

Abstract
Neurohumoral remodeling is fundamental to the evolution of heart disease. This study examined the effects of chronic treatment with an ACE inhibitor (captopril, 3 mg·kg−1·day−1), AT1 receptor antagonist (losartan, 3 mg·kg−1·day−1), or AT2 receptor agonist (CGP42112A, 0.14 mg·kg−1·day−1) on remodeling of the guinea pig intrinsic cardiac plexus following chronic myocardial infarction (MI). MI was surgically induced and animals recovered for 6 or 7 wk, with or without drug treatment. Intracellular voltage recordings from whole mounts of the cardiac plexus were used to monitor changes in neuronal responses to norepinephrine (NE), muscarinic agonists (bethanechol), or ANG II. MI produced an increase in neuronal excitability with NE and a loss of sensitivity to ANG II. MI animals treated with captopril exhibited increased neuronal excitability with NE application, while MI animals treated with CGP42112A did not. Losartan treatment of MI animals did not alter excitability with NE compared with untreated MIs, but these animals did show an enhanced synaptic efficacy. This effect on synaptic function was likely due to presynaptic AT1 receptors, since ANG II was able to reduce output to nerve fiber stimulation in control animals, and this effect was prevented by inclusion of losartan in the bath solution. Analysis of AT receptor expression by Western blot showed a decrease in both AT1 and AT2 receptors with MI that was reversed by all three drug treatments. These data indicate that neuronal remodeling of the guinea pig cardiac plexus following MI is mediated, in part, by activation of both AT1 and AT2 receptors.
chronic heart disease induces remodeling of cardiac tissues and the elements of the cardiac nervous system that control it (2, 20). Much of this remodeling is due to alterations in the balance of autonomic and humoral factors that result from overstimulation of sympathetic efferent pathways (32) and the renin-angiotensin system (RAS), both local and systemic (25, 30), with a corresponding decrease in central parasympathetic drive (31). Increased sympathetic activity evokes elevated levels of norepinephrine (NE) release within the heart (7). An increase in the synthesis of ANG II from both enhanced renin release and increased protease activity within the heart interstitial tissues contributes to the hyperdynamic sympathetic response (6, 21, 24). Inhibition of adrenergic receptors (e.g., β-blockade) or treatment with drugs that target ANG II synthesis (ACE inhibitors) or receptor activation (AT1 inhibitors) has been demonstrated to alter adverse remodeling of the cardiac muscle (30).
ANG II has multiple receptor targets that include both AT1 and AT2 receptors. Overstimulation of AT1 receptors has been associated with many of the negative symptoms associated with chronic heart disease (10, 26), while stimulation of AT2 receptors can counteract many of these actions (17). Previous research suggests that it is the balance of AT1 vs. AT2 stimulation that is crucial in determining the outcome in chronic heart disease (16, 23). The present study was designed to investigate the role of altered angiotensin levels following a chronic ischemic event on intrinsic cardiac (IC) neuronal function, with particular focus on differential effects of AT1 vs AT2 receptors.
Previous studies in our laboratory have shown that chronic myocardial infarction (MI) induces remodeling of the neurons located within the IC neural plexus of the guinea pig (13). This cardiac plexus is a primary integration site for descending parasympathetic preganglionic inputs, sympathetic efferents, and sensory afferent information (3). In the guinea pig model, the majority of these neurons are cholinergic (19) and likely represent postganglionic parasympathetic neurons. Additionally, these neurons could also be acting as cholinergic local circuit neurons (3). Remodeling of this network with disease exerts profound effects on beat-to-beat modulation of regional cardiac function (3). Remodeling of the IC plexus with chronic MI includes an enhanced sensitivity to NE and a reduced response to ANG II (14). Prior research from our group has also shown that ANG II mediates direct effects on these neurons via AT2 receptors to potentiate both adrenergic and muscarinic responses (9). The hypothesis for this study was that chronic alterations in ANG II synthesis or receptor stimulation would alter the IC neuronal remodeling following MI. Specifically, we hypothesized that drugs that would increase the relative stimulation of AT2 vs. AT1 receptors would reverse the alterations in IC neuronal responses to ANG II and/or NE following MI.
MATERIALS AND METHODS
Animals.
Nine-week-old, male Hartley guinea pigs (Charles River), weighing between 500 and 650 g, were used in these studies. Animals of the same age and weight were used as controls. All procedures were approved by the Institutional Animal Care and Use Committees of East Tennessee State University and Ithaca College and were in accordance with the Guide for the Care and Use of Laboratory Animals (National Academy Press, Washington DC, 2010).
Induction of chronic myocardial infarction.
MI was induced in a total of 32 animals by surgical ligation of the ventral descending coronary artery just distal to its first diagonal branch, as previously described (13). This procedure produces an infarct region equivalent to ∼7–8% of the left ventricular tissue (13). Animals were treated with appropriate analgesics postoperatively and allowed to recover for ∼7 wk.
Drug treatments.
For chronic drug treatment, animals were anesthetized and surgically implanted with an Alzet osmotic pump (2ML4) under the skin (14) 1 wk after the MI surgery. The pumps contained either 3 mg·kg−1·day−1 of captopril (n = 6), 3 mg·kg−1·day−1 of losartan (n = 6), or 0.14 mg·kg−1·day−1 of CGP 42112A (n = 8). Pumps were replaced after 3 wk, for a total drug treatment time of 6 wk. A subset of control animals were surgically implanted with pumps containing either captopril, losartan, or CGP 42112A (n = 6 for each condition), as described above, and maintained for 3 or 4 wk. Previous studies (14) found no effect of sham MI surgery coupled with pump implantation (no drug) on intrinsic neuronal properties or responses to modulators, such as NE, bethanechol, or ANG II, so these experiments were not repeated.
