

In a clinically relevant porcine model of metabolic syndrome and peripheral vascular disease, treatment with a novel sustained-release nitrite formulation restored cardiac endothelial nitric oxide synthase, enhancing myocardial nitrite levels, reduced oxidative stress, and improved ex vivo coronary vascular function via endothelium-independent vasodilation mechanism in obese Ossabaw swine.
Keywords: Ossabaw swine, endothelial dysfunction, atherosclerosis, nitric oxide, sustained-release sodium nitrite
Abstract
Metabolic syndrome (MetS) reduces endothelial nitric oxide (NO) bioavailability and exacerbates vascular dysfunction in patients with preexisting vascular diseases. Nitrite, a storage form of NO, can mediate vascular function during pathological conditions when endogenous NO is reduced. The aims of the present study were to characterize the effects of severe MetS and obesity on dyslipidemia, myocardial oxidative stress, and endothelial NO synthase (eNOS) regulation in the obese Ossabaw swine (OS) model and to examine the effects of a novel, sustained-release formulation of sodium nitrite (SR-nitrite) on coronary vascular reactivity and myocardial redox status in obese OS subjected to critical limb ischemia (CLI). After 6 mo of an atherogenic diet, obese OS displayed a MetS phenotype. Obese OS had decreased eNOS functionality and NO bioavailability. In addition, obese OS exhibited increased oxidative stress and a significant reduction in antioxidant enzymes. The efficacy of SR-nitrite therapy was examined in obese OS subjected to CLI. After 3 wk of treatment, SR-nitrite (80 mg·kg−1·day−1 bid po) increased myocardial nitrite levels and eNOS function. Treatment with SR-nitrite reduced myocardial oxidative stress while increasing myocardial antioxidant capacity. Ex vivo assessment of vascular reactivity of left anterior descending coronary artery segments demonstrated marked improvement in vasoreactivity to sodium nitroprusside but not to substance P and bradykinin in SR-nitrite-treated animals compared with placebo-treated animals. In conclusion, in a clinically relevant, large-animal model of MetS and CLI, treatment with SR-nitrite enhanced myocardial NO bioavailability, attenuated oxidative stress, and improved ex vivo coronary artery vasorelaxation.
NEW & NOTEWORTHY
In a clinically relevant porcine model of metabolic syndrome and peripheral vascular disease, treatment with a novel sustained-release nitrite formulation restored cardiac endothelial nitric oxide synthase, enhancing myocardial nitrite levels, reduced oxidative stress, and improved ex vivo coronary vascular function via endothelium-independent vasodilation mechanism in obese Ossabaw swine.
metabolic syndrome (MetS) is a multifactorial disease that is characterized by a number of risk factors including obesity, hypertension, dyslipidemia, and hyperglycemia (27). Clinically, MetS is associated with an increased risk for the development of cardiovascular disease. In patients with preexisting atherosclerotic vascular disease, such as peripheral artery disease (PAD), MetS exacerbates vascular damage by impairing vascular endothelial and smooth muscle function (37). Furthermore, PAD patients with MetS have an increased incidence of acute coronary syndromes, resulting in significant cardiovascular morbidity and mortality (5, 16, 20, 21).
One of the earliest manifestations of cardiovascular diseases, including PAD, is a loss of endothelial function characterized by dysfunction of endothelial nitric oxide (NO) synthase (eNOS) and reduced NO bioavailability (7, 19, 44). Our laboratory has recently published data (40) demonstrating significant reductions in circulating NO bioavailability in patients with critical limb ischemia (CLI), a severe manifestation of PAD.
Nitrite is recognized as an important physiological storage reservoir of NO in the blood and tissues that protects various organs against ischemic injury (10, 12, 31). During pathological conditions, nitrite is rapidly reduced to NO to restore NO levels and promote organ homeostasis and survival (10, 12, 31). Moreover, endogenous nitrite has been shown to exert endocrine actions during ischemic injury via transport in the circulation and subsequent metabolism in distant organs to mediate cytoprotection (12). In rodent models of cardiovascular disease and aging, daily nitrite supplementation increases myocardial NO levels (2, 6), reverses vascular endothelial dysfunction (43), and attenuates oxidative stress (8). These data suggest a promising therapeutic potential of sodium nitrite in cardiovascular disease states. One of the major limitations with the use of sodium nitrite for the treatment of chronic cardiovascular diseases is the relatively short half-life of sodium nitrite and the inability to maintain pharmacologically relevant levels of nitrite and NO for sustained periods (30). To overcome this severe limitation, we recently investigated the effects of a novel, sustained-release formulation of sodium nitrite (SR-nitrite) in a clinically relevant model of CLI (40). In this study, obese Ossabaw swine (OS) were subjected to CLI for 35 days and treated with SR-nitrite starting at day 14. In this study, we failed to observe significant improvements in hindlimb perfusion, revascularization, or perfusion pressures. However, we did observe significant increases in NO bioavailability in the plasma and ischemic skeletal muscle at 35 days post-CLI injury coupled with reductions in oxidative stress and induction in proangiogenic signaling (40).
At present, there are no studies evaluating the effects of obesity/MetS in the presence of peripheral vascular disease on myocardial oxidative stress and coronary artery vasomotion. The goals of the present study were twofold. Initially, we sought to assess the extent of eNOS dysregulation, NO insufficiency, and myocardial oxidative stress in a clinically relevant model of obesity and MetS (i.e., obese OS model). Subsequently, we evaluated the effects of SR-nitrite on blood pressure, myocardial oxidative stress, NO bioavailability, and coronary artery vasoreactivity in obese OS subjected to CLI.
METHODS
Animals.
Male and female OS (6–8 mo of age) obtained from Indiana University and female Yorkshire (3 mo of age) obtained from Palmetto Research Swine (Reevesville, SC) were handled in compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. This protocol was approved by the Institutional Animal Care and Use Committee of Emory University.
Lean versus obese OS experiments.
OS (6–8 mo of age) were fed either 725 g/day lean diet (5L80, Lab Diet) (n = 17, 30% female) or 1 kg/day atherogenic obese diet (KT324, Test Diet) (n = 24; 22% female) composed of 2% cholesterol, 17% coconut oil, 2.5% corn oil, and 0.7% sodium cholate for 6 mo. After 6 mo of feeding, heparin (200 U/kg iv) was injected and allowed to circulate for ∼5 min, and animals were then euthanized while under deep inhalant anesthesia [isoflurane (5%)]. The heart and blood were collected.
Serum cholesterol and biomarkers.
Lipid profiles and asymmetric dimethylarginine (ADMA) quantification was performed by Atherotech Diagnostics Lab (Birmingham, AL).
CLI model.
Castrated male obese OS (6–8 mo of age) subjected to CLI investigated in the present study were a part of another experimental study that investigated the efficacy of SR-nitrite in skeletal muscle (40). Animals were sedated with medications that included ketamine (15 mg/kg) in combination with xylazine (1 mg/kg) for sedation, and, to aid induction, animals were endotracheally intubated and maintained on isoflurane in O2. Preoperative analgesic buprenorphine (0.15 mg for each pig) was given along with perioperative antibiotics [cefazolin (1,000 mg)]. The postoperative analgesics carprofen (10 mg) and buprenorphine (0.15–0.3 mg) were given immediately after the procedure. Postoperative day 1, pigs received the additional analgesics buprenorphine (0.3 mg) in combination with carprofen (100 mg). All pigs were monitored during the procedure for heart rate, respiration, O2 saturation, end-tidal CO2, fluid volume, and body temperature. Activated clotting time was measured with a Hemachron after the arterial sheath was placed and heparin was given.
