

Abstract
Chronic failure in maintenance and regeneration of skeletal muscles leads to lower muscle mass (sarcopenia), muscle weakness, and poor response to injury. Evidence suggests that aberrant p38 MAPK signaling undermines the repair process after injury in aged mice. Previous studies have shown that hyperhomocysteinemia (HHcy) has been associated with muscle weakness and lower than normal body weights. However, whether or not HHcy condition also compromises skeletal muscle regenerative capabilities is not clear. In the current study, we show that CBS−/+ mice, a model for HHcy condition, exhibited compromised regenerative function and cell proliferation upon injury. However, there was no significant difference in Pax7 expression levels in the satellite cells from CBS−/+ mouse skeletal muscles. Interestingly, the satellite cells from CBS−/+ mice not only exhibited diminished in vitro proliferative capabilities, but also there was heightened oxidative stress. In addition, there was enhanced p38 MAPK activation as well as p16 and p21 expression in the CBS−/+ mouse satellite cells. Moreover, the C2C12 myoblasts also exhibited higher p38 MAPK activation and p16 expression upon treatment with homocysteine in addition to enhanced ROS presence. Tissue engraftment potential and regeneration after injury were restored to some extent upon treatment with the p38-MAPK inhibitor, SB203580, in the CBS−/+ mice. These results together suggest that HHcy-induced diminished satellite cell proliferation involves excessive oxidative stress and p38 MAPK signaling. Our study further proposes that HHcy is a potential risk factor for elderly frailty, and need to be considered as a therapeutic target while designing the alleviation interventions/postinjury rehabilitation measures for adults with HHcy.
Keywords: hyperhomocysteinemia, skeletal muscle, p38 MAPK, p16, p21, reactive oxygen species, CBS
skeletal muscles exhibit amazing regenerative potential that sustains muscle health during exercise and injury. Regenerative capacity of skeletal muscles has been reported to be mainly dependent on the function and/or number of resident muscle precursor/stem cells, also known as satellite cells. However, the regenerative potential of skeletal muscles diminishes with age (1, 4). Chronic failure in maintenance and regeneration of skeletal muscles leads to lower muscle mass, muscle weakness, and poor response to injury. Several studies have been conducted to understand the mechanistic basis of age-dependent decline in physical function and physiological maintenance of satellite cell pool in aged animals. These studies have highlighted various molecules/factors that cause decline in satellite cell number and/or proliferative potential. Earlier studies have identified Pax7, a crucial transcription factor that signifies satellite cell stemness, a requirement in self-renewal and in prevention of precocious differentiation of satellite cells (1, 4, 27). Further, these studies have identified Pax7 as a viable marker for satellite cell identity. It was also demonstrated that asymmetric localization of par complex is crucial in maintenance and perpetuation of satellite cells by allowing asymmetric activation of p38 alpha/beta MAPK signaling and subsequent myogenesis only in the committed daughter cells that eventually form myofibers (11, 22). Recent studies elegantly demonstrated that despite having a normal satellite cell population, aged mouse satellite cells exhibited enhanced cell cycle inhibitor presence, notably the p16 (1, 4, 21). Further, these studies also noted that excessive MAPK activation (p38 alpha/beta MAPK) is involved in inhibition of satellite cell self-renewal upon injury in aged mice (1, 4). Age-dependent enhancement of ambient FGF-2 production and attenuation of cell intrinsic FGFR1 signaling, specifically in satellite cells, were associated with loss of Pax7-expressing cells and resulted in p38 alpha/beta MAPK activation (1). Consequently, pharmacological inhibition of p38 alpha/beta MAPK and ectopic activation of FGFR1 were validated to enhance satellite cell function (1, 4). In addition, it was reported that certain growth factors, especially myostatin, and certain inflammatory mediators are also involved in enhancement of cell cycle inhibitors such as p21 and p16, which further mediate satellite cell quiescence and thereby inhibit satellite cell recruitment/activation upon injury and potentially impair muscle regeneration (6). Apart from cytokine changes, changes in satellite cell environment were also shown to cause impairment of satellite cell function and regeneration. Specifically, it was demonstrated that heightened oxidative stress might hinder satellite cell proliferative function (17). While several factors that cause impaired satellite cell function were identified, the metabolic factors/conditions that potentially derail satellite cell proliferation leading to muscle weakness are not characterized well.
Several of the age-induced changes in skeletal muscles such as reduced body weight and muscle weakness were also reported during hyperhomocysteinemia (HHcy), a metabolic condition that results from accumulation of the noncoding intermediary amino acid, homocysteine (Hcy), due to defective metabolism (24). Furthermore, HHcy was reported to aggravate elderly frailty (24). Earlier studies reported that children born with severe HHcy condition, which often results in homocystinuria, exhibited lower than normal body weights (12). Moreover, pups (2 wk postpartum) carrying homozygous deletion for cystathionine beta synthase (CBS) gene exhibited significant reduction in body weights (29). However, the pups with heterozygous deletion for Cbs gene (CBS−/+ pups), which exhibit mild to moderate HHcy, did not show considerable reduction in overall body weights, suggesting the chronic nature of moderate HHcy effects on muscle mass (29). In adult mouse models of HHcy, the CBS−/+ mouse and CSE-deficient mouse, there was reduction in overall body weight (10, 14, 29). Several parallels suggested HHcy mediated inhibition of satellite cell proliferation: 1) HHcy was shown to enhance p38 MAPK signaling in different tissue types (19, 28); 2) HHcy was demonstrated to enhance oxidative stress in skeletal muscles (10, 25); and 3) HHcy was shown to induce myostatin, an inhibitor of myogenesis, in skeletal muscles (7). However, it was not known whether HHcy also compromises satellite cell function and impairs the repair process after injury in skeletal muscles.
