

Abstract
The BRCA1 tumor suppressor protein is a central constituent of several distinct macromolecular protein complexes that execute homology-directed DNA damage repair and cell cycle checkpoints. Recent years have borne witness to an exciting phase of discovery at the basic molecular level for how this network of DNA repair proteins acts to maintain genome stability and suppress cancer. The clinical dividends of this investment are now being realized with the approval of first-in-class BRCA-targeted therapies for ovarian cancer and identification of molecular events that determine responsiveness to these agents. Further delineation of the basic science underlying BRCA network function holds promise to maximally exploit genome instability for hereditary and sporadic cancer therapy.
Keywords: BRCA1, BRCA2, breast cancer, DNA damage response, DNA repair
Introduction
The breast cancer early-onset genes BRCA1 and BRCA2 were discovered by positional cloning approaches in kindreds with a high prevalence of breast and ovarian cancer. The initial lack of clarity presented by the domain structure of the proteins was remedied by a series of discoveries that revealed the proteins biochemically interact in large nuclear foci in response to DNA damage and are required to execute homology-directed DNA repair and cell cycle checkpoints (1). These observations strongly suggested that a common function in genome integrity maintenance is necessary to suppress cancer. Subsequent advances in protein purification and mass spectrometry methodologies have led to the expansion of this concept with the discovery that at least 13 different tumor suppressor proteins interact with BRCA1 and BRCA2. Despite these striking similarities, a linear model of BRCA-dependent DNA repair and tumor suppression is challenged by multiple other observations, namely, only ∼5% of each protein exists in association with the BRCA1-BRCA2 complex, and breast cancers occurring in individuals with germ-line BRCA1 or BRCA2 mutations typically display different histopathologies and gene expression profiles. Coupled with the realization that chemoresistance mechanisms in BRCA1 and BRCA2 mutant cancers also differ, these features have inspired a network model relating BRCA molecular function in DNA repair to tumor suppression. We highlight recent insights into the BRCA tumor suppressor network and stress the connections between basic molecular knowledge of these proteins and their roles in genome integrity, tumor suppression, and response to therapy.
Overview of BRCA1 Structure and Cancer Susceptibility
The BRCA1 protein has several definable structural domains that suggest it uses modular and potentially cooperative interactions to execute its DNA damage response (DDR)2 functions (Fig. 1). The N-terminal RING (really interesting new gene) domain enables E3 ubiquitin ligase activity through its interaction with E2 enzymes. Clinical mutations to the RING domain are associated with cancer susceptibility. However, knock-in of a rationally designed BRCA1 RING mutant (I26A) that disrupts interaction with E2 enzymes does not abrogate genome stability or confer cancer predisposition in mice (2). Conversely, a known clinical mutant (C61G) that also disrupts E3 activity causes genome instability and cancer in mice (3). Complicating interpretation of this phenotype as proof that BRCA1 E3 activity suppresses cancer is the confounding issue that the C61G mutation disrupts RING architecture and association with BARD1, a stoichiometric binding partner of BRCA1 (4). The C61G mutation may impart cancer susceptibility by loss of E3 ligase activity, diminished BARD1 interaction, or a combination of these deleterious events.
FIGURE 1.
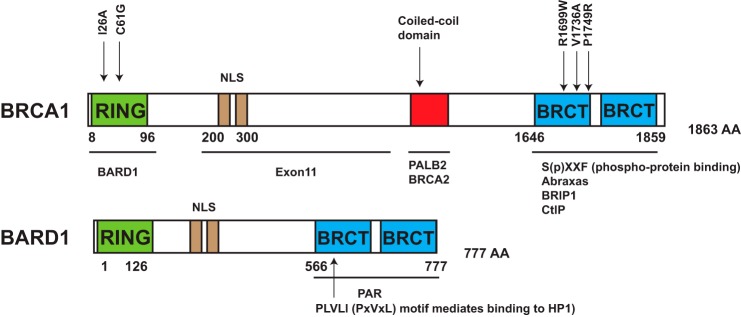
Domain structure of BRCA1-BARD1 and key interacting partners in the BRCA network. Key interacting partners and mutations referred to in text are indicated. R1699W and V1736A were found in patients with BRCA1 biallelic mutations. K619A, C645R, and V695L mutations within the BARD1 BRCT repeats fail to interact with poly(ADP-ribose) (PAR). NLS, nuclear localization sequence.
Approximately 60% of the BRCA1 protein is composed of the centrally located exon 11-encoded region. This poorly conserved region lacks definitive domain elements or interacting partners yet is required for full homologous recombination (HR) and checkpoint function as well as tumor suppression (5, 6). Nonetheless, exon 11 mutations allow an in-frame splice between exons 10 and 12 and the production of a partially active protein that localizes to DNA damage sites. Downstream of exon 11, BRCA1 harbors a coiled-coil region near its C terminus that interacts with PALB2, which biochemically bridges BRCA1 to BRCA2 in a tumor suppressor supercomplex (Fig. 1). PALB2 mutations confer high-penetrance breast cancer phenotypes (7), similar to BRCA1 and BRCA2. C-terminal to this region are the BRCA1 C-terminal (BRCT) repeats, which bind to phosphopeptides. Mutually exclusive interactions with three bona fide BRCT-interacting proteins allow segregation into at least three different protein complexes. The distinct BRCA1-containing complexes, which are discussed in detail below, are thought to work together in response to double-strand breaks (DSBs) (8). The BRCT domain of BRCA1 contributes to most of its functional interaction with different protein complexes. BRIP1, Abraxas, and CtIP all contain a consensus BRCT-interacting motif (SXXF), which is phosphorylated at serine to mediate the interaction (9, 10).
Connecting BRCA Network Biochemistry to the DDR
BARD1 has a similar domain structure to BRCA1, with an N-terminal RING domain and BRCT repeats. Unlike BRCA1, the BARD1 RING domain does not interact with E2 enzymes, and its BRCT repeats do not bind phosphopeptides. The stoichiometric interaction between BRCA1 and BARD1 instead provides stability to both proteins, enhances DNA damage site recognition, and increases BRCA1 E3 ubiquitin ligase activity in vitro. The BRCA1-BARD1 RING domains ubiquitylate histone H2A in vitro and in vivo at Lys127–129 (11), although the relationship of this activity to DNA repair is unknown. The crystal structure of the most similar E3 ubiquitin ligase heterodimer (Ring1B/Bmi1) bound to a nucleosome suggests a possible mode for how BRCA1-BARD1 could directly interact with nucleosomes. A high level of conservation shared between the Ring1B nucleosome-binding loop and the corresponding BRCA1 region predicts that BRCA1 targets to Lys127–129 by binding to a nucleosomal H2A-H2B acidic patch using its own basic residues within the nucleosome-binding loop (12). It will thus be interesting to understand how BRCA1 ubiquitinates different lysines on H2A than Ring1B/Bmi1 and how Lys127–129 ubiquitination contributes to BRCA1 function.
