

Abstract
Suppression of type 17 immunity by type I interferon (IFN) during influenza A infection has been shown to enhance susceptibility to secondary bacterial pneumonia. Although this mechanism has been described in coinfection with gram-positive bacteria, it is unclear whether similar mechanisms may impair lung defense against gram-negative infections. Furthermore, precise delineation of the duration of type I IFN-associated susceptibility to bacterial infection remains underexplored. Therefore, we investigated the effects of preceding influenza A virus infection on subsequent challenge with the gram-negative bacteria Escherichia coli or Pseudomonas aeruginosa and the temporal association between IFN expression with susceptibility to Staphylococcus aureus challenge in a mouse model of influenza and bacterial coinfection. Here we demonstrate that preceding influenza A virus led to increased lung E. coli and P. aeruginosa bacterial burden, which was associated with suppression of type 17 immunity and attenuation of antimicrobial peptide expression. Enhanced susceptibility to S. aureus coinfection ceased at day 14 of influenza infection, when influenza-associated type I IFN levels had returned to baseline levels, further suggesting a key role for type I IFN in coinfection pathogenesis. These findings further implicate type I IFN-associated suppression of type 17 immunity and antimicrobial peptide production as a conserved mechanism for enhanced susceptibility to both gram-positive and gram-negative bacterial coinfection during influenza infection.
Keywords: influenza A, Escherichia coli, Staphylococcus aureus, coinfection, type I interferon
influenza, a highly contagious group of RNA viruses of the family Orthomyxoviridae, is one of the most important causes of seasonal respiratory illness worldwide. The majority of infected patients resolve influenza infection without difficulty. However, a subset of patients suffers from severe disease or mortality. In the United States, influenza-related mortality can reach nearly 50,000 deaths in some seasons (1). In addition to the burden posed by seasonal influenza epidemics, the potential for catastrophic pandemic influenza A is a constant threat. Despite the availability of influenza vaccines, suboptimal vaccine efficacy, uptake, and strain matching make influenza infection an ongoing public health problem.
The association between influenza A virus (IAV) infection and secondary bacterial pneumonia, particularly during pandemic disease, is now well established. During the 1918 Spanish influenza pandemic, nearly all influenza-associated deaths were associated with evidence of bacterial superinfection, predominantly with upper respiratory tract bacterial pathogens (32). In recent years, an increasing role for bacterial superinfections as a cause of influenza-associated mortality during seasonal influenza epidemics, particularly due to community-acquired methicillin-resistant Staphylococcus aureus (CA-MRSA), has been appreciated (14, 18, 36). Therefore, increasing focus has been paid toward understanding the immunological mechanisms that enhance host susceptibility to secondary bacterial pneumonia during IAV infection, including the role of type 17 immunity.
Activation of type 17 immunity requires stimulation by IL-1β and IL-23, leading to IL-17 and IL-22 secretion by type 17 T cells following induction of the transcription factors ROR-α and ROR-γt (2). This signaling cascade results in two major effector functions: the proliferation and recruitment of inflammatory cells, and the elaboration of antimicrobial peptides (AMP) by the mucosal epithelium. We have previously demonstrated that attenuation of type 17 immune responses by preceding IAV infection is an important component of increased susceptibility to secondary gram-positive bacterial pneumonia in mice and that this mechanism was mediated at least in part by viral-induced type I interferon (IFN) via suppression of IL-23 (23). In humans, coinfection with gram-negative bacterial species occurs less frequently than with gram-positive organisms, but it is unclear whether this is due to differences in host responses or bacteria-specific factors. Although the importance of type 17 immune responses to gram-negative bacterial lung infections, such as Klebsiella pneumoniae and E. coli, has previously been demonstrated (3, 4, 19, 45, 46), it is unknown whether similar mechanisms could suppress type 17 immune responses to pulmonary challenge with gram-negative bacteria during IAV infection. Furthermore, the kinetics of type I IFN-mediated suppression of type 17 immunity remains underexplored. It would seem likely that a critical window period exists for susceptibility to enhanced secondary bacterial infection, a window that closes as type I IFN production during IAV infection resolves upon viral clearance.
To examine type 17 immune responses to gram-negative bacteria during IAV coinfection and the kinetics of type 17 immune suppression by IAV, we subjected both wild-type (WT) and IFN-α receptor (IFNAR)−/− mice to bacterial challenge with the gram-negative bacteria Escherichia coli and Pseudomonas aeruginosa following IAV infection. In addition, we assessed the kinetics of influenza-associated type I IFN induction in relation to type 17 immune inhibition and susceptibility to pneumonia following challenge with S. aureus.
METHODS
Animals.
WT 6- to 8-wk-old male C57BL/6 mice were purchased from Taconic Farms (Germantown, NY). IFNAR−/− mice were generated as previously reported (16, 21). Mice were maintained under pathogen-free conditions by the University of Pittsburgh, Division of Laboratory Animal Resources. All studies were conducted on age- and sex-matched mice. All experiments were approved by the University of Pittsburgh Institutional Animal Care and Use Committee.
Bacterial and viral infections.
Panton-Valentine leukocidin and γ-hemolysin-producing methicillin-susceptible S. aureus [American Type Culture Collection (ATCC) 49775] was grown overnight to stationary growth phase in CCY-modified medium according to ATCC instructions. Mice received 1 × 108 colony-forming units (cfu) of bacteria in 50 μl of sterile PBS. E. coli (DH5α genotype containing the pMAL-p2 plasmid, expressing ampicillin resistance) was obtained as a gift from Dr. Marc Jenkins (University of Minnesota) (10). This strain is a widely used nonenteropathogenic laboratory strain. E. coli was grown overnight to stationary growth phase in Luria broth supplemented with 2 mg/ml glucose. Mice were infected with 1 × 107 cfu of bacteria in 50 μl of sterile PBS. P. aeruginosa strain PAO1 (ATCC BAA-47), a widely used laboratory strain descended from a pathogenic clinical isolate, was grown in 100 ml Luria-Bertani (LB) broth for 12 h. After 12 h, 1 ml of culture was added to fresh LB broth and grown for an additional 12 h. Bacteria were then pelleted, washed, and diluted to give a target inoculum in 50 μl PBS. Serial dilutions of bacteria were grown on LB plates and back calculated to confirm the inocula administered. In three separate experiments, mice received inocula of 4.5 × 105, 1.7 × 106, and 2.7 × 106 cfu. Influenza A/PR/8/34 was propagated in chicken eggs as described (8). Mice were infected with 100 plaque-forming units of virus in 50 μl of sterile PBS. All infections were delivered via oropharyngeal aspiration. Mice were sequentially challenged with influenza or vehicle for 6 or 14 days followed by infection with bacteria or vehicle for 1 additional day. Resulting treatments groups consisted of influenza alone, bacteria alone, or coinfection.