Using sterile conditions and under fluoroscopic guidance, an 8-Fr introducer (Maximum Hemostasis Introducer 23 cm ACT Sheath, St. Jude Medical, St. Paul, MN) was inserted into the right external carotid artery, advanced into the abdominal aorta, and placed just above the aortoiliac bifurcation. Heparin (300 U/kg) was administered, and activated clotting time was monitored throughout the procedure to ensure clotting times of at least 250 s. A percutaneous guidewire (0.035 × 230 cm, Rosen Starter, Boston Scientific, Natick, MA) was positioned in the right external iliac artery. A 7-Fr delivery guide catheter (MACH 1 MP1, Boston Scientific) containing a self-expanding endoluminal endoprosthesis consisting of an expanded polytetrafluoroethylene (ePTFE) lining with an external nitinol stent (6 mm × 15 cm, Viabahn, W.L. Gore and Associates, Flagstaff, AZ) was advanced, positioned within the right external iliac artery, and deployed. Contrast arteriography was performed to evaluate vessel patency. After placement of the ePTFE-lined endoprosthesis, a self-expanding nitinol mesh occlusion device (8 × 7 mm, Amplatzer Vascular Plug II, St. Jude Medical) was loaded onto the delivery guide catheter, advanced, positioned within the right external iliac artery, and deployed within the proximal portion of the ePTFE-lined stent. An arteriogram was performed to verify occlusion. The guide wire, guide catheter, and introducer sheath were removed, and the left external carotid artery was ligated. The incision was closed, the animal was allowed to recover, and buprenorphine (0.05 mg/kg) was administered for analgesia. The ePTFE-lined endoprosthesis served to inhibit recanalization of the artery by collateral vessels, typically observed in animal models of CLI.
Sodium nitrite therapy.
Sustained-release sodium nitrite tablets and placebo tablets were obtained from TheraVasc (Cleveland, OH). Animals received placebo (n = 6) or 80 mg SR-nitrite (n = 7) by mouth (po) twice daily (bid). Treatment was initiated at 14 days after the induction of CLI and continued for 21 days.
DSI radiotelemetry implantation.
Before the induction of CLI, obese OS (n = 8) underwent surgical implantation of an arterial blood pressure (BP) radiotelemeter (TA11PA-D70, Data Sciences, St Paul, MN) to assess the extent of hypertension in obese OS. Anesthesia was induced and maintained as described above. The BP telemeter body was implanted subcutaneously dorsally in the neck, with the telemeter catheter inserted 10 cm into the left carotid artery.
Telemetry equipment (Data Sciences) was set up to monitor hemodynamics. The BP telemeter signal was received by a receiver (RLA1020) positioned in a protective case on the floor of the holding pen. The strongest signal was selected by a multiplexer (RMX10) and passed to an analog adapter (R11CPA), which provided a calibrated voltage output after correcting for atmospheric pressure using an ambient pressure monitor (APR-1). The calibrated signal was then recorded using a computerized data-acquisition system. The BP signal was sampled for 10 s at 30-s intervals; mean arterial BPs (MABPs) during this sample period were computed and stored for offline analysis.
Euthanasia and tissue collection.
After 35 days of CLI, obese OS were euthanized, and blood, myocardial tissue, and coronary artery segments were obtained. Heparin (200 U/kg iv) was injected and allowed to circulate for ∼5 min, and animals were then euthanized while under deep inhalant anesthesia [isoflurane (5%)]. The heart and coronary arteries were collected.
Measurement of NO metabolites.
Nitrite concentrations were quantified in ventricular tissue biopsies by ion chromatography (ENO20 Analyzer, Eicom, Kyoto, Japan).
cGMP radioimmunoassay.
Coronary artery cGMP concentrations were quantified as previously described (2).
Western blot analysis.
Ventricular tissue was used for Western blot analysis, which was performed as previously described (23). The following primary antibodies were used: VEGF, CD31 (Novus Biologicals, Littleton, CO), eNOS (Ser1177; Abcam, Cambridge, MA), eNOS, eNOS (Thr495; Cell Signaling, Danvers, MA), and GAPDH (Santa Cruz Biotechnology, Santa Cruz, CA).
Real-time PCR.
Ventricular mRNA was quantified using TaqMan primers from Life Technologies and performed as previously described (36).
Determination of protein carbonyl content.
Protein carbonyl content from ventricular tissue was measured as previously described (23).
Measurement of malondialdehyde levels.
Malondialdehyde (MDA) levels were measured in ventricular tissue as previously described (23).
Functional assessment of isolated coronary rings.
Isolated coronary artery experiments were performed in obese OS and lean, nonischemic Yorkshire control pigs. CLI experiments involved obese OS that received either placebo or SR-nitrite (80 mg po bid). Obese OS vascular reactivity responses were compared with nonischemic lean Yorkshire responses. Coronary arteries from Yorkshire pigs (3 mo, Palmetto Farms) were used as a reference for a nonischemic control. Left anterior descending (LAD) arteries were dissected, cleaned of surrounding perivascular adipose tissue, and collected into to ice-cold Krebs buffer. Arteries were cut into 3-mm rings and mounted in organ baths with warm Krebs buffer for isometric tension experiments. Coronary arteries were placed under 0.5 g of tension for 90–120 min to obtain a stable and optimal baseline passive tension. Arteries were stimulated initially with 40 mM and then 100 mM KCl (n = 2 nonischemic control, n = 6 CLI + placebo, and n = 6 CLI + SR-nitrite) to assess vessel viability and contractile responsiveness. After a series of washes, rings were precontracted with PGF2α (30 μM). At peak constriction, vascular function was assess by the addition of graded concentrations of sodium nitroprusside (SNP; 10−9–10−5 M, n = 2 nonischemic control, n = 6 CLI + placebo, and n = 5 CLI + SR-nitrite), bradykinin (10−9–10−5 M, n = 2 nonischemic control, n = 6 CLI + placebo, and n = 5 CLI + SR-nitrite), or substance P (10−11–10−8 M, n = 2 nonischemic control, n = 6 CLI + placebo, and n = 4 CLI + SR-nitrite), to the tissue bath.
Statistical analysis.
All data in the present study are expressed as means ± SE. Differences in data between groups were compared using Prism 6 (GraphPad Software). A Student's unpaired, two-tailed t-test was used when comparison was between two groups (i.e., lean OS vs. obese OS or CLI + placebo vs. CLI + SR-nitrite). For ambulatory MABP, two-way ANOVA was used to test the effects of placebo and SR-nitrite therapy over time. For isometric tension dose-dependent experiments, two-way ANOVA was used to test the effect of treatment and various vasodilators on the coronary relaxation response. For maximal relaxation, one-way ANOVA was used to test the effect of the treatment and the maximum dose of vasodilator on maximum vasorelaxation. When statistical significance was found with ANOVA, a Tukey multiple-comparison test was performed. P values of <0.05 were considered statistically significant.
RESULTS
The obese OS is a well-characterized model of central obesity and MetS (11, 35). The data reported in the present study represent data collected from two independent experimental studies. Initial experiments evaluated the cardiovascular and MetS phenotype in obese OS compared with lean OS. The results from the first study are shown in Figs. 1–5. The second study reported in the present report is a subset of a study in which we investigated the efficacy of a novel extended-release formulation of nitrite (i.e., SR-nitrite) in a CLI model (40). The present study is focused on the effects of SR-nitrite on myocardial oxidative stress, coronary vascular reactivity, and systemic hemodynamics in obese OS subjected to CLI. These results are shown in Figs. 6–11.
Fig. 1.

Ossabaw swine (OS) fed a high-fat, high-cholesterol diet exhibit a metabolic syndrome (MetS) phenotype. A: body weight. B: plasma glucose. C: insulin. D: alanine transaminase (ALT). Values are means ± SE; numbers inside bars are numbers of animals per group.
Fig. 5.