The BARD1 BRCT repeats were reported to interact with poly(ADP) ribose (13) and more recently to specifically recognize histone H3 dimethylated at Lys9 (H3K9me2) through its interaction with HP1 proteins.14 This interaction was reported to anchor BRCA1-BARD1 at DNA damage sites (14). Genetically, the BRCA1-BARD1 interaction is unique in the BRCA1 network in that it appears to be necessary for the majority of BRCA1 in vivo function. BARD1 deficiency fully recapitulates BRCA1 nullizygosity, with Bard1 knock-outs displaying embryonic lethality, genome instability, and cancer susceptibility (15, 16).
Abraxas resides in a five-member complex (RAP80, Abraxas, MERIT40, BRCC45, and BRCC36) that preferentially binds to Lys63-linked ubiquitin through the RAP80 tandem ubiquitin-interacting motifs. The finding that BRCA1-RAP80 interaction is required for focus formation first implicated non-degradative ubiquitin as a DNA damage recognition platform during DSB signaling (17–19). DSB ubiquitination occurs as a result of a γH2AX-initiated signaling cascade that recruits RNF168 to perform large-scale DSB chromatin ubiquitination at H2A Lys13 and Lys15 (20). Several other DDR proteins rely on RNF168 E3 activity for DSB localization, including 53BP1, which is a specific reader of the H2A Lys15-ubiquitin mark (21). In addition to ubiquitin, SUMOylation also contributes to BRCA1 DSB recruitment. RAP80 contains SUMO-interacting motifs N-terminal to its tandem ubiquitin-interacting motifs and shows ∼80-fold higher affinity for hybrid SUMO-ubiquitin chains than Lys63-linked ubiquitin alone, suggesting a mixed SUMO-ubiquitin targeting signal (22). The RAP80 complex specifically deubiquitinates the Lys63-ubiquitin chains through the actions of its associated Zn2+-dependent deubiquitinating enzyme, BRCC36. Unresolved questions remain as to whether deubiquitinating enzyme activity serves to terminate DNA damage association by removing the Lys63-ubiquitin recognition signal for RAP80 or, alternatively, in a ubiquitin-editing capacity, whereby it removes Lys63-ubiquitin, thus allowing accumulation of either monoubiquitin or other ubiquitin topologies that have been reported at DSBs (23). That loss of any member of the RAP80 complex eliminates observable BRCA1 focus formation at DSBs raises the question of whether the RAP80 complex accounts for BRCA1 function in HR. Interestingly, loss of RAP80 leads to over-resection and increased sister chromatid exchanges in response to DSB-inducing agents, indicating that the RAP80 complex is required to fine-tune the HR efficiency by controlling the resection level (24, 25).
In contrast to the embryonic lethality of Brca1 knock-out mice, Rap80, Abraxas, or Merit40 knock-out mice are viable and do not exhibit apparent developmental defects (26–29). Indeed, Rap80 was recently shown to target BRCA1 to chromatin regions that are ∼1 kb from break sites and to affect checkpoint responses and not DSB repair (30). Despite this relatively mild DNA repair phenotype, germ-line mutations exist in RAP80 and Abraxas in familial breast cancer, and numerous somatic mutations are observed in all members of the complex (28, 31, 32). Moreover, Rap80 and Abraxas knock-out mice are tumor-prone, with ∼20% of mice developing lymphomas at 1 year of age (26–28). These observations are consistent with the concept that complete loss of BRCA function in the DDR is not necessary for cancer susceptibility.
BRIP1 (also known as BACH1/FANCJ) was first cloned and identified as a putative DNA helicase protein interacting with the BRCT domain of BRCA1 (33). Phosphorylation at Ser990 of BRIP1 allows its interaction with the BRCA1 BRCT repeats (9). BRIP1-deficient cells show reduced HR efficiency in the pDR-GFP assay (35). Interestingly, BRIP1 contributes to the DNA cross-link repair pathway independent of its interaction with BRCA1 (36). BRIP1 deficiency causes severe DNA cross-link sensitivity, and BRIP1 is biallelically mutated in Fanconi anemia (FA). However, loss of interaction with BRCA1 does not confer sensitivity to DNA interstrand cross-link (ICL) inducing agent mitomycin C (36, 37). Instead, this is thought to affect a balance between HR and translesion synthesis at cross-links. BRIP1 has bona fide in vitro helicase activity and resolves G-quadraplex structures with 5′–3′ polarity. Concordant with this in vitro specificity, both Caenorhabditis elegans and human cells deficient in BRIP1 show loss of repetitive G-rich DNA and telomere abnormalities (38, 39). However, whether BRIP1 helicase activity contributes to maintenance of GC-rich regions through its association with BRCA1 remains an unresolved question.
CtIP (also known as RBBP8) interacts with the Mre11-Rad50-Nbs1 complex and stimulates its nuclease activity, in turn mediating end resection in the S and G2 phases of the cell cycle, which initiate HR (40). The interaction between CtIP and BRCA1 depends on cyclin-dependent kinase phosphorylation at Ser327. Knock-in of the human phospho-deficient Ctip mutant S327A (equivalent to Ser326 in mice) revealed no impact on resection in chicken and mouse cells and is not essential for resection-mediated repair, tumor suppression, or viability (41–43). Although BRCA1-CtIP interaction is not required for end resection, it enhances the speed of this process (44). Therefore, BRCA1 might interact with CtIP to ensure optimum end resection timing, a subtlety that may not be easily detected by other assays. Similarly, Palb2 knock-in mice that disrupt interaction with BRCA1 have been generated. In contrast to either Brca1 or Palb2 knock-out mice, these Palb2 mutants are viable, indicating that although the interaction between BRCA1 and PALB2 contributes to HR, either protein is largely functional in the absence of this interaction (45). This again signifies that interaction with BRCA1 is not equivalent to function and, together with data showing that disruption of BRCA1-BRIP1 also does not recapitulate knock-out phenotypes, suggests caution when interpreting the importance of BRCA1 protein-protein interactions. Guilt by association is clearly an oversimplification.
Transcription Influences DNA Damage Recognition by BRCA1 Complexes
Questions remain as to where in the genome the BRCA1 network executes its DDR functions. Recent reports reveal strong connections between BRCA1 genome integrity maintenance and transcription. BRCA1 forms a complex with the mRNA-splicing machinery in a DNA damage-dependent manner to regulate pre-mRNA splicing of genes involved in DNA damage signaling (46). BRCA1 has also been shown to participate in transcription-associated damage control, where it functions at stalled or defective transcription sites to assist transcription restart and resolve RNA-DNA hybrids (R-loop) that are known to promote DSB formation (47). This link to transcription is buttressed by the finding that BRCA1, PALB2, and other HR proteins co-reside at transcriptionally active regions throughout the genome (48–50). Moreover, active transcription has been directly implicated in BRCA1 targeting to DNA damage sites, revealing RNA- and transcription-associated chromatin modifications in BRCA1 DNA damage recognition and repair. In support of this assertion, disruption of BRCA1 interaction with senataxin leads to R-loop-driven DNA damage (51). BRCA2 also prevents R-loop accumulation to prevent transcriptional stress (52), implicating R-loop stability as a common mechanism for both proteins to maintain genome stability. These data also raise the intriguing possibility that the transcriptional profile of a cell could influence genome instability in the context of BRCA deficiency. Perhaps this could play a role in tissue specificity of its tumor suppression.