Measurement of lung inflammation.
One day following bacterial or vehicle challenge, mouse lungs were lavaged with 1 ml of sterile PBS. Cell differential counts were performed on bronchoalveolar lavage (BAL) fluid by cytospin and Protocol Hema 3 (Fisher Scientific, Kalamazoo, MI) staining. In BAL, lactate dehydrogenase (LDH) was measured by Cyto-Tox 96 Non-radioactive Cytotoxicity assay (Promega, Madison, WI) and total protein by Pierce BCA Protein Assay (Pierce Biotechnology, Rockford, IL), per manufacturer's instructions. The cranial lobe of the right lung was homogenized in sterile PBS by mechanical grinding for quantification of bacterial burden by serial dilution plating and for cytokine production measurement by Lincoplex (Millipore, Billerica, MA) or Bio-plex (Bio-Rad, Hercules, CA, for P. aeruginosa experiments). The middle and caudal lobes of the lung were snap frozen in liquid nitrogen for later purification of RNA by use of the Absolutely RNA Miniprep Kit (Agilent Technologies, Santa Clara, CA). Gene expression was analyzed by RT-PCR utilizing Assay on Demand TaqMan primer and probe sets (Applied Biosystems, Foster City, CA), by using the ΔΔCT method relative to the endogenous control gene hypoxanthine-guanine phosphoribosyltransferase.
Measurement of antimicrobial peptide production.
Human bronchial epithelia (HBE) were cultured from excess pathological tissue after lung transplantation and organ donation under protocols approved by the Universities of Pittsburgh Investigational Review Boards (IRB970946). Primary cultures of HBE cells were prepared by enzymatic dispersion by established methods as previously described (13, 26). Epithelial cells were dissociated and seeded onto collagen-coated, semipermeable membranes with a 0.4-μm pore size (Millicell-HA; surface area, 0.6 cm2; EMD Millipore, Billerica, MA). Cells were maintained in 2% Ultroser G medium at 37°C with 5% CO2. Twenty-four hours after seeding, the mucosal medium was removed and the cells were allowed to grow at the air-liquid interface. Only well-differentiated cultures (>4 wk old) were used in these studies. The presence of tight junctions between differentiated epithelial cells was confirmed by transepithelial resistance using an electrical resistance system (ERS) meter (EMD Millipore; resistance >500 Ω·cm2). Cells were stimulated for 24 h with IL-17A (10 ng/ml) and/or TNF-α (1 ng/ml) (R&D Systems, Minneapolis, MN). Gene expression was analyzed by TaqMan-based RT-PCR (ABI7700, Applied Biosystems, Foster City, CA) by using the ΔΔCT method relative to the endogenous control gene β-glucuronidase for the target BPIFA1, formerly known as PLUNC or SPLUNC1 (7). Human lipocalin 2 (LCN2) was assessed utilizing Assay on Demand TaqMan primer and probe sets (Applied Biosystems, Foster City, CA), relative to the endogenous control gene β-actin. BPIFA1 concentration was measured in HBE cell culture supernatant following 24 h of stimulation with cytokine by ELISA (Hycult Biotech, Plymouth Meeting, PA). In the mouse model, lung expression of mouse BPIFA1 and LCN2 was measured as described above.
Statistical analyses.
All data are presented as means ± standard error of the mean. Significance was determined by unpaired t-test or one-way ANOVA followed by post hoc Tukey's test for multiple comparisons, as appropriate. A P value ≤ 0.05 was considered to be statistically significant. Data were processed by use of the Microsoft Excel and GraphPad Prism (GraphPad Software, La Jolla, CA) software packages.
RESULTS
To examine the effects of IAV on type 17 immune responses during subsequent infection with gram-negative bacteria, mice were challenged with influenza A/PR/8/34 or vehicle for 6 days prior to challenge with E. coli or vehicle. Twenty-four hours later, tissues were harvested and bacterial burden and inflammation were assessed. Preceding IAV significantly increased E. coli burden in the lung and resulted in increased BAL neutrophil counts compared with single pathogen-challenged mice (Fig. 1, A and B). Furthermore, coinfection resulted in significantly increased levels of the granulocyte promoting cytokines IL-6, G-CSF, KC, and MIP-1α compared with either challenge alone (Fig. 1C). Similar to data found with S. aureus coinfection, IAV significantly attenuated subsequent IL-1β production in response to E. coli (Fig. 1D). TNF-α was significantly increased by coinfection, whereas IFN-γ production was significantly increased during influenza and during coinfection compared with bacterial infection alone. Coinfected mice also had evidence of greater lung damage compared with mice infected with bacteria alone, with increased LDH (mean OD490 1.3 vs. 0.28, P < 0.0001, data not shown) and total protein as measured by BCA assay (mean 3,999 vs. 592.2 μg/ml, P < 0.0001, data not shown). These data show that E. coli burden is increased during IAV coinfection despite elevated innate immune cell recruitment to the lung and that coinfected mice have greater lung damage than mice with bacterial pneumonia alone.
Fig. 1.