Terminal experiments.
Guinea pigs were euthanized by CO2 inhalation and exsanguination. The heart was removed and placed into ice-cold Krebs-Ringer solution (in mM: 121 NaCl, 5.9 KCl, 2.5 CaCl2, 1.2 MgCl2, 1.2 NaH2PO4, 25 NaHCO3, 8 glucose, aerated with 95% O2-5% CO2 for a pH of 7.4). The cardiac plexus, located in the epicardium of the atria and underlying the area of the coronary sinus, was dissected as previously described (11). The tissue was pinned to a Sylgard-lined 60-mm Petri dish and continuously superfused (6–8 ml/min) with 35–37°C Krebs Ringer. NE (Sigma, 10−3 M) and bethanechol (a muscarinic agonist, Sigma, 10−3 M) were applied by local pressure ejection (6–9 psi; Picospritzer, General Valve) through small-tip diameter (5–10 μm) glass micropipettes positioned 50–100 μm from the individual neuron. For multiple tests of responses in the same cell, the cells were allowed to wash (via the circulating Krebs solution) for several minutes between applications, until the responses returned to control levels. ANG II was applied by inclusion in the circulating bath solution (100 nM, Sigma) and was applied for a total of 3–5 min. Losartan (1 μM) alone, or in combination with ANG II, was also applied by inclusion in the bath solution for some studies.
Electrophysiological methods.
Intracellular voltage recordings from intracardiac neurons were obtained with an AxoClamp 2B amplifier (Axon Instruments) from cells impaled with 2 M KCl-filled microelectrodes (40–80 MΩ), as previously described (13). Input resistance was determined with small 0.1- or −0.1-nA pulses (500 ms), and single action potentials were stimulated with short, depolarizing pulses (0.3–0.9 nA, 5 ms).
Neuronal soma excitability was monitored by observing the response to a series of long depolarizing current pulses (0.1–0.6 nA, 500 ms). The number of evoked action potentials (APs) vs. stimulus intensity was determined to assess relative changes in excitability.
For each cell, following characterization of the basic electrophysiological properties, induced changes in evoked AP frequency were assessed immediately following a 1–2-s application of NE. Changes in frequency/intensity curves (both the amplitude of the responses and the slope of the curves) were determined to assess relative drug-induced changes in excitability.
To assess synaptic efficacy, an extracellular electrode (concentric bipolar electrode) was placed on nerve fiber bundles leading to the ganglion containing the neuron of interest. Stimuli (1.5 ms, 0.1–10 V) were first given at a frequency of 0.5 Hz to elicit an orthodromic action potential from the intracardiac neuron. Orthodromic responses were determined either by the ability to generate a subthreshold response or by the presence of sufficient time delay between the stimulus artifact and the neuronal response, since it was not possible to obtain subthreshold excitatory postsynaptic potentials in all cells. Suprathreshold stimuli were then given in 2-s trains at frequencies of 10, 20, and 30 Hz, and the number of action potentials produced by the neuron of interest was determined.
Western blot analysis.
Tissue samples of the cardiac plexus were frozen in liquid nitrogen after the completion of the electrophysiological studies. Tissues were homogenized in lysis buffer containing 1% SDS and Mini Complete Protease inhibitor (Roche). Samples (40 μg of protein) were loaded onto 4–12% gels for electrophoresis, and the proteins were then transferred to polyvinylidene difluoride membranes. Blots were stained with antibodies to AT1 receptors (1:200; Santa Cruz), AT2 receptors (1:1,000; Alomone Labs), and glyceraldeyde-3-phosphate dehydrogenase (1:10,000; GAPDH, Millipore). Antibody staining was visualized with HRP-linked species-specific secondary antibodies (1:10,000; GE Healthcare) and chemiluminescent substrate [Luminata Forte (Millipore) or SuperSignal (Thermo Scientific)]. Blots were stripped with Restore (Thermo Scientific) for 15 min between antibody probes. Results were analyzed with ImageJ. The density of AT1 and AT2 receptor bands was normalized to GAPDH levels, and all samples were normalized to values from control tissues within the same blot.
Statistical analysis.
Values are expressed as the means ± SE. All statistical analyses included an initial Shapiro-Wilk test for normal distribution. Normally distributed data were then analyzed by ANOVA or ANOVA with repeated measures (if the data included multiple tests on a given cell). If the data were not normally distributed, Friedman's repeated-measures ANOVA on ranks was used for measurement of different treatments in a single cell, followed by post hoc comparisons with Holm-Sidak or Dunn's post hoc tests. For comparisons of treatments between different animals, Kruskal-Wallis ANOVA on ranks was used with appropriate post hoc nonparametric analysis. In all cases, a P value <0.05 was considered significant.
RESULTS
Effects of ANG II modulators in healthy animals.
Since alterations in ANG II synthesis or receptor stimulation could alter neuronal responses in healthy animals, guinea pigs were implanted with Alzet osmotic pumps containing either captopril (ACE inhibitor; n = 6 animals), losartan (AT1 receptor antagonist; n = 6), or CGP 42112A (AT2 receptor agonist; n = 6) and were maintained for 3 or 4 wk prior to experimentation. Previous studies (14) showed that implantation of empty pumps, in either control or sham MI surgery, had no effect on neuronal responses. Intracellular voltage recordings were obtained from individual neurons in whole-mount preparations of the cardiac plexus. A total of ∼100 neurons were sampled from control preparations (n = 20 animals) and ∼30 neurons each from losartan-, captopril-, and CGP 42112A-treated preparations. Resting membrane potentials, input resistance, and single action potentials were monitored, and no significant differences were found among the different treatments compared with untreated animals (data not shown). The average resting membrane potentials among the different treatments was −47.8 mV ± 1.7 (means range from −45.4 mV to −50.6 mV for the different groups). Neuronal excitability was assessed by examining the frequency of action potentials produced by increasing stimulus intensities both prior to and then following a brief application of NE or bethanechol. Chronic drug treatments did not alter neuronal excitability in the absence of NE application (baseline, Fig. 1A). Following NE or bethanechol application, there was an increase in both the number of action potentials produced with increasing stimulus intensities and an increase in the slope of the frequency/intensity (F/I) curves for all treatments. There were no differences in the responses between the different treatments and control tissues (Fig. 1, B and C).