Multifactorial Responses of the BRCA Network to Replication Stress
PALB2 bridges the interaction between BRCA1 and BRCA2. The BRCA1-BRCA2-PALB2 complex promotes RAD51 nucleofilament formation, thus initiating homology-directed repair (53). It has more recently become apparent that BRCA1 and BRCA2 also participate in replication fork protection and restart. BRCA2 was found to prevent degradation of nascent strands at stalled replication forks by MRE11, and the C-terminal site of BRCA2 is critical for replication fork protection (54, 55). BRCA1 also contributes to fork protection in a similar manner to BRCA2, suggesting that BRCA1-BRCA2-PALB2 maintains genome stability by contributing to both HR and replication fork maintenance (56).
These concerted activities are interesting in light of the hypothesis that genome instability in BRCA-deficient cells arises primarily in S phase as a consequence of replication stress. By adapting the bacterial replication terminator Tus-Ter complex to induce site-specific replication fork stalling in mouse cells, it was shown that both the tandem BRCT repeats and exon 11 regions of BRCA1 are required for HR at stalled replication forks in mammalian cells (57). BRCA1 also regulates multiple aspects of replication to promote ICL repair independent of RAD51 nucleofilament formation. Studies using Xenopus egg extracts revealed that BRCA1, possibly through its interaction with RAP80, is required to unload the replicative Cdc45-MCM-GINS helicase complex when bidirectional replication forks collide with an ICL (58), thus affording access to cross-link recognition proteins. These data are in agreement with genetic studies implicating BRCA1 in early stages of ICL repair that occur prior to HR.
Consistent with replication stress being the limiting aspect of BRCA1 genome integrity control, primary mammary epithelial cells from patients with heterozygous BRCA1 or PALB2 status experience higher replication stress compared with cells with two wild-type copies of either gene (59, 60). Conversely, other BRCA1 functions in DSB repair and checkpoint activation are proficient in BRCA1mut/+ cells (60). This observation supports the hypothesis that BRCA haploinsufficiency in resolving replication stress might contribute to high risk of cancer in mutation carriers. Genetically engineered mouse models of pancreatic and ovarian cancer are consistent with this hypothesis, as BRCA1 or BRCA2 heterozygosity accelerated tumor formation in both instances (61, 62).
FA Provides a Window into Dysfunction of the BRCA Network
FA is a rare hereditary syndrome characterized by developmental defects, progressive bone marrow failure, and cancer susceptibility and is associated with DNA cross-link repair deficiency and sensitivity to endogenous aldehydes (63). In contrast to heterozygous mutations that cause hereditary breast and ovarian cancer, FA requires biallelic mutations within a network of 17 genes dedicated to DNA cross-link repair. Mutations within several members of the BRCA1 network are causative for FA. BRCA2 (FANCD1) and PALB2 (FANCN) interact with the BRCA1 coiled-coil domains, whereas BRIP1 (FANCJ) interacts with the BRCA1 BRCT repeats (Fig. 1). More recently, two patients have been identified with missense mutations (R1699W and V1736A) within the first BRCA1 BRCT domain and a FA-like syndrome, establishing BRCA1 as a Fanconi gene (FANCS) (64, 65). Both patients had one allele with a missense mutation in BRCT repeats and another deleterious mutation in either exon 10 or 11. Patient-derived BRCA1/FANCS cells were obtained and found to have reduced BRCA1 BRCT interactions, diminished RAD51 focus formation, and mitomycin C and poly(ADP-ribose) polymerase inhibitor (PARPi) hypersensitivity, in contrast to a sibling control, who had only one mutated BRCA1 allele (65). These findings thus mimic the clinical context of a biallelically mutant tumor and heterozygous patient. Indeed, BRCA mutant tumors are hypersensitive to platinum salts or PARPi, whereas increased systemic toxicity in noncancerous tissues is not seen in BRCA carriers.
Comparison of FA phenotypes derived from mutations to different genes within the BRCA network has also been illuminating (66). BRCA2/FANCD1 and PALB2/FANCN patients displayed bone marrow failure and severe FA phenotypes associated with solid tumors outside of the typical BRCA spectrum, including Wilms tumor and medulloblastoma. BRIP1 FA patients displayed bone marrow failure and leukemia. In contrast, neither of the BRCA1/FANCS patients developed spontaneous bone marrow failure, and both maintained breast or ovarian cancer predilection, with early-onset ovarian cancer at age 28 and breast cancer at age 23. The difference in patient phenotype is especially interesting in relation to BRIP1/FANCJ, as both BRCA1/FANCS patient mutations occurred in the BRCT region and disrupted the BRCA1-BRIP1 interaction. The BRCT BRCA1/FANCS mutations also abrogated interaction with CtIP and the RAP80 complex, possibly accounting for these differences. In addition, it is clear that BRIP1/FANCJ has many BRCA1-independent activities.
Determinants of Synthetic Lethality with Dysfunction of the BRCA Network
PARPs are highly abundant proteins that are responsible for poly(ADP-ribosylation) during the DDR. PARP1/2 can bind to DNA single-strand breaks (SSBs) and facilitate resolution of these lesions. Collision of active replication forks with SSBs in S phase is thought to lead to one-sided DSBs that necessitate BRCA-RAD51-mediated HR for repair (see Fig. 3). Indeed, landmark studies showed that PARPi is synthetic lethal in the context of BRCA deficiency, ushering in a new era of targeted therapy for patients with mutant BRCA (67, 68). PARPi (olaparib, trade name Lynparza) was been rapidly translated to the clinic, with successful phase I (69) and phase II (70–72) clinical trials leading to Food and Drug Administration approval in December 2014 for advanced mutant BRCA ovarian cancer. However, resistance mechanisms for PARPi treatment have arisen in patients and in the laboratory setting in cells exposed to chronic PARPi, indicating that a more thorough understanding of resistance mechanisms and alternative targets is needed.
FIGURE 3.

Determinants of PARPi response. PARP inhibition creates trapped PARP-DNA complexes that require BRCA HR activity during DNA replication. Resistance mechanisms occur due to events that restore HR in BRCA1 mutant cells. This entails secondary reversion mutations that lead to production of partially functional BRCA1 protein. Alternatively, restoration of HR and resistance can occur due to loss of 53BP1 or several of its interacting partners. RPA, replication protein A.
PARP recognizes SSBs and exerts its function by autoribosylation and by poly(ADP-ribose)-dependent recruitment of other DNA repair proteins. Negatively charged ADP-ribose polymers facilitate dissociation of PARP1 from the DNA. In turn, PARPi traps PARP1/2 on DNA, leading to the generation of trapped PARP-DNA complexes, which are thought to be more cytotoxic than unrepaired SSBs (73, 74). It is likely that trapped PARP-DNA complexes create cytotoxic complexes when encountered by replication forks, increasing the burden on HR protein to fully resolve the lesions (see Fig. 3). This proposed mechanism is supported by the observation that the ability of PARPi to trap PARP-DNA complexes correlates with its effectiveness in killing BRCA-deficient cells (73).