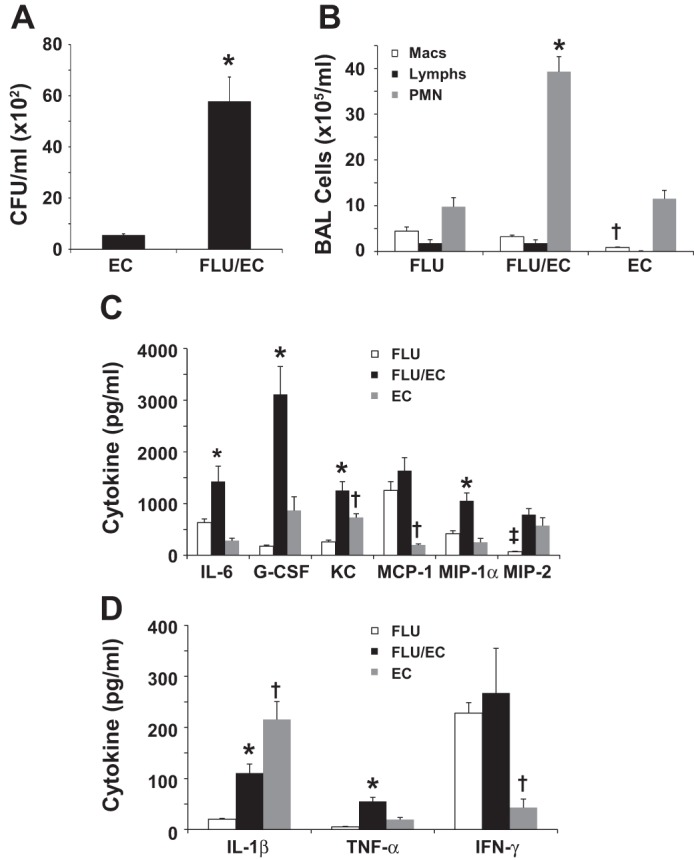
Preceding influenza infection exacerbates secondary gram-negative pneumonia with Escherichia coli. Mice were challenged with influenza A/PR/8/34 [100 plaque-forming units (pfu)] or vehicle for 6 days followed by infection with E. coli [1 × 107 colony-forming units (cfu)] for 24 h. EC, E. coli only; FLU/EC, influenza and E. coli; FLU, influenza only. A: bacterial burden in the lung (n = 7, 8). B: bronchoalveolar lavage (BAL) inflammatory cells (n = 8). Macs, macrophages; Lymphs, lymphocytes; PMN, neutrophils. C and D: inflammatory cytokines in lung homogenate (n = 8). *P < 0.05 vs. FLU and EC; †P < 0.05 vs. FLU and FLU/EC; ‡P < 0.05 vs. FLU/EC and EC.
The initial findings in the IAV-E. coli coinfection model were thus similar to those reported with S. aureus (23). We next examined whether type 17 immune activation by E. coli was suppressed by preceding IAV infection. IL-17 and IL-22 expression were markedly induced by E. coli challenge and this production was significantly attenuated by preceding IAV (Fig. 2A). Similarly, IL-17 protein concentration was significantly increased in E. coli infection alone but not during coinfection (Fig. 2B). Furthermore, E. coli significantly induced expression of the type 17 immune cell transcription factors ROR-γt and ROR-α, as well as the promoting cytokine IL-23. Preceding IAV infection inhibited expression of all three type 17 markers, whereas type I IFN induction was significantly exacerbated by coinfection (Fig. 2C).
Fig. 2.

Preceding influenza infection attenuates type 17 immunity during secondary gram-negative pneumonia with E. coli. Mice were challenged with influenza A/PR/8/34 (100 pfu) or vehicle for 6 days followed by infection with E. coli (1 × 107 cfu) for 24 h. EC, E. coli only; FLU/EC, influenza and E. coli; FLU, influenza only. A and C: cytokine gene expression in lung (n = 4). B: inflammatory cytokine in lung homogenate (n = 8). *P < 0.05 vs. FLU and EC; †P < 0.05 vs. FLU and FLU/EC; ‡P < 0.05 vs. FLU.
To confirm that host responses were similar in the context of other gram-negative bacterial infections, we challenged mice with influenza A/PR/8/34 or vehicle for 6 days prior to challenge with P. aeruginosa, an important, clinically relevant opportunistic human respiratory pathogen. We again observed significant exacerbation of lung bacterial burden in coinfected mice (Fig. 3A), which was associated with increased BAL neutrophil counts (Fig. 3B). The inflammatory cytokines IL-6, MCP-1, MIP-1α, and MIP-1β were significantly elevated in the lungs of coinfected mice, although no differences were observed for KC, G-CSF, or IL-1β (Fig. 3, C and D), and TNF-α was not detected (data not shown). IFN-γ was again significantly elevated in coinfected mice (Fig. 3D). Similar to the E. coli model, coinfected mice had increased lung damage compared with mice infected with P. aeruginosa alone, with increased LDH (mean OD490 0.69 vs. 0.26, P = 0.013, data not shown) and total protein as measured by BCA assay (mean 3,525 vs. 1,767 μg/ml, P < 0.0001, data not shown). Protein concentration of IL-23 (Fig. 3E) and gene expression of ROR-α and ROR-γt (Fig. 3F) were all decreased in the lungs of coinfected mice, although no differences were observed at this time point in IL-17, IL-22, or IFN-β (data not shown). Overall, consistent results were seen with either P. aeruginosa or E. coli, with coinfection leading to increased bacterial lung burden, inflammation, lung damage, and attenuated type 17 responses as measured by IL-23, ROR-α, and ROR-γt.
Fig. 3.

Preceding influenza infection exacerbates secondary gram-negative pneumonia with Pseudomonas aeruginosa. Mice were challenged with influenza A/PR/8/34 (100 pfu) or vehicle for 6 days followed by infection with P. aeruginosa (range, 4.5 × 105. 2.7 × 106 cfu) for 24 h. PA, P. aeruginosa only; Flu/PA, influenza and P. aeruginosa. A: bacterial burden in the lung (n = 12). B: bronchoalveolar lavage inflammatory cells (n = 12, 11). C–E: inflammatory cytokines in lung homogenate (n = 12). F: cytokine gene expression in lung (n = 4, representative data from 1 experiment shown, repeated twice). *P < 0.05 vs. PA; †P < 0.05 vs. Flu/PA Mac and Flu/PA Lymph.
We previously demonstrated that AMP expression was attenuated in IAV-S. aureus coinfection compared with S. aureus infection alone (35). To see whether similar effects could be observed with gram-negative infection, we examined expression of AMPs with known activity against gram-negative bacteria. First, we confirmed in vitro that IL-17 acts synergistically with TNF-α to augment expression of both BPIFA1 (Fig. 4A) and LCN2 (Fig. 4B) as well as to stimulate the secretion of BPIF1A (Fig. 4C) in HBE cells. In vivo, E. coli significantly induced BPIFA1 expression, which was inhibited by preceding IAV infection (Fig. 4D), whereas LCN2 expression was unchanged (data not shown). P. aeruginosa significantly induced both BPIFA1 and LCN2 expression, which were both inhibited by preceding IAV infection (Fig. 4E). These findings demonstrate that IL-17 is required for optimal induction of BPIFA1 and LCN2, but that preceding IAV attenuates production of these AMPs by gram-negative bacteria.
Fig. 4.