Fig. 1.
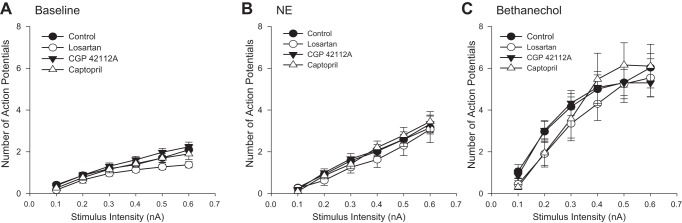
Neuronal excitability is unchanged by ANG II modulators in control animals. Frequency-intensity curves of evoked action potentials were determined with and without either norepinephrine (NE) or bethanechol application in control animals, and animals were treated with captropril, losartan, or the AT2 receptor agonist, CGP42112A for 3 wk. Points represent the means ± SE for each respective stimulus intensity (n ≥ 29 cells for each point). Chronic treatment with the ANG II modulators does not alter soma excitability as assessed from control neurons (baseline, A). NE and bethanechol each increased the number of action potentials, as well as the slope of the frequency-intensity (F/I) curve. There was no difference in evoked responses between control animals and animals chronically treated with ANG II receptor modulators following NE or bethanechol application (B and C).
Effects of ANG II modulators following MI.
Myocardial infarction was induced in guinea pigs by ligation of the descending coronary artery. Previous research found that this intervention produces an infarct that corresponds to ∼8% of the left ventricular tissue (13). The neurons of the cardiac plexus used in this study are located primarily in the left atrium, in an area not directly impacted by the infarction. A total of ∼50 neurons were sampled from MI preparations (n = 10 animals), and ∼30 neurons, each, were sampled from MI preparations treated with captopril (n = 6 animals), losartan (n = 6 animals), or CGP42112A (n = 6 animals). Neurons from animals treated with captopril, losartan, or CGP 42112A following MI showed no changes in passive membrane properties or single action potentials (data not shown). Additionally, there was no change in the baseline excitability in neurons from control, MI, and MI with drug treatments. Previous studies demonstrated that chronic MI induces an increased sensitivity to NE in the F/I curves (14). This result was confirmed in the present study (Fig. 2A). In animals treated with captopril, following MI, the increase in neuronal excitability with NE application was still seen (Fig. 2B). In MI animals treated with AT1 inhibitor losartan for 6 wk following MI, NE still increased neuronal excitability over baseline at the higher stimulus intensities (Fig. 2C). Conversely, in MI animals treated with the AT2 agonist CGP 42112A for 6 wk NE no longer produced a significant increase in neuronal firing over baseline controls (Fig. 2D). These effects can be seen in both the slope of the frequency/intensity curve following NE application and the responses at higher stimulus levels (Fig. 2).
Fig. 2.

Neuronal excitability to NE following myocardial infarction (MI) is altered by ANG II modulators. F-I curves were determined in animals 7 wk after surgical MI with and without NE application in animals with MI alone (n = 56 cells), or combined with 6 wk of treatment with captopril (n = 32 cells), losartan (n = 28 cells), or CGP42112A (n = 42 cells). NE increases neuronal firing over baseline in both control and MI preparations (A; *P < 0.05 vs. control at that stimulus intensity). In addition, in MI animals, the effect of NE was significantly greater than NE in control animals at the higher stimulus intensities (A; #P < 0.05 vs. control NE at that stimulus intensity). The same MI with NE data are shown in B–D for comparison. There were no changes in baseline excitability (in the absence of NE) with the different drug treatment (control with drug vs. MI with drug, B–D). Captopril-treated MI animals showed the same increase in excitability with NE as untreated MI with NE (B; *P < 0.05 vs. captopril control at that stimulus intensity). With losartan treatment, NE still increased excitability over baseline in MI animals treated with losartan (C; *P < 0.05 vs. losartan control at that stimulus intensity). There was no significant difference between untreated MI with NE and losartan-treated MI with NE at any stimulus intensity. MI animals treated with CGP42112A no longer showed a significant increase in excitability with NE vs. CGP42112A controls (D; *P < 0.05 vs. CGP 42112A control at that stimulus intensity). Significant differences were determined by Kruskal-Wallis ANOVA on ranks for each stimulus intensity with nonparametric post hoc comparisons.
Contrary to the changes in adrenergic sensitivity, there were no significant changes in the sensitivity to muscarinic agonists with chronic MI with or without drug treatment (Fig. 3). Bethanechol significantly increases neuronal excitability, as shown previously (9), but this response is not altered following MI and is likewise unaffected by angiotensin receptor modulation.
Fig. 3.