Resistance Mechanisms to PARP Inhibition
HR proficiency is a key determinant of cellular sensitivity to PARPi. This indicates that restoring HR capacity would produce PARPi resistance. Indeed, genetic reversion of mutant BRCA1 or BRCA2 has been found in human tumors. Secondary mutations that restore reading frames lead to generation of partially functional BRCA proteins and render these cells HR-proficient and resistant to PARPi or cisplatin (75, 76). A reported 46.2% of platinum-resistant recurrences have secondary mutations restoring BRCA1/2 (77), indicating that genetic reversion is a common resistance mechanism.
Competition between repair pathways also appears to be a key determinant of PARPi sensitivity (Figs. 2 and 3). Loss of 53BP1 suppresses embryonic lethality, HR deficiency, and PARPi hypersensitivity in BRCA1 knock-out mice (78, 79). Remarkably, double knock-out mice are also not cancer-prone, firmly connecting loss of HR to BRCA1 cancer susceptibility (80). A putative mechanism underlying this observation is that 53BP1 blocks end resection in BRCA1-deficient cells and promotes toxic non-homologous end joining (NHEJ) in S phase (Fig. 3). In turn, loss of 53BP1 renders these cells HR-proficient in the absence of BRCA1 by permitting excessive single-stranded DNA, the initial substrate of RAD51-dependent HR (78). Similarly, deficiencies in several 53BP1-interacting partners (RIF1, PTIP, REV7, and Artemis) participate in blocking end resection. Loss of these four effector proteins also promotes PARPi resistance to varying extents in BRCA1 mutant cells (81–84). So how is this competition between the BRCA1 and 53BP1 networks regulated? The cell cycle seems to be a key determinant, with BRCA1 exclusion from foci in G1, whereas RIF1 and PTIP have limited association with damage-induced 53BP1 foci in S/G2, when HR is favored. Acetylation also appears to play a role in this balance, in part through reducing 53BP1 affinity for histone H4 tails containing methylated Lys20 (48, 85).
FIGURE 2.

Alternative DNA repair pathways compensate for BRCA deficiency. NHEJ and HR are the primary DSB repair pathways. End resection produces single-stranded DNA overhangs, which are required for HR. Alternative DSB repair pathways, single-strand annealing (SSA), and microhomology-mediated end joining (MMEJ) also necessitate end resection and represent compensatory DNA repair mechanisms when BRCA-dependent HR is impaired. Proteins that execute alternative DNA repair mechanisms represent potential targets to selectively kill BRCA mutant cells. RPA, replication protein A.
Novel Targets for BRCA Network Dysfunctional Cancers
Given the numerous possibilities of acquired PARPi resistance, it would be prudent to develop alternative strategies that selectively kill BRCA mutant cells. An attractive target is the HR protein RAD52, which is synthetically lethal with combined deficiency in BRCA1, PALB2, or BRCA2 (86, 87). RAD52 was proposed to execute alternative repair pathways that perform RAD51-mediated HR independent of the BRCA1-BRCA2-PALB2 complex. Alternatively, RAD52 is also known to catalyze the process of single-strand annealing and may function in a salvage pathway in the absence of BRCA proteins (88). A second exciting potential target to selectively treat HR-deficient cancers is DNA polymerase θ (pol θ), which mediates error-prone alternative NHEJ (also called microhomology-mediated end joining) (89–92). pol θ promotes microhomology-mediated end joining and restricts RAD51-mediated recombination. Combined FANCD2 and pol θ nullizygosity produces embryonic lethality in mice, and loss of pol θ in BRCA1- or BRCA2-deficient cells results in significantly reduced plating efficiency, indicating a synthetic lethal relationship with HR deficiency (90, 91). Both pol θ and RAD52 are up-regulated in HR-deficient cancers and are potentially druggable targets. Although the clinical utility of such agents is to be ascertained, it would be interesting and feasible to determine whether RAD52 or pol θ inhibition restores PARPi sensitivity in BRCA1 and 53BP1 double-mutant cells. If either of these modalities is toxic to double knock-out cells, they may potentially be used to reverse the resistance of BRCA1 mutant cancers to PARPi.
Elevated replication stress is frequently experienced in rapidly replicating cancer cells, particularly in those experiencing HR deficiency. Inhibition of replication stress-activated kinase ATR or Chk1 sensitizes ovarian cancer cells with defective HR to commonly used chemotherapy agents, and ATR and Chk1 inhibition sensitizes BRCA mutant cancer cells (93). In a possible example of proof of concept, Chk1 inhibition caused synthetic lethality with Mre11-Rad50-Nbs1 mutation and showed a curative clinical response in a tumor harboring a Rad50 mutation (94).
Concluding Thoughts
The past 5 years has brought forward a revolutionary increase in the basic understanding of BRCA molecular function in the DDR, the first clinically approved therapies to selectively treat BRCA mutant cancers, and a host of resistance mechanisms that connect restoration of HR to therapeutic response. Still, there are many questions remaining. As detailed above, most BRCA1 protein-protein interactions do not satisfactorily explain the roles of BRCA1 in DNA repair or tumor suppression. This in part relates to a lack of knowledge regarding how BRCA1 functions in DNA repair. In stark contrast to seminal studies describing BRCA2-dependent RAD51 nucleofilament formation by displacement of replication protein A (34), meaningful in vitro assays to dissect BRCA1 repair function have not been reported. The development of such approaches would represent an important advance because it remains a matter of debate as to whether the primary function of BRCA1 resides in its ability to promote DNA repair transactions or to act as a competitor of 53BP1 in counteracting toxic NHEJ. Close links between basic discovery and clinical observations should help resolve these questions and pose new and unanticipated issues. They should also continue to enable translation of new and existing therapies and further refine our knowledge of the BRCA1 tumor suppressor network.
Author Contributions
Both authors conceived of the direction of the minireview, wrote the manuscript, and composed the figures.
This work was supported, in whole or in part, by National Institutes of Health Grants CA138835 and CA17494 (to R. A. G.). This work was also supported by the Abramson Family Cancer Research Institute and the Basser Research Center for BRCA (to R. A. G.). The authors declare that they have no conflicts of interest with the contents of this article.
- DDR
- DNA damage response
- HR
- homologous recombination
- BRCT
- BRCA1 C-terminal
- DSB
- double-strand break
- SUMO
- small ubiquitin-like modifier
- FA
- Fanconi anemia
- ICL
- interstrand cross-link
- PARPi
- poly(ADP-ribose) polymerase inhibitor
- SSB
- single-strand break
- NHEJ
- non-homologous end joining
- pol θ
- DNA polymerase θ.