In the current study, we investigated whether or not HHcy impairs skeletal muscle repair after injury and if so what are the molecular mechanisms. The current study utilized the CBS−/+ mouse, a model for HHcy, to examine the HHcy-associated molecular changes in skeletal muscle regenerative capacity. We measured specific muscle weights, in vivo proliferation after muscle injury, and various stress markers that regulate satellite cell function and also attempted to reverse these changes using pharmacological means.
MATERIALS AND METHODS
Animal care and tissue collection.
WT (C57BL/6J), CBS−/+ (B6.129P2-Cbstm1Unc/J 002853), and C57BL/6-Tg-EGFP mice were reared on regular chow and water as reported previously (23). To generate a severe HHcy condition, we used 2-mo-old mice that were fed with a high-methionine and low-folate, B6 and B12 diet (HM-LF diet) for 8 wk as reported earlier (9). All the animal studies were approved by the Institutional Animal Care and Use Committee and are in conformity with the prescribed institutional standards. Standard procedures were followed for collection of tissues after induction of general anesthesia using tribromoethanol as described before (26). We used male mice of around 5–6 mo in our study. Genotyping for CBS−/+ heterozygotes was performed as described earlier (25).
C2C12 cell culture.
The C2C12 cell culture was maintained as reported before (26). When the myoblasts were at the confluence of 80%, they were subjected to Hcy treatment for 24 h. At the end of the treatment period, cells were lysed and the cell lysates were prepared using RIPA lysis buffer supplemented with protease and phosphatase inhibitors.
Masson's trichrome staining.
Cryo sections were stained with Masson's trichrome stain (Chromaview, no. KTRA87019, Fisher Scientific, Pittsburgh, PA) by following the manufacturer's recommendations. At the end of the staining procedure, slides were dehydrated and were mounted with Permount (Fisher Scientific). Images were obtained with the QC-capture software.
Immunohistochemistry.
Tissue harvesting was done at the end of the treatment period (28 days after initial cardiotoxin injury). Tibialis anterior (TA) muscles were collected and frozen in liquid nitrogen using tissue freezing medium (Triangle Biomedical Sciences, Durham, NC). Tissues were stored at −80°C. Cryosections of 8-μm thickness were sectioned using a Leica CM1850 cryostat (Leica Microsystems). Tissue sections were probed with specific antibodies by following the previously described protocol (23). Briefly, sections were fixed with 4% paraformaldehyde for 15 min, blocked, and permeabilized with PBS containing 3% BSA and 0.3% Triton X-100 for 1 h, incubated overnight with primary antibody, incubated for 1 h with secondary antibody, and stained for 15 min with 4′,6-diamidino-2-phenylindole. Slides were washed in between steps for three times each with 5 min duration. Stained slides were mounted, and images were obtained with a laser scanning microscope (Olympus FluoView1000, B&B Microscope). Wherever necessary, antigen retrieval was performed as described previously (8).
Western blotting.
Cells were lysed using RIPA lysis buffer containing protease and phosphatase inhibitors and brief sonication. The protein concentration was estimated using the Bio-Rad Bradford reagent. Equal amounts of total proteins were resolved using SDS-PAGE and were transferred to polyvinylidene difluoride (PVDF) membranes. Membranes were probed sequentially with overnight primary antibody and 1 h of secondary antibody. The peroxidase signal was developed using luminata forte (Millipore, Billerica, MA), and signal capturing was obtained with the Bio-Rad ChemiDoc XRS+ system and Image Lab software (Bio-Rad) as described before (25).
Antibodies and reagents.
The antibody sources are anti-p16 and anti-p38 MAPK from Santa Cruz (Paso Robles, CA); anti-BrdU from Sigma (St. Louis, MO); anti-Pax7 and anti-Laminin-2α from Abcam (Cambridge, MA); anti-phospho-p44/42 MAPK (Thr202/Ty204) and anti-phospho-p38 MAPK (Thr180/Ty182) from Cell signaling (Boston, MA); and anti-GAPDH from Millipore (Billerica, MA). Horseradish peroxidase (HRP)-conjugated secondary antibodies were purchased from Santa Cruz Biotechnology (Dallas, TX). Alexa fluor dye conjugated secondary antibodies were from Life Technologies (Grand Island, NY). The reactive oxygen species (ROS) detecting dye, DCFDA, was from Life technologies (Grand Island, NY). The 5-bromo-2′-deoxyuridine (BrdU) and cardiotoxin were purchased from Sigma (St. Louis, MO), and the stock solutions were prepared as per manufacturer's recommendations.
Satellite cell isolation.
Satellite cells from freshly prepared hindlimb muscles were isolated by using Miltenyi Biotech satellite cell isolation kit (San Diego, CA) and following the manufacturer's recommended protocol. Briefly, isolated and minced muscles were incubated with the enzyme mix in DMEM on a gentleMACS dissociator for 1.5 h at 37°C. After the incubation, the suspension was cleared by using a cell strainer and centrifugation. The cell pellet was cleared from RBCs and was incubated with a mixture of antibodies (CD45, CD31, CD11b, and Sca1) conjugated with magnetic beads. The labeled nontarget cells were removed by passing the cell suspension through a column attached to a strong magnet. The purified satellite cells were used for further analysis or for cell culture.
Q-PCR.
Total RNA was isolated using the TRIzol reagent. The quantity of RNA was assayed using a nanodrop spectrophotometer. Equal amounts of total RNA were used for cDNA synthesis by using a miScript II RT kit (Quiagen, Valencia, CA). Real-time PCR was performed using SYBR green chemistry (Quiagen, Valencia, CA) and a light cycler (Roche, Indianapolis, IN). The following primer pairs (5′ to 3′) were used to quantify specific gene expression: beta-Actin, forward GGCTGTATTCCCCTCCATCG and reverse CCAGTTGGTAACAATGCCATGT; p21 forward CCTGGTGATGTCCGACCTG and reverse CCATGAGCGCATCGCAATC.
Crystal violet staining.