Neuronal excitability to bethanechol following MI is unchanged by ANG II modulators. F-I curves were determined in animals 7 wk after surgical MI induction with and without bethanechol application in animals with MI alone (n = 37 cells), or combined with 6 wk of treatment with captopril (n = 25 cells), losartan (n = 30 cells), or CGP42112A (n = 37 cells). MI does not alter neuronal excitability with bethanechol application over control animals (A; *P < 0.05 vs. both control and MI control at that stimulus intensity). The same MI with bethanechol data is shown in B–D for comparison. None of the AT-related drug treatments produced any significant differences in bethanechol sensitivity vs. MI alone (*P < 0.05 vs. control at each stimulus intensity using Kruskal-Wallis ANOVA on ranks).
Confirming the results seen in previous studies (9), in control animals, the addition of 100 nM ANG II to the circulating bath solution potentiated the NE-induced increase in neuronal excitability (Fig. 4). Following MI, the addition of ANG II no longer produced a significant increase in the maximal NE response (Fig. 4A1). None of the treatments tested in this study restored the ANG II-induced potentiation of the NE response to levels seen in control tissues. In healthy animals treated with captopril, losartan, or CGP 42112A, the addition of ANG II to the bath circulation did not induce a significant increase in the NE-induced neuronal excitability, as it did in untreated animals (Fig. 4A2–4). Similarly, following MI, there was no significant increase in the NE response in the presence of ANG II in MI animals treated with captopril, losartan, or CGP 42112A (Fig. 4A2–4).
Fig. 4.

Loss of ANG II potentiation following MI and ANG II treatments. In untreated animals, the maximal excitability response (at 0.6 nA) to either NE application (A1) or bethanechol application (B1) is increased over baseline levels (*P < 0.05 vs. baseline), an effect that was enhanced with the inclusion of 100 nM ANG II in the bath solution [Control: #P < 0.05 vs. NE (n = 19 cells) or bethanechol alone (n = 15 cells)]. Following MI, the response to NE was significantly increased over untreated animals (A1, **P < 0.05 vs. Control + NE); however, the addition of ANG II no longer produced a significant increase (n = 24 cells). In the untreated group, ANG II was still able to increase the response to bethanechol post-MI (n = 16 cells, B1, #P < 0.05 vs. MI bethanechol alone). However, in animals treated with CGP 42112A (n = 19 cells), captopril (n = 12 cells), or losartan (n = 12 cells, A2, A3, A4), the addition of ANG II no longer increased the NE response in either healthy animals or MI animals. The ability of ANG II to enhance the bethanechol response was retained in the control animals treated with losartan (n = 15 cells) or CGP 421121A (n = 18 cells, B2, B3), but not with captopril (n = 13 cells, B4). Following MI, none of the drug-treated animals showed an increase in excitability with ANG II over control bethanechol levels. Significance was determined by Kruskal-Wallis ANOVA with nonparametric post hoc comparisons.
The effects of muscarinic receptor activation were also altered by some of the drug treatments. In control animals, bethanechol increases neuronal excitability, and the addition of ANG II to the bath further increases excitability (Fig. 4B1), as was seen previously (9). These responses were unchanged with MI. Treatment of control animals with losartan or CGP 42112A did not alter the bethanechol responses (Fig. 4B3–4). However, the increase in excitability with ANG II was no longer seen in MI animals treated with these drugs. Treatment of either control or MI animals with captopril eliminated the ANG II potentiation under both conditions (Fig. 4B2).
Modulation of synaptic efficacy.
Stimulation of fibers leading to intracardiac neurons with a focal extracellular electrode was used to monitor synaptic efficacy of the IC network. In both control and MI tissues, intracardiac neurons are able to fire action potentials in response to nerve fiber stimulation at frequencies up to ∼6–12 Hz (Fig. 5). At higher frequencies, the postsynaptic neuron was not able to follow the input drive frequency. In animals treated with losartan following MI, there was a significant increase in the output frequency of intracardiac neurons at higher stimulus frequencies (Fig. 5). A sample recording at the 20-Hz stimulus from an MI preparation and a losartan-treated MI preparation is shown in Fig. 6. This increase in output frequency was not seen in healthy animals treated with losartan, nor in any of the animals treated with captopril or CGP 42112A (either control or MI).
Fig. 5.

Synaptic efficacy following MI and ANG II modulators. Synaptic efficacy was determined in neurons from control (n = 28 cells) and MI (n = 39 cells) preparations both with and without chronic treatment with captopril (n = 24 cells without MI, n = 18 cells with MI), losartan (n = 15 cells without MI, n = 18 cells with MI), or CGP 42112A (n = 11 cells without MI, n = 15 cells with MI). Nerve fibers leading to the neurons were stimulated with a 2-s train at increasing frequencies. In healthy animals, the output from neurons was similar with and without drug treatment, producing a maximal output frequency of ∼6–12 Hz. In MI animals, this output frequency was similar, except in MI animals treated with losartan. In losartan-treated MI animals, the neurons showed a significant increase in output frequency as the stimulus frequency increased (*P < 0.01, **P < 0.001 by ANOVA).
Fig. 6.

Responses to 20-Hz stimulation in MI vs. MI with losartan treatment. Voltage recordings from intrinsic cardiac neurons with a nerve fiber stimulation of 20 Hz for 2 s in a preparation from an animal with chronic MI vs. a preparation from an MI animal with chronic losartan treatment. Resting membrane potentials were −60 mV for MI and −55 mV for MI + losartan.
Recent studies in rats demonstrated evidence for presynaptic AT1 receptors that can reduce parasympathetic responses to vagal stimulation by inhibiting ACh release (28). To test for the presence of this mechanism in guinea pigs, we determined the output frequency of intracardiac neurons from control animals with and without 100 nM ANG II in the bath solution. The addition of ANG II to the bath solution reduced the number of action potentials with 20-Hz stimulation for 2 s (P < 0.05 by Friedman's ANOVA with repeated measures; Fig. 7, n = 12). When 1 μM of losartan was added to the bath solution, the ability of ANG II to reduce synaptic efficacy was inhibited (Fig. 7, n = 4).