IL-17 augments antimicrobial peptide (AMP) expression, but preceding influenza attenuates AMP expression during gram-negative bacterial challenge. EC, E. coli only; FLU/EC, influenza and E. coli; FLU, influenza only; PA, P. aeruginosa only; Flu/PA, influenza and P. aeruginosa, LCN, lipocalin-2. A and B: AMP gene expression in cultured human bronchial epithelial cells (n = 2 donors, each condition performed in triplicate). C: AMP protein in cultured human bronchial epithelial cells (representative data from 1 human donor, each condition in triplicate, repeated once). D: AMP gene expression in lung (n = 8). E: AMP gene expression in lung (n = 4, representative data from 1 experiment shown, repeated twice). *P < 0.05 vs. Control; †P < 0.05 vs. TNF-α + IL-17A; ‡P < 0.05 vs. FLU and FLU/EC; §P < 0.05 vs. PA.
We have previously implicated type I IFN signaling in the exacerbation of secondary S. aureus infection following IAV (23). Since we observed increased type I IFN production in IAV-E. coli coinfected mice, we investigated the role of type I IFN in susceptibility to bacterial infection. WT or IFNAR−/− mice were challenged with influenza A/PR/8/34 or vehicle for 6 days prior to challenge with E. coli or vehicle for 1 additional day. IAV failed to significantly exacerbate E. coli bacterial burden in the lung in IFNAR−/− mice (Fig. 5A). Neutrophils in the BAL were significantly increased in coinfected WT mice compared with single-pathogen infection and were similarly increased in IFNAR−/− mice (Fig. 5B). IFN-γ and TNF-α production did not differ between WT and IFNAR−/− mice (Fig. 5C). IL-1β production was significantly suppressed during coinfection in WT mice compared with E. coli alone but was similar in coinfected and E. coli-infected IFNAR−/− mice. Finally, IL-17 gene expression was clearly elevated in IFNAR−/− mice compared with WT during influenza and coinfection, although these results did not reach statistical significance, whereas WT mice showed significant attenuation of IL-17 expression during coinfection compared with E. coli alone (Fig. 5D). These data suggest that type I IFN is important in the exacerbation of E. coli infection by IAV, and the absence of IFN signaling may potentially rescue type 17 immunity similar to what has been observed with gram-positive bacteria.
Fig. 5.

Bacterial clearance is improved in IFN-α receptor (IFNAR)−/− mice during coinfection. Wild-type (WT) or IFNAR−/− mice were challenged with influenza A/PR/8/34 (100 pfu) or vehicle for 6 days followed by infection with E. coli (1 × 107 cfu) for 24 h. EC, E. coli only; FLU/EC or F/E, influenza and E. coli; FLU, influenza only. A: bacterial burden in the lung (n = 6). B: bronchoalveolar lavage inflammatory cells (n = 4–6). C: inflammatory cytokines in lung homogenate (n = 4–6). D: cytokine gene expression in lung (n = 4–6). *P < 0.05 vs. FLU and EC; †P < 0.05 vs. FLU and FLU/EC; ‡P < 0.05 vs. FLU/EC and EC; **P < 0.05 vs. EC.
These data suggest that, during IAV infection, impaired host defenses against both gram-negative and gram-positive bacteria are due to similar mechanisms, of which type I IFN signaling plays an important role. We thus sought to more precisely characterize the contribution of type I IFN during the course of IAV infection. We hypothesized that the window period of susceptibility to bacterial coinfection following IAV infection was transient and linked to the IFN-related antiviral response. Since the majority of coinfection studies in the mouse model have focused on gram-positive bacterial infection 3–7 days following IAV infection (22–25, 30, 31, 34, 35, 38, 39), we again returned to the IAV-S. aureus coinfection model to test this hypothesis. Mice were challenged with influenza A/PR/8/34 or vehicle for 14 days prior to challenge with S. aureus or vehicle. Bacterial burden and inflammation were then assessed 1 day after bacterial challenge. Preceding IAV failed to exacerbate S. aureus bacterial burden in the lung (Fig. 6A). Neutrophil recruitment to the lung was still significantly elevated in coinfected animals vs. either infection alone, with higher neutrophil counts observed during bacterial vs. influenza infection (Fig. 6B). Interestingly, coinfection no longer exacerbated production of cytokines (Fig. 6, C and D). In fact, production of IL-1α, IL-1β, IL-6, KC, IFN-γ, TNF-α, IP-10, MIP-1α, MIP-2, and G-CSF were decreased in coinfection compared with S. aureus infection alone. These data indicate that IAV no longer attenuated S. aureus clearance when challenged 14 days after infection and that coinfection resulted in decreased lung cytokine production at this time point compared with bacterial infection alone.
Fig. 6.
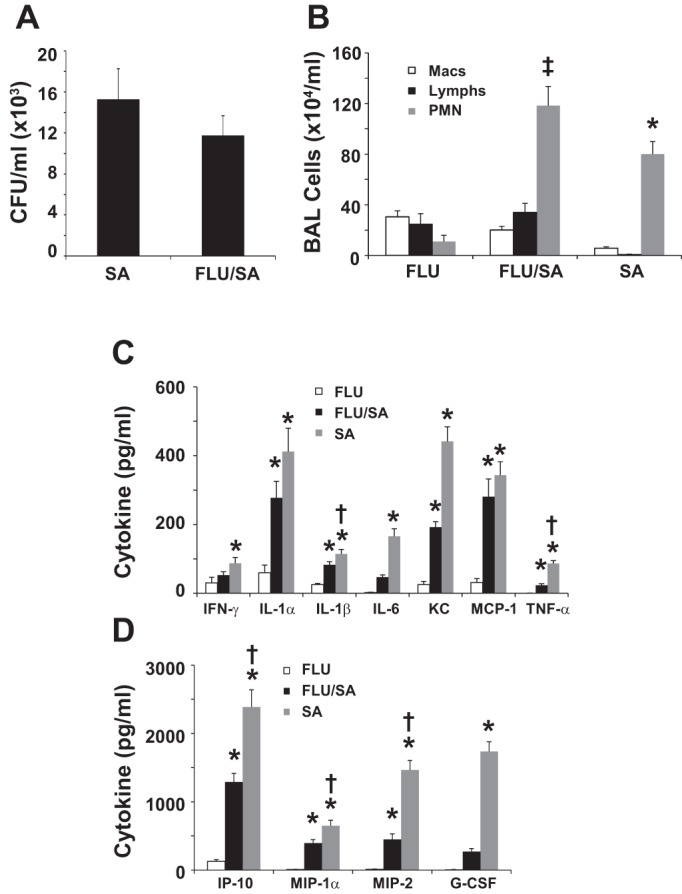
Fourteen days postchallenge, influenza fails to exacerbate Staphylococcus aureus coinfection. Mice were challenged with influenza A/PR/8/34 (100 pfu) or vehicle for 14 days followed by infection with S. aureus (1 × 108 cfu) for 24 h. SA, S. aureus only; FLU/SA, influenza and S. aureus; FLU, influenza only. A: bacterial burden in the lung (n = 12). B: bronchoalveolar lavage inflammatory cells (n = 12). C and D: inflammatory cytokines in lung homogenate (n = 12). *P < 0.05 vs. FLU; †P < 0.05 vs. FLU/SA; ‡P < 0.05 vs. FLU and SA.