MetS alters the expression of antioxidant enzymes in the heart. A–D: mRNA levels of glutathione peroxidase (GPx)-1 (A), catalase (B), SOD2 (C), and heme oxygenase (HO)-1 (D) in myocardial tissue from lean and obese OS. Values are means ± SE; numbers inside bars are numbers of animals per group.
Fig. 6.
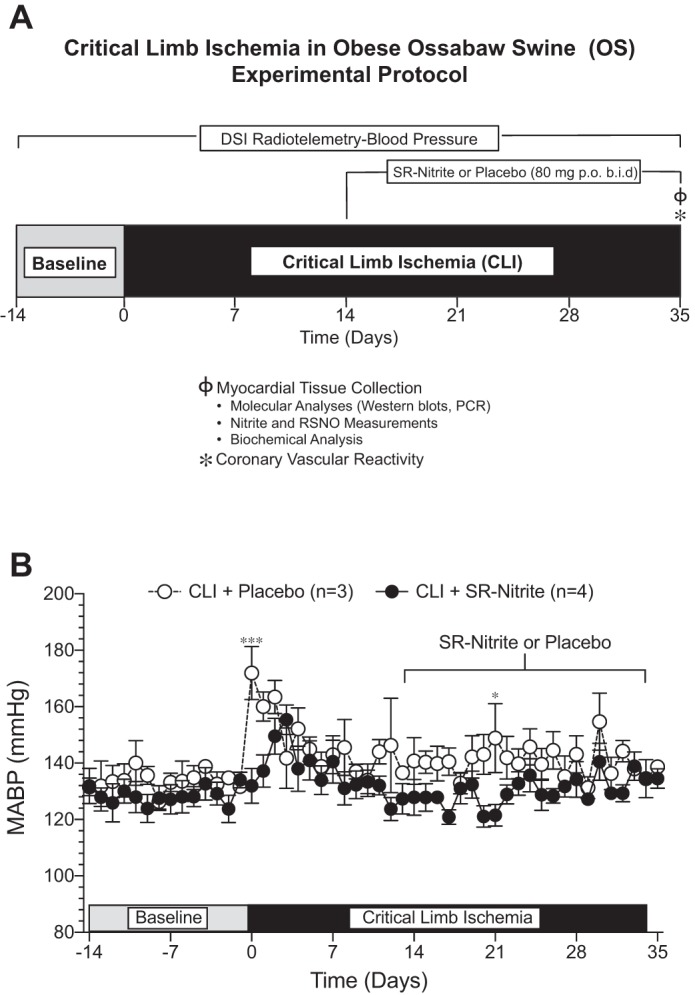
Experimental protocol for experiments of critical limb ischemia (CLI) in obese OS. A: obese OS were subjected to CLI for 35 days. Sustained-release sodium nitrite (SR-nitrite; 80 mg po bid) or placebo therapy was initiated at 14 days after CLI injury. On day 35 after CLI, myocardial tissues were collected to evaluate eNOS functionality, NO bioavailability, and oxidative stress after CLI. In addition, epicardial coronary artery segments were harvested, and in vitro coronary vascular reactivity was evaluated. We also investigated the effect of CLI in obese OS on systemic blood pressure. B: AM and PM averages of mean arterial blood pressure (MABP). Values are means ± SE. *P < 0.05 vs. the CLI + placebo-treated group. ***P < 0.001 vs. the CLI + placebo-treated group.
Fig. 11.

SR-nitrite therapy had no effect on coronary vascular reactivity to endothelium-dependent vasodilators. Freshly isolated coronary arteries were precontracted with PGF2α. A and B: dose-dependent relaxation (%relaxation) of the left anterior descending coronary artery in response to substance P (A) and bradykinin (B). C and D: maximum relaxation at 10−8 M substance P (C) and 10−5 M bradykinin (D). Values are means ± SE; numbers inside bars are numbers of animals per group.
Characterization of MetS in obese OS.
After 6 mo of a high-fat, high-cholesterol diet, obese OS exhibited characteristics of a MetS phenotype (Figs. 1 and 2) compared with lean controls. Obese OS displayed a significant (P < 0.01) increase in body weight (Fig. 1A) compared with lean OS (73.0 ± 5.0 vs. 40.0 ± 2.0 kg). There was no significant difference in plasma insulin levels (Fig. 1C); however, blood glucose (Fig. 1B) was elevated (P < 0.05) in obese OS (80.2 ± 4.0 mg/dl) compared with lean OS (67.4 ± 4.6 mg/dl). Obese OS exhibited twofold higher circulating levels (53.5 ± 9.1 U/l) of alanine aminotransferase (ALT; P < 0.05; Fig. 1D) compared with lean OS (24.3 ± 1.2 U/l). Lee et al. (24) reported that after 6 mo of feeding with a very similar high-fat, high-cholesterol diet, OS displayed abnormal fatty liver histology that is similar to human nonalcoholic steatohepatitis. Therefore, we suspect that the increases in ALT that we observed in the present study are related to steatohepatitis.
Fig. 2.

Obese OS display dyslipidemia after a high-fat, high-cholesterol diet. A: cholesterol. B: triglycerides. C: LDL-cholesterol. D: HDL-cholesterol. E: VLDL-cholesterol. Values are means ± SE; numbers inside bars are numbers of animals per group.
The atherogenic diet resulted in significant dyslipidemia in obese OS, as shown in Fig. 2. Circulating cholesterol levels were significantly (P < 0.0001) elevated in obese OS (508.5 ± 78.9 mg/dl) compared with lean OS (66.4 ± 2.7 mg/dl). Similarly, obese OS exhibited significant (P < 0.05) elevations in plasma triglycerides (Fig. 2B) versus lean OS (59.1 ± 13.4 vs. 25.4 ± 2.7 mg/dl). Although there was no significant difference between lean OS and obese OS in terms of HDL-cholesterol (Fig. 2D), circulating LDL-cholesterol (Fig. 2C) was increased (P < 0.01) in obese OS (290.0 ± 51.0 mg/dl) compared with lean OS (26.6 ± 2.1 mg/dl). VLDL-cholesterol levels (Fig. 2E) demonstrated significantly (P < 0.01) higher VLDL in obese OS (26.0 ± 4.0 mg/dl) versus lean OS (10.5 ± 0.2 mg/dl).
OS, myocardial eNOS dysregulation, and NO deficiency.
Experiments were performed to investigate the extent of myocardial eNOS coupling in both lean and obese OS using Western blot techniques. It has been well appreciated that phosphorylation of Ser1177 on eNOS activates the enzyme to increase NO production, whereas phosphorylation of Thr495 inactivates the enzyme and attenuates NO production (4, 26, 34). Western blot analysis of ventricular tissue biopsies obtained from lean and obese OS were similar in total eNOS protein expression (Fig. 3B). Obesity and MetS resulted in a significant reduction in phosphorylation of eNOS at the primary activation site, Ser1177 (Fig. 3C), in obese OS hearts compared with lean OS hearts (0.5 ± 0.1 vs. 1.0 ± 0.1 RI, obese OS vs. lean OS, respectively, P < 0.01). Moreover, obese OS exhibited a marked increase in phosphorylation of the eNOS inhibitory site, Thr495 (2.6 ± 0.2 vs. 1.1 ± 0.3 RI, obese OS vs. lean OS, respectively, P < 0.05; Fig. 3D). Myocardial tissue nitrite levels were measured as a surrogate index of NO bioavailability in the heart (Fig. 3E). Obese OS displayed reduced cardiac nitrite levels compared with lean OS (0.8 ± 0.08 vs. 2.2 ± 0.3 μM, obese OS vs. lean OS, respectively, P < 0.01). In addition, NO-mediated activation of guanylyl cyclase and downstream signaling as measured by cGMP (Fig. 3F) was significantly (P < 0.05) decreased in coronary arteries of obese OS (2.0 ± 2.0 pmol/g) compared with lean OS (8.9 ± 2.5 pmol/g).