References
- 1. Jasin M. (2002) Homologous repair of DNA damage and tumorigenesis: the BRCA connection. Oncogene 21, 8981–8993 [DOI] [PubMed] [Google Scholar]
- 2. Shakya R., Reid L. J., Reczek C. R., Cole F., Egli D., Lin C.-S., deRooij D. G., Hirsch S., Ravi K., Hicks J. B., Szabolcs M., Jasin M., Baer R., Ludwig T. (2011) BRCA1 tumor suppression depends on BRCT phosphoprotein binding, but not its E3 ligase activity. Science 334, 525–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Drost R., Bouwman P., Rottenberg S., Boon U., Schut E., Klarenbeek S., Klijn C., van der Heijden I., van der Gulden H., Wientjens E., Pieterse M., Catteau A., Green P., Solomon E., Morris J. R., Jonkers J. (2011) BRCA1 RING function is essential for tumor suppression but dispensable for therapy resistance. Cancer Cell 20, 797–809 [DOI] [PubMed] [Google Scholar]
- 4. Wu L. C., Wang Z. W., Tsan J. T., Spillman M. A., Phung A., Xu X. L., Yang M. C., Hwang L. Y., Bowcock A. M., Baer R. (1996) Identification of a RING protein that can interact in vivo with the BRCA1 gene product. Nat. Genet. 14, 430–440 [DOI] [PubMed] [Google Scholar]
- 5. Xu X., Weaver Z., Linke S. P., Li C., Gotay J., Wang X.-W., Harris C. C., Ried T., Deng C.-X. (1999) Centrosome amplification and a defective G2-M cell cycle checkpoint induce genetic instability in BRCA1 exon 11 isoform-deficient cells. Mol. Cell 3, 389–395 [DOI] [PubMed] [Google Scholar]
- 6. Xu X., Wagner K. U., Larson D., Weaver Z., Li C., Ried T., Hennighausen L., Wynshaw-Boris A., Deng C.-X. (1999) Conditional mutation of Brca1 in mammary epithelial cells results in blunted ductal morphogenesis and tumour formation. Nat. Genet. 22, 37–43 [DOI] [PubMed] [Google Scholar]
- 7. Antoniou A. C., Casadei S., Heikkinen T., Barrowdale D., Pylkäs K., Roberts J., Lee A., Subramanian D., De Leeneer K., Fostira F., Tomiak E., Neuhausen S. L., Teo Z. L., Khan S., Aittomäki K., Moilanen J. S., Turnbull C., Seal S., Mannermaa A., Kallioniemi A., Lindeman G. J., Buys S. S., Andrulis I. L., Radice P., Tondini C., Manoukian S., Toland A. E., Miron P., Weitzel J. N., Domchek S. M., Poppe B., Claes K. B. M., Yannoukakos D., Concannon P., Bernstein J. L., James P. A., Easton D. F., Goldgar D. E., Hopper J. L., Rahman N., Peterlongo P., Nevanlinna H., King M.-C., Couch F. J., Southey M. C., Winqvist R., Foulkes W. D., Tischkowitz M. (2014) Breast-cancer risk in families with mutations in PALB2. N. Engl. J. Med. 371, 497–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Greenberg R. A., Sobhian B., Pathania S., Cantor S. B., Nakatani Y., Livingston D. M. (2006) Multifactorial contributions to an acute DNA damage response by BRCA1/BARD1-containing complexes. Genes Dev. 20, 34–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yu X., Chini C. C. S., He M., Mer G., Chen J. (2003) The BRCT domain is a phospho-protein binding domain. Science 302, 639–642 [DOI] [PubMed] [Google Scholar]
- 10. Manke I. A., Lowery D. M., Nguyen A., Yaffe M. B. (2003) BRCT repeats as phosphopeptide-binding modules involved in protein targeting. Science 302, 636–639 [DOI] [PubMed] [Google Scholar]
- 11. Kalb R., Mallery D. L., Larkin C., Huang J. T. J., Hiom K. (2014) BRCA1 is a histone-H2A-specific ubiquitin ligase. Cell Rep. 8, 999–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. McGinty R. K., Henrici R. C., Tan S. (2014) Crystal structure of the PRC1 ubiquitylation module bound to the nucleosome. Nature 514, 591–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li M., Yu X. (2013) Function of BRCA1 in the DNA damage response is mediated by ADP-ribosylation. Cancer Cell 23, 693–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wu W., Nishikawa H., Fukuda T., Vittal V., Asano M., Miyoshi Y., Klevit R. E., Ohta T. (2015) Interaction of BARD1 and HP1 is required for BRCA1 retention at sites of DNA damage. Cancer Res. 75, 1311–1321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Joukov V., Chen J., Fox E. A., Green J. B., Livingston D. M. (2001) Functional communication between endogenous BRCA1 and its partner, BARD1, during Xenopus laevis development. Proc. Natl. Acad. Sci. U.S.A. 98, 12078–12083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shakya R., Szabolcs M., McCarthy E., Ospina E., Basso K., Nandula S., Murty V., Baer R., Ludwig T. (2008) The basal-like mammary carcinomas induced by Brca1 or Bard1 inactivation implicate the BRCA1/BARD1 heterodimer in tumor suppression. Proc. Natl. Acad. Sci. U.S.A. 105, 7040–7045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sobhian B., Shao G., Lilli D. R., Culhane A. C., Moreau L. A., Xia B., Livingston D. M., Greenberg R. A. (2007) RAP80 targets BRCA1 to specific ubiquitin structures at DNA damage sites. Science 316, 1198–1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kim H., Chen J., Yu X. (2007) Ubiquitin-binding protein RAP80 mediates BRCA1-dependent DNA damage response. Science 316, 1202–1205 [DOI] [PubMed] [Google Scholar]
- 19. Wang B., Matsuoka S., Ballif B. A., Zhang D., Smogorzewska A., Gygi S. P., Elledge S. J. (2007) Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science 316, 1194–1198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mattiroli F., Vissers J. H. A., van Dijk W. J., Ikpa P., Citterio E., Vermeulen W., Marteijn J. A., Sixma T. K. (2012) RNF168 ubiquitinates K13–15 on H2A/H2AX to drive DNA damage signaling. Cell 150, 1182–1195 [DOI] [PubMed] [Google Scholar]
- 21. Fradet-Turcotte A., Canny M. D., Escribano-Díaz C., Orthwein A., Leung C. C. Y., Huang H., Landry M.-C., Kitevski-LeBlanc J., Noordermeer S. M., Sicheri F., Durocher D. (2013) 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature 499, 50–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Guzzo C. M., Berndsen C. E., Zhu J., Gupta V., Datta A., Greenberg R. A., Wolberger C., Matunis M. J. (2012) RNF4-dependent hybrid SUMO-ubiquitin chains are signals for RAP80 and thereby mediate the recruitment of BRCA1 to sites of DNA damage. Sci. Signal. 5, ra88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gatti M., Pinato S., Maiolica A., Rocchio F., Prato M. G., Aebersold R., Penengo L. (2015) RNF168 promotes noncanonical K27 ubiquitination to signal DNA damage. Cell Rep. 10, 226–238 [DOI] [PubMed] [Google Scholar]
