

Abstract
Purpose
To identify tumor suppressor genes epigenetically silenced by promoter hypermethylation in extranodal natural killer cell lymphoma (NKCL).
Experimental Design
Promoter methylation was analyzed with global and locus-specific methylation assays in NKCL cases and NK cell lines. Gene expression profiles were used to identify genes for which aberrant promoter methylation was associated with transcriptional silencing. Selected DNA methylations were validated by RRBS, pyrosequencing, or q-MSP. Decitabine treatment was performed to evaluate reactivation of methylated genes. The tumor suppressor effect of silenced genes was evaluated functionally by reintroducing them into NK cell lines.
Results
We observed significant promoter hypermethylation in most NKCL samples compared with normal NK cells. Correlation of global promoter methylation with gene expression profiles identified 95 genes with strong evidence for being silenced because of promoter methylation, including BCL2L11 (BIM), DAPK1, PTPN6 (SHP1), TET2, SOCS6, and ASNS. Known tumor suppressor genes were significantly overrepresented in this set of genes. Decitabine treatment of NK cell lines was associated with reexpression of all 10 selected methylated and silenced genes. Ectopic expression of frequently silenced BIM in two BIM-nonexpressing NK cell lines led to increased apoptosis and eventual elimination of BIM-transduced cells. It also sensitized these cell lines to chemotherapy-induced apoptosis. Similarly, reintroduction of SOCS6 significantly inhibited growth in SOCS6-nonexpressing NK cell lines. NK cell lines lacking ASNS expression showed increased sensitivity to treatment with l-asparaginase. Reintroduction of ASNS reduced drug sensitivity.
Conclusion
Promoter region hypermethylation is frequent in NKCL, and aberrantly methylated genes are pathologically and clinically significant.
Introduction
Natural killer (NK) cell malignancies are aggressive neoplasms and represent 1% to 2% of all non-Hodgkin lymphomas (NHL; ref. 1). They are divided into 2 clinic subtypes based on the WHO classification: aggressive NK cell leukemia (ANKL) and extranodal NK cell lymphoma (NKCL) of nasal type (2). NKCL prevalence is higher in Central and South America and East Asia than in other regions (1). NKCLs are associated with Epstein–Barr virus (EBV) infections (3), and patients with localized nasal or paranasal disease respond significantly to combined radiotherapy and chemotherapy (4); however, NKCLs are resistant to chemotherapy in advanced stages and when the disease is extranasal. An l-aspar-aginase–containing multiagent chemotherapeutic regimen containing steroid (dexamethasone), methotrexate, ifosfamide, and etoposide (SMILE) is currently the best option for refractory NKCLs and has shown promising clinical response in patients with advanced disease (5, 6). Recently, the combination of genomic and functional analysis has identified PRDM1 as a candidate tumor suppressor gene (TSG) in NKCLs (7–9). However, few aberrant genes that contribute to NKCL pathogenesis and can potentially serve as therapeutic targets have been identified and characterized.
Aberrant promoter methylation is a major mechanism contributing to neoplastic transformation by deregulating expression of oncogenes and TSGs (10). Transcriptional repression mediated by CpG island/promoter hypermethylation has been detected for PRDM1 (7, 9), SHP1 (PTPN6; ref. 11), DAPK1 (12), and HACE1 (13) in NKCL, suggesting that aberrant promoter methylation is an important mechanism of TSG silencing in NKCL as in other malignancies (14, 15). However, only locus-specific assays were used for the assessment of promoter hypermethylation in NKCL samples, and the global promoter methylation changes have not been reported. To more comprehensively evaluate the inactivation of potential TSGs in NKCLs, we applied genetic, epigenetic, and functional approaches to study a series of NKCL cases and cell lines and have detected promoter hypermethylation and transcriptional silencing of SHP1, BIM, SOCS6, TET2 and other novel candidate TSGs that may serve as therapeutic targets in NKCLs. In addition, we showed frequent silencing of asparagine synthetase (ASNS) and an association between l-asparaginase-induced cell death and ASNS expression, suggesting that ASNS methylation may serve as a biomarker for response to l-aspar-aginase treatment.
Materials and Methods
Cell lines and tumor specimens
Twelve NKCL cases and 7 NK cell lines (NK92, KHYG1, YT, SNK1, SNK6, NKYS, and KAI3) were used in this study. The characteristics of NK cell tumor cases and NK cell lines have been described previously (3) and are summarized in Supplementary Table S1. KHYG1 and KAI3 cell lines were obtained from the Health Science Research Resource Bank (Osaka, Japan). NKYS, SNK1, and SNK6 cell lines were provided by Dr. Norio Shimuzu. NK92 and YT cell lines were obtained from the German Collection of Microorganism and Cell Culture (GCMCC; DSMZ). HEK293T and DHL16 cells were obtained from ATCC. All cell lines were expanded, frozen, and used for experiments within 6 months of cell culture after receiving them with the assumption that authentication was performed by the original provider. All NK cell lines were cultured in RPMI-1640 (Gibco-Invitrogen) supplemented with 10% FBS, penicillin G (100 units/mL), streptomycin (100 μg/mL), 4 mmol/L l-glutamine (Life Technologies Inc.), and 5 to 7 ng/mL IL2 (R&D Bioscience) at 37°C in 5% CO2. 293T cells were cultured in DMEM (Gibco-Invitrogen) supplemented with same culture components used for NK cell lines apart from IL2.
Methyl-sensitive cut counting
Global methylation analysis of 12 NKCL cases and 2 NK cell lines (KHYG1 and NK92) was performed using the methyl-sensitive cut counting (MSCC) procedure as previously described (13, 16). Forty eight–hour IL2-activated human peripheral blood NK cells (n = 3) were used as the normal NK cell standard; normal human tonsil provided a second normal control. The MSCC protocol generates a library on the basis of the cleavage that occurs when DNA is treated with a restriction enzyme, HpaII, which cuts only unmethylated CCGG sequences. The MSCC procedure is briefly described as follows: 2 μg gDNA was digested with 20 U of the restriction enzyme HpaII (NEB). An adapter containing a recognition site for the restriction enzyme MmeI was then ligated to DNA. Adapter-ligated DNA was nick-repaired with Bst DNA polymerase (NEB). The DNA was digested with 2 U MmeI to capture the 18 bases adjacent to HpaII sites, and the fragments were subsequently ligated to a second adaptor to allow PCR amplification using iProof high-fidelity polymerase (BioRad) and final high-throughput sequencing. A 10% PAGE gel was used for tag size purification. Final tags were evaluated for proper size and concentration using a Bioanalyzer High Sensitivity DNA chip (Agilent).
Library preparation and high-throughput sequencing were performed at the UNMC epigenetic core facility using the Illumina Genome Analyzer IIx. The 18-bp sequence tags generated were aligned with Bowtie (17). Perl scripts (16) were used to align the sequences of the unique tags and the genes in the NCBI reference hg19. The basic MSCC statistics are shown in Supplementary Table S2. The sequencing data will be submitted to SRA database of PubMed. (http:/www.ncbi.nlm.nih.gov/sra/).
Decitabine treatment
Decitabine (5′-aza-2′-deoxycytidine) treatment was performed as previously described (7). Briefly, 4 × 105 cells from KHYG1, NK92, or SNK6 cell lines were seeded in 2 mL in 6-well plates in the presence of 0, 0.25, 0.5, or 0.75 μmol/L of decitabine (Sigma-Aldrich). After 2 days, cells were spun down at 300 × g for 10 minutes, and fresh culture medium including the same decitabine concentrations was added. Cells were harvested for qRT-PCR after 4 total days of treatment.
l-Asparaginase treatment of NK cell lines and cell survival assay
A total of 5 × 104 cells from NK cell lines (n = 6) in 1-mL medium were seeded into 24-well plates in duplicates. Cells were treated with 0, 0.01, 0.05, 0.075, 0.1, or 0.5 IU of l-asparaginase (Sigma-Aldrich) for 24 or 48 hours. Cell survival was determined after the treatment using Cell Titer Glo Luminescent Cell Viability Assay (Promega Inc.) or Vi-CELL XR Cell Viability Analyzer (Beckman Coulter Inc.) according to the manufacturers' recommendations.
