

Background: ZIP transporters increase the cytosolic concentration of first row transition metals.
Results: We have developed a structural model of hZIP4 by combining protein prediction methods with in situ experiments.
Conclusion: Analysis of our experiments provides insight into the permeation pathway of hZIP4.
Significance: Comparison of this model to membrane transporter crystal structures provides a structural linkage to MFS proteins.
Keywords: computer modeling, membrane biophysics, membrane protein, metal homeostasis, metal ion-protein interaction, protein evolution, transport metal, transporter, zinc
Abstract
Members of the Zrt and Irt protein (ZIP) family are a central participant in transition metal homeostasis as they function to increase the cytosolic concentration of zinc and/or iron. However, the lack of a crystal structure hinders elucidation of the molecular mechanism of ZIP proteins. Here, we employed GREMLIN, a co-evolution-based contact prediction approach in conjunction with the Rosetta structure prediction program to construct a structural model of the human (h) ZIP4 transporter. The predicted contact data are best fit by modeling hZIP4 as a dimer. Mutagenesis of residues that comprise a central putative hZIP4 transmembrane transition metal coordination site in the structural model alter the kinetics and specificity of hZIP4. Comparison of the hZIP4 dimer model to all known membrane protein structures identifies the 12-transmembrane monomeric Piriformospora indica phosphate transporter (PiPT), a member of the major facilitator superfamily (MFS), as a likely structural homolog.
Introduction
The zinc- and iron-regulated transport protein (ZIP)3 family functions to increase the cytosolic concentration of first row transition metals (1, 2). Although mutations to these proteins result in cellular zinc and/or iron deficiency, analysis of in situ experiments have demonstrated that ZIP transporters can also transport Ni2+, Cu2+, and/or Cd2+ (3, 4). ZIP member proteins have eight transmembrane domains and can be further classified into subfamilies (ZIPI, ZIPII, gufA, and LIV-1) based on sequence alignments (5). The largest subfamily, LIV-1, is distinct from other ZIP proteins as its members encode a highly conserved sequence (HSVFEGLAVGIQ) in transmembrane domain (TM) 4 that has been proposed to be important for transition metal transport (6). The plasma membrane human (h) ZIP4 protein was the first human member of the LIV-1 subfamily of proteins to be identified as mutations within this protein lead to the zinc deficiency disease acrodermatitis enteropathica (6–9).
The Znt family of proteins functions in opposition to ZIP member proteins as they act to decrease the cytosolic concentration of zinc (10). Elucidation of the crystal structure of the bacterial Znt protein, YiiP, and subsequent functional studies have provided insight into the molecular details of transition metal transport mediated by Znt proteins (11–13). The six TM YiiP protein encodes a TM transition metal coordination site and is dimeric, and both termini are cytoplasmic.
Znt transporters belong to the cation diffusion facilitator (CDF) family. CDF proteins are found in bacteria, archaea, and eukaryotes (14). Member transporters translocate first and/or second row transition metals (2, 10). Both Znt and ZIP proteins are part of a larger group of transport proteins, named solute carrier (SLC) proteins. This group of proteins includes 52 families that function as facilitative or secondary active transporters, such as the major facilitator superfamily (MFS) (15, 16). MFS transporters encode 12 TMs and transport small solutes in response to chemiosmotic gradients. Although mechanistic linkages among family members are being studied, structural linkages between families of SLC group proteins are sparse.
The absence of structural information for ZIP transporters has limited the scope and impact of mechanistic studies as it is not possible to decipher whether mutations that affect the kinetics and/or cation selectivity of ZIP proteins directly or indirectly affect transport function. Thus, the molecular mechanism of transition metal transport mediated by ZIP proteins is not resolved. In this study, we take advantage of recent progress in predicting residue pair contacts in a protein structure using co-evolution data to generate the first molecular model of hZIP4 using Rosetta in combination with co-evolution data (17, 18). Mutagenesis studies that probe residues important for transition metal translocation and specificity are consistent with the model of hZIP4. Furthermore, the model has a similarity to the central transition metal coordination site of the Znt exporter, YiiP (11, 13). Comparison of these structures suggests that the Znt and ZIP families may share a common metal selectivity for zinc.