- 24. Coleman K. A., Greenberg R. A. (2011) The BRCA1-RAP80 complex regulates DNA repair mechanism utilization by restricting end resection. J. Biol. Chem. 286, 13669–13680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hu Y., Scully R., Sobhian B., Xie A., Shestakova E., Livingston D. M. (2011) RAP80-directed tuning of BRCA1 homologous recombination function at ionizing radiation-induced nuclear foci. Genes Dev. 25, 685–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yin Z., Menendez D., Resnick M. A., French J. E., Janardhan K. S., Jetten A. M. (2012) RAP80 is critical in maintaining genomic stability and suppressing tumor development. Cancer Res. 72, 5080–5090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wu J., Liu C., Chen J., Yu X. (2012) RAP80 protein is important for genomic stability and is required for stabilizing BRCA1-A complex at DNA damage sites in vivo. J. Biol. Chem. 287, 22919–22926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Castillo A., Paul A., Sun B., Huang T. H., Wang Y., Yazinski S. A., Tyler J., Li L., You M. J., Zou L., Yao J., Wang B. (2014) The BRCA1-interacting protein Abraxas is required for genomic stability and tumor suppression. Cell Rep. 8, 807–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rozenova K., Jiang J., Donaghy R., Aressy B., Greenberg R. A., Tong W. (2015) MERIT40 deficiency expands hematopoietic stem cell pools by regulating thrombopoietin receptor signaling. Blood. 125, 1730–1738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Goldstein M., Kastan M. B. (2015) Repair versus checkpoint functions of Brca1 are differentially regulated by site of chromatin binding. Cancer Res. canres.0400.2015 10.1158/0008-5472.CAN-15-0400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nikkilä J., Coleman K. A., Morrissey D., Pylkäs K., Erkko H., Messick T. E., Karppinen S.-M., Amelina A., Winqvist R., Greenberg R. A. (2009) Familial breast cancer screening reveals an alteration in the RAP80 UIM domain that impairs DNA damage response function. Oncogene 28, 1843–1852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Solyom S., Aressy B., Pylkäs K., Patterson-Fortin J., Hartikainen J. M., Kallioniemi A., Kauppila S., Nikkilä J., Kosma V.-M., Mannermaa A., Greenberg R. A., Winqvist R. (2012) Breast cancer-associated Abraxas mutation disrupts nuclear localization and DNA damage response functions. Sci. Transl. Med. 4, 122ra23–122ra23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cantor S. B., Bell D. W., Ganesan S., Kass E. M., Drapkin R., Grossman S., Wahrer D. C., Sgroi D. C., Lane W. S., Haber D. A., Livingston D. M. (2001) BACH1, a novel helicase-like protein, interacts directly with BRCA1 and contributes to its DNA repair function. Cell 105, 149–160 [DOI] [PubMed] [Google Scholar]
- 34. Yang H., Li Q., Fan J., Holloman W. K., Pavletich N. P. (2005) The BRCA2 homologue Brh2 nucleates RAD51 filament formation at a dsDNA-ssDNA junction. Nature 433, 653–657 [DOI] [PubMed] [Google Scholar]
- 35. Litman R., Peng M., Jin Z., Zhang F., Zhang J., Powell S., Andreassen P. R., Cantor S. B. (2005) BACH1 is critical for homologous recombination and appears to be the Fanconi anemia gene product FANCJ. Cancer Cell 8, 255–265 [DOI] [PubMed] [Google Scholar]
- 36. Bridge W. L., Vandenberg C. J., Franklin R. J., Hiom K. (2005) The BRIP1 helicase functions independently of BRCA1 in the Fanconi anemia pathway for DNA crosslink repair. Nat. Genet. 37, 953–957 [DOI] [PubMed] [Google Scholar]
- 37. Peng M., Litman R., Xie J., Sharma S., Brosh R. M., Jr., Cantor S. B. (2007) The FANCJ/MutLα interaction is required for correction of the cross-link response in FA-J cells. EMBO J. 26, 3238–3249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cheung I., Schertzer M., Rose A., Lansdorp P. M. (2002) Disruption of dog-1 in Caenorhabditis elegans triggers deletions upstream of guanine-rich DNA. Nat. Genet. 31, 405–409 [DOI] [PubMed] [Google Scholar]
- 39. London T. B. C., Barber L. J., Mosedale G., Kelly G. P., Balasubramanian S., Hickson I. D., Boulton S. J., Hiom K. (2008) FANCJ is a structure-specific DNA helicase associated with the maintenance of genomic G/C tracts. J. Biol. Chem. 283, 36132–36139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sartori A. A., Lukas C., Coates J., Mistrik M., Fu S., Bartek J., Baer R., Lukas J., Jackson S. P. (2007) Human CtIP promotes DNA end resection. Nature 450, 509–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Reczek C. R., Szabolcs M., Stark J. M., Ludwig T., Baer R. (2013) The interaction between CtIP and BRCA1 is not essential for resection-mediated DNA repair or tumor suppression. J. Cell Biol. 201, 693–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Polato F., Callen E., Wong N., Faryabi R., Bunting S., Chen H.-T., Kozak M., Kruhlak M. J., Reczek C. R., Lee W. H., Ludwig T., Baer R., Feigenbaum L., Jackson S., Nussenzweig A. (2014) CtIP-mediated resection is essential for viability and can operate independently of BRCA1. J. Exp. Med. 211, 1027–1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yun M. H., Hiom K. (2009) CtIP-BRCA1 modulates the choice of DNA double-strand-break repair pathway throughout the cell cycle. Nature 459, 460–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cruz-García A., López-Saavedra A., Huertas P. (2014) BRCA1 accelerates CtIP-mediated DNA-end resection. Cell Rep. 9, 451–459 [DOI] [PubMed] [Google Scholar]
- 45. Simhadri S., Peterson S., Patel D. S., Huo Y., Cai H., Bowman-Colin C., Miller S., Ludwig T., Ganesan S., Bhaumik M., Bunting S. F., Jasin M., Xia B. (2014) Male fertility defect associated with disrupted BRCA1-PALB2 interaction in mice. J. Biol. Chem. 289, 24617–24629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Savage K. I., Gorski J. J., Barros E. M., Irwin G. W., Manti L., Powell A. J., Pellagatti A., Lukashchuk N., McCance D. J., McCluggage W. G., Schettino G., Salto-Tellez M., Boultwood J., Richard D. J., McDade S. S., Harkin D. P. (2014) Identification of a BRCA1-mRNA splicing complex required for efficient DNA repair and maintenance of genomic stability. Mol. Cell 54, 445–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hill S. J., Rolland T., Adelmant G., Xia X., Owen M. S., Dricot A., Zack T. I., Sahni N., Jacob Y., Hao T., McKinney K. M., Clark A. P., Reyon D., Tsai S. Q., Joung J. K., Beroukhim R., Marto J. A., Vidal M., Gaudet S., Hill D. E., Livingston D. M. (2014) Systematic screening reveals a role for BRCA1 in the response to transcription-associated DNA damage. Genes Dev. 28, 1957–1975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tang J., Cho N. W., Cui G., Manion E. M., Shanbhag N. M., Botuyan M. V., Mer G., Greenberg R. A. (2013) Acetylation limits 53BP1 association with damaged chromatin to promote homologous recombination. Nat. Struct. Mol. Biol. 20, 317–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gardini A., Baillat