Statistical and computational analysis of epigenetically silenced genes
R language scripts (http://www.R-project.org) were used for statistical analysis of genome-wide methylation data and to identify genes showing concurrent promoter methylation and transcriptional repression.
Additional experimental methods of (i) sample isolation and NK cell activation, (ii) GEP, (iii) pyrosequencing, (iv) Western blotting, (v) RRBS, (vi) q-MSP, (vii) qRT-PCR, (viii) expression of BIM-EL, SOCS6, and ASNS in NK cell lines, (ix) apoptosis assay, (x) transfection of 293T cells with BIM, and (xi) additional statistical methods are described in the Supplementary Methods.
Results
Most NKCL samples have global promoter hypermethylation compared with normal NK cells
We compared global DNA methylation in malignant NK samples (12 NKCL cases and 2 NK cell lines) with normal NK cells and normal tonsil. Each mappable HpaII site was uniquely assigned to the closest TSS. We calculated the number of HpaII cuts per site for each sample, arbitrarily treating the promoter as the region within 1 kb of the TSS. The overall distribution of HpaII sites and of MSCC reads in normal NK cells is shown in Supplementary Fig. S1. HpaII sites are strongly associated with CpG islands (CGIs) and most promoters overlap with CGIs. Consistent with the normal absence of methylation at most promoter-associated CGIs (18), the average number of cuts per site was higher in CGI-associated promoters than elsewhere in both normal and malignant NK samples (mean, 13.97 vs. 3.58) and the number of cuts per site varied little among CGI-associated promoters in normal NK cells (3.76 mean log2 cuts per site, 0.70 SD for promoters with at least 5 sites). However, the corresponding mean value was lower and the SD higher, for all malignant NK samples (range, 3.09–3.69; mean, 0.75–1.34, SD), consistent with frequent aberrant promoter methylation in malignant samples. Graphing the average number of cuts per site at various distances from the TSS revealed a clear global pattern of promoter hypermethylation, hypomethylation distal to the promoter, or usually both in 11 of the 14 malignant samples (Fig. 1A), which suggests widespread dysregulation of DNA methylation in malignant NK samples. The 3 exceptional cases (#3, 9, and 12) likely either have low tumor content or more normal regulation of DNA methylation; they were therefore not included in additional analyses.
Figure 1.
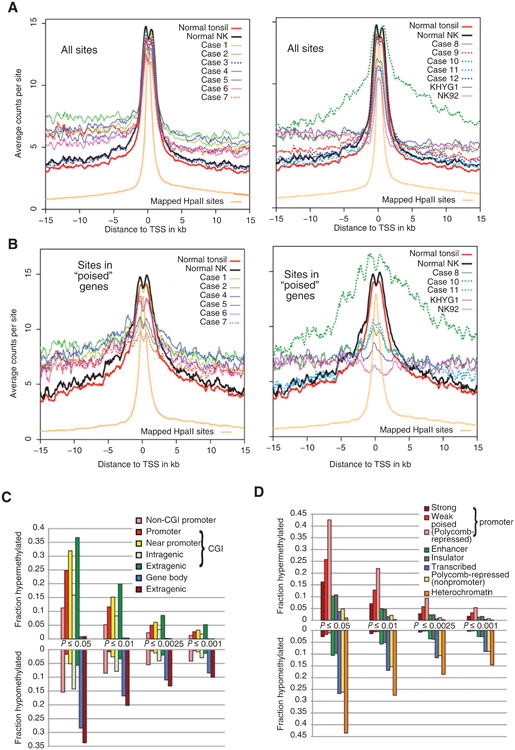
Compared with normal cells, most malignant NK samples show promoter hypermethylation, which affects a large fraction of genes. A, average MSCC counts were calculated for each of 12 NKCL cases, 2 NK cell lines, normal NK cells (average of 3 technical replicates), and tonsil (average of 5 technical replicates) and plotted against the relative distance to the TSS at 0.1-kb intervals using a 0.5-kb sliding window. B, average MSCC counts at poised genes for normal NK cells and tonsil and malignant NK samples (9 NKCL cases and 2 NK cell lines). Three cases with modest methylation abnormalities (#3, 9, 12) were not included. Note that case 10 lacks obvious global promoter hypermethylation and has very striking distal hypomethylation. C, a t test was used to compare the 2 normal samples (tonsil and NK cells) to 9 cases and 2 cell lines with clear aberrant methylation. Cut sites for each gene were clustered into categories as shown (sites ± 1 kb of the TSS are considered promoter sites). Only genes with ≥3 cut sites for a given category were included, yielding 2,343, 13,842, 2,002, 1,774, 1,669, 17,627, and 14,994 genes in the order shown. The fractions of hypermethylated and hypomethyated gene regions at various levels of significance are shown. D, as in C, except that sites were annotated based on the chromatin assignments in hESCs (available at www.genome.ucsc.edu; ref. 21).
Previous analysis showed that genes hypermethylated in cancer frequently have a “poised” promoter as in embryonic stem cells (ESC; refs. 19, 20); that is, one with histone marks of both activation (H3K4me3) and repression (H3K27me3), the latter associated with Polycomb complexes. Strikingly, most malignant NK samples show a much more prominent pattern of promoter hypermethylation at poised genes than at other promoters (Fig. 1B).
Cut site reads were summed for each gene after assigning sites to various categories. Log2 values were compared between 11 malignant samples (9 tumors and 2 cell lines) and the 2 normal samples (NK cells and tonsil) using 2-sided t tests. A large fraction of promoters were significantly hypermethylated in malignant samples (Fig. 1C); most categories of CGIs were hypermethylated as well. In contrast, distal gene bodies and extragenic regions, which are highly methylated in normal NK cells, tend to be relatively hypomethylated in malignant samples. Comparing normal and malignant samples after summing cut site reads based on the pattern of chromatin modification in human embryonic stem cells (hESC; refs. 21) demonstrated that a much larger fraction of “poised” promoters (Polycomb-repressed in hESCs) were significantly hypermethylated compared with other promoters (Fig. 1D). In contrast, distal transcribed regions, heterochromatin, and Poly-comb-repressed regions not associated with promoters are highly methylated in normal samples (Supplementary Fig. S1B) and frequently are relatively hypomethylated in malignant samples.
Relationship between promoter methylation and gene expression
Because the transcriptional effects of altered DNA methylation at distal sites are hard to predict and are likely variable, further analysis centered on promoter methylation. We compared methylation and gene expression for the 10,912 genes that had both gene expression data and at least one mappable HpaII site ± 1kb of the TSS. Most malignant samples display globally increased promoter methylation (Fig. 1). In general, genes with low expression were more hypermethylated than those with high expression (Supplementary Results). Because promoter methylation generally silences gene expression, there will be selection for methylation of tumor suppressor genes but negative selection against methylation at genes required for survival and proliferation such as housekeeping genes. No selection for or against hypermethylation is expected for genes not expressed in NK cells because they are functionally silent. Indeed, genes with little or no expression in normal NK cells are significantly more likely than other genes to be hypermethylated in tumors as a manifestation of the abnormal methylation; their gene bodies are also more likely to be hypo-methylated as often seen in cancer although the detailed pattern varies among the malignant samples (Supplementary Fig. S2).
Part of the explanation for the tendency of promoters poised in hESCs to become hypermethylated is that such genes are especially likely to have low or no expression in NK cells; however, this is unlikely to be a full explanation because even when the analyzed genes are in the top half of mean normal expression, “poised” genes are more frequently hypermethylated (for genes with at least 5 promoter cut sites, a mean of 2.17 samples per gene with significantly decreased cut counts compared with 1.55 for the remaining genes; Wilcoxon test, P = 1.1 × 10−6).