Fig. 7.
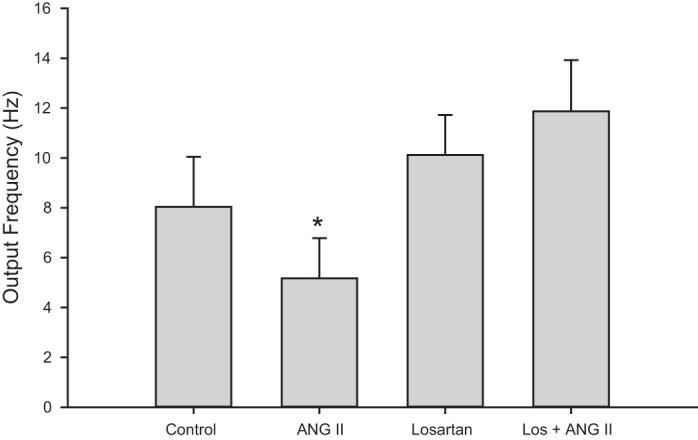
ANG II modulates output frequency by AT1R stimulation. Synaptic efficacy was determined at 20-Hz stimulation for 2 s in control animals with and without 100 nM ANG II in the bath solution. ANG II significantly reduced the output frequency (*P < 0.05, Friedmans repeated-measures ANOVA on ranks). Losartan (1 μM) in the bath solution inhibited the effects of ANG II on output frequency (n = 4). Bars represent the means ± SE.
Receptor protein expression.
Western blot analysis of the expression of the AT1 and AT2 receptors in the cardiac plexus showed a significant decrease in the expression of both receptor subtypes following MI (Fig. 8), with the reduction in AT1 receptors greater than the reduction in AT2 receptors. In control animals treated with losartan, the expression of AT1 receptors was significantly increased over untreated controls (Fig. 8). Neither captopril nor CGP 42112A treatments in control animals significantly altered protein expression levels. In MI animals, treatment with losartan, captopril, or CGP 42112A resulted in the return to control levels of expression for both AT1 and AT2 receptors, although the high variability in the density of the protein bands in both control and MI with captopril make it difficult to draw definitive conclusions for this drug treatment. Analysis of the ratio of AT2R/AT1R expression shows a significant decrease in the expression of AT1 relative to AT2 following MI (Fig. 9). In control tissues, there is a relative 2:1 ratio of AT2 receptors to AT1 receptors. With MI, this ratio increases to almost 5:1. Conversely, these ratios returned to control levels with all three drug treatments in both control and MI animals (Fig. 9).
Fig. 8.

Expression of AT1 and AT2 receptors. The relative protein expression levels of AT1R and AT2R were determined by Western blot analysis from homogenates of cardiac plexus samples. The images are from the same blots labeled with antibodies to AT1R, AT2R, and GAPDH. Not all lanes from the blots are shown, and the white lines indicate regions of the blot that were rearranged for clarity. The graphs show the quantification of relative density from several blots. Each sample was run on at least two different blots for technical replicates. The means of technical replicates were then averaged for each treatment (Control: n = 5, MI: n = 6, all other conditions: n = 3). The sizes of the bands observed were consistent with predicted sizes (AT1R ∼43 kDa, AT2R ∼48 kDa, GAPDH ∼38 kDa) with no other major bands present on the blots. Individual band densities were quantified with ImageJ and normalized to GAPDH levels for that sample. Within each blot, values were normalized to control levels of each receptor. The levels of both AT1R and AT2R were significantly reduced with MI (*P < 0.05, **P < 0.01 vs. Control by ANOVA). Losartan treatment in control animals results in a significant increase in AT1R expression over controls (#P < 0.05 vs. Control by ANOVA).
Fig. 9.
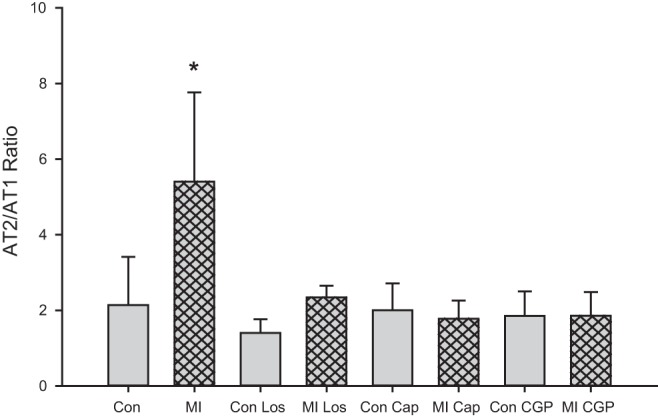
Changes in AT2R/AT1R ratios with MI. The ratio of AT2R/AT1R was determined (using values normalized to GAPDH) for each treatment. MI resulted in a significant increase in the relative abundance of AT2R vs. AT2R (*P < 0.01 by ANOVA). All drug treatments, either in control or MI animals, resulted in an AT2R/AT1R ratio equivalent to control ratios.