D., Cesaroni M., Shiekhattar R. (2014) Genome-wide analysis reveals a role for BRCA1 and PALB2 in transcriptional co-activation. EMBO J. 33, 890–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Aymard F., Bugler B., Schmidt C. K., Guillou E., Caron P., Briois S., Iacovoni J. S., Daburon V., Miller K. M., Jackson S. P., Legube G. (2014) Transcriptionally active chromatin recruits homologous recombination at DNA double-strand breaks. Nat. Struct. Mol. Biol. 21, 366–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hatchi E., Skourti-Stathaki K., Ventz S., Pinello L., Yen A., Kamieniarz-Gdula K., Dimitrov S., Pathania S., McKinney K. M., Eaton M. L., Kellis M., Hill S. J., Parmigiani G., Proudfoot N. J., Livingston D. M. (2015) BRCA1 recruitment to transcriptional pause sites is required for R-loop-driven DNA damage repair. Mol. Cell 57, 636–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bhatia V., Barroso S. I., García-Rubio M. L., Tumini E., Herrera-Moyano E., Aguilera A. (2014) BRCA2 prevents R-loop accumulation and associates with TREX-2 mRNA export factor PCID2. Nature 511, 362–365 [DOI] [PubMed] [Google Scholar]
- 53. Xia B., Sheng Q., Nakanishi K., Ohashi A., Wu J., Christ N., Liu X., Jasin M., Couch F. J., Livingston D. M. (2006) Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol. Cell 22, 719–729 [DOI] [PubMed] [Google Scholar]
- 54. Schlacher K., Christ N., Siaud N., Egashira A., Wu H., Jasin M. (2011) Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 145, 529–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ying S., Hamdy F. C., Helleday T. (2012) Mre11-dependent degradation of stalled DNA replication forks is prevented by BRCA2 and PARP1. Cancer Res. 72, 2814–2821 [DOI] [PubMed] [Google Scholar]
- 56. Schlacher K., Wu H., Jasin M. (2012) A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 22, 106–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Willis N. A., Chandramouly G., Huang B., Kwok A., Follonier C., Deng C., Scully R. (2014) BRCA1 controls homologous recombination at Tus/Ter-stalled mammalian replication forks. Nature 510, 556–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Long D. T., Joukov V., Budzowska M., Walter J. C. (2014) BRCA1 promotes unloading of the CMG helicase from a stalled DNA replication fork. Mol. Cell 56, 174–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Nikkilä J., Parplys A. C., Pylkäs K., Bose M., Huo Y., Borgmann K., Rapakko K., Nieminen P., Xia B., Pospiech H., Winqvist R. (2013) Heterozygous mutations in PALB2 cause DNA replication and damage response defects. Nat. Commun. 4, 2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Pathania S., Bade S., Le Guillou M., Burke K., Reed R., Bowman-Colin C., Su Y., Ting D. T., Polyak K., Richardson A. L., Feunteun J., Garber J. E., Livingston D. M. (2014) BRCA1 haploinsufficiency for replication stress suppression in primary cells. Nat. Commun. 5, 5496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Perets R., Wyant G. A., Muto K. W., Bijron J. G., Poole B. B., Chin K. T., Chen J. Y. H., Ohman A. W., Stepule C. D., Kwak S., Karst A. M., Hirsch M. S., Setlur S. R., Crum C. P., Dinulescu D. M., Drapkin R. (2013) Transformation of the fallopian tube secretory epithelium leads to high-grade serous ovarian cancer in Brca;Tp53;Pten models. Cancer Cell 24, 751–765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Skoulidis F., Cassidy L. D., Pisupati V., Jonasson J. G., Bjarnason H., Eyfjord J. E., Karreth F. A., Lim M., Barber L. M., Clatworthy S. A., Davies S. E., Olive K. P., Tuveson D. A., Venkitaraman A. R. (2010) Germline Brca2 heterozygosity promotes KrasG12D-driven carcinogenesis in a murine model of familial pancreatic cancer. Cancer Cell 18, 499–509 [DOI] [PubMed] [Google Scholar]
- 63. Joenje H., Patel K. J. (2001) The emerging genetic and molecular basis of Fanconi anaemia. Nat. Rev. Genet. 2, 446–457 [DOI] [PubMed] [Google Scholar]
- 64. Domchek S. M., Tang J., Stopfer J., Lilli D. R., Hamel N., Tischkowitz M., Monteiro A. N. A., Messick T. E., Powers J., Yonker A., Couch F. J., Goldgar D. E., Davidson H. R., Nathanson K. L., Foulkes W. D., Greenberg R. A. (2013) Biallelic deleterious BRCA1 mutations in a woman with early-onset ovarian cancer. Cancer Discov. 3, 399–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sawyer S. L., Tian L., Kähkönen M., Schwartzentruber J., Kircher M., University of Washington Centre for Mendelian Genomics, FORGE Canada Consortium, Majewski J., Dyment D. A., Innes A. M., Boycott K. M., Moreau L. A., Moilanen J. S., Greenberg R. A. (2015) Biallelic mutations in BRCA1 cause a new Fanconi anemia subtype. Cancer Discov. 5, 135–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kottemann M. C., Smogorzewska A. (2013) Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature 493, 356–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Farmer H., McCabe N., Lord C. J., Tutt A. N. J., Johnson D. A., Richardson T. B., Santarosa M., Dillon K. J., Hickson I., Knights C., Martin N. M. B., Jackson S. P., Smith G. C. M., Ashworth A. (2005) Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434, 917–921 [DOI] [PubMed] [Google Scholar]
- 68. Bryant H. E., Schultz N., Thomas H. D., Parker K. M., Flower D., Lopez E., Kyle S., Meuth M., Curtin N. J., Helleday T. (2005) Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434, 913–917 [DOI] [PubMed] [Google Scholar]
- 69. Fong P. C., Boss D. S., Yap T. A., Tutt A., Wu P., Mergui-Roelvink M., Mortimer P., Swaisland H., Lau A., O'Connor M. J., Ashworth A., Carmichael J., Kaye S. B., Schellens J. H. M., de Bono J. S. (2009) Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med. 361, 123–134 [DOI] [PubMed] [Google Scholar]
- 70. Tutt A., Robson M., Garber J. E., Domchek S. M., Audeh M. W., Weitzel J. N., Friedlander M., Arun B., Loman N., Schmutzler R. K., Wardley A., Mitchell G., Earl H., Wickens M., Carmichael J. (2010) Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet 376, 235–244 [DOI] [PubMed] [Google Scholar]
- 71. Audeh M. W., Carmichael J., Penson R. T., Friedlander M., Powell B., Bell-McGuinn K. M., Scott C., Weitzel J. N., Oaknin A., Loman N., Lu K., Schmutzler R. K., Matulonis U., Wickens M., Tutt A. (2010) Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet 376, 245–251 [DOI] [PubMed] [Google Scholar]
- 72. Gelmon K. A., Tischkowitz M., Mackay H., Swenerton K., Robidoux A., Tonkin K., Hirte H., Huntsman D., Clemons M., Gilks B., Yerushalmi R., Macpherson E., Carmichael J., Oza A. (2011) Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. Lancet Oncol. 12, 852–861 [DOI] [PubMed] [Google Scholar]