Materials and Methods
Reagents
The SP6 mMESSAGE mMACHINE kit was acquired from Invitrogen. Restriction enzymes were purchased from New England Biolabs (Ipswich, MA). The XL-1 Blue supercompetent cells were obtained from Agilent (Santa Clara, CA). Pfu polymerase was procured from Stratagene (La Jolla, CA). The radioisotopes 65ZnCl2 and 59FeCl3 were purchased from PerkinElmer. All transition metal chloride salts were purchased from Alfa Aesar (Ward Hill, MA). Additional chemicals were purchased from Sigma-Aldrich unless otherwise indicated.
Contact Predictions from Co-evolution
GREMLIN, a contact prediction method that utilizes co-evolution, was used to predict residue-residue contacts in hZIP4. GREMLIN constructs a global statistical model that simultaneously captures the conservation and co-evolution patterns in the input alignment. Strongly co-evolving residue pairs as identified by this approach are highly likely to be in contact with each other in the three-dimensional structure (18).
The input sequence of hZIP4 was trimmed to only include the following residues: 328–427 and 487–642. This trimmed sequence excludes the extracellular N-terminal domain and the only significant intracellular domain between TM3 and TM4. Both of these domains have substantial variation between species. A multiple sequence alignment was generated using HHblits (19) with the following options: n, 8; e, 1 × 10−20; maxfilt, ∞; neffmax, 20; nodiff, realign_max ∞. The resulting alignment was then filtered to exclude any sequence that did not cover at least 50% of the query and to reduce the sequence redundancy to 90%. GREMLIN was then run with default parameters.
Rosetta Membrane ab Initio
The standard Rosetta ab initio structure prediction method was used to model the three-dimensional structure of hZIP4 trimmed to only include residues 328–648. The default Rosetta energy function was modified to enable membrane specific terms with the following weights: fa_sol, 0.0; fa_mbenv, 0.3; fa_mbsolv, 0.35; and Menv_smooth, 0.5 (20, 21). Transmembrane spans (residues 2–23, 33–53, 74–95, 167–191, 197–222, 228–252, 259–282, and 290–314) were defined using the consensus output of the MESSA server (22). To reduce the sampling space, sigmoidal restraints that modified the energy function were introduced. The shape of the sigmoid is defined using a distance cutoff, the slope, the intercept, and the strength of the weight parameter (Fig. 1A).
FIGURE 1.
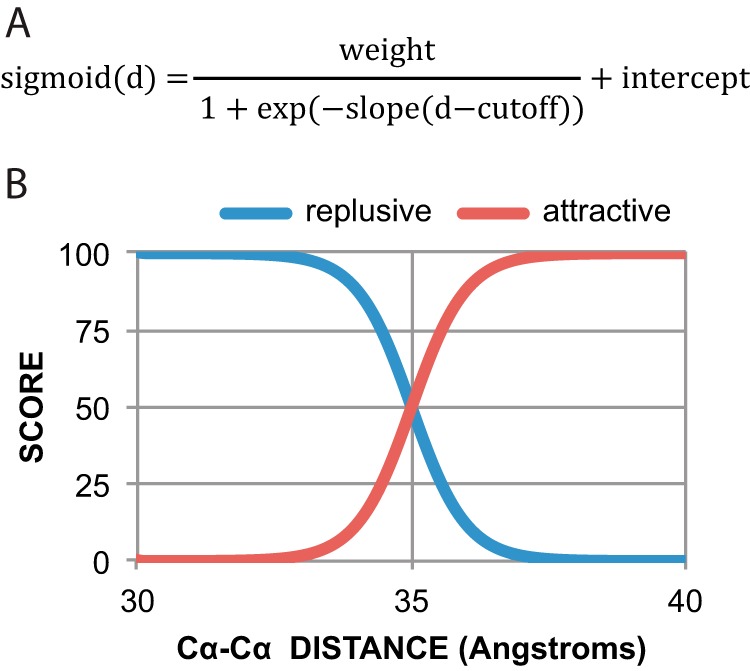
A, sigmoidal distance restraint function. d is the distance between given distance evaluated. B, strong repulsive distance restraints were added between extracellular regions and intracellular regions, and strong attractive restraints were added within intracellular regions and extracellular regions, effectively constructing a membrane-like sampling space.