Since bacterial clearance was not affected by IAV 14 days after challenge, we examined activation of type 17 immunity at this time. S. aureus induced similar expression of IL-17 and ROR-γt in S. aureus and coinfected animals (Fig. 7A). IL-17 protein production was not different between S. aureus and coinfected mice (Fig. 7B). However, production of IL-22 and IL-23 were statistically lower in coinfected mice compared with S. aureus alone (Fig. 7C). These data indicate that type 17 immunity may only be partially functional during coinfection at this later time point. Finally, we examined IFN-β and IFN-γ production in the lung during the IAV infection time course. IFN-β levels peaked at day 4 postinfection and remained elevated through day 10 (Fig. 7D). By day 14 after IAV infection, IFN-β expression was similar to sham-treated mice. IFN-γ protein levels were highest on day 6 postinfection and were not significantly elevated at any other time point (Fig. 7E). These data suggest that the lack of type I IFN at 14 days postinfluenza may explain the lack of exacerbation of S. aureus clearance.
Fig. 7.

In the absence of increased IFN-β induction, influenza infection only partially inhibits type 17 immunity. Mice were challenged with influenza A/PR/8/34 (100 pfu) or vehicle for 14 days (d) followed by infection with S. aureus (1 × 108 cfu) for 24 h. SA, S. aureus only; FLU/SA, influenza and S. aureus; FLU, influenza only. A and C: cytokine gene expression in lung (n = 11–12). B: inflammatory cytokine in lung homogenate (n = 8). D: cytokine gene expression in lung (n = 3–4). E: cytokine concentration in lung homogenate (n = 4). *P < 0.05 vs. all other groups; †P < 0.05 vs. all groups except day 8.
DISCUSSION
These data demonstrate that preceding IAV infection impairs the host response to bacterial challenge with gram-negative bacteria, similar to the attenuation previously described during IAV and gram-positive bacterial coinfection (23, 25). Importantly, bacterial challenge in the setting of elevated type I IFN expression (i.e., during a specific window period during influenza infection) was associated with attenuation of type 17 immunity in WT but not IFNAR−/− mice. To our knowledge these are the first data that demonstrate attenuation of type 17 immunity against gram-negative bacteria in the setting of IAV infection. These findings support previously reported data suggesting the importance of type I IFN in the subversion of type 17 immunity, increasing host susceptibility to infection from a broad range of extracellular bacteria, including bacterial coinfections complicating influenza.
In this study, the major effect of preceding influenza infection on bacterial infection was delayed clearance from the lung, since lethality was not assessed and neither E. coli nor P. aeruginosa exhibited increases in bacterial burden beyond the infecting inoculum that would suggest uncontrolled or overwhelming infection. A previous study showed similar delays in bacterial clearance in the context of influenza and K. pneumoniae coinfection, which was also associated with increased subsequent mortality (20). We previously demonstrated that bacterial clearance from the lung during influenza-S. aureus coinfection was significantly delayed compared with bacterial infection alone (35). In our present study, coinfection was associated with significantly increased inflammation and evidence of increased lung injury compared with bacterial infection alone, suggesting that delayed clearance might expose the host to a prolonged duration of inflammation and associated risk of greater lung injury. Similar to our previous findings, increased neutrophils and inflammatory cytokines and chemokines were observed in coinfected mice, suggesting that impaired bacterial clearance was not due to a defect in inflammatory cell recruitment to the lung (23, 34). Furthermore, neutrophil depletion has not been shown to affect S. aureus lung bacterial burden, suggesting an alternate mechanism for impaired bacterial clearance (22, 35). This is in contrast to reports of neutrophil-dependent mechanisms during IAV-Streptococcus pneumoniae coinfection (31, 38). In one study, decreased levels of KC and MIP-2 were observed in coinfection and exogenous KC and MIP-2 rescued bacterial clearance, but restoration of neutrophil recruitment was not explicitly demonstrated (38). Furthermore, no attenuation of KC or MIP-2 was observed in our model. It is possible that species-specific bacterial effects may play some role, with differences in the murine response to S. pneumoniae and S. aureus.
As expected, infection with E. coli or P. aeruginosa alone was associated with increased IL-23, ROR-α, and ROR-γt, consistent with type 17 immune activation. In E. coli infection alone, increased protein levels of IL-1β and IL-17A and increased expression of IL-17A and IL-22 were also detected. These responses were attenuated in mice infected with IAV alone and in coinfected mice, consistent with IAV-associated inhibition of type 17 activity. Because no defects in inflammatory cell recruitment were observed despite suppressed type 17 activity, we sought to determine whether AMP production was attenuated (35). In vitro, IL-17 alone was not sufficient for AMP expression, but IL-17 in the context of low levels of TNF-α clearly augmented AMP expression by HBE. In vivo, expression of BPIFA1 and LCN2 were suppressed during IAV-P. aeruginosa coinfection, with similar results seen for BPIFA1 during IAV-E. coli coinfection. Importantly, AMP expression in vivo did not appear to be modulated solely by TNF-α, which was significantly elevated in IAV-E. coli coinfection but was not detected at this time point in IAV-P. aeruginosa coinfection. BPIFA1 is secreted by secretory cells of the airway epithelium and in mice has been shown to contribute to lung defense against gram-negative bacteria, mainly owing to inhibition of biofilm formation (6, 12, 26–28, 37). To our knowledge, this is the first assessment of the role of BPIFA1 in response to E. coli pneumonia in vivo. The role of LCN2 in mucosal defense against both gram-positive and gram-negative bacteria is well established (3, 9, 15, 35, 44), where LCN2 exerts bacteriostatic activity via interference of siderophore-mediated bacterial iron acquisition (17). Because neither BPIFA1 nor LCN2 is bactericidal, they alone are unlikely to be sufficient effectors of antimicrobial killing. Rather, they likely reflect activation of host responses that synergize to promote bacterial eradication. We have previously shown that exogenous LCN2 rescued the bacterial clearance defect in IAV-S. aureus coinfection, demonstrating the importance of AMP production in bacterial clearance, despite lack of direct bactericidal activity (35). These results further implicate inhibition of AMP as an important mechanism for IAV-mediated attenuation of bacterial lung defense, with type 17 suppression a likely contributor to this effect.