Fig. 3.

MetS decreases phosphorylation of endothelial nitric oxide (NO) synthase (eNOS) at Ser1177 and increases phosphorylation of eNOS at Thr495. These alterations in eNOS phosphorylation result in profound inhibition of eNOS and reduced myocardial NO bioavailability. A: representative immunoblots of cardiac protein expression of phosphorylated eNOS (Ser1177), eNOS (Thr495), and total eNOS. B–D: densitometry analysis of total eNOS (B), phosphorylated eNOS (Ser1177; C), and phosphorylated eNOS (Thr495; D). E: cardiac nitrite levels in lean and obese OS. F: coronary artery cGMP levels. Values are means ± SE; numbers inside bars are numbers of animals per group. NS, not significant.
Obese OS exhibit altered redox status in the heart.
The reductions in eNOS functionality along with a concomitant reduction in nitrite in obese OS may be mediated by several mechanisms, such as excessive oxidative stress and/or increased levels of endogenous eNOS inhibitors. To determine the overall redox status in obese OS hearts, we measured the extent of oxidative stress in myocardial tissue samples using MDA and carbonyl as overall indexes of oxidative stress. These data are shown in Fig. 4. Cardiac MDA levels were similar between obese OS and lean OS (Fig. 4A). Obese OS (3.1 ± 0.5 nmol/protein) did exhibit significantly (P < 0.05) elevated myocardial carbonyl protein levels (Fig. 4B) compared with lean OS (2.1 ± 0.2 nmol/protein). ADMA is an endogenous competitive inhibitor of eNOS function via its interaction with the l-arginine-binding site and thereby inhibits NO production (9). Interestingly, plasma ADMA (Fig. 4C) levels were significantly (P < 0.05) higher in obese OS (2.3 ± 0.1 μM/l) compared with lean OS (1.6 ± 0.2 μM/l).
Fig. 4.

MetS increases oxidative stress in the heart. A and B: myocardial malondialdehyde (MDA) levels (in nmol/g protein; A) and carbonyl protein cardiac levels (B) in lean and obese OS. C: plasma levels (in μM) of asymmetric dimethylarginine (ADMA). Values are means ± SE; numbers inside bars are numbers of animals per group.
We also measured mRNA levels of critical antioxidant enzymes in myocardial tissue from lean and obese OS (Fig. 5) to further evaluate the extent of oxidative stress. Glutathione peroxidase (GPx)-1 (Fig. 5A) and catalase (Fig. 5B) mRNA levels were not statistically different between the study groups; however, obese OS had a significant reduction in hemeoxygenase (HO)-1 (Fig. 5D) mRNA in the heart compared with lean OS (2.1 ± 0.3 vs. 5.8 ± 0.9 arbitrary units, P < 0.01). In contrast, quantitative PCR analysis of SOD2 mRNA (P < 0.05; Fig. 5C) revealed a marked increase in obese OS hearts (1.2 ± 0.1 arbitrary units) compared with lean OS (0.7 ± 0.1 arbitrary units).
Investigation of hypertension, myocardial redox status, and coronary vascular reactivity in obese OS after nitrite therapy.
By using a severe model of peripheral vascular disease, we sought to investigate the effects of MetS in the setting of CLI on hypertension, myocardial redox status, and coronary vascular function and also to evaluate the potential cytoprotective effects of nitrite in this severe model of cardiovascular disease. The results of these experiments are shown in Figs. 6–11.
Hypertension in obese OS and effects of SR-nitrite.
Using obese OS, CLI injury was surgically induced in the right external iliac artery and sustained for 35 days, as shown in the experimental protocol in Fig. 6. SR-nitrite (80 mg po bid) or placebo therapy was initiated on day 14 after CLI surgery. To ensure that SR-nitrite therapy did not induce systemic hypotension, MABP was assessed by radiotelemetry. Ambulatory AM and PM averages of MABP are shown in Fig. 6B and revealed that obese OS exhibited a significant hypertensive phenotype. A major concern with the chronic use of nitrite and NO-based therapeutics is the potential for significant and potentially dangerous reductions in systemic BP related to increased circulating NO levels and profound vasodilation. SR-nitrite therapy at the dosage investigated in the present study failed to exert a significant effect on MABP compared with placebo, indicating that this 80 mg po bid dose did not result in significant hypotension.
SR-nitrite therapy enhances myocardial eNOS function and NO bioavailability in obese OS after CLI.
Obesity and MetS in obese OS results in dysregulation of eNOS with increased phosphorylation of the enzyme's inhibitory site and reduction at the active site. This profound inhibition of eNOS results in reductions in NO bioavailability and acceleration in vascular and myocardial pathology. We next evaluated whether SR-nitrite therapy restored physiological regulation of myocardial eNOS via alterations in phosphorylation and potentially increased myocardial NO bioavailability in obese OS after the surgical induction of CLI (Fig. 7). Western blot analysis of heart tissue revealed that SR-nitrite therapy had no significant effect on total eNOS compared with placebo (Fig. 7B). Twenty-one days of SR-nitrite resulted in a significant increase in phosphorylation at eNOS Ser1177 (Fig. 7C) compared with placebo (1.5 ± 0.1 vs. 0.9 ± 0.2 RI, CLI + SR-nitrite vs. CLI + placebo, respectively, P < 0.05). Furthermore, SR-nitrite increased myocardial nitrite levels (P < 0.05; Fig. 7E) in the CLI + SR-nitrite-treated group (1.9 ± 0.2 nmol/mg protein) compared with the CLI + placebo-treated group (1.2 ± 0.3 nmol/mg protein). Interestingly, there was no change in eNOS Thr495 phosphorylation between CLI + SR-nitrite- and CLI + placebo-treated groups (Fig. 7D).
Fig. 7.

SR-nitrite therapy induces eNOS (Ser1177) active site phosphorylation and increases NO levels in the heart in obese OS after CLI. A: representative immunoblots of cardiac protein expression of phosphorylated eNOS (Ser1177), eNOS (Thr495), and total eNOS. B–D: densitometry analysis of total eNOS (B), phosphorylated eNOS (Ser1177; C), and phosphorylated eNOS (Thr495; D). E: cardiac nitrite levels in CLI + placebo- and CLI + SR-nitrite-treated groups. Values are means ± SE; numbers inside bars are numbers of animals per group.
SR-nitrite therapy reduced oxidative stress in obese OS after CLI.
We next evaluated the effects of chronic SR-nitrite treatment on myocardial oxidative stress in obese OS after CLI injury (Fig. 8). Cardiac levels of MDA (Fig. 8A) were significantly (P < 0.05) reduced in the CLI + SR-nitrite-treated group (3.3 ± 0.2 nmol/mg protein) compared with the CLI + placebo-treated group (4.1 ± 0.3 nmol/mg protein). Similarly, carbonyl protein levels (Fig. 8B) in CLI + SR-nitrite-treated hearts (20.5 ± 0.8 nmol/mg protein) were significantly (P < 0.05) attenuated compared with CLI + placebo-treated hearts (25.9 ± 1.5 nmol/mg protein). SR-nitrite therapy (23.7 ± 2.9 arbitrary units) increased myocardial GPx-1 (P < 0.05; Fig. 8C) levels in the CLI + SR-nitrite-treated group compared with the CLI + placebo-treated control group (14.2 ± 1.7 arbitrary units). Although there was a trend toward an increase, there was no difference in myocardial catalase, SOD1, or SOD2 (Fig. 8, D–F) between CLI + SR-nitrite- and CLI + placebo-treated groups (P = not significant for all between study groups).
Fig. 8.