The Rosetta ab initio protocol consists of two stages. In the initial stage (“centroid”), side chains are represented by fixed center-of-mass atoms allowing for rapid generation and evaluation of various protein-like topologies; the second stage (“full-atom”) places all-atom side chains into a starting topology and iteratively refines the model until a low energy structure is found (17, 23).
To favor sampling of topologies consistent with GREMLIN predictions, sigmoidal distance restraints (Fig. 1A) were introduced between residue pairs predicted to be in contact by GREMLIN (Fig. 2). When used in the centroid stage of Rosetta, these restraints were introduced between carbon-β atoms (carbon-α in the case of glycine), at amino acid pair-specific Cβ-Cβ cutoff and slopes, as described in Supporting Information Table 3 within Ref. 18; in the full-atom stage, these were replaced with ambiguous distance restraints between side-chain heavy atoms (cutoff of 5.5 and slope of 4) (24). The relative weight of each restraint was based on the GREMLIN score. The total atom pair restraint score was scaled to be roughly one-half the total Rosetta score.
FIGURE 2.

A, contact map, left to right, top to bottom (N to C terminus) showing the top 384 co-evolving residues between transmembrane helices represented as a contact map. The darker and larger the blue dots, the higher strength in covariance. B, table of interhelical residue pairs, within the top 64 predictions. The matching labels are shown in the bottom left portion of the contact map (A).
In addition to these restraints, additional strong repulsive distance restraints (weight 100, cutoff 35, slope 2, and intercept of 100) were added between extracellular regions (defined by residues 329, 391,522, 582, and 648) and intracellular regions (defined by residues 353, 425, 490, 553, and 615), and strong attractive restraints (weight 100, cutoff 35, slope 2 and intercept of 0) were added within intracellular regions and extracellular regions, effectively constructing a membrane-like sampling space (Fig. 1B). These restraints were introduced between pairs of Cα atoms.
The top 1010 models ranked by distance restraints score, with z-score ≥2 (Fig. 3), were clustered based on structural similarity as calculated by TM-score (25) after excluding the disordered regions (positions 428–486 and 643–648) (26). Clusters were defined as connected components of a network, where each edge is between models with TM-score ≥0.7. The selected model was then further energy-minimized with Rosetta to remove clashes, while respecting structural symmetry and GREMLIN restraints (27).
FIGURE 3.
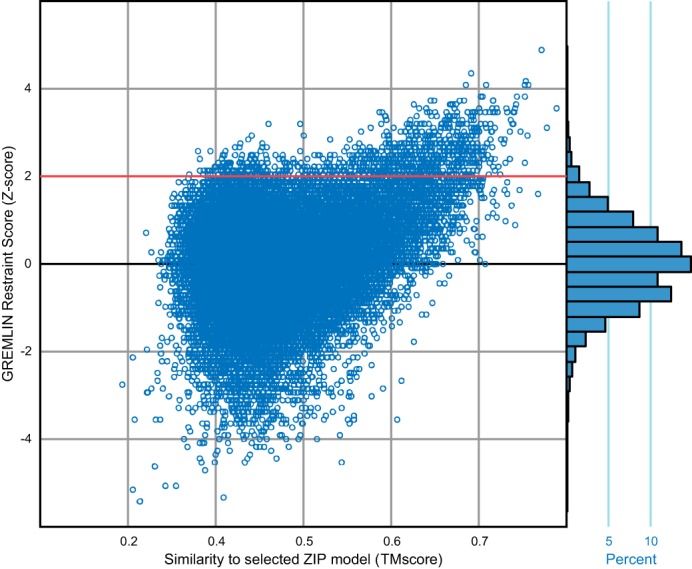
Distribution of GREMLIN restraint scores for Rosetta ab initio models. Models above the red line (with z-score ≥2) were selected for the clustering analysis. The selected model is the refined model from cluster A (Fig. 4A). The ab initio models closer to the selected model tend to have a higher GREMLIN restraint score.