For the 9 malignant samples (7 tumors and 2 cell lines) with both MSCC and GEP data, the Pearson correlation coefficient (RMSCC-GEP) was calculated for each gene, using log2 MSCC and GEP values. The median RMSCC-GEP was 0.146 for all evaluable genes; this value is low partly due to the inclusion of genes that are either not expressed in NK cells or are never hypermethylated; variation in their apparent expression, MSCC values, or both would result from experimental noise, yielding no true correlation.
To narrow our further analysis to genes with the strongest evidence for silencing due to promoter hypermethylation, we devised 6 weak filters (Fig. 2), described in more detail in Supplementary Methods, which together yield a small tractable list of genes (Table 1 and Supplementary Table S3). (i) The gene must be expressed in NK cells. (ii) Cut counts and (iii) expression must vary among the samples. Three or more samples show evidence for a decrease in (iv) cut counts or (v) expression compared with normal NK cells. (vi) RMSCC-GEP must be at least 0.5 or 0.65, yielding 174 or 95 genes, respectively (Fig. 2). Each successive filter increased the mean RMSCC-GEP among the remaining genes (Fig. 2). We developed a linear predictor to prioritize additional genes that barely failed one or more filters, as described in Supplementary Methods. Table 1 and Supplementary Table S3 show the genes with the best evidence for being recurrently silenced in NKCL due to promoter methylation. In addition, Table 1 shows the pattern of hypermethylation of the 95 genes among 11 malignant samples. Supplementary Figure S3 illustrates examples of the strong negative correlation between promoter methylation and mRNA expression of the genes on the short list.
Figure 2.

Diagram showing successive steps in identifying a set of genes with strong evidence of downregulated expression by promoter methylation. On the right is a flowchart illustrating the filtering steps and the number of genes remaining after each filter, their mean RMSCC-GEP. The filters are designated as in the text. The numbers of genes with various 0.05 width intervals of Pearson correlation RMSCC-GEP (e.g., from −1 to −0.95) are graphed on the left. RMSCC-GEP increasingly skews toward higher values as the gene list is narrowed. We focused most of our further analysis on the 95 genes in box F, with occasional reference to a larger list of 174 genes within E with an RMSCC-GEP > 0.5 cutoff.
Table 1. Promoter methylation pattern of 95 epigenetically silenced genes detected by MSCC in NKCL samples.
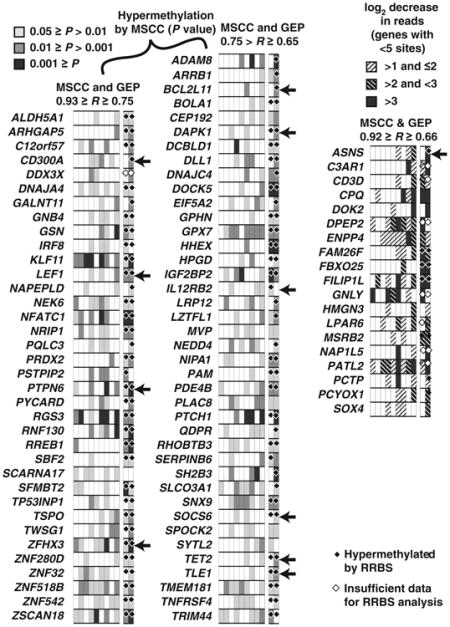
|
Note: MSCC evidence for promoter hypermethylation is shown for 9 tumor specimens and 2 cell lines (right 2 columns). For 76 genes with ≥5 MSCC cut sites, the levels of significance are shown from a 1-sided Wilcoxon test for decreased cut counts for these sites compared with normal NK cells. For 19 genes with <5 cut sites (right), the log2 fold decrease in cut counts is indicated. RRBS results were also obtained for normal NK cells and the 2 cell lines. Genes with significant differential methylation identified by the BiSeq program are shown (black diamonds); white diamonds indicate genes with insufficient RRBS data, that is, genes for which the minimum (in the 3 samples) total number of reads (C or T) within 1 kb was <100. Arrows on the right indicate genes experimentally analyzed individually in this study.
To validate our approach here to identify methylated genes using MSCC data, we performed reduced representation bisulfite sequencing (RRBS) on 2 NK cell lines with MSCC data (NK92 and KHYG1) and 3-day IL2-activated normal NK cells. Side-by-side comparison of the MSCC and RRBS data for the promoters of 95 methylated genes showed high concordance between these 2 methodologies (Table 1). In addition, a global comparison of pairwise (NK cell line vs. normal NK) methylation differences (Supplementary Fig. S4) showed high (R > 0.7) correlations between MSCC and RBSS results.
The 174-gene list was evaluated by Ingenuity Pathway Analysis (IPA; Table 2 and Supplementary Table S4). The genes were very significantly enriched in several functional groups of interest that involve cell death, cell activation, homeostasis and differentiation, proliferation and cancer, motility, and regulation of transcription. “Cellular homeostasis” and “cell death of immune cells” showed the greatest significance, at 5.7 × 10−9 and 1.4 × 10−8, respectively. Of note, many known tumor suppressor genes were enriched in our list (Supplementary Results).
Table 2. Selected functional groups for 174 recurrently hypermethylated genes in IPA.
| Diseases or functions annotation | P | Gene number |
|---|---|---|
| Cell death | ||
| Cell death of immune cells | 1.37E-08 | 25 |
| Necrosis | 4.84E-06 | 51 |
| Cell death | 3.30E-05 | 59 |
| Cell activation | ||
| Activation of T lymphocytes | 4.75E-07 | 16 |
| Activation of leukocytes | 1.25E-05 | 20 |
| Cell surface receptor–linked signal transduction | 8.84E-05 | 10 |
| Protein kinase cascade | 9.55E-04 | 12 |
| Homeostasis and differentiation | ||
| Cellular homeostasis | 5.73E-09 | 40 |
| T-cell development | 1.08E-07 | 20 |
| Homeostasis of leukocytes | 1.12E-07 | 21 |
| Proliferation and cancer | ||
| Proliferation of lymphocytes | 1.19E-06 | 23 |
| Proliferation of cells | 7.71E-06 | 64 |
| Cell-cycle progression | 1.72E-05 | 26 |
| Hyperplasia | 1.26E-04 | 16 |
| Cancer | 8.53E-03 | 126 |
| Motility | ||
| Cell movement | 5.76E-06 | 43 |
| Chemotaxis | 2.43E-04 | 15 |
| Leukocyte migration | 1.84E-03 | 19 |
| Regulation of transcription | ||
| Activation of DNA endogenous promoter | 5.07E-04 | 22 |
| Expression of RNA | 7.20E-03 | 33 |
Note: The name of the genes in these groups is indicated in Supplementary Table S4.
Methylated candidate tumor suppressor genes are silenced in NK cell lines
We performed qRT-PCR on NK cell lines to determine whether the expression of methylated candidate tumor suppressor genes was silenced. mRNA expression of 2 proapoptotic genes, BIM and DAPK1, was low in NK-cell lines compared with normal NK cells (Supplementary Fig. S5A). Similarly, we observed silencing of regulators of the JAK-STAT pathway, SOCS6, IL12RB2, and ZFHX3 (ATBF1), in NK cell lines compared with normal NK cells (Supplementary Fig. S5B). The expression of 2 other candidate genes, TET2 and TLE1, was in general low in NK cell lines compared with activated NK cells (Supplementary Fig. S5C). We observed low expression of ASNS in 5 of 6 NK cell lines compared with normal NK cells (Supplementary Fig. S5D, left); however, the KHYG1 cell line showed unusually high mRNA expression based on qRT-PCR, an observation consistent with MSCC and GEP results. Finally, we observed silencing of CD300A, which encodes an inhibitory NK cell receptor, in NK cell lines (Supplementary Fig. S5D, right). In general, the 2 NK cell lines found to be hypermethylated by MSCC showed low gene expression for candidate TSGs assayed with qRT-PCR. qMSP was performed on NK cell lines (n = 6) for the genes with qRT-PCR data available using unmethylated (DAPK1, ZFHX3, TLE1, SOCS6) or methylated (IL12RB2, TET2 and CD300A) DNA-specific primers and cell lines with low expression of these genes showed promoter hypermethylation compared with normal NK cells (Supplementary Fig. S5). For BIM and ASNS qMSP experiments in NK cell lines, both methylated and unmethylated DNA-specific primers were used as described below.