Nitrite therapy with sustained release SR-nitrite significantly reduced myocardial oxidative stress and increased antioxidant enzyme mRNA levels in obese OS after CLI. A–F: MDA (A), carbonyl protein (B), GPx-1 (C), SOD1 (D), catalase (E), and SOD2 (F) mRNA levels in the hearts of CLI + placebo- and CLI + SR-nitrite-treated pigs. Values are means ± SE; numbers inside bars are numbers of animals per group.
SR-nitrite therapy improves coronary vascular reactivity in the setting of obese MetS and CLI.
One of the primary goals of the present study was to examine the effects of chronic administration of SR-nitrite on coronary vascular reactivity in vitro. LAD arteries were removed from lean nonischemic control pigs (Yorkshire domestic farm pigs) and from CLI-treated obese OS that received placebo or SR-nitrite for 21 days. These data are shown in Figs. 9–11. The contraction response (Fig. 9) to KCl and PGF2α demonstrated differences between nonischemic control swine and CLI-treated obese OS. Using lean nonischemic Yorkshire pigs as a reference, we observed a 49% reduction in contraction of CLI-treated obese OS coronaries at concentrations of 40 mM (Fig. 9A) and 100 mM (Fig. 9B) KCl compared with nonischemic control swine. The LAD contraction response to PGF2α (Fig. 9C) was attenuated by 72% in CLI-treated obese OS compared with nonischemic controls (P < 0.001). There was no significant difference between the CLI + placebo-treated group compared with the CLI + SR-nitrite-treated group (P = not significant between groups).
Fig. 9.

Obese OS subjected to CLI exhibit impaired coronary vascular contraction compared with nonischemic, lean Yorkshire control animals. A–C: left anterior descending coronary artery vasoconstriction with KCl at 40 mM (A) and 100 mM (B) or PGF2α at 30 mM (C). Values are means ± SE; numbers inside bars are numbers of animals per group.
Administration of SNP resulted in a dose-dependent relaxation from PGF2α-induced contraction in nonischemic control, CLI + placebo-treated, and CLI + SR-nitrite-treated isolated LAD coronary arteries (Fig. 10A). LAD coronary arteries from the CLI + SR-nitrite-treated group demonstrated a significantly (P < 0.05) greater relaxation at 10−6 and 10−5 M SNP compared with the CLI + placebo-treated group. Maximum relaxation in response to SNP (Fig. 10B) revealed a significantly greater (60% increase) vasorelaxation of CLI + SR-nitrite-treated LAD arteries compared with CLI + placebo-treated LAD arteries (P < 0.05). However, the relaxation was impaired compared with LAD arteries from lean nonischemic controls (Fig. 10), with the CLI + SR-nitrite-treated group have a 30% decrease in maximum relaxation (P < 0.05).
Fig. 10.
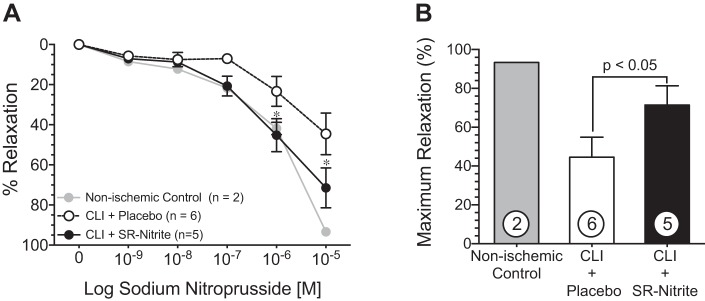
SR-nitrite therapy improves coronary vascular reactivity by an NO-dependent mechanism in the setting of MetS and CLI. Freshly isolated coronary arteries were precontracted with PGF2α. A: dose-dependent relaxation (%relaxation) of the left anterior descending coronary artery in response to sodium nitroprusside. B: maximum relaxation at 10−5 M sodium nitroprusside. Values are means ± SE; numbers inside bars are numbers of animals per group. *P < 0.05 vs. the CLI + placebo-treated group.
Substance P and bradykinin were studied to assess endothelium-dependent relaxation in isolated LAD coronary arteries. Although lean nonischemic coronary arteries demonstrated a dose-dependent response to substance P (Fig. 11A) and bradykinin (Fig. 11B), the relaxation of CLI + placebo- and CLI + SR-nitrite-treated coronary arteries was impaired. There was no significant difference in the maximum relaxation response (percentage of contraction) between CLI + placebo- and CLI + SR-nitrite-treated groups in response to 10−8 M substance P or 10−5 M bradykinin (Fig. 11, C and D).
DISCUSSION
MetS, with a rising prevalence worldwide, is associated with increases in the risk of cardiovascular morbidity and mortality (32). In the present study, we have provided evidence that the obese OS is a clinically relevant model of MetS that exhibits profound coronary vascular dysfunction that appears to be mediated via reduction in NO bioavailability and increased oxidative stress.
The OS has a “thrifty genotype” that enables the storage of excess food when available and thereby promotes survival during times of famine. In agreement with previously reported findings (11, 35), we observed that 6 mo of a high-fat, high-cholesterol diet resulted in central obesity, elevated blood glucose, increased cholesterol, and increased triglycerides compared with lean OS fed a standard diet. As defined by Adult Treatment Panel III (28), our data clearly demonstrate a MetS phenotype in the obese OS fed a high-calorie, atherogenic diet.
The combination of cardiovascular risk factors associated with MetS has a significant impact on vascular integrity, with many MetS patients presenting vascular dysfunction in conduits and small arteries (18, 42). It has been shown in clinical studies in patients with coronary artery disease (7, 19, 44) that virtually every cardiovascular disease risk factor identified to date promotes endothelial dysfunction and attenuates NO bioavailability, thus leading us to postulate that the link between MetS and vascular disease, specifically coronary artery disease, is mediated by loss of eNOS functionality and reduced endogenous NO production. NO derived from coronary arteries and cardiac myocytes plays a significant role in vessel reactivity and cardiac myocyte homeostasis, helping to protect against pathological insult (29). NO synthesis in the heart and vessels is regulated primarily by eNOS. The generation of NO by eNOS is mediated by multisite phosphorylation at Ser1177 and Thr495 amino acids sites either enhancing or inhibiting eNOS generation of NO, respectively (4, 26, 34). Endothelial cells from MetS patients display increases in phosphorylation at Thr495 and decreases in NO production (1). Furthermore, Marchesi et al. (33) have demonstrated eNOS uncoupling in New Zealand obese mice with MetS impairing vascular function. In the present study, we observed a marked reduction in phosphorylation at Ser1177 and an increase in phosphorylation at Thr495 in the hearts of obese OS compared with lean OS. These findings were coupled with concomitant reductions in myocardial nitrite and coronary cGMP, confirming decreased NO bioavailability in the heart and signaling in the coronaries.
The loss of eNOS function and reduction in NO bioavailability in response to MetS may be due to several possible mechanisms including excessive oxidative stress and/or increased levels of endogenous eNOS inhibitors. Oxidative stress, as measured by urinary 8-epi-PGF2α, correlates to an increasing body mass index in humans (14, 15). The Framingham study (22) reported that both body mass index and diabetes are associated with systemic oxidative stress. In the present study, we observed significant increases in protein carbonyl content, a marker of oxidative stress, in myocardial tissue from obese OS compared with lean OS. In Fischer rats fed a high-fat, high-refined sugar diet, Roberts and colleagues (41) reported reductions in the expression of the powerful antioxidant enzymes GPx-1 and HO-2 in the aorta. Although we observed no statistical change in GPx-1, obese OS had a significantly lower antioxidant expression of myocardial HO-1 mRNA compared with lean OS. Interestingly, we observed an increase in myocardial SOD2 mRNA levels in the obese OS group, suggesting a possible compensatory response to increased oxidative stress resulting in increased levels of this antioxidant enzyme. Obese OS also had marked increases in plasma ADMA, an endogenous inhibitor of eNOS. Several investigators have reported increases in ADMA levels in rats and humans with various risk factors of MetS, including hyperglycemia (25) and hypercholesterolemia (3). Moreover, a number of cardiovascular diseases correlate with increases in circulating ADMA and oxidative stress (45, 46).