DISCUSSION
Myocardial infarction leads to increased sympathetic activity and increased stimulation of the RAS, leading to elevated NE release and increased levels of ANG II (18, 25, 27, 30). The current study examined the effects of either inhibiting ANG II-generating enzymes, or altering stimulation of specific ANG II receptors during the remodeling phase following chronic MI in guinea pigs (12). Specifically, we looked for alterations in the remodeling of neurons in the intrinsic cardiac plexus, since our previous studies have shown that chronic MI leads to an increase in neuronal responses to NE and a decrease in responses to ANG II (14). Within the guinea pig intrinsic cardiac plexus, the majority of neurons are cholinergic (19) and are either postganglionic parasympathetic neurons or local circuit neurons (3). These neurons are primarily excited by stimulation of nicotinic receptors and also can be modulated by muscarinic receptor activation (1). The presence of sympathetic postganglionic innervation of these neurons likely represents a local reflex circuit to provide rapid feedback of altered sympathetic activity (4). NE acts as a neuromodulator on these cells, altering neuronal excitability to other stimuli, rather than producing direct action potential activity (14). Similarly, ANG II also seems to act as a neuromodulator to alter the neuronal sensitivity to other neurotransmitters (15). As a result, changes in neuronal activation of either AT1R or AT2R could lead to altered signal processing within the cardiac plexus. Since chronic heart disease induces upregulation in both NE release and ANG II production, this could result in significant alterations in signal regulation and output within the intrinsic cardiac nervous system.
Overall, none of the drug treatments alone, the surgically induced heart disease, or the combination of the two, altered the passive membrane properties or basal firing levels of the intracardiac neurons. This suggests that any changes observed were due to changes in sensitivity to other chemical signals and/or changes in signal transduction mechanisms.
Inhibition of ACE by captopril did not alter the remodeling of intrinsic cardiac neurons following MI. The increased sensitivity to NE was still observed in the neurons, and there was no ANG II-induced potentiation of adrenergic or muscarinic responses in the MI animals treated with captopril for 6 wk. Although captopril inhibits ACE1, it does not affect the activity of other proteases, such as chymase, which have been shown to increase in cardiac tissues with MI (6, 22). Thus, although the normal pathways for ANG II production are inhibited by captopril, the alternative pathways for generating ANG II are unaffected by drug treatment, and may be of greater importance in elevating local ANG II levels within the cardiac interstitium (6, 22). This suggests that inhibition of ACE1 activity by captopril is insufficient to alter the effects of disease-induced increases in ANG II levels within the guinea pig cardiac plexus.
Several studies have highlighted the importance of the balance between stimulation of AT1 and AT2 receptors in response to heart disease (6, 23, 26, 30). Studies of AT1 receptor knockout or inhibition in combination with studies of AT2 receptor knockout or upregulation all point toward better outcomes with chronic heart disease when there is an increase in AT2 vs. AT1 stimulation (5, 10, 17, 29). For the current study, we compared the effects on remodeling with either inhibition of AT1 receptors with losartan, or stimulation of AT2 receptors with the agonist CGP 42112A. Inhibition of AT1 receptors with losartan did not significantly alter neuronal sensitivity to NE or restore the sensitivity to ANG II. However, losartan treatment following MI enhanced synaptic efficacy within the IC neural network. This enhanced synaptic function is not seen in control animals treated with losartan, or in untreated MI animals. Previous studies in a pressure overload model of chronic heart disease in guinea pigs (14) did show an increase in synaptic function. The pressure overload model also showed evidence of pulmonary edema and may indicate that chronic changes in synaptic function correlate with advanced cardiac disease. In agreement with a recent study in rats (28), we found evidence for presynaptic AT1R inhibition of IC synaptic function in the normal guinea pig. In control animals, the addition of ANG II to the bath solution resulted in a significant decrease in the number of action potentials produced with nerve fiber stimulation. The addition of losartan to the bath solution prevented this decrease. Our analysis of AT1 expression in the cardiac plexus showed a significant decrease in this receptor following MI. The elevation in cardiac ANG II levels with MI may induce a decrease in AT1 expression to prevent excessive inhibition of ganglionic function with the chronic elevation of ANG II. In addition, with the increased sympathetic efferent activity following MI, other changes within the ganglion may develop to retain normal synaptic output. With losartan treatment, downregulation of the AT1R is inhibited along with activation of these receptors by circulating ANG II. Thus, the balance of inputs to the ganglion may be altered without a compensatory change in presynaptic AT1 receptors. This could result in an overall imbalance of inputs resulting in the observed increase in synaptic efficacy with MI and losartan treatment. In contrast, stimulation with AT2 agonists or inhibition of ACE would not alter AT1R stimulation, so the synaptic efficacy would be unchanged in these animals. Further experiments are needed to identify the specific changes in neural inputs within the ganglion induced with chronic heart disease.
Chronic stimulation of AT2 receptors following MI prevented the increase in neuronal sensitivity to NE. With CGP 42112A treatment in MI animals, the soma excitability response to NE remained at control levels. This suggests that the increase in NE sensitivity is prevented by increased AT2 receptor activation. In MI animals treated with losartan, NE-induced excitability was still elevated over control levels, but it was consistently lower than that seen in untreated MI preparations. Other research has suggested that inhibition of AT1 receptors leads to an increase in the levels of ANG II, which then leads to an increase in AT2 receptor stimulation (30). In this case, the indirect increase in AT2 receptor stimulation may not have been sufficient to completely inhibit the change in NE sensitivity, but did show a trend in that direction. Again, inhibition of ACE would not alter the relative balance of AT1 to AT2 receptor stimulation and, thus, did not alter NE sensitivity. It is unclear at this point how increased AT2 activation with CGP 42112A leads to an inhibition of the increase in NE sensitivity. However, it is known that chronic treatment with AT2 agonists leads to increases in AT2R expression (16). Our analysis of AT2R expression demonstrated that CGP 42112A treatment restored receptor expression back to control levels in MI animals. Thus, if stimulation of AT2 receptors can reduce the NE effects, treatments with AT2 agonist could amplify this response. Future studies will need to determine the specific mechanisms that underlie the enhanced NE responses to understand the mechanisms that control neuronal remodeling.