Finally, we evaluated BIM expression in NK cell lines by Western blotting and detected very low or no BIM protein in accordance with the qRT-PCR result (Supplementary Fig. S6).
Locus-specific validation of BIM, ASNS, and DAPK1 promoter methylation with pyrosequencing and qMSP
We evaluated the validity of the MSCC technique by comparing the promoter methylation profile of the NKCL cases with previously published Biotage pyrosequencing results (9) for the HpaII site at 747-bp upstream of the PRDM1α TSS. MSCC and pyrosequencing results were both available for this site in 13 malignant NK samples and 2 (resting and IL2-activated NK cells for Biotage pyrosequencing) or 3 (technical triplicate of IL2-activated NK cells for MSCC) normal NK samples. Comparison of the methylation difference between normal and malignant NK samples revealed a high inverse correlation (R = −0.75) between MSCC and pyrosequencing results (Supplementary Fig. S7B).
Next, we screened 2 separate regions in each of BIM, ASNS, or DAPK1 promoters within ±1 kb of the TSSs in 2 NK cell lines and normal NK cells using pyrosequencing. Side-by-side comparison of the 8 CpG sites with both MSCC and pyrosequencing data available showed a high inverse Pearson correlation between MSCC cut counts and pyrosequencing (R = −0.81 and −0.97 for BIM; R = −0.72, −0.53 and −1.00 for ASNS; R = −1.00, −1.00, −1.00 for DAPK1; Supplementary Figs. S7C and S8). No promoter methylation was detected in normal NK cells, whereas NK92 cells showed promoter hypermethylation in all 3 genes (Supplementary Fig. S8A–S8C). KHYG1 cells showed hypermethylation in the promoter of DAPK1 (Supplementary Fig. S8C). We then screened the BIM promoter (−510 to −360 bp of the TSS) with qMSP in NK cell lines (n = 7) using primers specific for methylated (M) or unmethylated (U) DNA (Supplementary Fig. S9A, left). We observed hypermethylation in 6 of 7 NK cell lines (Supplementary Fig. S9B, left). Screening the same BIM promoter region in 8 NKCL cases with qMSP using methylation-specific primers revealed increased BIM methylation in all NKCL cases compared with normal NK cells, suggesting that the BIM promoter is frequently hypermethylated in NKCL (Supplementary Figs. S9B, middle). Next, we applied the qMSP methodology on 7 malignant NK cell lines to examine methylation between 305 and 16 bp of the ASNS TSS (Supplementary Fig. S9A, right). We detected promoter hypermethylation in 6 of 7 (86%) NK cell lines (Supplementary Fig. S9C, left) and in 5 of 8 (62.5%) NKCL cases (Supplementary Fig. S9C, middle). As a control, we observed amplification of enzymatically methylated DNA only with BIM or ASNS methylation-specific qMSP primers but not with primers specific for unmethylated DNA (Supplementary Fig. S9D).
Decitabine treatment is associated with reexpression of silenced candidate TSGs in NK cell lines
Next, we wanted to test whether silenced tumor suppressor gene candidates could be reactivated using decitabine, a DNA methyltransferase inhibitor commonly used to evaluate the association between methylation and gene expression (7). NK cell lines with MSCC (i.e., NK92 and KHYG1) or qMSP data (i.e., SNK6) on promoter hypermethylation of the candidate tumor suppressor gene were treated for 4 days with Decitabine. We observed >2-fold upregulation of BIM mRNA expression in decitabine-treated NK92 and SNK6 cell lines, in which the BIM promoter is methylated (Fig. 3A, left). Consistent with qRT-PCR, we observed induction of BIM protein expression by Western blotting in NK92 cells (Supplementary Fig. S10). Similarly, DAPK1 was induced in response to decitabine in DAPK1-null NK92 and KHYG1 cells (Fig. 3A, right). A robust and decitabine dose-dependent increase in mRNA expression was observed for SOCS6, ZFHX3, SHP1, and TET2 in KHYG1 and NK92 cell lines (Fig. 3B). CD300A showed more than 200-fold induction of gene expression in all doses of decitabine tested in NK92 cells (Fig. 3C, left). TLE1, LEF1, and ASNS similarly showed more than 2-fold upregulation of mRNA expression in NK92 cells (Fig. 3C). Finally, we evaluated GAPDH and IL12RB2 expression in decitabine-treated NK92 cell line and ASNS and CD300A expression in decitabine-treated KHYG1 cells as negative controls and observed no significant changes in gene expression, as expected (Fig. 3D).
Figure 3.

Decitabine treatment reactivates expression of hypermethylated genes. A-C, BIM, DAPK1, SOCS6, ZFHX3, PTPN6, TET2, CD300A, TLE1, LEF1, and ASNS expression was determined by qRT-PCR in NK cell lines treated with decitabine for 4 days. D, GAPDH, IL12RB2, ASNS, or CD300A mRNA expression was determined by qRT-PCR in decitabine-treated NK92 or KHYG1 cells as negative controls. The mRNA levels of decitabine-treated NK cell lines were normalized to that of the untreated cells. Data are shown as mean ± SD. The data are representative of 2 biological replicates, each with 2 technical replicates (n = 4).
Reintroduction of BIM leads to negative selection pressure and causes apoptosis in BIM-null NK cell lines
To address whether decreased BIM expression contributes to enhanced survival of malignant NK cells, we reintroduced BIM-EL into 2 BIM-null NK cell lines, NKYS and KAI3, using the retrovirus murine stem cell virus (MSCV)-GFP (pMIG). GFP+ cells represented the transduced population, and tracking the changes in the percentage of GFP+ cells over time was used to evaluate whether there is any selection pressure on transduced cells. We observed a significant decrease in the percentage of GFP+NKYS cells between 2 and 3 days after transduction (Fig. 4A, left). Similarly, we observed a significant decrease in the percentage of GFP+ KAI3 cells 3 days after transduction compared with the percentage 2 days after transduction (Fig. 4A, right), suggesting that there is selection against NK cell lines with ectopic BIM expression. Ectopic expression of BIM was confirmed in BIM-EL–transduced NKYS cells by Western blotting (Supplementary Fig. S11).
Figure 4.