Previous studies from our laboratory have shown that daily sodium nitrite supplementation enhances myocardial nitrite levels in murine cardiac disease models of NO deficiency (2, 6). Nitrite therapy reverses age-induced vascular endothelial dysfunction in mice (43). Because there is a high prevalence of MetS patients that present with clinical manifestations of atherosclerotic vascular disease, specifically PAD (17), we developed a model that would represent this high-risk population. Similar to coronary artery disease, PAD patients exhibit impaired NO synthesis (3) and endothelial dysfunction (5, 16). The vascular dysfunctions associated with PAD increase the risk of vascular complications in other area, including the coronary circulation (13). Since endogenous nitrite has been shown to elicit endocrine effects and mediate cytoprotection in remote organs (12), we hypothesized that chronic administration of SR-nitrite would attenuate myocardial oxidative stress and improve coronary vascular reactivity in the setting of MetS and CLI. In the present study, obese OS were treated daily with a SR-nitrite formulation (80 mg po bid) starting at 14 days after the onset of CLI injury. SR-nitrite restored myocardial phosphorylation of the eNOS Ser1177 active site in the SR-nitrite-treated OS group, resulting in increased NO bioavailability in the heart. We determined that the dosage of SR-nitrite that was used in the present study did not reduce systemic BP, suggesting a favorable safety profile and possible clinical utility for peripheral vascular and other cardiovascular diseases.
Under certain physiological conditions, NO is a potent scavenger of superoxide anion that modulates the redox balance in the circulation and tissues. Thus, the balance between NO formation and superoxide is critical for cardiovascular homeostasis (8). In the present study, SR-nitrite treatment attenuated MetS-induced increases in oxidative stress as the CLI + SR-nitrite-treated group exhibited significantly lower levels of the oxidative stress markers MDA and protein carbonyl. SR-nitrite also resulted in significant elevations of GPx-1 and catalase. These effects may have occurred by the scavenging of the superoxide anion by nitrite-derived NO.
Previous studies have evaluated vascular reactivity in the obese OS model. Payne et al. (39) demonstrated that coronary arteries obtained from obese OS were associated with endothelial dysfunction, which was exacerbated in the presence of perivascular adipose tissue. In the present study, LAD coronary arteries obtained from obese OS had attenuated contraction to KCl and PGF2α compared with Yorkshire nonischemic lean control swine with no significant difference between CLI + SR-nitrite- and CLI + placebo-treated groups. Similar to our findings, Owen et al. (38) concluded that obesity augments Ca2+ cycling and smooth muscle vasoconstriction after observing an attenuated contraction in obese OS coronaries compared with lean control arteries. Using vasodilators that induce relaxation through endothelium-independent (SNP) and endothelium-dependent (substance P and bradykinin) mechanisms, we examined the vascular reactivity of LAD coronary arteries obtained from CLI + SR-nitrite-treated pigs, CLI + placebo-treated pigs, and nonischemic lean control pigs. PGF2α-induced precontracted LAD coronary arteries displayed impaired relaxation in response to SNP, substance P, and bradykinin in obese OS compared with nonischemic lean control swine, clearly indicating that vasodilatory properties of the vessel are impaired in the setting of obesity and MetS. In obese OS, SR-nitrite therapy improved relaxation in response to SNP, indicating the partial restoration of smooth muscle-dependent vasorelaxation. However, we did not observe improved vasoreactivity in response to an endothelium-dependent vasorelaxation (i.e., substance P or bradykinin) mechanism, signifying that the combination of MetS and CLI induces endothelial dysfunction. Although chronic nitrite treatment has been shown to reverse age-dependent endothelial dysfunction (43), we did not observe improvements in endothelium-dependent vasorelaxation. We believe that the primary reason for the difference between the two experimental studies is that obese OS subjected to CLI represent a significantly more severe form of cardiovascular disease compared with aging alone. Obese OS exhibit severe hyperlipidemia coupled with coronary artery atherosclerotic lesions, and, therefore, restoration of coronary endothelium-dependent vascular reactivity may not be possible in these animals in this 35-day treatment protocol. Longer durations of SR-nitrite therapy may be required to partially restore endothelium-dependent coronary vascular reactivity.
In conclusion, our findings demonstrate that OS fed a high-fat, high-cholesterol diet result in a MetS phenotype that has significant impact on NO bioavailability by impairing eNOS function in the heart. Concomitant with reduced NO bioavailability, we observed a significant increase in myocardial oxidative stress in obese OS. SR-nitrite therapy restored cardiac eNOS function in the setting of severe CLI by enhancing myocardial nitrite levels and attenuating oxidative stress. Although CLI in the setting of MetS was associated with marked oxidative stress and profound endothelial dysfunction, SR-nitrite therapy improved ex vivo vascular function of coronary arteries via direct effects on coronary vascular smooth muscle. SR-nitrite therapy may prove beneficial for MetS patients with preexisting chronic cardiovascular disease states, such as CLI, that are characterized by an upregulation of oxidative stress and vascular dysfunction.
GRANTS
This work was supported by National Institutes of Health (NIH) Grants 1-R01-HL-092141 (to D. J. Lefer), 1-R01-HL-093579 (to D. J. Lefer), 1-U24-HL-094373 (to D. J. Lefer), 1-P20-HL-113452 (to D. J. Lefer), and 1-KO8-HL-119592 (to L. P. Brewster) and supported in part by NIH Grant UL1-TR-000454 from the Clinical and Translational Science Award Program (to L. P. Brewster) and American Heart Association Grant 13IRG14740001 (to L. P. Brewster). The authors are also grateful for the generous funding from the Carlyle Fraser Heart Center at Emory University Midtown Hospital, the Emory University Department of Surgery, and the Louisiana State University Medical School Alumni Association.
DISCLOSURES
D. J. Lefer is on the scientific advisory board of Theravasc. Theravasc is currently developing sodium nitrite for the treatment of cardiovascular diseases. D. J. Lefer is a participant of a pending United States patent filed on October 14, 2003 (patent no. 60/511244), regarding the use of sodium nitrite in cardiovascular disease. D. J. Lefer is a participant of a pending United States patent filed on November 15, 2007 (patent no. 61/003150), regarding the use of nitrite salts in chronic ischemia.
AUTHOR CONTRIBUTIONS
Author contributions: J.M.B., K.N.I., L.P.B., T.T.G., and D.J.L. conception and design of research; J.M.B., K.N.I., D.J.P., E.D., L.P.B., Y.T., T.T.G., and D.J.L. performed experiments; J.M.B., K.N.I., D.J.P., E.D., and Y.T. analyzed data; J.M.B., K.N.I., D.J.P., and D.J.L. interpreted results of experiments; J.M.B., K.N.I., and D.J.P. prepared figures; J.M.B. and K.N.I. drafted manuscript; J.M.B., K.N.I., T.T.G., and D.J.L. edited and revised manuscript; J.M.B., K.N.I., D.J.P., E.D., L.P.B., Y.T., T.T.G., and D.J.L. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors are grateful to TheraVasc (Cleveland, OH) for the generous supply of SR Nitrite for these experiments. The authors thank W. L. Gore and Associates (Flagstaff, AZ) for providing the GORE VIABAHN Endoprosthesis used in these experiments. The authors thank Liposcience, Inc. (Raleigh, NC) for the generous and skillful advanced lipid testing. The authors also thank Shashi Bhushan, Hiroyuki Otsuka, Scott Robinson, Kevin Brown, Michael Sweet, and Amy Souder for the expert technical assistance during the course of these experiments.