Perspectives and Significance
This study supports the growing evidence for the role of angiotensin receptors in the autonomic remodeling process that follows myocardial infarction. Current therapies for chronic heart disease often include ACE inhibitors and AT1 antagonists (8). These pharmacotherapies have been shown to reduce some of the negative symptomology associated with these disorders. This study expands our understanding of the effects of these clinical treatments on other tissues that may impact the overall cardiac function, specifically the intrinsic cardiac nervous system. In the case of the guinea pig intrinsic plexus, ACE inhibitors appeared to have little effect on neuronal function, while direct inhibition (losartan) or activation (CGP 42112A) of ANG II receptors produced significant changes in intracardiac neuronal function. As has been shown in other systems, it is the relative balance between AT1 and AT2 receptor activation that determines the overall outcome with chronic heart disease. In the case of remodeling within the guinea pig cardiac plexus, the current study demonstrates that regulation of presynaptic AT1 receptors can modulate synaptic efficacy in the cardiac nervous system. In addition, increased stimulation of AT2 receptors can prevent the upregulation in adrenergic sensitivity of the intrinsic cardiac neurons produced with myocardial infarction. Changes in synaptic processing may be important in determining reflex stimulation of parasympathetic output. Taken together, these results demonstrate the importance of balancing the stimulation of AT2 vs. AT1 receptors as we develop more effective treatments for chronic heart disease.
GRANTS
This work was supported by National Institutes of Health Grants R01 HL-098589 to J. C. Hardwick, and E. M. Southerland and HL-71830 to J. L. Ardell.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: J.C.H., E.M.S., and J.L.A. conception and design of research; J.C.H., S.E.R., E.N.P., and E.M.S. performed experiments; J.C.H., S.E.R., and E.N.P. analyzed data; J.C.H. and J.L.A. interpreted results of experiments; J.C.H. prepared figures; J.C.H. drafted manuscript; J.C.H. and J.L.A. edited and revised manuscript; J.C.H., S.E.R., E.N.P., E.M.S., and J.L.A. approved final version of manuscript.
REFERENCES
- 1.Allen TG, Burnstock G. M1 and M2 muscarinic receptors mediate excitation and inhibition of guinea-pig intracardiac neurones in culture. J Physiol 422: 463–480, 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Armour JA. Cardiac neuronal hierarchy in health and disease. Am J Physiol Regul Integr Comp Physiol 287: R262–R271, 2004. [DOI] [PubMed] [Google Scholar]
- 3.Armour JA. Potential clinical relevance of the ‘little brain’ on the mammalian heart. Exp Physiol 93: 165–176, 2008. [DOI] [PubMed] [Google Scholar]
- 4.Beaumont E, Salavatian S, Southerland EM, Vinet A, Jacquemet V, Armour JA, Ardell JL. Network interactions within the canine intrinsic cardiac nervous system: implications for reflex control of regional cardiac function. J Physiol 591: 4515–4533, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bove CM, Gilson WD, Scott CD, Epstein FH, Yang Z, Dimaria JM, Berr SS, French BA, Bishop SP, Kramer CM. The angiotensin II type 2 receptor and improved adjacent region function post-MI. J Cardiovasc Magn Reson 7: 459–464, 2005. [DOI] [PubMed] [Google Scholar]
- 6.Dell'Italia LJ. Translational success stories: angiotensin receptor 1 antagonists in heart failure. Circ Res 109: 437–452, 2011. [DOI] [PubMed] [Google Scholar]
- 7.Farrell DM, Wei CC, Tallaj J, Ardell JL, Armour JA, Hageman GR, Bradley WE, Dell'Italia LJ. Angiotensin II modulates catecholamine release into interstitial fluid of canine myocardium in vivo. Am J Physiol Heart Circ Physiol 281: H813–H822, 2001. [DOI] [PubMed] [Google Scholar]
- 8.Florea VG, Cohn JN. The autonomic nervous system and heart failure. Circ Res 114: 1815–1826, 2014. [DOI] [PubMed] [Google Scholar]
- 9.Girasole AE, Palmer CP, Corrado SL, Marie SE, Ardell JL, Hardwick JC. Angiotensin II potentiates adrenergic and muscarinic modulation of guinea pig intracardiac neurons. Am J Physiol Regul Integr Comp Physiol 301: R1391–R1399, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gonzalez GE, Seropian IM, Krieger ML, Palleiro J, Lopez Verrilli MA, Gironacci MM, Cavallero S, Wilensky L, Tomasi VH, Gelpi RJ, Morales C. Effect of early versus late AT1 receptor blockade with losartan on postmyocardial infarction ventricular remodeling in rabbits. Am J Physiol Heart Circ Physiol 297: H375–H386, 2009. [DOI] [PubMed] [Google Scholar]
- 11.Hardwick JC, Mawe GM, Parsons RL. Evidence for afferent fiber innervation of parasympathetic neurons of the guinea-pig cardiac ganglion. J Auton Nerv Syst 53: 166–174, 1995. [DOI] [PubMed] [Google Scholar]