Expression of hZIP4 in Xenopus laevis Oocytes
Preparation of plasmid construct and mRNA was performed as described previously (3). Site-directed mutagenesis was performed using QuikChange according to the manufacturer's instructions. Following mutagenesis, the entire gene was sequenced.
Oocytes were extracted from X. laevis in accordance with Worcester Polytechnic Institute Animal Care and Use committee-approved protocol (3, 28). Oocytes were digested with collagenase (3 mg/ml) for 3 h. Following enzymatic digestion, oocytes were rinsed with ORI−Ca2+ buffer (90 mm NaCl, 2 mm KCl, and 5 mm MOPS at pH 7.4) three times and stored in ORI+Ca2+ (ORI−Ca2+ buffer with 2 mm CaCl2 and 20 μg/ml gentamycin) buffer overnight at 16 °C. mRNA or diethyl pyrocarbonate-treated water was injected into each oocyte using a micro-injector and stored in ORI+Ca2+ buffer for 3 days at 16 °C.
Radioisotope Uptake Assay
Radioisotope uptake assays were performed 72 h following injection (3). Briefly, oocytes were washed with uptake buffer (90 mm NaCl, 10 mm HEPES, pH 7.4, and 1 mm ascorbic acid). Oocytes were incubated in uptake buffer containing 65ZnCl2 for 1 h. At the conclusion of the transport assay, oocytes were washed three times with uptake buffer and solubilized in 200 μl of 1% w/v SDS solution. Solubilized oocytes were mixed with scintillation fluid (Scintisafe-30%, Fisher Scientific) and subjected to radioactivity measurement using a Beckman LS6500 multi-purpose scintillation counter. For iron uptake assays, 59FeCl2 was added as 59FeCl3 in the presence of 1 mm ascorbic acid. For competition experiments, 3 μm 65ZnCl2 was added to 600 μm of various transition metals. For control, no additional transition metal was added. In these experiments, it is necessary to subtract diethyl pyrocarbonate/water-injected controls at each concentration of zinc during the uptake assay to eliminate background radioisotope uptake. The resulting data points were fit with the equation: y = (Vmax × [Zn2+]n)/Kmn + [Zn2+]n where Vmax is the maximal velocity, [Zn2+] is the concentration of divalent metal ion, Km is the concentration of divalent metal at one-half Vmax, and n is the Hill co-efficient.