REFERENCES
- 1.Agouni A, Lagrue-Lak-Hal AH, Ducluzeau PH, Mostefai HA, Draunet-Busson C, Leftheriotis G, Heymes C, Martinez MC, Andriantsitohaina R. Endothelial dysfunction caused by circulating microparticles from patients with metabolic syndrome. Am J Pathol 173: 1210–1219, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bhushan S, Kondo K, Polhemus DJ, Otsuka H, Nicholson CK, Tao YX, Huang H, Georgiopoulou VV, Murohara T, Calvert JW, Butler J, Lefer DJ. Nitrite therapy improves left ventricular function during heart failure via restoration of nitric oxide-mediated cytoprotective signaling. Circ Res 114: 1281–1291, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boger RH, Bode-Boger SM, Szuba A, Tsao PS, Chan JR, Tangphao O, Blaschke TF, Cooke JP. Asymmetric dimethylarginine (ADMA): a novel risk factor for endothelial dysfunction: its role in hypercholesterolemia. Circulation 98: 1842–1847, 1998. [DOI] [PubMed] [Google Scholar]
- 4.Boo YC, Sorescu G, Boyd N, Shiojima I, Walsh K, Du J, Jo H. Shear stress stimulates phosphorylation of endothelial nitric-oxide synthase at Ser1179 by Akt-independent mechanisms: role of protein kinase A. J Biol Chem 277: 3388–3396, 2002. [DOI] [PubMed] [Google Scholar]
- 5.Brevetti G, Silvestro A, Schiano V, Chiariello M. Endothelial dysfunction and cardiovascular risk prediction in peripheral arterial disease: additive value of flow-mediated dilation to ankle-brachial pressure index. Circulation 108: 2093–2098, 2003. [DOI] [PubMed] [Google Scholar]
- 6.Bryan NS, Calvert JW, Elrod JW, Gundewar S, Ji SY, Lefer DJ. Dietary nitrite supplementation protects against myocardial ischemia-reperfusion injury. Proc Natl Acad Sci USA 104: 19144–19149, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res 87: 840–844, 2000. [DOI] [PubMed] [Google Scholar]
- 8.Carlstrom M, Larsen FJ, Nystrom T, Hezel M, Borniquel S, Weitzberg E, Lundberg JO. Dietary inorganic nitrate reverses features of metabolic syndrome in endothelial nitric oxide synthase-deficient mice. Proc Natl Acad Sci USA 107: 17716–17720, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cooke JP. Does ADMA cause endothelial dysfunction? Arterioscler Thromb Vasc Biol 20: 2032–2037, 2000. [DOI] [PubMed] [Google Scholar]
- 10.Cosby K, Partovi KS, Crawford JH, Patel RP, Reiter CD, Martyr S, Yang BK, Waclawiw MA, Zalos G, Xu X, Huang KT, Shields H, Kim-Shapiro DB, Schechter AN, Cannon RO 3rd, Gladwin MT. Nitrite reduction to nitric oxide by deoxyhemoglobin vasodilates the human circulation. Nat Med 9: 1498–1505, 2003. [DOI] [PubMed] [Google Scholar]
- 11.Dyson MC, Alloosh M, Vuchetich JP, Mokelke EA, Sturek M. Components of metabolic syndrome and coronary artery disease in female Ossabaw swine fed excess atherogenic diet. Comp Med 56: 35–45, 2006. [PubMed] [Google Scholar]
- 12.Elrod JW, Calvert JW, Gundewar S, Bryan NS, Lefer DJ. Nitric oxide promotes distant organ protection: evidence for an endocrine role of nitric oxide. Proc Natl Acad Sci USA 105: 11430–11435, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Froehlich JB, Mukherjee D, Avezum A, Budaj A, Kline-Rogers EM, Lopez-Sendon J, Allegrone J, Eagle KA, Mehta RH, Goldberg RJ; GRACE Investigators. Association of peripheral artery disease with treatment and outcomes in acute coronary syndromes. The Global Registry of Acute Coronary Events (GRACE). Am Heart J 151: 1123–1128, 2006. [DOI] [PubMed] [Google Scholar]
- 14.Fujita K, Nishizawa H, Funahashi T, Shimomura I, Shimabukuro M. Systemic oxidative stress is associated with visceral fat accumulation and the metabolic syndrome. Circ J 70: 1437–1442, 2006. [DOI] [PubMed] [Google Scholar]
- 15.Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y, Nakayama O, Makishima M, Matsuda M, Shimomura I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest 114: 1752–1761, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gokce N, Keaney JF Jr, Hunter LM, Watkins MT, Nedeljkovic ZS, Menzoian JO, Vita JA. Predictive value of noninvasively determined endothelial dysfunction for long-term cardiovascular events in patients with peripheral vascular disease. J Am Coll Cardiol 41: 1769–1775, 2003. [DOI] [PubMed] [Google Scholar]
- 17.Gorter PM, Olijhoek JK, van der Graaf Y, Algra A, Rabelink TJ, Visseren FL, Group SS. Prevalence of the metabolic syndrome in patients with coronary heart disease, cerebrovascular disease, peripheral arterial disease or abdominal aortic aneurysm. Atherosclerosis 173: 363–369, 2004. [DOI] [PubMed] [Google Scholar]
- 18.Greenstein AS, Khavandi K, Withers SB, Sonoyama K, Clancy O, Jeziorska M, Laing I, Yates AP, Pemberton PW, Malik RA, Heagerty AM. Local inflammation and hypoxia abolish the protective anticontractile properties of perivascular fat in obese patients. Circulation 119: 1661–1670, 2009. [DOI] [PubMed] [Google Scholar]
- 19.Heitzer T, Schlinzig T, Krohn K, Meinertz T, Munzel T. Endothelial dysfunction, oxidative stress, and risk of cardiovascular events in patients with coronary artery disease. Circulation 104: 2673–2678, 2001. [DOI] [PubMed] [Google Scholar]
- 20.Hooi JD, Kester AD, Stoffers HE, Rinkens PE, Knottnerus JA, van Ree JW. Asymptomatic peripheral arterial occlusive disease predicted cardiovascular morbidity and mortality in a 7-year follow-up study. J Clin Epidemiol 57: 294–300, 2004. [DOI] [PubMed] [Google Scholar]
- 21.Hooi JD, Stoffers HE, Knottnerus JA, van Ree JW. The prognosis of non-critical limb ischaemia: a systematic review of population-based evidence. Br J Gen Pract 49: 49–55, 1999. [PMC free article] [PubMed] [Google Scholar]
- 22.Keaney JF Jr, Larson MG, Vasan RS, Wilson PW, Lipinska I, Corey D, Massaro JM, Sutherland P, Vita JA, Benjamin EJ, Framingham S. Obesity and systemic oxidative stress: clinical correlates of oxidative stress in the Framingham study. Arterioscler Thromb Vasc Biol 23: 434–439, 2003. [DOI] [PubMed] [Google Scholar]
- 23.King AL, Polhemus DJ, Bhushan S, Otsuka H, Kondo K, Nicholson CK, Bradley JM, Islam KN, Calvert JW, Tao YX, Dugas TR, Kelley EE, Elrod JW, Huang PL, Wang R, Lefer DJ. Hydrogen sulfide cytoprotective signaling is endothelial nitric oxide synthase-nitric oxide dependent. Proc Natl Acad Sci USA 111: 3182–3187, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee L, Alloosh M, Saxena R, Van Alstine W, Watkins BA, Klaunig JE, Sturek M, Chalasani N. Nutritional model of steatohepatitis and metabolic syndrome in the Ossabaw miniature swine. Hepatology 50: 56–67, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin KY, Ito A, Asagami T, Tsao PS, Adimoolam S, Kimoto M, Tsuji H, Reaven GM, Cooke JP. Impaired nitric oxide synthase pathway in diabetes mellitus: role of asymmetric dimethylarginine and dimethylarginine dimethylaminohydrolase. Circulation 106: 987–992, 2002. [DOI] [PubMed] [Google Scholar]