Equal number of satellite cells were cultured on the laminin-coated cell-culture dishes for 5 days with the DMEM medium containing 10% FBS and 1% penicillin and streptomycin solution. Medium was changed on every other day. At the end of the cell culture, the cells were gently washed with PBS and were fixed with 1% paraformaldehyde for 10 min. After appropriate washes, the plates were further incubated with 0.1% crystal violet stain for 15 min. The excess stain was removed with 3 separate washes and the plates were dried. Cell growth pattern and colony morphology images were obtained with the help of Bio-Rad ChemiDoc XRS+ system.
Flow analysis.
Purified satellite cells were prepared for flow analysis using indirect labeling method. Briefly, cells were fixed with 0.03% paraformaldehyde for 10 min and were washed three times before the permeabilization with 1% BSA and 0.5% Triton X in PBS. The cells were incubated overnight with specific antibodies and were further incubated with an appropriate combination of secondary antibodies conjugated with Alexa fluor dyes for 30 min. After thorough washes, cells were analyzed using Accuri C6 flow cytometer (BD Biosciences, San Jose, CA).
DCFDA staining and ROS level determination.
The C2C12 cells were harvested after treatment with homocysteine for 24 h using trypsin-EDTA. An equal number of cells was incubated in dark with the 0.2 μM working DCFDA solution for 30 min at 37°C. After incubation, cells were collected and washed three times with PBS and were analyzed using Accuri C6 flow cytometer (BD Biosciences, San Jose, CA) at a wavelength of 488 nm.
BrdU labeling.
We employed the protocol that was described before (18). Briefly, to measure cell proliferation rate in the cardiotoxin-injured muscles, BrdU dissolved in sterile saline was injected via intraperitoneal route for 5 days beginning 4 h before the muscle injury. After 5 days of cardiotoxin injury, the TA muscles were surgically separated and were analyzed for the extent of BrdU labeling. Injured muscles were surgically isolated at the end of the treatment and were frozen in liquid nitrogen after embedding in tissue freezing medium.
Satellite cell engraftment.
Age- and sex-matched mice were divided into two groups and were treated with either saline or SB202190 (Sigma-Aldrich, St. Louis, MO) for 3 days with a dose of 5 mg·kg−1·day−1 by administering the drug intraperitoneally. On the first day of SB202190 administration, TA muscles on both the legs were also injected with a single dose of 15 μg of cardiotoxin (Sigma-Aldrich) into each muscle at three different sites. On the third day, all mice received a single injection of purified satellite cells (∼40,000 live cells) isolated from the GFP mouse (a transgenic mouse with C57 background that expresses enhanced GFP protein under chicken beta-actin promoter; stock number 006567, The Jackson Laboratory, Bar Harbor, ME) hindlimb muscles using the Miltenyi Biotech satellite isolation kit. GFP-satellite cells from different mice were pooled, and live satellite cell count was determined by using trypan blue dye. All the TA muscle engraftments received 40,000 live satellite cells at three different locations. All the engraftment procedures were carried out after administration of general anesthesia using 3% isoflurane. After 4 wk of the engraftment, mice were killed, and the TA muscles with and without engraftments were collected from different groups for analysis.
Statistical analysis.
Student's t-test was used to enumerate the degree of significance between the observed means from different groups. A P value of <0.05 was considered significant. Images from the Western blotting were obtained and analyzed using Image lab (Bio-Rad, Hercules, CA) software. For quantification and analysis of Q-PCR data, we used light cycler software from Roche. Unless otherwise mentioned a minimum of 3 replicates was used for the studies. Values are presented as means ± SE.
RESULTS
Reduced muscle mass in CBS−/+ mice.
In the current study, we directly measured the individual muscle weights at the age of 6 mo in CBS−/+ mice compared with the age- and sex-matched WT control mice to know if the HHcy condition causes sarcopenia. As shown in Fig. 1, A and B, only the gastrocnemius (GN) muscles from CBS−/+ mice weighed significantly lower compared with the GN muscles from WT, but there is no difference in the tibialis anterior (TA) muscle weights between the two groups. In addition, when the CBS−/+ mice were fed with high-methionine and low-folate diet (HM-LF diet) for 8 wk, which creates a severe HHcy condition, both the GN and TA muscles showed considerable sarcopenia. These results together with the previous reports suggest that the HHcy condition is associated with sarcopenia.
Fig. 1.
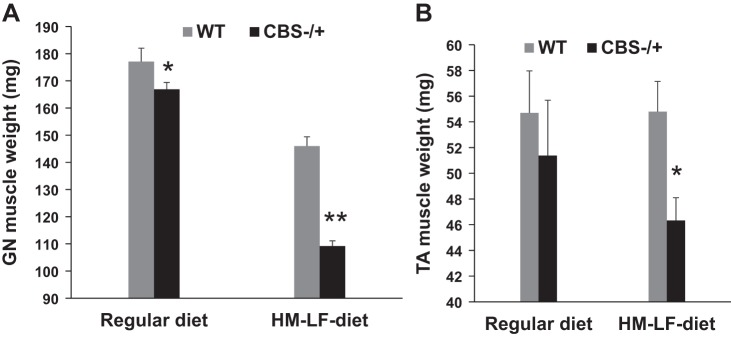
Hyperhomocysteinemia (HHcy) induces sarcopenia. A: bar graph shows the gastrocnemius (GN) muscle weights from age- and sex-matched wild-type (WT) and CBS−/+ mice given with either normal diet or with a hyperhomocysteinemic diet containing high methionine (HM) and low folate (LF) levels. Muscles were harvested surgically and immediately weighed. CBS, cystathionine beta synthase. *P < 0.05 vs. WT. **P < 0.01 vs. WT. B: bar graph displays tibialis anterior (TA) muscle weights from the above groups.
Compromised in vivo cell proliferation/activation after injury in CBS−/+ mouse muscles.