Detection of hZIP4 Surface Expression in Oocytes
hZIP4 surface expression was detected using EZ-Link Sulfo-NHS-SS-Biotin and biotinylation kit (Thermo Scientific) according to the manufacturer's instructions. In brief, 35 oocytes 72 h after injection were washed three times with ice-cold PBS (0.1 m sodium phosphate, 0.15 m NaCl, pH 7.2) before incubating them with 2 ml of biotinylation buffer. Sulfo-NHS-SS-Biotin was dissolved in PBS to a final concentration of 0.11 mg/ml, and oocytes were incubated with biotinylation buffer for 90 min at 4 °C with gentle shaking. The reaction was stopped upon the addition of 200 μl of quenching solution. Biotin-labeled oocytes were washed four times with TBS (0.025 m Tris, 0.15 m NaCl, pH 7.2). Finally, oocytes were resuspended in 500 μl of solubilization buffer (TBS buffer containing 1% (w/v) n-dodecyl-β-d-maltoside and 1 mm PMSF). Oocytes were homogenized by passing them through a 25-gauge needle. The lysate was centrifuged at 14,000 × g for 20 min at 4 °C. The supernatant was collected into microcentrifuge tubes. The pellet was resuspended in solubilization buffer and centrifuged at 14,000 × g for 20 min at 4 °C. The supernatants were mixed and incubated with 50 μl of NeutrAvidin resin at room temperature for 60 min with gentle shaking. The resin was washed four times with solubilization buffer. Finally, biotinylated proteins were eluted by boiling resin with Laemmli buffer for 20 min. The eluted biotinylated proteins were separated on SDS-PAGE. hZIP4 was detected by Western blot with a rabbit polyclonal antibody raised against the N terminus of hZIP4 (Aviva Systems Biology, San Diego, CA).
Oligomeric State of hZIP4
Loosely adapting the protocol from Dürr et al. (29), 50 oocytes were added to breaking buffer (20 mm HEPES, pH 7.4, 150 mm NaCl and 1 mm PMSF) and passed through a 25-gauge needle followed by 1 min of low speed centrifugation (100 × g and 4 °C). The supernatant was collected in a separate microcentrifuge tube and centrifuged (1 min at 100 × g and 4 °C). The procedure was repeated until no pellet was observed upon centrifugation. The supernatant was then spun down at 14,000 × g for 30 min at 4 °C. The membrane pellet was resuspended in solubilizing buffer (20 mm HEPES, pH 7.4, 150 mm NaCl, 1% w/v n-dodecyl-β-d-maltoside, and 1 mm PMSF) and incubated at room temperature for 5 min. The insoluble fraction was separated by high speed centrifugation (14,000 × g for 30 min at 4 °C). The supernatant was added into Laemmli buffer in the absence of reducing agent. The solubilized total membrane proteins were separated on SDS-PAGE. The Western blot was developed using rabbit polyclonal antibody raised against N terminus of hZIP4 (Aviva Systems Biology).
Results
A model of hZIP4 was generated using predicted contacts and Rosetta (18). Functional studies support this model. The structure comparison method was then used to compare the hZIP4 structural model with non-redundant Protein Data Bank (PDB) membrane protein structures (30, 31).
Residue-Residue Contacts Predicted from Co-evolution
Membrane structures have been modeled using contact prediction based on co-evolutionary patterns in large multiple sequence alignments of homologous proteins (32, 33). To see whether such an analysis was possible for hZIP4, we constructed an alignment over the conserved transmembrane region by querying the UniProt database with HHblits (19, 34). The resulting non-redundant alignment contained 1731 homologous sequences, 6.8 sequences per length of 255, which is more than the minimum sequences (5 sequences per length) required to predict residue pairs in contact in the three-dimensional structure of hZIP4 using GREMLIN, a co-evolution-based contact prediction approach that is more accurate than other extant approaches (18). These predictions were then used as distance restraints in the Rosetta ab initio protocol along with other membrane-specific terms to generate over 100,000 structural models of hZIP4 using a distributed computing network, Rosetta@home. Of these, the top 1010 models based on restraint score were extracted. These models formed three large clusters (Fig. 4), of which one cluster agreed with GREMLIN predictions and formed physically realistic structures.
FIGURE 4.

Contact maps showing contacts made within each cluster (top) and cartoons of the top three models colored blue to red (N to C terminus) for each cluster (bottom). The intensity of the blue (in the contact map) is indicative of the percentage of models within the cluster that make those contacts. Intensity of blue ranges from light blue (10%) to blue (100%). x- and y-axis numbering (1–8) in contact maps represent transmembrane helices 1–8. Red boxes indicate helices predicted to be in contact but are not in the given cluster. A, cluster A makes contacts between all the top co-evolving helices, but there is a disagreement between models in the regions boxed in green. Further analysis reveals that variation within cluster A is explained within the context of dimer helix swapping (Fig. 6). B and C, both clusters B and C have missing helical contacts in regions with strong co-evolution signal. Additionally, they both expose histidines into the membrane, which is highly improbable for membrane proteins.