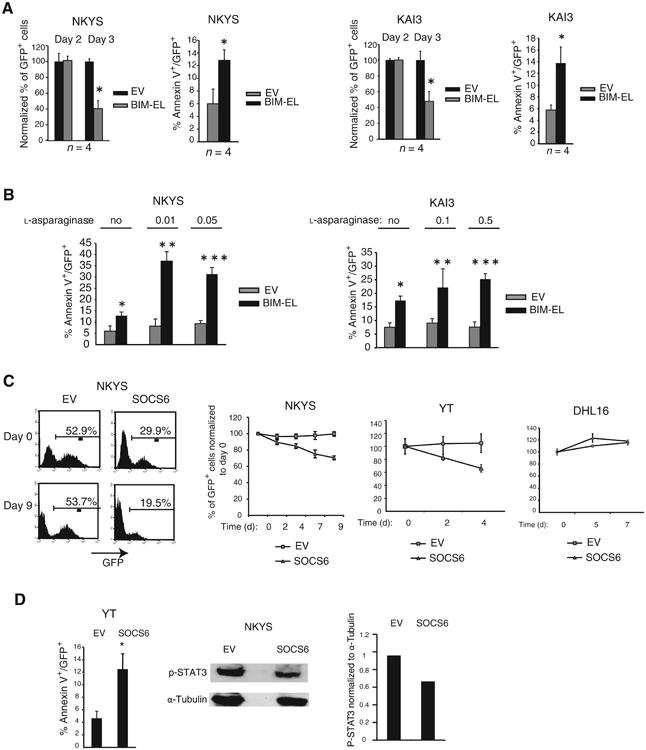
Ectopic expression of BIM or SOCS6 in methylated NK cell lines results in apoptosis, and reintroduction of BIM sensitizes malignant NK cells to asparaginase-induced apoptosis. A, quantification of the percentage of GFP+ cells 2 and 3 days after transduction of NKYS cells with empty vector or BIM-EL (left). The percentage of GFP+ cells in empty vector or BIM-EL-transduced NKYS cells 3 days after transduction were normalized to the percentage of GFP+ cells 2 days after transduction. Data are mean ± SD of 2 independent experiments with 2 biologic replicates (n = 4). *, P = 0.0075 versus percent GFP+ at day 2. Quantification of the rate of apoptosis in empty vector or BIM-EL-transduced NKYS cells by determining the percentage of Annexin-V/GFP double-positive NKYS cells (left). The percentage of apoptotic cells for each sample was calculated as follows: (% Annexin-V/GFP double-positive cells)/(Total % GFP-positive cells). Data are mean ± SD of 2 independent experiments with 2 biological replicates (n = 4). *, P = 0.037 versus vector-only. The percentage of GFP+ cells in empty vector or BIM-EL-transduced KAI3 cells 3 days after transduction normalized to the percentage of GFP+ cells 2 days after transduction (right). *, P = 0.0018 versus day 2 (n = 4). Quantification of the rate of apoptosis in empty vector or BIM-EL-transduced KAI3 cells as in (A, left) by identifying the percentage of Annexin-V/GFP double-positive KAI3 cells (right). Data are mean ± SD of 2 independent experiments with 2 biological replicates (n = 4).*, P = 0.017 versus vector only. B, comparison of the percentage of Annexin-V-PE+/GFP+ vector or BIM-EL-transduced NKYS cells in the presence or absence of 0.01 or 0.05 IU of l-asparaginase (left). Data are mean ± SD of a combination of 2 independent experiments (n ≥ 3). Comparison of the percentage of Annexin-V-PE+/GFP+ vector or BIM-EL-transduced KAI3 cells in the presence or absence of 0.1 or 0.5 IU of l-asparaginase treatment (right). Data are mean ± SD of 3 independent experiments (n = 4). *, P = 6.4 × 10−6; **, P = 2.6 × 10−3; and ***, P = 2.5 × 10−3 versus vector. C, representative FACS plots showing the percentage of GFP+ cells in empty vector or SOCS6-transduced NKYS cells 3 days (day 0) and 12 days (day 9) after transduction (left). Quantification of the percentage of GFP+ cells by FACS in NKYS or YT cells transduced with empty vector or SOCS6 in 2- or 3-day intervals after transduction (middle). Quantification of the percentage of the GFP+ population in empty vector or SOCS6-transduced DHL16 cells (right). Mean (±SD) is shown. Each data point represents 2 to 4 biologic replicates. D, quantification of the percentage of the Annexin-V+/GFP+ population in YT cells transduced with empty vector or SOCS6 3 days after transduction (left). Each data point represents a combination of 2 biological replicates with 2 technical replicates (n = 4). *, P = 0.001. NKYS cells were transduced with empty vector or SOCS6. Two days after transduction, IL2 was removed from the culture medium. Three days after transduction, GFP+ cells were sorted side by side, and Western blotting was performed to evaluate p-STAT3 (Y705) expression (middle). A representative gel image of 2 Western blots is shown. Normalized p-STAT3 expression quantified with the ImageJ program (http://rsb.info.nih.gov/ij/) is shown (right).
Next, we stained BIM-EL–transduced NKYS cells with Annexin V to determine whether reintroduction of BIM-EL increases the rate of apoptosis. We observed a significant increase in Annexin V staining in the GFP-gated cellular population 2 days post-transduction (Fig. 4A, left). Similarly, we observed a significant increase in Annexin V staining in BIM-EL–transduced KAI3 cells 2 days after transduction (Fig. 4A, right) suggesting that methylation-mediated silencing of BIM enhances survival of malignant NK cells.
Reintroduction of BIM increases the sensitivity of malignant NK cells to asparaginase-induced apoptosis
We treated empty vector (EV) or BIM-EL–transduced BIM-null NK cell lines with l-asparaginase to test whether there is an increase of sensitivity in induction of apoptosis. BIM-transduced NKYS cells treated with 0.01 or 0.05 IU of l-asparaginase showed a higher percentage of Annexin V+ cells compared with cells transduced with BIM-EL but not treated with l-asparaginase or empty vector–transduced, l-asparaginase–treated cells (Fig. 4B, left). Next, we treated another BIM-transduced BIM-null NK cell line, KAI3, with 0.1 or 0.5 IU of l-asparaginase and observed an increase in induction of apoptosis in l-asparaginase–treated, BIM-transduced KAI3 cells compared with l-asparaginase–treated, vector-only transduced cells (Fig. 4B, right).
Ectopic expression of SOCS6 in NK cell lines leads to negative selection pressure and induces apoptosis under limiting IL2 concentrations
To address whether epigenetic silencing of SOCS6 contributes to the neoplastic transformation of NK cells through more sustained activation of the JAK/STAT pathway, we transduced the SOCS6-null NKYS cell line with a SOCS6 expression vector or with empty vector. We then quantified the percentage of GFP+ cells at regular time intervals to assess whether SOCS6-transduced NKYS cells are negatively selected. IL2 is known to activate STAT3 and STAT5 (22). Therefore, IL2 was removed from the cell culture medium of transduced cells 6 days after transduction. The percentage of GFP+ cells declined about 30% in the SOCS6-transduced population between 3 and 12 days after transduction, whereas empty vector transduced cells did not show any decrease (Fig. 4C, left). We repeated the experiment with the SOCS6-null YT cell line and again observed a significant reduction in GFP+ cells between 4 and 8 days after transduction in SOCS6-transduced YT cells but not those transduced with empty vector (Fig. 4C, middle). SOCS6-transduced DHL16 cell line, which is a germinal center B-cell–like diffuse large B-cell lymphoma (GCB-DLBCL) cell line with no JAK-STAT3 pathway activation, was not negatively selected (Fig. 4C, right). Ectopic expression of SOCS6 was confirmed in SOCS6-transduced NKYS and YT cells by qRT-PCR (Supplementary Fig. S12B and S12C).
To address whether the negative selection observed in SOCS6-transduced NK cell lines is due to increased apoptosis, we transduced YT cells with the SOCS6 expression vector or empty vector and observed a significantly higher percentage of Annexin-V+/GFP+ cells in SOCS6-transduced YT cells than in empty vector–transduced cells (Fig. 4D, left). In addition, we observed moderately lower levels of phospho-STAT3 (Tyr705) expression in SOCS6-transduced NKYS cells than in empty vector–transduced cells (Fig. 4D, middle and right), suggesting that SOCS6-mediated inhibition of NK cell growth may be due to JAK/STAT3 pathway inhibition.
NK cell lines with low ASNS expression are more sensitive to l-asparaginase treatment
To address whether the effectiveness of l-asparaginase treatment correlated with the ASNS mRNA levels in the malignant NK cells, we treated NK cell lines (n = 6) with progressively increasing doses of l-asparaginase. Initially, we treated KHYG1, SNK6, NKYS, and YT cells with 0.1 or 0.5 IU of asparaginase and observed greater asparaginase sensitivity in SNK6, NKYS, and YT cells than in KHYG1 cells (Fig. 5A). In the second experiment, we treated KHYG1, KAI3, NK92, and YT cell lines with lower concentrations (0.01, 0.05, or 0.075 IU) of asparaginase and observed high sensitivity in KAI3, NK92, and YT cell lines, which have low ASNS expression due to promoter methylation (Fig. 5B). As expected, the KHYG1 cell line, with no promoter methylation and high ASNS expression, was least sensitive to asparaginase treatment (Fig. 5B, left). In fact, ASNS mRNA expression was highly correlated with normalized cell survival in response to 0.01, 0.05, and 0.075 IU asparaginase (R = 0.77, 0.84, and 0.88, respectively; Fig. 5C).
Figure 5.