Next, to directly assess if HHcy condition compromises the repair process after injury, we used the cardiotoxin muscle injury model in combination with BrdU labeling as described before (18). After 5 days of cardiotoxin injury, the TA muscles were surgically separated and were analyzed for the extent of BrdU labeling. As depicted in Fig. 2, A and B, the relative cell proliferation as evidenced by the BrdU-labeled nuclei within the skeletal muscles sections was compromised in CBS−/+ mouse compared with that of the WT mouse. These results together with the lower muscle mass indicated that there was a certain degree of satellite cell, the primary cells that participate in muscle repair after injury, functional compromise in the HHcy mouse.
Fig. 2.

Compromised cell proliferation in response to cardiotoxin injury. A: confocal microscopy images from the skeletal muscle sections (60×) shows labeling for nuclei (DAPI), BrdU (green), laminin-2α (magenta) and merged (to show BrdU staining in the nuclei: marked with white arrows). B: bar graph depicts average number of nuclei stained with BrdU label per unit area. *P < 0.05 vs. WT.
No significant difference in Pax7 expression levels in satellite cells from CBS−/+ mouse.
To test if there is any difference in the level of Pax7 protein in the satellite cells from WT and CBS−/+ mouse muscles, we have purified satellite cells from hindlimb muscles and assessed the levels of Pax7 through flow cytometry. As measured in Fig. 3, the satellite cells from both the CBS−/+ and WT mice exhibited similar levels of Pax7 expression, suggesting that the proliferative defect and chronic loss in muscle mass observed in CBS−/+ mice are not due to changes in the Pax7 expression levels.
Fig. 3.

No difference in Pax7 expression levels in satellites from WT and CBS−/+ mouse muscles. Flow cytogram depicts Pax7-stained satellite cells.
Satellite cells from CBS−/+ mouse exhibit compromised in vitro proliferation capacity.
Next, to further confirm whether the defect in injury-induced cell proliferation with the CBS−/+ mouse muscles is due to defective satellite cell proliferation, we purified satellite cells, cultured an equal number of cells on laminin-coated cell-culture plates, and assessed the colony forming capacity. As shown in Fig. 4A, satellite cells from CBS−/+ mouse displayed attenuated proliferative function after 5 days of culture. Further, there was a significant difference in the total number of detectable colonies as well as in the average colony color density staining (Fig. 4, B and C) between the satellite cells from WT and CBS−/+ mice. These results further indicated that there is a detectable deficiency in the proliferative capacity of the satellite cells from CBS−/+ mice.
Fig. 4.
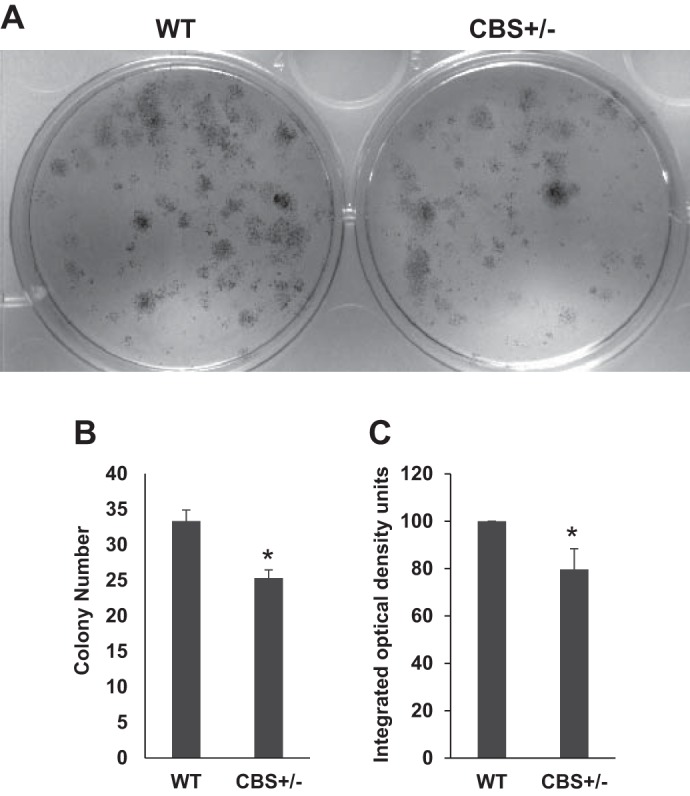
Compromised colony forming ability of the satellite cells from CBS−/+ mouse muscles. A: representative culture wells show crystal violet staining of colonies formed after 5 days of culture of equal number of purified satellite cells from WT and CBS−/+ mouse muscles. B: bar graph depicts average number of colonies from each well. C: the bar graph displays average crystal violet staining density across the well from the three different experiments as measured with ImageJ software. *P < 0.05 vs. WT.
Upregulation of p16 and p21 levels in satellite cells from CBS−/+ mice.
As there was a measurable defect in proliferative capacity of the isolated satellite cells, next we checked if there is any inhibitory mechanism in place that prevents efficient satellite cell proliferation and activation after injury. Earlier studies have demonstrated that increased presence of cell cycle inhibitors such as p21, p16 and p27 might undermine cell proliferative function (1, 4, 6). As shown in Fig. 5A, the satellite cells from CBS−/+ mouse muscle sections exhibited higher amounts of p16 levels that were colocalized with Pax7-containing nuclei, whereas the Pax7-expressing nuclei from WT muscle sections showed minimal amounts of p16 levels. Further, we measured p16 expression levels through flow analysis in the isolated satellite cells (Fig. 5B). The results from the flow analysis further supported the findings from the colocalization studies and suggest that satellite cells from HHcy mouse model exhibited compromised cell replication potential due to elevated p16 levels. Next, we also examined p21 levels, another inhibitor of CDK and cell cycle, through Q-PCR and found that there was a significant elevation in the p21 mRNA levels as well in the satellite cells from CBS−/+ mice (Fig. 5C). These findings together implied that enhanced presence of cell cycle inhibitors (p21 and p16) could be attributable to the derailed satellite cell function in HHcy mouse.
Fig. 5.