- 12.Hardwick JC, Ryan SE, Beaumont E, Ardell JL, Southerland EM. Dynamic remodeling of the guinea pig intrinsic cardiac plexus induced by chronic myocardial infarction. Auton Neurosci 181: 4–12, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hardwick JC, Southerland EM, Ardell JL. Chronic myocardial infarction induces phenotypic and functional remodeling in the guinea pig cardiac plexus. Am J Physiol Regul Integr Comp Physiol 295: R1926–R1933, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hardwick JC, Southerland EM, Girasole AE, Ryan SE, Negrotto S, Ardell JL. Remodeling of intrinsic cardiac neurons: effects of β-adrenergic receptor blockade in guinea pig models of chronic heart disease. Am J Physiol Regul Integr Comp Physiol 303: R950–R958, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Horackova M, Armour JA. ANG II modifies cardiomyocyte function via extracardiac and intracardiac neurons: in situ and in vitro studies. Am J Physiol Regul Integr Comp Physiol 272: R766–R775, 1997. [DOI] [PubMed] [Google Scholar]
- 16.Jehle AB, Xu Y, Dimaria JM, French BA, Epstein FH, Berr SS, Roy RJ, Kemp BA, Carey RM, Kramer CM. A nonpeptide angiotensin II type 2 receptor agonist does not attenuate postmyocardial infarction left ventricular remodeling in mice. J Cardiovasc Pharmacol 59: 363–368, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaschina E, Grzesiak A, Li J, Foryst-Ludwig A, Timm M, Rompe F, Sommerfeld M, Kemnitz UR, Curato C, Namsolleck P, Tschope C, Hallberg A, Alterman M, Hucko T, Paetsch I, Dietrich T, Schnackenburg B, Graf K, Dahlof B, Kintscher U, Unger T, Steckelings UM. Angiotensin II type 2 receptor stimulation: a novel option of therapeutic interference with the renin-angiotensin system in myocardial infarction? Circulation 118: 2523–2532, 2008. [DOI] [PubMed] [Google Scholar]
- 18.Malpas SC. Sympathetic nervous system overactivity and its role in the development of cardiovascular disease. Physiol Rev 90: 513–557, 2010. [DOI] [PubMed] [Google Scholar]
- 19.Mawe GM, Talmage EK, Lee KP, Parsons RL. Expression of choline acetyltransferase immunoreactivity in guinea pig cardiac ganglia. Cell Tissue Res 285: 281–286, 1996. [DOI] [PubMed] [Google Scholar]
- 20.Mill JG, Stefanon I, dos SL, Baldo MP. Remodeling in the ischemic heart: the stepwise progression for heart failure. Braz J Med Biol Res 44: 890–898, 2011. [DOI] [PubMed] [Google Scholar]
- 21.Miyazaki M, Takai S, Jin D, Muramatsu M. Pathological roles of angiotensin II produced by mast cell chymase and the effects of chymase inhibition in animal models. Pharmacol Ther 112: 668–676, 2006. [DOI] [PubMed] [Google Scholar]
- 22.Miyazaki M, Takai S, Jin D, Muramatsu M. Pathological roles of angiotensin II produced by mast cell chymase and the effects of chymase inhibition in animal models. Pharmacol Ther 112: 668–676, 2006. [DOI] [PubMed] [Google Scholar]
- 23.Parlakpinar H, Ozer MK, Acet A. Effects of captopril and angiotensin II receptor blockers (AT1, AT2) on myocardial ischemia-reperfusion induced infarct size. Cytokine 56: 688–694, 2011. [DOI] [PubMed] [Google Scholar]
- 24.Silver RB, Reid AC, Mackins CJ, Askwith T, Schaefer U, Herzlinger D, Levi R. Mast cells: a unique source of renin. Proc Natl Acad Sci USA 101: 13, 607–13, 612, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Triposkiadis F, Karayannis G, Giamouzis G, Skoularigis J, Louridas G, Butler J. The sympathetic nervous system in heart failure physiology, pathophysiology, and clinical implications. J Am Coll Cardiol 54: 1747–1762, 2009. [DOI] [PubMed] [Google Scholar]
- 26.Voros S, Yang Z, Bove CM, Gilson WD, Epstein FH, French BA, Berr SS, Bishop SP, Conaway MR, Matsubara H, Carey RM, Kramer CM. Interaction between AT1 and AT2 receptors during postinfarction left ventricular remodeling. Am J Physiol Heart Circ Physiol 290: H1004–H1010, 2006. [DOI] [PubMed] [Google Scholar]
- 27.Wang WZ, Gao L, Wang HJ, Zucker IH, Wang W. Interaction between cardiac sympathetic afferent reflex and chemoreflex is mediated by the NTS AT1 receptors in heart failure. Am J Physiol Heart Circ Physiol 295: H1216–H1226, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yamaki F, Arai T, Aoyama M, Watanabe A, Takata Y. Angiotensin AT1-receptor blockers enhance cardiac responses to parasympathetic nerve stimulation via presynaptic AT1 receptors in pithed rats. J Pharmacol Sci 122: 28–33, 2013. [DOI] [PubMed] [Google Scholar]
- 29.Yang Z, Bove CM, French BA, Epstein FH, Berr SS, Dimaria JM, Gibson JJ, Carey RM, Kramer CM. Angiotensin II type 2 receptor overexpression preserves left ventricular function after myocardial infarction. Circulation 106: 106–111, 2002. [DOI] [PubMed] [Google Scholar]
- 30.Zhu YC, Zhu YZ, Lu N, Wang MJ, Wang YX, Yao T. Role of angiotensin AT1 and AT2 receptors in cardiac hypertrophy and cardiac remodelling. Clin Exp Pharmacol Physiol 30: 911–918, 2003. [DOI] [PubMed] [Google Scholar]
- 31.Zucker IH, Patel KP, Schultz HD. Neurohumoral stimulation. Heart Fail Clin 8: 87–99, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zucker IH, Schultz HD, Patel KP, Wang W, Gao L. Regulation of central angiotensin type 1 receptors and sympathetic outflow in heart failure. Am J Physiol Heart Circ Physiol 297: H1557–H1566, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]