NK cell lines with silenced ASNS expression are more sensitive to l-asparaginase treatment, and reintroduction of ASNS reduces drug sensitivity. NK cel ines were treated with l-asparaginase for 24 hours using 0.1 or 0.5 IU (A) or 0.01, 0.05, 0.075 IU (B) of l-asparaginase, and cell survival was determined as a percentage of the untreated control for each cell line. KHYG1 cells, whipromoter methylation in NKCL ch do not have promoter methylation and high ASNS expression, were used as the control during asparaginase treatment of NK cell lines. Relative ASNS mRNA expression based on qRT-PCR is shown below each panel. The numeric values below each panel represent the ASNS expression fold difference relative to resting NK cells. ASNS promoter methylation is shown for each NK cell line based on the qMSP Ct value difference between each malignant NK cell line and 3-day IL2-activated NK cells, as shown in Supplementary Figs. S5D and S9C. No, no hypermethylation; ++, highly hypermethylated, using the cutoff values described in Supplementary Fig. S5. C, Pearson correlation coefficient (R) between mRNA expression based on qRT-PCR, and percent normalized cell survival was determined for NK cell lines (n = 4) treated with 0.01, 0.05, or 0.075 IU of l-asparaginase (left). Tables show normalized ASNS mRNA expression and percentage (%) survival in NK cell lines (n = 4) after 0.01, 0.05, or 0.075 IU of asparaginase treatment (right). Data are mean ± SD of 2 independent experiments. D, absolute (left) or normalized (middle) cell numbers of empty vector (EV) or ASNS-transduced NKYS cells treated with the indicated concentrations of asparaginase concentrations are shown. GFP+ cells were sorted 8 days after transduction. Three days later, sorted cells were seeded for asparaginase treatment, and 48 hours after treatment, viable cell number was determined as described in Materials and Methods. Each column shows mean ± SD of 3 biological replicates. Ectopic ASNS expression is shown by qRT-PCR 2 days after transduction of NKYS cells (right). Each data point shows mean ± SD of triplicate of ASNS calibrated to RPL13A (housekeeping gene). ASNS levels in ASNS-transduced cells normalized to empty vector-transduced cells are shown. *, P < 0.01 versus vector only.
Next, we asked whether ASNS reexpression can reduce cell death after asparaginase treatment or not. To address this question, we transduced ASNS-nonexpressing NKYS cell line with an empty vector or ASNS expression vector. Then, we sorted GFP+ cells and treated empty vector or ASNS-transduced NKYS cells with 0, 0.01, 0.05, or 0.075 IU asparaginase for 48 hours. Ectopic expression of ASNS in ASNS-transduced NKYS cells was demonstrated by qRT-PCR (Fig. 5D, right). There was marked reduction in cell viability in empty vector–transduced cells for all tested concentrations, and no cytotoxicity was observed in ASNS-transduced cells (Fig. 5D, left). However, there was significantly more normalized cell survival after asparaginase treatment in ASNS-transduced cells compared with empty vector–transduced cells, which supports the idea that epigenetic silencing of ASNS in NKCLs increases asparaginase sensitivity (Fig. 5D, middle).
Discussion
Using the MSCC technique to analyze DNA methylation throughout the genome revealed that promoter hypermethylation and distal DNA hypomethylation were widespread in most malignant NK samples. Selection for inactivation at individual loci cannot entirely explain this hypermethylation because it was also frequent at promoters of genes with little or no expression in normal NK cells. Several categories of genes appear to be prone to hypermethylation in malignant NK samples including the category of genes “poised” in hESCs and genes expressed but functionless in NK cells (e.g., CD3D). We therefore used a number of strategies to filter out noise and to identify a set of genes that are most likely to be functionally significant. Known TSGs and genes important in normal NK function are enriched among these hypermethylated genes. We also determined the profile of promoter methylation in 2 cell lines using another global technique, RRBS, which yielded very comparable results. This increased our confidence in our previous analysis and the set of recurrently hypermethylated and silenced genes, many of which likely function as TSGs in these NK cell tumors.
Inactivation of TSGs through genetic or epigenetic mechanisms is a critical mediator of the neoplastic transformation of normal cells. Hypermethylation of the promoters of TSGs has commonly been observed in a variety of lymphoid malignancies (14) including NKCLs (9, 11). Epigenetic silencing of multiple TSGs may have additive or synergistic effects leading to the evasion of normal growth control mechanisms.
The genes found to be silenced through promoter methylation in malignant NK cells can limit the growth and survival of NK cells through regulation of multiple pathways (Table 2). Silencing of BIM may enhance NK cell survival by conferring resistance to proapoptotic signals, as observed in Burkitt lymphoma cells (14). EBV infection may contribute to BIM promoter methylation and silencing (23), and interestingly, NKCL is almost invariably EBV infected. Downregulation of DAPK1, a serine/threonine kinase known to activate a p19ARF/p53-dependent apoptotic checkpoint (24), was observed in chronic lymphocytic leukemia (25) in addition to NKCLs (12).
SOCS6 and ZNFX3 negatively regulate the JAK/STAT pathway (26, 27) and are downregulated in a variety of solid tumors including gastric, pancreatic, and lung cancers (28–31). As previously observed (11), we found SHP1 (PTPN6) promoter methylation in NKCL cases (Table 1 and Supplementary Table S3) and now showed its reactivation after decitabine treatment (Fig. 3B, right). Interestingly, aside from its role as a negative regulator of signaling through activating NK-cell receptors, SHP1 has been shown to inactivate STAT3 through dephosphorylation of tyro-sine-705 (32), which suggests that epigenetic silencing of SHP1 may promote activation of the JAK/STAT3 pathway. Epigenetic silencing of the negative regulators of this pathway may be associated with the neoplastic transformation of NK cells as constitutive activation of the JAK/STAT pathway is observed in NKCLs (33).
Alterations in epigenetic regulators are frequent in lymphomas and likely of importance in NKCL. TET2 loss-of-function mutations are frequent in myelodysplastic syndrome (MDS; ref. 34) and T-cell lymphoma (35). Silencing of TET2 may contribute to aberrant promoter methylation of genes that contribute to the development of NKCL (36). In fact, early epigenetic inactivation of TET2 may contribute to the establishment of global promoter hypermethylation observed in NKCL samples.
TLE1 is a groucho homolog, and its epigenetic inactivation was observed in hematologic malignancies (37) including acute myelogenous leukemia (38). IL12RB2 is a TSG in B-cell malignancies (39, 40). The consequences of silencing of IL12RB2 in NK cells are less clear; however, a recent study showed induction of cell death during prolonged IL12 receptor signaling in NK cells (41), suggesting that silencing of IL12RB2 may contribute to NK cell survival in the presence of prolonged IL12 stimulation from the microenvironment.
PRDM1 was not in our list because the available HpaII sites around the TSS (−747 and +99) are not in the critical hypermethylated genomic locus of PRDM1α (9) we previously identified in NKCLs. The lists failed to include HACE1 only because the SD for gene expression fell below the threshold, which may reflect suboptimal microarray probe sets.
Methylation-mediated silencing of ASNS in malignant NK samples is a significant observation from the clinical point of view; a similar observation was made in acute lymphoblastic leukemia (42). The mechanism responsible for the apparent selection for loss of ASNS expression in tumors is unclear. Nevertheless, the high correlation between ASNS expression and cell survival in response to l-asparaginase treatment may be useful for predicting which patients with NKCL will likely respond to l-asparaginase-containing chemotherapeutic regimens. High ASNS expression in some patients with NKCL may account for the resistance to l-asparaginase–containing regimens (43). In addition, l-asparaginase treatment-associated side effects (44, 45) may be minimized by using lower concentrations of l-asparaginase for treating patients with NKCL with low ASNS expression.
In summary, we integrated global promoter methylation and gene expression profiling data to identify candidate TSGs silenced because of promoter hypermethylation. Selected candidates were validated by functional assays. Our findings improve our understanding of the pathogenesis of NKCL and may suggest novel potential therapeutic targets.
Supplementary Material
Translational Relevance.