CBS−/+ mouse satellite cells expressed elevated cell cycle inhibitors. A: representative confocal images (60×) showing the staining for nuclei (DAPI-blue); Pax7 (green); and p16 (red) and merged image showing the colocalization of p16 and Pax7 in the nuclei. Inset depicts the representative nucleus with all the three colors. B: flow cytogram showing the p16 intensity levels in the Pax7-expressing satellite cells from WT and CBS−/+ mouse muscles. C: bar graph displaying the relative p21 mRNA levels as assayed by the Q-PCR. Level of beta-actin was used to normalize the mRNA levels in the samples. *P < 0.05 vs. WT.
CBS−/+ mouse satellite cells exhibit elevated phospho-p38 MAPK signaling.
Enhanced presence of cyclin-dependent kinase (CDK) inhibitors (p21 and p16) in the satellite cells from aged muscles was reported mainly due to excessive activation of p38 alpha/beta MAPK (1, 4). Moreover, earlier studies have independently demonstrated that HHcy condition induces inadvertent p38 MAPK activation in both the cardiomyocytes and glomerular mesangial cells (19, 28). Hence, we elected to test if satellite cells from CBS−/+ mice also exhibit enhanced p38 MAPK activation. To this end, we studied the extent of p38 MAPK activation by assaying for phosphorylated p38 alpha/beta MAPK levels in satellite cells through flow cytometry. As exhibited in Fig. 6, A and B, satellite cells from CBS−/+ mice displayed significantly elevated presence of activated p38 MAPK levels. Based on these results, it is plausible that excess p38 MAPK activation might be involved in upregulation of cell cycle inhibitors, p21 and p16, and in inhibition of satellite cell proliferation.
Fig. 6.

Enhanced phosphorylation of p38-MAPK in CBS−/+ mouse satellite cells. A: representative flow cytogram shows activated p38 MAPK intensity levels in the Pax7-expressing satellite cells from WT and CBS−/+ mouse muscles. B: bar graph displays the quantification of the satellite cells (% of total number of cells analyzed) expressing the highest levels of phosphorylated p38 MAPK. *P < 0.05 vs. WT.
Heightened oxidative stress in CBS−/+ mouse satellite cells.
Since HHcy condition in general and specifically in different muscle tissues [cardiac and skeletal muscles (25, 28)] was implicated in causing ROS accumulation and enhancement of oxidative stress, and oxidative stress has been implicated in aberrant activation of p38 MAPK activation (3, 13, 20), we verified whether there is any heightened ROS accumulation concurrent with aberrant p38 MAPK activation in satellite cells from CBS−/+ mice. As displayed in Fig. 7, we measured the levels of ROS accumulation in purified satellite cells using cell-permeant reagent 2′,7′-dichlorofluorescin diacetate (DCFDA). The satellite cells from CBS−/+ mouse muscles displayed enhanced ROS presence as evidenced by the enhanced fluorescence intensity (Fig. 7), further suggesting enhanced ROS presence involvement in aberrant activation of p38 MAPK and elevated expression of cell cycle inhibitors thereby causing attenuated self-renewal capacity in the satellite cells from CBS−/+ mouse.
Fig. 7.

Enhanced oxidative stress in satellite cells from CBS−/+ mouse muscles. Flow cytogram shows the levels of reactive oxygen species (ROS) accumulation as an intensity of DCF staining (DCFDA is converted into a fluorescent DCF in the presence of ROS) in the satellite cells from WT and CBS−/+ mouse muscles.
Hcy treatment increases oxidative stress, p38 MAPK activation and p16 levels in C2C12 myoblasts.
To further confirm our results and to establish the cause and effect relationship, we treated C2C12 myoblasts with homocysteine (Hcy) for 24 h and assayed for ROS levels through flow cytometry (Fig. 8, A and B) and p38 MAPK activation and p16 levels through Western probing (Fig. 8, C and D). As presented in Fig. 8, treatment of C2C12 myoblasts with Hcy not only caused ROS accumulation but also increased concomitant p38 MAPK activation as well as p16 levels. These findings further corroborated our earlier findings and suggested that HHcy is inhibiting satellite cell proliferation/self-renewal through enhancing ROS, p38 MAPK activation, and p16 and p21 levels. We also assayed for activation of ERK (phosphorylated p44/42 MAPK) levels after Hcy treatment in C2C12 cells (Fig. 8C). Although we were able to observe a tendency for enhanced ERK activation, the activation was not significantly different.
Fig. 8.

Hcy treatment enhances ROS accumulation and p38 MAPK phosphorylation as well as p16 levels in C2C12 cells. The C2C12 cells were treated with (Hcy) or without (Cont) homocysteine. A: flow cytogram displays the levels of ROS accumulation as measured with DCF staining intensity in C2C12 cell. B: bar graph depicts number of C2C12 cells expressing the highest levels of ROS accumulation. *P < 0.05 vs. Cont. C: Western blot images show relative levels of different proteins in the C2C12 lysate. AU, arbitrary units. D: bar graphs displays the relative quantities of different protein levels from the two groups. *P < 0.05, **P < 0.01 vs. Cont.
Inhibition of p38 MAPK results in amelioration of muscle repair failure after injury.
Next, we tested whether CBS−/+ mouse is more prone to muscle injury and if so whether or not it can be ameliorated by pharmacological inhibition of p38 MAPK. For this purpose, we used cardiotoxin injury model with a combination of highly specific p38 MAPK inhibitor (SB203580) administration as described before (1, 4). As displayed in Fig. 9, there was a considerable degree of attenuation of muscle repair after cardiotoxin injury in CBS−/+ mouse muscles as evidenced by enhanced collagen deposition. However, with simultaneous administration of p38 MAPK inhibitor, we were able to notice significant amelioration of muscle injury and the repair process that is close to normal (WT control and WT treated) mouse muscles.
Fig. 9.