- 73. Murai J., Huang S. N., Das B. B., Renaud A., Zhang Y., Doroshow J. H., Ji J., Takeda S., Pommier Y. (2012) Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 72, 5588–5599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ström C. E., Johansson F., Uhlén M., Szigyarto C. A.-K., Erixon K., Helleday T. (2011) Poly (ADP-ribose) polymerase (PARP) is not involved in base excision repair but PARP inhibition traps a single-strand intermediate. Nucleic Acids Res. 39, 3166–3175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Edwards S. L., Brough R., Lord C. J., Natrajan R., Vatcheva R., Levine D. A., Boyd J., Reis-Filho J. S., Ashworth A. (2008) Resistance to therapy caused by intragenic deletion in BRCA2. Nature 451, 1111–1115 [DOI] [PubMed] [Google Scholar]
- 76. Sakai W., Swisher E. M., Karlan B. Y., Agarwal M. K., Higgins J., Friedman C., Villegas E., Jacquemont C., Farrugia D. J., Couch F. J., Urban N., Taniguchi T. (2008) Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature 451, 1116–1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Norquist B., Wurz K. A., Pennil C. C., Garcia R., Gross J., Sakai W., Karlan B. Y., Taniguchi T., Swisher E. M. (2011) Secondary somatic mutations restoring BRCA1/2 predict chemotherapy resistance in hereditary ovarian carcinomas. J. Clin. Oncol. 29, 3008–3015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Bunting S. F., Callén E., Wong N., Chen H.-T., Polato F., Gunn A., Bothmer A., Feldhahn N., Fernandez-Capetillo O., Cao L., Xu X., Deng C.-X., Finkel T., Nussenzweig M., Stark J. M., Nussenzweig A. (2010) 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 141, 243–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Bouwman P., Aly A., Escandell J. M., Pieterse M., Bartkova J., van der Gulden H., Hiddingh S., Thanasoula M., Kulkarni A., Yang Q., Haffty B. G., Tommiska J., Blomqvist C., Drapkin R., Adams D. J., Nevanlinna H., Bartek J., Tarsounas M., Ganesan S., Jonkers J. (2010) 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat. Struct. Mol. Biol. 17, 688–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Jaspers J. E., Kersbergen A., Boon U., Sol W., van Deemter L., Zander S. A., Drost R., Wientjens E., Ji J., Aly A., Doroshow J. H., Cranston A., Martin N. M. B., Lau A., O'Connor M. J., Ganesan S., Borst P., Jonkers J., Rottenberg S. (2013) Loss of 53BP1 causes PARP inhibitor resistance in Brca1-mutated mouse mammary tumors. Cancer Discov. 3, 68–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Xu G., Chapman J. R., Brandsma I., Yuan J., Mistrik M., Bouwman P., Bartkova J., Gogola E., Warmerdam D., Barazas M., Jaspers J. E., Watanabe K., Pieterse M., Kersbergen A., Sol W., Celie P. H. N., Schouten P. C., van den Broek B., Salman A., Nieuwland M., de Rink I., de Ronde J., Jalink K., Boulton S. J., Chen J., van Gent D. C., Bartek J., Jonkers J., Borst P., Rottenberg S. (2015) REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature 521, 541–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Boersma V., Moatti N., Segura-Bayona S., Peuscher M. H., van der Torre J., Wevers B. A., Orthwein A., Durocher D., Jacobs J. J. L. (2015) MAD2L2 controls DNA repair at telomeres and DNA breaks by inhibiting 5′ end resection. Nature 521, 537–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Wang J., Aroumougame A., Lobrich M., Li Y., Chen D., Chen J., Gong Z. (2014) PTIP associates with Artemis to dictate DNA repair pathway choice. Genes Dev. 28, 2693–2698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Panier S., Boulton S. J. (2014) Double-strand break repair: 53BP1 comes into focus. Nat. Rev. Mol. Cell Biol. 15, 7–18 [DOI] [PubMed] [Google Scholar]
- 85. Hsiao K. Y., Mizzen C. A. (2013) Histone H4 deacetylation facilitates 53BP1 DNA damage signaling and double-strand break repair. J. Mol. Cell Biol. 5, 157–165 [DOI] [PubMed] [Google Scholar]
- 86. Feng Z., Scott S. P., Bussen W., Sharma G. G., Guo G., Pandita T. K., Powell S. N. (2011) Rad52 inactivation is synthetically lethal with BRCA2 deficiency. Proc. Natl. Acad. Sci. U.S.A. 108, 686–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Lok B. H., Carley A. C., Tchang B., Powell S. N. (2013) RAD52 inactivation is synthetically lethal with deficiencies in BRCA1 and PALB2 in addition to BRCA2 through RAD51-mediated homologous recombination. Oncogene 32, 3552–3558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Lok B. H., Powell S. N. (2012) Molecular pathways: understanding the role of Rad52 in homologous recombination for therapeutic advancement. Clin. Cancer Res. 18, 6400–6406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Yousefzadeh M. J., Wyatt D. W., Takata K., Mu Y., Hensley S. C., Tomida J., Bylund G. O., Doublié S., Johansson E., Ramsden D. A., McBride K. M., Wood R. D. (2014) Mechanism of suppression of chromosomal instability by DNA polymerase POLQ. PLoS Genet. 10, e1004654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Mateos-Gomez P. A., Gong F., Nair N., Miller K. M., Lazzerini-Denchi E., Sfeir A. (2015) Mammalian polymerase θ promotes alternative NHEJ and suppresses recombination. Nature 518, 254–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Ceccaldi R., Liu J. C., Amunugama R., Hajdu I., Primack B., Petalcorin M. I. R., O'Connor K. W., Konstantinopoulos P. A., Elledge S. J., Boulton S. J., Yusufzai T., D'Andrea A. D. (2015) Homologous-recombination-deficient tumours are dependent on Polθ-mediated repair. Nature 518, 258–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Kent T., Chandramouly G., McDevitt S. M., Ozdemir A. Y., Pomerantz R. T. (2015) Mechanism of microhomology-mediated end-joining promoted by human DNA polymerase θ. Nat. Struct. Mol. Biol. 22, 230–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Huntoon C. J., Flatten K. S., Wahner Hendrickson A. E., Huehls A. M., Sutor S. L., Kaufmann S. H., Karnitz L. M. (2013) ATR inhibition broadly sensitizes ovarian cancer cells to chemotherapy independent of BRCA status. Cancer Res. 73, 3683–3691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Al-Ahmadie H., Iyer G., Hohl M., Asthana S., Inagaki A., Schultz N., Hanrahan A. J., Scott S. N., Brannon A. R., McDermott G. C., Pirun M., Ostrovnaya I., Kim P., Socci N. D., Viale A., Schwartz G. K., Reuter V., Bochner B. H., Rosenberg J. E., Bajorin D. F., Berger M. F., Petrini J. H. J., Solit D. B., Taylor B. S. (2014) Synthetic lethality in ATM-deficient RAD50-mutant tumors underlies outlier response to cancer therapy. Cancer Discov. 4, 1014–1021 [DOI] [PMC free article] [PubMed] [Google Scholar]