This is the first study to analyze genome-wide promoter methylation in natural killer cell lymphoma (NKCL). Reactivation of the candidate tumor suppressor genes found to be silenced through promoter methylation can be used as a therapeutic strategy for patients with NKCL. This therapeutic potential was clearly observed by the reintroduction of BIM and SOCS6 into nonexpressing cells. The therapeutic significance of the identification of BIM silencing in NKCL is further supported by the fact that BIM sensitizes NK cell lines to chemotherapy-induced apoptosis. In addition, ASNS methylation can be used as a biomarker for response to asparaginase-based regimens. Asparaginase-related side effects may be reduced by decreasing the dosage of asparaginase administered to patients with NKCL with high levels of ASNS hypermethylation.
Acknowledgments
The authors thank Dr. Dean Anthony Lee (MD Anderson Cancer Center, Houston, TX) for kindly providing the K562-Clone9-mbIL21 cells, a K562 cell line engineered to express 4-1BBL, CD86, and mbIL21 on the cell surface to achieve higher levels of NK cell activation.
Grant Support: The UNMC DNA Sequencing Core receives partial support from the NCRR (1S10RR027754-01, 5P20RR016469, RR018788-08) and the National Institute for General Medical Science (NIGMS; 8P20GM103427, GM103471-09).
Footnotes
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Disclosure of Potential Conflicts of Interest: No potential conflicts of interest were disclosed.
Authors' Contributions: Conception and design: C. Küçük, J. Iqbal, W.C. Chan
Development of methodology: T.W. McKeithan Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): J. Iqbal, P. Gaulard, W.C. Chan
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): C. Küçük, X. Hu, B. Jiang, D. Klinkebiel, H. Geng, Q. Gong, A. Bouska, J. Iqbal, T.W. McKeithan, W.C. Chan
Writing, review, and/or revision of the manuscript: C. Küçük, A. Bouska, T.W. McKeithan, W.C. Chan
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): X. Hu, B. Jiang
Study supervision: W.C. Chan
Other (performed in vitro experiments): X. Hu
Other (performed experiments): C. Küçük
References
- 1.Vose J, Armitage J, Weisenburger D. International peripheral T-cell and natural killer/T-cell lymphoma study: pathology findings and clinical outcomes. J Clin Oncol. 2008;26:4124–30. doi: 10.1200/JCO.2008.16.4558. [DOI] [PubMed] [Google Scholar]
- 2.Jaffe ES, Nicolae A, Pittaluga S. Peripheral T-cell and NK-cell lymphomas in the WHO classification: pearls and pitfalls. Mod Pathol. 2013;26(Suppl 1):S71–87. doi: 10.1038/modpathol.2012.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Iqbal J, Weisenburger DD, Chowdhury A, Tsai MY, Srivastava G, Greiner TC, et al. Natural killer cell lymphoma shares strikingly similar molecular features with a group of non-hepatosplenic gammadelta T-cell lymphoma and is highly sensitive to a novel aurora kinase A inhibitor in vitro. Leukemia. 2011;25:348–58. doi: 10.1038/leu.2010.255. [DOI] [PubMed] [Google Scholar]
- 4.Kim SJ, Kim K, Kim BS, Kim CY, Suh C, Huh J, et al. Phase II trial of concurrent radiation and weekly cisplatin followed by VIPD chemotherapy in newly diagnosed, stage IE to IIE, nasal, extranodal NK/T-Cell Lymphoma: Consortium for Improving Survival of Lymphoma study. J Clin Oncol. 2009;27:6027–32. doi: 10.1200/JCO.2009.23.8592. [DOI] [PubMed] [Google Scholar]
- 5.Jaccard A, Petit B, Girault S, Suarez F, Gressin R, Zini JM, et al. L-aspar-aginase-based treatment of 15 western patients with extranodal NK/T-cell lymphoma and leukemia and a review of the literature. Ann Oncol. 2009;20:110–6. doi: 10.1093/annonc/mdn542. [DOI] [PubMed] [Google Scholar]
- 6.Yamaguchi M, Kwong YL, Kim WS, Maeda Y, Hashimoto C, Suh C, et al. Phase II study of SMILE chemotherapy for newly diagnosed stage IV, relapsed, or refractory extranodal natural killer (NK)/T-cell lymphoma, nasal type: the NK-Cell Tumor Study Group study. J Clin Oncol. 2011;29:4410–6. doi: 10.1200/JCO.2011.35.6287. [DOI] [PubMed] [Google Scholar]
- 7.Iqbal J, Kucuk C, Deleeuw RJ, Srivastava G, Tam W, Geng H, et al. Genomic analyses reveal global functional alterations that promote tumor growth and novel tumor suppressor genes in natural killer-cell malignancies. Leukemia. 2009;23:1139–51. doi: 10.1038/leu.2009.3. [DOI] [PubMed] [Google Scholar]
- 8.Karube K, Nakagawa M, Tsuzuki S, Takeuchi I, Honma K, Nakashima Y, et al. Identification of FOXO3 and PRDM1 as tumor-suppressor gene candidates in NK-cell neoplasms by genomic and functional analyses. Blood. 2011;118:3195–204. doi: 10.1182/blood-2011-04-346890. [DOI] [PubMed] [Google Scholar]
- 9.Kucuk C, Iqbal J, Hu X, Gaulard P, De Leval L, Srivastava G, et al. PRDM1 is a tumor suppressor gene in natural killer cell malignancies. Proc Natl Acad Sci U S A. 2011;108:20119–24. doi: 10.1073/pnas.1115128108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3:415–28. doi: 10.1038/nrg816. [DOI] [PubMed] [Google Scholar]
- 11.Oka T, Ouchida M, Koyama M, Ogama Y, Takada S, Nakatani Y, et al. Gene silencing of the tyrosine phosphatase SHP1 gene by aberrant methylation in leukemias/lymphomas. Cancer Res. 2002;62:6390–4. [PubMed] [Google Scholar]
- 12.Rohrs S, Romani J, Zaborski M, Drexler HG, Quentmeier H. Hypermethylation of Death-Associated Protein Kinase 1 differentiates natural killer cell lines from cell lines derived from T-acute lymphoblastic leukemia. Leukemia. 2009;23:1174–6. doi: 10.1038/leu.2009.5. [DOI] [PubMed] [Google Scholar]
- 13.Kucuk C, Hu X, Iqbal J, Gaulard P, Klinkebiel D, Cornish A, et al. HACE1 is a tumor suppressor gene candidate in natural killer cell neoplasms. Am J Pathol. 2013;182:49–55. doi: 10.1016/j.ajpath.2012.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Richter-Larrea JA, Robles EF, Fresquet V, Beltran E, Rullan AJ, Agirre X, et al. Reversion of epigenetically mediated BIM silencing overcomes chemoresistance in Burkitt lymphoma. Blood. 2010;116:2531–42. doi: 10.1182/blood-2010-02-268003. [DOI] [PubMed] [Google Scholar]
- 15.Chim CS, Wong KY, Loong F, Srivastava G. SOCS1 and SHP1 hypermethylation in mantle cell lymphoma and follicular lymphoma: implications for epigenetic activation of the Jak/STAT pathway. Leukemia. 2004;18:356–8. doi: 10.1038/sj.leu.2403216. [DOI] [PubMed] [Google Scholar]