Amelioration of attenuated muscle repair in the CBS−/+ mouse muscles with p38 MAPK inhibitor treatment. A: bright field microscopy images (10×) display damage recovery and collagen deposition after cardiotoxin injury in the TA muscles of WT and CBS−/+ mouse with (SB203580) or without (Saline) administration of p38 MAPK inhibitor (SB203580). B: bar graph depicts the quantitation of % area with collagen in different groups. *P < 0.05, WT vs. CBS−/+ plus saline; *P < 0.05, CBS−/+ plus saline vs. CBS−/+ plus SB203580.
Inhibition of p38 MAPK results in better satellite cell engraftment after injury.
In addition to extent of muscle damage after injury, we also evaluated engraftment potential of the exogenously delivered GFP expressing WT satellite cells in the injured muscles with and without pharmacological inhibition of p38 MAPK. As presented in Fig. 10, there is a considerable degree of improvement in engraftment of exogenously delivered satellite cells in CBS−/+ mouse only with SB203580 administration after injury. These results together imply that HHcy not only attenuates the repair process by inhibiting the satellite cell activation/proliferation, but also produces a hostile environment that is not conducive for exogenously implanted satellite cells. All these HHcy-induced aberrances can be ameliorated by inhibiting p38 MAPK activity.
Fig. 10.

Restoration of compromised satellite cell engraftment in the CBS−/+ mouse muscles with p38 MAPK inhibitor treatment. A: confocal microscopy images (60×) shows GFP-expressing muscle fibers formed after delivery of GFP-expressing satellite cells into the injured TA muscles of WT and CBS−/+ mouse with (SB203580) or without (saline) administration of p38 MAPK inhibitor (SB203580). Nuclei (DAPI) and engrafted muscle fibers (GFP) are presented. B: bar graph depicts the quantitation of number of muscle fibers with GFP expression in different groups. *P < 0.05, WT vs. CBS−/+ plus saline; *P < 0.05, CBS−/+ plus saline vs. CBS−/+ plus SB203580.
DISCUSSION
In the current study we demonstrated that HHcy is associated with 1) sarcopenia, 2) reduced cell proliferation after muscle injury, 3) diminished proliferation capacity of satellite cells in vitro, 4) increased ROS accumulation, 5) aberrant p38 MAPK activation, and 6) elevated CDK inhibitor presence in the satellite cells. Further, by using specific p38 MAPK inhibitor administration, we also showed that the HHcy-induced deficiencies in the muscle repair and engraftment potential can be ameliorated. Although it needs to be further established in the case of human scenario with the HHcy condition, our study identified several similarities that indicate compromised satellite cell function in humans as well. 1) Association between HHcy and muscle weakness was reported both in the humans and in mouse models (24, 26). 2) HHcy was associated with progressive decline in body weights both in humans and in mouse models (12, 14, 29). Notably, the CBS−/+ mice do not exhibit significant reduction in the body weights within 14 days of postpartum, but do exhibit significant reduction at around 3 mo of age. Moreover, when the CBS−/+ mice were fed with the high-methionine and low-folate diet, which further enhances Hcy levels, there was further acceleration in sarcopenia (Fig. 1). Hence, our study indicates that severity of the satellite cell dysfunction is dependent on the Hcy plasma levels, the duration of the HHcy condition, and the intake/availability of the dietary factors that modulate Hcy levels. The similarities between human and mouse HHcy muscle phenotypes together with our current findings imply that the HHcy condition may chronically inhibit satellite cell activation and may derail recovery after injury. The study further proposes that either correction of the HHcy condition or targeting p38 MAPK activation might benefit alleviation interventions/postinjury rehabilitation measures for adults with HHcy.
Apart from cell cycle arrest in the satellite cells, sarcopenia could also result from excessive cell death or activation of atrophic transcription through FOXO transcription factors (15, 23) in the muscle fibers. However, our previous study ruled out the induction of atrophic genes such as atrogin-1 and MuRF-1 in the intact muscles from CBS−/+ mice (23). Furthermore, histological sections from CBS−/+ mice neither exhibited abnormal cellular infiltration nor showed centronuclear muscle fibers that are suggestive of cell death or inflammation (26). These findings together strengthen the current finding that HHcy-associated sarcopenia could be due to compromised regenerative potential and may not be due to excessive muscle atrophy (through activation of atrogin and MuRF) or cell death. However, the previous study also indicated that there was significant reduction in the muscle fiber size in the CBS−/+ mouse muscles as evidenced by the presence of relatively more intermediate-size muscle fibers instead of large-size muscle fibers (26). The overall reduction in muscle mass and muscle fiber size could be due to other types of atrophy independent of FOXO transcription as well as defective satellite function as described in the current report. Although speculative, it would also be interesting to further investigate whether there are any muscle type-specific (location in the body or usage or vascular intensity) or fiber type-specific (oxidative vs. glycolytic) changes associated with HHcy.
One potential outcome from the current study is that oxidative stress induced by HHcy is involved in the inhibition of satellite cell function. Although previous studies have implicated the damaging effects of oxidative stress or ROS accumulation over the satellite cell function (17), the relevant metabolic factors that cause oxidative stress are not identified. Our study indicates that pathological elevation in the homocysteine levels prevents satellite cell self-renewal/proliferation upon injury by potentially elevating ROS levels. Although the mouse muscle scenario is different compared with human skeletal muscles in that the Hcy metabolizing enzymes are not expressed in the mouse skeletal muscles (25), the potential harmful effects of HHcy on skeletal muscles are still noticeable in humans, albeit, at later stages of life (aged population) (24). Our study further suggests that amelioration of oxidative stress by locally enhancing antioxidants or inhibition of p38 MAPK by pharmacological means are the efficient strategies in targeting rehabilitation/injury recovery and in prevention of elderly frailty observed in HHcy individuals.
One limitation of our study is that we could not perform functional studies as we lack the equipment to measure the in vivo functional parameters of the TA muscles in different groups. Nonetheless, previous studies have well established that pharmacological inhibition of p38 MAPK is the efficient way of recovery of function after injury in aged animals (1, 4). Another shortcoming is that we could not create extensive multiple-muscle damage (whole limb damage) on ethical grounds; hence we performed restricted damage only to the TA muscles. Another potential weakness is that we could not employ irradiation strategy (unable to get access to an irradiator and radioisotopes) to completely deplete the endogenous satellite cell pool before the engraftment of GFP-expressing exogenous satellite cells. Nonetheless, our results that measured collagen deposition after injury not only complemented the engraftment studies but also supported the conclusions made from the engraftment experiments.
