGAR, (2) decreases in NADPH and GSH, (3) ROS production, and (4) ROS-mediated late apoptosis/necrosis. The first-in-human MUC1-C inhibitor has entered phase 1 evaluation in patients with AML to establish a maximum tolerated dose that could be used for treatment of patients with relapsed/refractory CTCL. Our finding of MUC1 inhibition leading to increased sensitivity of the CTCL cells to stress-induced apoptosis has led us to explore synergistic combination strategies of GO-203 with other drugs with marked anti–T-cell lymphoma activity.
Acknowledgments
Imaging studies were performed in collaboration with the Small Animal Imaging Shared Resource within the Beth Israel Deaconess Medical Center.
This work was supported by National Institutes of Health National Cancer Institute, MM Specialized Program of Research Excellence (grant P50CA100707); the Conquer Cancer Foundation Young Investigator Grant; National Institutes of Health National Institute of Arthritis and Musculoskeletal and Skin Diseases (grants R01AR063962 and R01AR056720) (R.A.C.); a Damon Runyon Clinical Investigator Award (R.A.C.); the National Institutes of Health National Cancer Institute, Specialized Program of Research Excellence in Skin Cancer (P50CA9368305) (T.S.K.); and National Institutes of Health National Institute of Allergy and Infectious Diseases (R01AI097128) (T.S.K. and R.A.C.).
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: S.J., D.K., and D.A. designed the research; S.J., D.S., and L.Y. performed the research; S.J., D.S., L.Y., P.B., J.R., D.K., and D.A. analyzed data; S.J., J.R., D.K., and D.A. wrote the manuscript; T.S.K. obtained CTCL patient samples; R.A.C. isolated CTCL T cells and identified the clonal malignant T cells; and M.A., H.R., R.A.C., T.S.K., K.P., M.D.C., A.P., M.B.-N., K.L., J.A., and R.J. advised in research design.
Conflict-of-interest disclosure: D.K. has consultancy with and equity ownership in Genus Oncology. The remaining authors declare no competing financial interests.
Correspondence: Salvia Jain, Division of Malignant Hematology and Bone Marrow Transplantation, Beth Israel Deaconess Medical Center, Harvard Medical School, 330 Brookline Ave, Boston, MA 02215; e-mail: ssjain@bidmc.harvard.edu.
References
- 1.Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105(10):3768–3785. doi: 10.1182/blood-2004-09-3502. [DOI] [PubMed] [Google Scholar]
- 2.Agrawal B, Krantz MJ, Parker J, Longenecker BM. Expression of MUC1 mucin on activated human T cells: implications for a role of MUC1 in normal immune regulation. Cancer Res. 1998;58(18):4079–4081. [PubMed] [Google Scholar]
- 3.Klemke CD, Brenner D, Weiss EM, et al. Lack of T-cell receptor-induced signaling is crucial for CD95 ligand up-regulation and protects cutaneous T-cell lymphoma cells from activation-induced cell death. Cancer Res. 2009;69(10):4175–4183. doi: 10.1158/0008-5472.CAN-08-4631. [DOI] [PubMed] [Google Scholar]
- 4.Takahashi T, Makiguchi Y, Hinoda Y, et al. Expression of MUC1 on myeloma cells and induction of HLA-unrestricted CTL against MUC1 from a multiple myeloma patient. J Immunol. 1994;153(5):2102–2109. [PubMed] [Google Scholar]
- 5.Burton J, Mishina D, Cardillo T, et al. Epithelial mucin-1 (MUC1) expression and MA5 anti-MUC1 monoclonal antibody targeting in multiple myeloma. Clin Cancer Res. 1999;5(10, suppl):3065s–3072s. [PubMed] [Google Scholar]
- 6.Treon SP, Mollick JA, Urashima M, et al. Muc-1 core protein is expressed on multiple myeloma cells and is induced by dexamethasone. Blood. 1999;93(4):1287–1298. [PubMed] [Google Scholar]
- 7.Li Y, Liu D, Chen D, Kharbanda S, Kufe D. Human DF3/MUC1 carcinoma-associated protein functions as an oncogene. Oncogene. 2003;22(38):6107–6110. doi: 10.1038/sj.onc.1206732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brossart P, Schneider A, Dill P, et al. The epithelial tumor antigen MUC1 is expressed in hematological malignancies and is recognized by MUC1-specific cytotoxic T-lymphocytes. Cancer Res. 2001;61(18):6846–6850. [PubMed] [Google Scholar]