- 16.Hlady RA, Novakova S, Opavska J, Klinkebiel D, Peters SL, Bies J, et al. Loss of Dnmt3b function upregulates the tumor modifier Ment and accelerates mouse lymphomagenesis. J Clin Invest. 2012;122:163–77. doi: 10.1172/JCI57292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eckhardt F, Lewin J, Cortese R, Rakyan VK, Attwood J, Burger M, et al. DNA methylation profiling of human chromosomes 6, 20 and 22. Nat Genet. 2006;38:1378–85. doi: 10.1038/ng1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Easwaran H, Johnstone SE, Van Neste L, Ohm J, Mosbruger T, Wang Q, et al. A DNA hypermethylation module for the stem/progenitor cell signature of cancer. Genome Res. 2012;22:837–49. doi: 10.1101/gr.131169.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ohm JE, McGarvey KM, Yu X, Cheng L, Schuebel KE, Cope L, et al. A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat Genet. 2007;39:237–42. doi: 10.1038/ng1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43–9. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johnston JA, Bacon CM, Finbloom DS, Rees RC, Kaplan D, Shibuya K, et al. Tyrosine phosphorylation and activation of STAT5, STAT3, and Janus kinases by interleukins 2 and 15. Proc Natl Acad Sci U S A. 1995;92:8705–9. doi: 10.1073/pnas.92.19.8705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Paschos K, Parker GA, Watanatanasup E, White RE, Allday MJ. BIM promoter directly targeted by EBNA3C in polycomb-mediated repression by EBV. Nucleic Acids Res. 2012;40:7233–46. doi: 10.1093/nar/gks391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martoriati A, Doumont G, Alcalay M, Bellefroid E, Pelicci PG, Marine JC. dapk1, encoding an activator of a p19ARF-p53-mediated apoptotic checkpoint, is a transcription target of p53. Oncogene. 2005;24:1461–6. doi: 10.1038/sj.onc.1208256. [DOI] [PubMed] [Google Scholar]
- 25.Raval A, Tanner SM, Byrd JC, Angerman EB, Perko JD, Chen SS, et al. Downregulation of death-associated protein kinase 1 (DAPK1) in chronic lymphocytic leukemia. Cell. 2007;129:879–90. doi: 10.1016/j.cell.2007.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hwang MN, Min CH, Kim HS, Lee H, Yoon KA, Park SY, et al. The nuclear localization of SOCS6 requires the N-terminal region and negatively regulates Stat3 protein levels. Biochem Biophys Res Commun. 2007;360:333–8. doi: 10.1016/j.bbrc.2007.06.062. [DOI] [PubMed] [Google Scholar]
- 27.Nojiri S, Joh T, Miura Y, Sakata N, Nomura T, Nakao H, et al. ATBF1 enhances the suppression of STAT3 signaling by interaction with PIAS3. Biochem Biophys Res Commun. 2004;314:97–103. doi: 10.1016/j.bbrc.2003.12.054. [DOI] [PubMed] [Google Scholar]
- 28.Wu K, Hu G, He X, Zhou P, Li J, He B, et al. MicroRNA-424-5p suppresses the expression of SOCS6 in pancreatic cancer. Pathol Oncol Res. 2013;19:739–48. doi: 10.1007/s12253-013-9637-x. [DOI] [PubMed] [Google Scholar]
- 29.Sriram KB, Larsen JE, Savarimuthu Francis SM, Wright CM, Clarke BE, Duhig EE, et al. Array-comparative genomic hybridization reveals loss of SOCS6 is associated with poor prognosis in primary lung squamous cell carcinoma. PLoS One. 2012;7:e30398. doi: 10.1371/journal.pone.0030398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Minamiya Y, Saito H, Ito M, Imai K, Konno H, Takahashi N, et al. Suppression of Zinc Finger Homeobox 3 expression in tumor cells decreases the survival rate among non-small cell lung cancer patients. Cancer Biomark. 2012;11:139–46. doi: 10.3233/CBM-2012-00272. [DOI] [PubMed] [Google Scholar]
- 31.Lai RH, Hsiao YW, Wang MJ, Lin HY, Wu CW, Chi CW, et al. SOCS6, down-regulated in gastric cancer, inhibits cell proliferation and colony formation. Cancer Lett. 2010;288:75–85. doi: 10.1016/j.canlet.2009.06.025. [DOI] [PubMed] [Google Scholar]
- 32.Wang P, Xue Y, Han Y, Lin L, Wu C, Xu S, et al. The STAT3-binding long noncoding RNA lnc-DC controls human dendritic cell differentiation. Science. 2014;344:310–3. doi: 10.1126/science.1251456. [DOI] [PubMed] [Google Scholar]
- 33.Huang Y, de Reynies A, de Leval L, Ghazi B, Martin-Garcia N, Travert M, et al. Gene expression profiling identifies emerging oncogenic pathways operating in extranodal NK/T-cell lymphoma, nasal type. Blood. 2010;115:1226–37. doi: 10.1182/blood-2009-05-221275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Langemeijer SM, Kuiper RP, Berends M, Knops R, Aslanyan MG, Massop M, et al. Acquired mutations in TET2 are common in myelodysplastic syndromes. Nat Genet. 2009;41:838–42. doi: 10.1038/ng.391. [DOI] [PubMed] [Google Scholar]
- 35.Quivoron C, Couronne L, Della Valle V, Lopez CK, Plo I, Wagner-Ballon O, et al. TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell. 2011;20:25–38. doi: 10.1016/j.ccr.2011.06.003. [DOI] [PubMed] [Google Scholar]
- 36.Ko M, Huang Y, Jankowska AM, Pape UJ, Tahiliani M, Bandukwala HS, et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature. 2010;468:839–43. doi: 10.1038/nature09586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fraga MF, Berdasco M, Ballestar E, Ropero S, Lopez-Nieva P, Lopez-Serra L, et al. Epigenetic inactivation of the Groucho homologue gene TLE1 in hematologic malignancies. Cancer Res. 2008;68:4116–22. doi: 10.1158/0008-5472.CAN-08-0085. [DOI] [PubMed] [Google Scholar]
- 38.Dayyani F, Wang J, Yeh JR, Ahn EY, Tobey E, Zhang DE, et al. Loss of TLE1 and TLE4 from the del (9q) commonly deleted region in AML cooperates with AML1-ETO to affect myeloid cell proliferation and survival. Blood. 2008;111:4338–47. doi: 10.1182/blood-2007-07-103291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Airoldi I, Di Carlo E, Cocco C, Sorrentino C, Fais F, Cilli M, et al. Lack of Il12rb2 signaling predisposes to spontaneous autoimmunity and malignancy. Blood. 2005;106:3846–53. doi: 10.1182/blood-2005-05-2034. [DOI] [PubMed] [Google Scholar]
- 40.Pistoia V, Cocco C, Airoldi I. Interleukin-12 receptor beta2: from cytokine receptor to gatekeeper gene in human B-cell malignancies. J Clin Oncol. 2009;27:4809–16. doi: 10.1200/JCO.2008.21.3579. [DOI] [PubMed] [Google Scholar]
- 41.Huang Y, Lei Y, Zhang H, Zhang M, Dayton A. Interleukin-12 treatment down-regulates STAT4 and induces apoptosis with increasing ROS production in human natural killer cells. J Leukoc Biol. 2011;90:87–97. doi: 10.1189/jlb.1210674. [DOI] [PubMed] [Google Scholar]
- 42.Stams WA, den Boer ML, Holleman A, Appel IM, Beverloo HB, van Wering ER, et al. Asparagine synthetase expression is linked with L-asparaginase resistance in TEL-AML1-negative but not TEL-AML1-positive pediatric acute lymphoblastic leukemia. Blood. 2005;105:4223–5. doi: 10.1182/blood-2004-10-3892. [DOI] [PubMed] [Google Scholar]
- 43.Jaccard A, Gachard N, Marin B, Rogez S, Audrain M, Suarez F, et al. Efficacy of L-asparaginase with methotrexate and dexamethasone (AspaMetDex regimen) in patients with refractory or relapsing extranodal NK/T-cell lymphoma, a phase 2 study. Blood. 2011;117:1834–9. doi: 10.1182/blood-2010-09-307454. [DOI] [PubMed] [Google Scholar]
- 44.Haskell CM, Canellos GP, Leventhal BG, Carbone PP, Block JB, Serpick AA, et al. L-asparaginase: therapeutic and toxic effects in patients with neoplastic disease. N Engl J Med. 1969;281:1028–34. doi: 10.1056/NEJM196911062811902. [DOI] [PubMed] [Google Scholar]
- 45.Yong W, Zheng W, Zhu J, Zhang Y, Wang X, Xie Y, et al. L-asparaginase in the treatment of refractory and relapsed extranodal NK/T-cell lymphoma, nasal type. Ann Hematol. 2009;88:647–52. doi: 10.1007/s00277-008-0669-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.