Although not tested in the current study, other potential factors that further aggravate the oxidative stress-induced inhibition of satellite cell proliferation need to be considered in the efficient design of interventions to counter HHcy-induced inhibition of satellite cell function. For example, HHcy was shown to induce myostatin in CBS−/+ skeletal muscles (7). Previously, it was demonstrated that imbalance between Notch and Smad3 activation could also induce CDK inhibitors such as p21 and p16 (2), as also reported here. Moreover, myostatin depends on the p38 MAPK signaling in induction of CDK inhibitors especially p21 (16). Future studies are needed to further unravel the relative contribution of elevated myostatin and ROS levels in induction of p16 and p21 levels through p38 MAPK signaling during HHcy. Although not verified in the current study, excessive induction of p16 and p21 has been noted to cause an irreversible senescence state in the resting satellite cells as evidenced by senescence-associated β-galactosidase staining (21). As p53 has been implicated to play a pivotal role in induction of p16 and p21, it is plausible HHcy may alter the p53 function or level as well, apart from excessive p38 MAPK activation. Further support for this possibility comes from the literature that suggested that homocysteine inhibits hepatocyte proliferation through elevation of p53 and p21 levels (30). Future studies are needed to test these possibilities in the satellite cells from HHcy.
As oxidative stress was also reported to induce MEK/ERK pathway (20) and activation of MEK/ERK pathway also induces cell cycle inhibitors and thereby causes cellular quiescence or senescence (5), we tested if Hcy treatment also causes induction of ERK (p44/42 MAPK) phosphorylation in C2C12 cells. Although we could not observe significant elevation of phosphorylated ERK after 24 h of 100 μm Hcy treatment, it is plausible that either ERK activation may occur at different time points not tested in the current study or the relative strength of the ERK signaling might be limited due to the feedback activation of ERK-specific phosphatases. It is also possible that Hcy-mediated oxidative stress in C2C12 cells may differentially activate ERK and p38 MAPK proteins as observed in different cell lines (31). Future studies are necessary to clarify these possibilities.
In summary, the results from the current study support the notion that the CBS−/+ mouse, a model for the HHcy condition, exhibited compromised regenerative function and cell proliferation upon injury. However, there was no difference in Pax7 expression levels in the satellite cells from CBS−/+ mouse skeletal muscles. Interestingly, the satellite cells from the CBS−/+ mouse not only exhibited diminished in vitro proliferative capabilities, but also there was heightened oxidative stress. In addition, there was enhanced p38 MAPK activation as well as p16 and p21 expression in the CBS−/+ mouse satellite cells. Moreover, the C2C12 myoblasts also exhibited higher p38 MAPK activation and p16 expression upon treatment with homocysteine in addition to enhanced ROS presence. Tissue engraftment potential and regeneration after injury were restored upon treatment with p38 MAPK inhibitor, SB203580, in the CBS−/+ mouse. These results together suggest that HHcy-induced diminished satellite cell proliferation involves excessive oxidative stress and p38 MAPK signaling. Our study further proposes that HHcy is a potential risk factor for elderly frailty, and needs to be considered as a therapeutic target while designing the alleviation interventions/postinjury rehabilitation measures for adults with HHcy.
GRANTS
Part of this study was supported by National Institutes of Health Grants HL-74185 and HL-108621 to S. C. Tyagi and NS-84823 to S. C. Tyagi and D. Lominadze.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: S.V. and S.C.T. conception and design of research; S.V. performed experiments; S.V. analyzed data; S.V. interpreted results of experiments; S.V. prepared figures; S.V. drafted manuscript; S.V., D.L., and S.C.T. edited and revised manuscript; S.V., D.L., and S.C.T. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. James McCracken for the use of gentleMACS dissociator.
REFERENCES
- 1.Bernet JD, Doles JD, Hall JK, Tanaka KK, Carter TA, Olwin BB. p38 MAPK signaling underlies a cell-autonomous loss of stem cell self-renewal in skeletal muscle of aged mice. Nat Med 20: 265–271, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carlson ME, Hsu M, Conboy IM. Imbalance between pSmad3 and Notch induces CDK inhibitors in old muscle stem cells. Nature 454: U528–U529, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Choi WS, Eom DS, Han BS, Kim WK, Han BH, Choi EJ, Oh TH, Markelonis GJ, Cho JW, Oh YJ. Phosphorylation of p38 MAPK induced by oxidative stress is linked to activation of both caspase-8-and-9-mediated apoptotic pathways in dopaminergic neurons. J Biol Chem 279: 20451–20460, 2004. [DOI] [PubMed] [Google Scholar]
- 4.Cosgrove BD, Gilbert PM, Porpiglia E, Mourkioti F, Lee SP, Corbel SY, Llewellyn ME, Delp SL, Blau HM. Rejuvenation of the muscle stem cell population restores strength to injured aged muscles. Nat Med 20: 255–264, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ferbeyre G, de Stanchina E, Lin AW, Querido E, McCurrach ME, Hannon GJ, Lowe SW. Oncogenic ras and p53 cooperate to induce cellular senescence. Mol Cell Biol 22: 3497–3508, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fu X, Wang H, Hu P. Stem cell activation in skeletal muscle regeneration. Cell Mol Life Sci 72: 1663–1677, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Givvimani S, Narayanan N, Armaghan F, Pushpakumar S, Tyagi SC. Attenuation of conducted vasodilation in skeletal muscle arterioles during hyperhomocysteinemia. Pharmacology 91: 287–296, 2013. [DOI] [PubMed] [Google Scholar]