- 26.Lin MI, Fulton D, Babbitt R, Fleming I, Busse R, Pritchard KA Jr, Sessa WC. Phosphorylation of threonine 497 in endothelial nitric-oxide synthase coordinates the coupling of l-arginine metabolism to efficient nitric oxide production. J Biol Chem 278: 44719–44726, 2003. [DOI] [PubMed] [Google Scholar]
- 27.Lloyd-Jones D, Adams RJ, Brown TM, Carnethon M, Dai S, De Simone G, Ferguson TB, Ford E, Furie K, Gillespie C, Go A, Greenlund K, Haase N, Hailpern S, Ho PM, Howard V, Kissela B, Kittner S, Lackland D, Lisabeth L, Marelli A, McDermott MM, Meigs J, Mozaffarian D, Mussolino M, Nichol G, Roger VL, Rosamond W, Sacco R, Sorlie P, Stafford R, Thom T, Wasserthiel-Smoller S, Wong ND, Wylie-Rosett J; American Heart Association Statistics Committee; Stroke Statistics Subcommittee. Executive summary: heart disease and stroke statistics–2010 update: a report from the American Heart Association. Circulation 121: 948–954, 2010. [DOI] [PubMed] [Google Scholar]
- 28.Lorenzo C, Williams K, Hunt KJ, Haffner SM. The National Cholesterol Education Program–Adult Treatment Panel III, International Diabetes Federation, and World Health Organization definitions of the metabolic syndrome as predictors of incident cardiovascular disease and diabetes. Diabetes Care 30: 8–13, 2007. [DOI] [PubMed] [Google Scholar]
- 29.Loscalzo J, Welch G. Nitric oxide and its role in the cardiovascular system. Progr Cardiovasc Dis 38: 87–104, 1995. [DOI] [PubMed] [Google Scholar]
- 30.Lundberg JO, Gladwin MT, Ahluwalia A, Benjamin N, Bryan NS, Butler A, Cabrales P, Fago A, Feelisch M, Ford PC, Freeman BA, Frenneaux M, Friedman J, Kelm M, Kevil CG, Kim-Shapiro DB, Kozlov AV, Lancaster JR Jr, Lefer DJ, McColl K, McCurry K, Patel RP, Petersson J, Rassaf T, Reutov VP, Richter-Addo GB, Schechter A, Shiva S, Tsuchiya K, van Faassen EE, Webb AJ, Zuckerbraun BS, Zweier JL, Weitzberg E. Nitrate and nitrite in biology, nutrition and therapeutics. Nat Chem Biol 5: 865–869, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lundberg JO, Weitzberg E, Gladwin MT. The nitrate-nitrite-nitric oxide pathway in physiology and therapeutics. Nat Rev Drug Discov 7: 156–167, 2008. [DOI] [PubMed] [Google Scholar]
- 32.Malik S, Wong ND, Franklin SS, Kamath TV, L'Italien GJ, Pio JR, Williams GR. Impact of the metabolic syndrome on mortality from coronary heart disease, cardiovascular disease, and all causes in United States adults. Circulation 110: 1245–1250, 2004. [DOI] [PubMed] [Google Scholar]
- 33.Marchesi C, Ebrahimian T, Angulo O, Paradis P, Schiffrin EL. Endothelial nitric oxide synthase uncoupling and perivascular adipose oxidative stress and inflammation contribute to vascular dysfunction in a rodent model of metabolic syndrome. Hypertension 54: 1384–1392, 2009. [DOI] [PubMed] [Google Scholar]
- 34.Mount PF, Kemp BE, Power DA. Regulation of endothelial and myocardial NO synthesis by multi-site eNOS phosphorylation. J Mol Cell Cardiol 42: 271–279, 2007. [DOI] [PubMed] [Google Scholar]
- 35.Neeb ZP, Edwards JM, Alloosh M, Long X, Mokelke EA, Sturek M. Metabolic syndrome and coronary artery disease in Ossabaw compared with Yucatan swine. Comp Med 60: 300–315, 2010. [PMC free article] [PubMed] [Google Scholar]
- 36.Nicholson CK, Lambert JP, Chow CW, Lefer DJ, Calvert JW. Chronic exercise downregulates myocardial myoglobin and attenuates nitrite reductase capacity during ischemia-reperfusion. J Mol Cell Cardiol 64: 1–10, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Olijhoek JK, van der Graaf Y, Banga JD, Algra A, Rabelink TJ, Visseren FL, Group SS. The metabolic syndrome is associated with advanced vascular damage in patients with coronary heart disease, stroke, peripheral arterial disease or abdominal aortic aneurysm. Eur Heart J 25: 342–348, 2004. [DOI] [PubMed] [Google Scholar]
- 38.Owen MK, Witzmann FA, McKenney ML, Lai X, Berwick ZC, Moberly SP, Alloosh M, Sturek M, Tune JD. Perivascular adipose tissue potentiates contraction of coronary vascular smooth muscle: influence of obesity. Circulation 128: 9–18, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Payne GA, Borbouse L, Kumar S, Neeb Z, Alloosh M, Sturek M, Tune JD. Epicardial perivascular adipose-derived leptin exacerbates coronary endothelial dysfunction in metabolic syndrome via a protein kinase C-β pathway. Arterioscler Thromb Vasc Biol 30: 1711–1717, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Polhemus DJ, Bradley JM, Islam KI, Brewster LP, Calvert JW, Tao YX, Chang CC, Pipinos II, Goodchild TT, Lefer DJ. Therapeutic potental of sustained release sodium nitrite for critical limb ischemia in the setting of metabolic syndrome. Am J Physiol Heart Circ Physiol; doi: 10.1152/ajpheart.00115.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roberts CK, Barnard RJ, Sindhu RK, Jurczak M, Ehdaie A, Vaziri ND. Oxidative stress and dysregulation of NAD(P)H oxidase and antioxidant enzymes in diet-induced metabolic syndrome. Metabolism 55: 928–934, 2006. [DOI] [PubMed] [Google Scholar]
- 42.Schillaci G, Pirro M, Vaudo G, Mannarino MR, Savarese G, Pucci G, Franklin SS, Mannarino E. Metabolic syndrome is associated with aortic stiffness in untreated essential hypertension. Hypertension 45: 1078–1082, 2005. [DOI] [PubMed] [Google Scholar]
- 43.Sindler AL, Fleenor BS, Calvert JW, Marshall KD, Zigler ML, Lefer DJ, Seals DR. Nitrite supplementation reverses vascular endothelial dysfunction and large elastic artery stiffness with aging. Aging Cell 10: 429–437, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Suwaidi JA, Hamasaki S, Higano ST, Nishimura RA, Holmes DR Jr, Lerman A. Long-term follow-up of patients with mild coronary artery disease and endothelial dysfunction. Circulation 101: 948–954, 2000. [DOI] [PubMed] [Google Scholar]
- 45.Wang D, Strandgaard S, Iversen J, Wilcox CS. Asymmetric dimethylarginine, oxidative stress, and vascular nitric oxide synthase in essential hypertension. Am J Physiol Regul Integr Comp Physiol 296: R195–R200, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yilmaz MI, Saglam M, Caglar K, Cakir E, Sonmez A, Ozgurtas T, Aydin A, Eyileten T, Ozcan O, Acikel C, Tasar M, Genctoy G, Erbil K, Vural A, Zoccali C. The determinants of endothelial dysfunction in CKD: oxidative stress and asymmetric dimethylarginine. Am J Kidney Dis 47: 42–50, 2006. [DOI] [PubMed] [Google Scholar]