Type I IFN signaling is an important mediator in suppression of type 17 immunity following IAV infection (23, 25, 41). Here, IFNAR−/− mice were rescued from impaired lung clearance of E. coli during coinfection, which was associated with increased IL-1β and IL-17A. Although not tested directly (e.g., by using IFNAR/IL-17 double knockout mice or IL-17 neutralization), these results suggest rescue of type 17 immunity and further support a role for type I IFN signaling in inhibition of type 17 immunity. Previous studies in the S. pneumoniae model identified type II IFN (IFN-γ) as the main mediator of susceptibility to secondary bacterial infection (39). In our model, IFN-γ was elevated at 6 days post-IAV infection, suggesting a potential role in susceptibility. However, IFNAR−/− mice also had increased levels of IFN-γ during coinfection but efficiently cleared E. coli, suggesting that type I IFN signaling played a vital role.
The epidemiology of human influenza and bacterial coinfection demonstrates an overwhelming preponderance of infections with gram-positive bacteria. During the 1918 influenza pandemic, up to three-quarters of influenza-associated deaths involved coinfection with Streptococcus or Staphylococcus species (32). In contrast, gram-negative bacteria are very infrequently encountered as community-acquired pathogens during influenza coinfection (33). Although our results suggest that increased susceptibility to gram-negative bacterial lung infections proceeds through similar mechanisms, this may not translate to an increased risk of human disease, with infrequent exceptions. This most likely reflects the presumed pathogenesis of bacterial superinfection during IAV infection: contiguous spread or aspiration of bacteria from the upper to lower respiratory tract in the setting of a compromised respiratory epithelium (5, 29, 42). Nasopharyngeal acquisition and carriage of both Streptococci and Staphylococci are well established in humans and likely play a critical role in coinfection. Nasal carriage of S. aureus has previously been shown to be associated with pneumonia in critically ill patients (11). In contrast, E. coli is an enteric organism, whereas P. aeruginosa is an environmental organism that is an opportunistic, nosocomial human pathogen. Enteric or nosocomial gram-negative bacteria infrequently colonize the upper respiratory tract of otherwise healthy hosts, the population disproportionately affected by IAV and gram-positive bacterial coinfections, particularly those secondary to CA-MRSA (36). This makes them an unlikely source of natural lung infections in this population and likely explains the reduced frequency of community-acquired IAV-gram-negative bacterial coinfections. However, our findings suggest the potential for pathological synergy between influenza and a broad range of extracellular bacteria, including typically nonpathogenic strains. This may have particularly important implications regarding susceptibility to hospital-acquired coinfections, where medical interventions remove typical barriers for gram-negative pathogens to gain access to the respiratory tract. Recent data suggest that, in patients admitted to the intensive care unit for influenza, recovery of gram-negative bacteria rises substantially following admission (33). Although the iatrogenic risks of intensive care unit admission alone may contribute to this effect, preceding influenza likely modulates this risk.
Infection with S. aureus on day 14 of IAV infection showed no differences in bacterial burden 1 day following challenge. Surprisingly, whereas total inflammatory cells were elevated in the BAL of mice coinfected with S. aureus at this time point, levels of proinflammatory cytokines were paradoxically lower than in mice infected with S. aureus alone. The reasons for this finding remain unclear. It is conceivable that at this point during the influenza recovery period, the balance of pro- vs. anti-inflammatory signals had shifted toward suppression of inflammation while the cellular infiltrate had not yet resolved. Interestingly, IL-22 and IL-23 expression remained attenuated at this time point in coinfection, but expression of ROR-γt and IL-17 levels did not differ, suggesting only a partial defect in type 17 immunity. In contrast to earlier time points, IL-17 was preserved when type I IFN expression had returned to baseline, suggesting that the effect of type I IFN may be more specific to IL-17 than IL-22 and that restoration of IL-17 alone may be sufficient to rescue bacterial clearance.
Numerous studies have demonstrated that increased susceptibility to secondary bacterial pneumonia due to S. aureus or S. pneumoniae began following elevations in type I IFN production, typically 3–7 days following IAV infection, whereas IFNAR−/− were rescued from impaired bacterial clearance or mortality (23–25, 30, 31, 34, 35, 38–40). Few studies, however, have demonstrated when restoration of normal bacterial clearance resumes following IAV infection. Our findings support a window period of 3–14 days of enhanced susceptibility to secondary S. aureus challenge following IAV infection (22), which correlates precisely with the presence of increased type I IFN expression. Others have reported that enhanced susceptibility to S. pneumoniae still occurred 14 days following IAV infection (20, 30, 43). No type I IFN data were presented in these studies, so it is difficult to draw comparative conclusions. Although definitive data regarding type I IFN levels and risk of coinfection in humans are lacking, the striking clinical and epidemiological observations of the association between IAV and secondary bacterial pneumonia, particularly in cases of influenza-associated death, suggest that a similar, critical risk window exists in human populations.
In conclusion, these results provide evidence that supports a common mechanistic pathway for attenuation of host lung defense and type 17 immunity against both gram-positive and gram-negative bacteria during IAV coinfection. We show that enhanced susceptibility coincides with the period of peak type I IFN production, supporting a role for IAV-induced type I IFN as a key mediator in this attenuation. Further insights into the mechanisms underlying the ability of type I IFN to inhibit type 17 immunity are needed to further elucidate the pathogenesis of IAV and bacterial coinfection. These may yield novel strategies for the management and prevention of secondary bacterial pneumonia complicating IAV infection, reducing morbidity and mortality.
GRANTS
This work was supported by the Eunice Kennedy Shriver National Institute of Child Health & Human Development (T32 HD071834 to B. Lee), the Flight Attendant Medical Research Institute (CIA-123062 to Y. P. Di), the National Institutes of Health National Heart, Lung, and Blood Institute (T32 HL007563 to K. M. Robinson; RO1 HL091938 to Y. P. Di; R01 HL107380 to J. F. Alcorn), and a Parker B. Francis Foundation Fellowship (to J. F. Alcorn).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
B.L., Y.P.D., and J.F.A. conception and design of research; B.L., K.J.M., E.V.S., S.M., C.C., Y.P.D., M.E.C., R.I.E., P.J.D., and J.F.A. performed experiments; B.L., Y.P.D., and J.F.A. analyzed data; B.L., K.M.R., Y.P.D., and J.F.A. interpreted results of experiments; B.L. and J.F.A. prepared figures; B.L. and J.F.A. drafted manuscript; B.L., K.M.R., Y.P.D., R.I.E., P.J.D., and J.F.A. edited and revised manuscript; B.L., K.M.R., Y.P.D., and J.F.A. approved final version of manuscript.