- 8.Hayhurst M, Wagner AK, Cerletti M, Wagers AJ, Rubin LL. A cell-autonomous defect in skeletal muscle satellite cells expressing low levels of survival of motor neuron protein. Dev Biol 368: 323–334, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hofmann MA, Lalla E, Lu Y, Gleason MR, Wolf BM, Tanji N, Ferran LJ, Kohl B, Rao V, Kisiel W, Stern DM, Schmidt AM. Hyperhomocysteinemia enhances vascular inflammation and accelerates atherosclerosis in a murine model. J Clin Invest 107: 675–683, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ishii I, Akahoshi N, Yamada H, Nakano S, Izumi T, Suematsu M. Cystathionine gamma-lyase-deficient mice require dietary cysteine to protect against acute lethal myopathy and oxidative injury. J Biol Chem 285: 26358–26368, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jones NC, Tyner KJ, Nibarger L, Stanley HM, Cornelison DDW, Fedorov YV, Olwin BB. The p38 alpha/beta MAPK functions as a molecular switch to activate the quiescent satellite cell. J Cell Biol 169: 105–116, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kalra BR, Ghose S, Sood NN. Homocystinuria with bilateral absolute glaucoma. Indian J Ophthalmol 33: 195–197, 1985. [PubMed] [Google Scholar]
- 13.Matos TJ, Duarte CB, Goncalo M, Lopes MC. Role of oxidative stress in ERK and p38 MAPK activation induced by the chemical sensitizer DNFB in a fetal skin dendritic cell line. Immunol Cell Biol 83: 607–614, 2005. [DOI] [PubMed] [Google Scholar]
- 14.Narayanan N, Pushpakumar SB, Givvimani S, Kundu S, Metreveli N, James D, Bratcher AP, Tyagi SC. Epigenetic regulation of aortic remodeling in hyperhomocysteinemia. FASEB J 28: 3411–3422, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Narici MV, Maffulli N. Sarcopenia: characteristics, mechanisms and functional significance. Br Med Bull 95: 139–159, 2010. [DOI] [PubMed] [Google Scholar]
- 16.Philip B, Lu ZJ, Gao YJ. Regulation of GDF-8 signaling by the p38 MAPK. Cell Signal 17: 365–375, 2005. [DOI] [PubMed] [Google Scholar]
- 17.Renault V, Thornell LE, Butler-Browne G, Mouly V. Human skeletal muscle satellite cells: aging, oxidative stress and the mitotic clock. Exp Gerontol 37: 1229–1236, 2002. [DOI] [PubMed] [Google Scholar]
- 18.Senf SM, Howard TM, Ahn B, Ferreira LF, Judge AR. Loss of the inducible Hsp70 delays the inflammatory response to skeletal muscle injury and severely impairs muscle regeneration. Plos One 8: 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shastry S, James LR. Homocysteine-induced macrophage inflammatory protein-2 production by glomerular mesangial cells is mediated by PI3 kinase and p38 MAPK. J Inflamm Lond 6: 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Son Y, Cheong YK, Kim NH, Chung HT, Kang DG, Pae HO. Mitogen-activated protein kinases and reactive oxygen species: how can ROS activate MAPK pathways? J Signal Transduct 2011: 792639, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sousa-Victor P, Gutarra S, Garcia-Prat L, Rodriguez-Ubreva J, Ortet L, Ruiz-Bonilla V, Jardi M, Ballestar E, Gonzalez S, Serrano AL, Perdiguero E, Munoz-Canoves P. Geriatric muscle stem cells switch reversible quiescence into senescence. Nature 506: 316–321, 2014. [DOI] [PubMed] [Google Scholar]
- 22.Troy A, Cadwallader AB, Fedorov Y, Tyner K, Tanaka KK, Olwin BB. Coordination of satellite cell activation and self-renewal by Par-complex-dependent asymmetric activation of p38 alpha/beta MAPK. Cell Stem Cell 11: 541–553, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Veeranki S, Givvimani S, Pushpakumar S, Tyagi SC. Hyperhomocysteinemia attenuates angiogenesis through reduction of HIF-1alpha and PGC-1alpha levels in muscle fibers during hindlimb ischemia. Am J Physiol Heart Circ Physiol 306: H1116–H1127, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Veeranki S, Tyagi SC. Defective homocysteine metabolism: potential implications for skeletal muscle malfunction. Int J Mol Sci 14: 15074–15091, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Veeranki S, Tyagi SC. Mechanisms of hyperhomocysteinemia induced skeletal muscle myopathy after ischemia in the CBS−/+ mouse model. Int J Mol Sci 16: 1252–1265, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Veeranki S, Winchester LJ, Tyagi SC. Hyperhomocysteinemia associated skeletal muscle weakness involves mitochondrial dysfunction and epigenetic modifications. Biochim Biophys Acta 1852: 732–741, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.von Maltzahn J, Jones AE, Parks RJ, Rudnicki MA. Pax7 is critical for the normal function of satellite cells in adult skeletal muscle. Proc Natl Acad Sci USA 110: 16474–16479, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang X, Cui L, Joseph J, Jiang B, Pimental D, Handy DE, Liao R, Loscalzo J. Homocysteine induces cardiomyocyte dysfunction and apoptosis through p38 MAPK-mediated increase in oxidant stress. J Mol Cell Cardiol 52: 753–760, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Watanabe M, Osada J, Aratani Y, Kluckman K, Reddick R, Malinow MR, Maeda N. Mice deficient in cystathionine beta-synthase: animal models for mild and severe homocyst(e)inemia. Proc Natl Acad Sci USA 92: 1585–1589, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yu X, Lv JJ, Zhu YZ, Duan LP, Ma LQ. Homocysteine inhibits hepatocyte proliferation via endoplasmic reticulum stress. Plos One 8: 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang L, Jope RS. Oxidative stress differentially modulates phosphorylation of ERK, p38 and CREB induced by NGF or EGF in PC12 cells. Neurobiol Aging 20: 271–278, 1999. [DOI] [PubMed] [Google Scholar]