- 9.Fatrai S, Schepers H, Tadema H, Vellenga E, Daenen SM, Schuringa JJ. Mucin1 expression is enriched in the human stem cell fraction of cord blood and is upregulated in majority of the AML cases. Exp Hematol. 2008;36(10):1254–1265. doi: 10.1016/j.exphem.2008.04.015. [DOI] [PubMed] [Google Scholar]
- 10.Hasegawa H, Komoda M, Yamada Y, et al. Aberrant expression of membrane-associated mucin contributes to tumor progression in adult T-cell leukemia/lymphoma cells. Leuk Lymphoma. 2011;52(6):1108–1117. doi: 10.3109/10428194.2011.559671. [DOI] [PubMed] [Google Scholar]
- 11.Kufe DW. Mucins in cancer: function, prognosis and therapy. Nat Rev Cancer. 2009;9(12):874–885. doi: 10.1038/nrc2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ramasamy S, Duraisamy S, Barbashov S, Kawano T, Kharbanda S, Kufe D. The MUC1 and galectin-3 oncoproteins function in a microRNA-dependent regulatory loop. Mol Cell. 2007;27(6):992–1004. doi: 10.1016/j.molcel.2007.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leng Y, Cao C, Ren J, et al. Nuclear import of the MUC1-C oncoprotein is mediated by nucleoporin Nup62. J Biol Chem. 2007;282(27):19321–19330. doi: 10.1074/jbc.M703222200. [DOI] [PubMed] [Google Scholar]
- 14.Raina D, Ahmad R, Joshi MD, et al. Direct targeting of the mucin 1 oncoprotein blocks survival and tumorigenicity of human breast carcinoma cells. Cancer Res. 2009;69(12):5133–5141. doi: 10.1158/0008-5472.CAN-09-0854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yin L, Ahmad R, Kosugi M, et al. Survival of human multiple myeloma cells is dependent on MUC1 C-terminal transmembrane subunit oncoprotein function. Mol Pharmacol. 2010;78(2):166–174. doi: 10.1124/mol.110.065011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yin L, Kosugi M, Kufe D. Inhibition of the MUC1-C oncoprotein induces multiple myeloma cell death by down-regulating TIGAR expression and depleting NADPH. Blood. 2012;119(3):810–816. doi: 10.1182/blood-2011-07-369686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stroopinsky D, Rosenblatt J, Ito K, et al. MUC1 is a potential target for the treatment of acute myeloid leukemia stem cells. Cancer Res. 2013;73(17):5569–5579. doi: 10.1158/0008-5472.CAN-13-0677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Toner LE, Vrhovac R, Smith EA, et al. The schedule-dependent effects of the novel antifolate pralatrexate and gemcitabine are superior to methotrexate and cytarabine in models of human non-Hodgkin’s lymphoma. Clin Cancer Res. 2006;12(3, pt 1):924–932. doi: 10.1158/1078-0432.CCR-05-0331. [DOI] [PubMed] [Google Scholar]
- 19.Rajabi H, Ahmad R, Jin C, et al. MUC1-C oncoprotein induces TCF7L2 transcription factor activation and promotes cyclin D1 expression in human breast cancer cells. J Biol Chem. 2012;287(13):10703–10713. doi: 10.1074/jbc.M111.323311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Clark RA, Shackelton JB, Watanabe R, et al. High-scatter T cells: a reliable biomarker for malignant T cells in cutaneous T-cell lymphoma. Blood. 2011;117(6):1966–1976. doi: 10.1182/blood-2010-05-287664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yin L, Kufe T, Avigan D, Kufe D. Targeting MUC1-C is synergistic with bortezomib in downregulating TIGAR and inducing ROS-mediated myeloma cell death. Blood. 2014;123(19):2997–3006. doi: 10.1182/blood-2013-11-539395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Piekarz RL, Frye R, Turner M, et al. Phase II multi-institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T-cell lymphoma. J Clin Oncol. 2009;27(32):5410–5417. doi: 10.1200/JCO.2008.21.6150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Whittaker SJ, Demierre MF, Kim EJ, et al. Final results from a multicenter, international, pivotal study of romidepsin in refractory cutaneous T-cell lymphoma. J Clin Oncol. 2010;28(29):4485–4491. doi: 10.1200/JCO.2010.28.9066. [DOI] [PubMed] [Google Scholar]
- 24.Horwitz SM, Kim YH, Foss F, et al. Identification of an active, well-tolerated dose of pralatrexate in patients with relapsed or refractory cutaneous T-cell lymphoma. Blood. 2012;119(18):4115–4122. doi: 10.1182/blood-2011-11-390211. [DOI] [PubMed] [Google Scholar]