ACKNOWLEDGMENTS
Present address for P. J. Dubin: Department of Pediatrics, West Virginia University, Morgantown, WV.
REFERENCES
- 1.Estimates of deaths associated with seasonal influenza — United States, 1976–2007. MMWR Morb Mortal Wkly Rep 59: 1057–1062, 2010. [PubMed] [Google Scholar]
- 2.Aujla SJ, Alcorn JF. T(H)17 cells in asthma and inflammation. Biochim Biophys Acta 1810: 1066–1079, 2011. [DOI] [PubMed] [Google Scholar]
- 3.Aujla SJ, Chan YR, Zheng M, Fei M, Askew DJ, Pociask DA, Reinhart TA, McAllister F, Edeal J, Gaus K, Husain S, Kreindler JL, Dubin PJ, Pilewski JM, Myerburg MM, Mason CA, Iwakura Y, Kolls JK. IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nat Med 14: 275–281, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Balamayooran T, Batra S, Balamayooran G, Cai S, Kobayashi KS, Flavell RA, Jeyaseelan S. Receptor-interacting protein 2 controls pulmonary host defense to Escherichia coli infection via the regulation of interleukin-17A. Infect Immun 79: 4588–4599, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ballinger MN, Standiford TJ. Postinfluenza bacterial pneumonia: host defenses gone awry. J Interferon Cytokine Res 30: 643–652, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bartlett JA, Gakhar L, Penterman J, Singh PK, Mallampalli RK, Porter E, McCray PB Jr. PLUNC: a multifunctional surfactant of the airways. Biochem Soc Trans 39: 1012–1016, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bingle CD, Seal RL, Craven CJ. Systematic nomenclature for the PLUNC/PSP/BSP30/SMGB proteins as a subfamily of the BPI fold-containing superfamily. Biochem Soc Trans 39: 977–983, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Braciale TJ. Immunologic recognition of influenza virus-infected cells. I. Generation of a virus-strain specific and a cross-reactive subpopulation of cytotoxic T cells in the response to type A influenza viruses of different subtypes. Cell Immunol 33: 423–436, 1977. [DOI] [PubMed] [Google Scholar]
- 9.Chan YR, Liu JS, Pociask DA, Zheng M, Mietzner TA, Berger T, Mak TW, Clifton MC, Strong RK, Ray P, Kolls JK. Lipocalin 2 is required for pulmonary host defense against Klebsiella infection. J Immunol 182: 4947–4956, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen ZM, Jenkins MK. Revealing the in vivo behavior of CD4+ T cells specific for an antigen expressed in Escherichia coli. J Immunol 160: 3462–3470, 1998. [PubMed] [Google Scholar]
- 11.Corne P, Marchandin H, Jonquet O, Campos J, Banuls AL. Molecular evidence that nasal carriage of Staphylococcus aureus plays a role in respiratory tract infections of critically ill patients. J Clin Microbiol 43: 3491–3493, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Di YP. Functional roles of SPLUNC1 in the innate immune response against Gram-negative bacteria. Biochem Soc Trans 39: 1051–1055, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Di YP, Harper R, Zhao Y, Pahlavan N, Finkbeiner W, Wu R. Molecular cloning and characterization of spurt, a human novel gene that is retinoic acid-inducible and encodes a secretory protein specific in upper respiratory tracts. J Biol Chem 278: 1165–1173, 2003. [DOI] [PubMed] [Google Scholar]
- 14.Finelli L, Fiore A, Dhara R, Brammer L, Shay DK, Kamimoto L, Fry A, Hageman J, Gorwitz R, Bresee J, Uyeki T. Influenza-associated pediatric mortality in the United States: increase of Staphylococcus aureus coinfection. Pediatrics 122: 805–811, 2008. [DOI] [PubMed] [Google Scholar]
- 15.Flo TH, Smith KD, Sato S, Rodriguez DJ, Holmes MA, Strong RK, Akira S, Aderem A. Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature 432: 917–921, 2004. [DOI] [PubMed] [Google Scholar]
- 16.Garcia-Sastre A, Durbin RK, Zheng H, Palese P, Gertner R, Levy DE, Durbin JE. The role of interferon in influenza virus tissue tropism. J Virol 72: 8550–8558, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goetz DH, Holmes MA, Borregaard N, Bluhm ME, Raymond KN, Strong RK. The neutrophil lipocalin NGAL is a bacteriostatic agent that interferes with siderophore-mediated iron acquisition. Mol Cell 10: 1033–1043, 2002. [DOI] [PubMed] [Google Scholar]
- 18.Hall MW, Geyer SM, Guo CY, Panoskaltsis-Mortari A, Jouvet P, Ferdinands J, Shay DK, Nateri J, Greathouse K, Sullivan R, Tran T, Keisling S, Randolph AG. Innate immune function and mortality in critically ill children with influenza: a multicenter study. Crit Care Med 41: 224–236, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Happel KI, Dubin PJ, Zheng M, Ghilardi N, Lockhart C, Quinton LJ, Odden AR, Shellito JE, Bagby GJ, Nelson S, Kolls JK. Divergent roles of IL-23 and IL-12 in host defense against Klebsiella pneumoniae. J Exp Med 202: 761–769, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haynes L, Szaba FM, Eaton SM, Kummer LW, Lanthier PA, Petell AH, Duso DK, Luo D, Lin JS, Lefebvre JS, Randall TD, Johnson LL, Kohlmeier JE, Woodland DL, Smiley ST. Immunity to the conserved influenza nucleoprotein reduces susceptibility to secondary bacterial infections. J Immunol 189: 4921–4929, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hwang SY, Hertzog PJ, Holland KA, Sumarsono SH, Tymms MJ, Hamilton JA, Whitty G, Bertoncello I, Kola I. A null mutation in the gene encoding a type I interferon receptor component eliminates antiproliferative and antiviral responses to interferons alpha and beta and alters macrophage responses. Proc Natl Acad Sci USA 92: 11284–11288, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iverson AR, Boyd KL, McAuley JL, Plano LR, Hart ME, McCullers JA. Influenza virus primes mice for pneumonia from Staphylococcus aureus. J Infect Dis 203: 880–888, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kudva A, Scheller EV, Robinson KM, Crowe CR, Choi SM, Slight SR, Khader SA, Dubin PJ, Enelow RI, Kolls JK, Alcorn JF. Influenza A inhibits Th17-mediated host defense against bacterial pneumonia in mice. J Immunol 186: 1666–1674, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.LeVine AM, Koeningsknecht V, Stark JM. Decreased pulmonary clearance of S. pneumoniae following influenza A infection in mice. J Virol Methods 94: 173–186, 2001. [DOI] [PubMed] [Google Scholar]
- 25.Li W, Moltedo B, Moran TM. Type I interferon induction during influenza virus infection increases susceptibility to secondary Streptococcus pneumoniae infection by negative regulation of gammadelta T cells. J Virol 86: 12304–12312, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu Y, Bartlett JA, Di ME, Bomberger JM, Chan YR, Gakhar L, Mallampalli RK, McCray PB Jr, Di YP. SPLUNC1/BPIFA1 contributes to pulmonary host defense against Klebsiella pneumoniae respiratory infection. Am J Pathol 182: 1519–1531, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu Y, Di ME, Chu HW, Liu X, Wang L, Wenzel S, Di YP. Increased susceptibility to pulmonary Pseudomonas infection in Splunc1 knockout mice. J Immunol 191: 4259–4268, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lukinskiene L, Liu Y, Reynolds SD, Steele C, Stripp BR, Leikauf GD, Kolls JK, Di YP. Antimicrobial activity of PLUNC protects against Pseudomonas aeruginosa infection. J Immunol 187: 382–390, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McCullers JA. Insights into the interaction between influenza virus and pneumococcus. Clin Microbiol Rev 19: 571–582, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McCullers JA, Rehg JE. Lethal synergism between influenza virus and Streptococcus pneumoniae: characterization of a mouse model and the role of platelet-activating factor receptor. J Infect Dis 186: 341–350, 2002. [DOI] [PubMed] [Google Scholar]
- 31.McNamee LA, Harmsen AG. Both influenza-induced neutrophil dysfunction and neutrophil-independent mechanisms contribute to increased susceptibility to a secondary Streptococcus pneumoniae infection. Infect Immun 74: 6707–6721, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morens DM, Taubenberger JK, Fauci AS. Predominant role of bacterial pneumonia as a cause of death in pandemic influenza: implications for pandemic influenza preparedness. J Infect Dis 198: 962–970, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Muscedere J, Ofner M, Kumar A, Long J, Lamontagne F, Cook D, McGeer A, Chant C, Marshall J, Jouvet P, Fowler R. The occurrence and impact of bacterial organisms complicating critical care illness associated with 2009 influenza A(H1N1) infection. Chest 144: 39–47, 2013. [DOI] [PubMed] [Google Scholar]
- 34.Robinson KM, Choi SM, McHugh KJ, Mandalapu S, Enelow RI, Kolls JK, Alcorn JF. Influenza A exacerbates Staphylococcus aureus pneumonia by attenuating IL-1beta production in mice. J Immunol 191: 5153–5159, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Robinson KM, McHugh KJ, Mandalapu S, Clay ME, Lee B, Scheller EV, Enelow RI, Chan YR, Kolls JK, Alcorn JF. Influenza A virus exacerbates Staphylococcus aureus pneumonia in mice by attenuating antimicrobial peptide production. J Infect Dis 209: 865–875, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rubinstein E, Kollef MH, Nathwani D. Pneumonia caused by methicillin-resistant Staphylococcus aureus. Clin Infect Dis 46, Suppl 5: S378–S385, 2008. [DOI] [PubMed] [Google Scholar]
- 37.Sayeed S, Nistico L, St Croix C, Di YP. Multifunctional role of human SPLUNC1 in Pseudomonas aeruginosa infection. Infect Immun 81: 285–291, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shahangian A, Chow EK, Tian X, Kang JR, Ghaffari A, Liu SY, Belperio JA, Cheng G, Deng JC. Type I IFNs mediate development of postinfluenza bacterial pneumonia in mice. J Clin Invest 119: 1910–1920, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sun K, Metzger DW. Inhibition of pulmonary antibacterial defense by interferon-gamma during recovery from influenza infection. Nat Med 14: 558–564, 2008. [DOI] [PubMed] [Google Scholar]
- 40.Tian X, Xu F, Lung WY, Meyerson C, Ghaffari AA, Cheng G, Deng JC. Poly I:C enhances susceptibility to secondary pulmonary infections by gram-positive bacteria. PLoS One 7: e41879, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tilg H, Moschen AR, Kaser A. Suppression of interleukin-17 by type I interferons: a contributing factor in virus-induced immunosuppression? Eur Cytokine Netw 20: 1–6, 2009. [DOI] [PubMed] [Google Scholar]
- 42.van der Sluijs KF, van der Poll T, Lutter R, Juffermans NP, Schultz MJ. Bench-to-bedside review: bacterial pneumonia with influenza. Pathogenesis and clinical implications. Crit Care 14: 219, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.van der Sluijs KF, van Elden LJ, Nijhuis M, Schuurman R, Pater JM, Florquin S, Goldman M, Jansen HM, Lutter R, van der Poll T. IL-10 is an important mediator of the enhanced susceptibility to pneumococcal pneumonia after influenza infection. J Immunol 172: 7603–7609, 2004. [DOI] [PubMed] [Google Scholar]
- 44.Wu H, Santoni-Rugiu E, Ralfkiaer E, Porse BT, Moser C, Hoiby N, Borregaard N, Cowland JB. Lipocalin 2 is protective against E. coli pneumonia. Respir Res 11: 96, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ye P, Garvey PB, Zhang P, Nelson S, Bagby G, Summer WR, Schwarzenberger P, Shellito JE, Kolls JK. Interleukin-17 and lung host defense against Klebsiella pneumoniae infection. Am J Respir Cell Mol Biol 25: 335–340, 2001. [DOI] [PubMed] [Google Scholar]
- 46.Ye P, Rodriguez FH, Kanaly S, Stocking KL, Schurr J, Schwarzenberger P, Oliver P, Huang W, Zhang P, Zhang J, Shellito JE, Bagby GJ, Nelson S, Charrier K, Peschon JJ, Kolls JK. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J Exp Med 194: 519–527, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]