Despite this extensive sampling, we observed one set of predicted contacts that were consistently not made by the structural models (Fig. 4A). Further inspection revealed that it was physically impossible to make all these contacts in regions between TM2–3 and TM7–8, without breaking contacts between other helices in a monomeric state. The top 384 (1.5 × length of query) co-evolving pairs of residues that were at least three residues apart (Fig. 2) in the protein sequence were then selected to be used in subsequent structural modeling in Rosetta. Using these top co-evolving pairs, an oligomeric interface could be readily made (Figs. 5, B and C, and 6). Notable for these results is that each of the variants in cluster within Fig. 4A can make the dimer interface as shown in Fig. 6. Equally, these models have histidine residues lining the transmembrane core forming a potential permeating pathway (Fig. 5A).
FIGURE 5.

Models of hZIP4 using Rosetta structure prediction guided by co-evolution-based contacts. A, ribbon diagram of the monomer structure, colored blue to red (N to C terminus), shows the predicted location of the transmembrane histidines in white. For clarity, the loop between residues 428 and 486 is omitted. B, consistency of co-evolution predicted contacts with hZIP4 structural model. Black dots, predicted contacts; gray, contacts in the Rosetta model monomer; red, contacts across dimer interface. The top coevolving residues are made within the context of the dimer. x- and y-axis numbering (1–8) in the contact map represent transmembrane helices 1–8. C, dimer view of the Rosetta structure model of hZIP4. The top 25 contacts are shown as yellow dashes. The highlighted regions (red and dark blue) indicate the conserved LIV-1 sequence in the fourth transmembrane helix.
FIGURE 6.
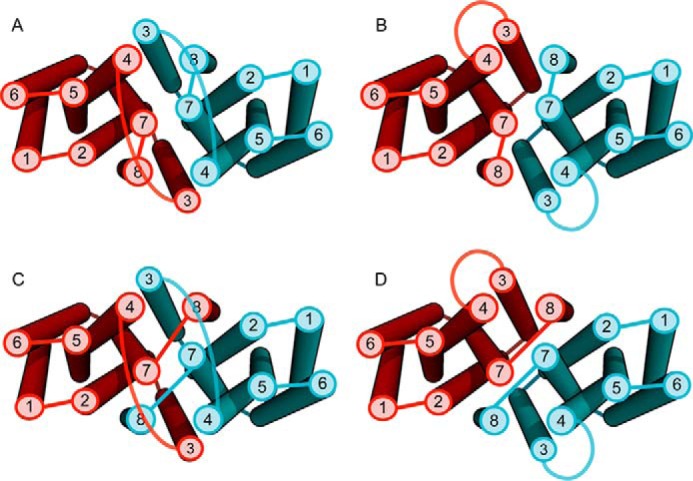
Variations within cluster A are consistent within context of a dimer. When we examine variation within cluster A, we find that the models are capable of making the remainder of the top co-evolving residues in the context of a dimer. For example, if we reconnect the loop regions between helices 2 and 3, swapping helix 3 between the homo-dimer (A ≥ B), this preserves all predicted helical contacts. Another example would be to reconnect the loop between helices 7 and 8, swapping helix 8 (A ≥ C), or a combination of both (A ≥ D). These swaps are represented in the variation of the top cluster (Fig. 4A).
The Effect of Histidine to Alanine Mutations on hZIP4 Zinc Transport
Our laboratory has demonstrated that covalent labeling of histidine, but not cysteine residues, decreases the velocity of Zn2+ transport (3). Independent of the modeling studies, we examined the functional role of each transmembrane histidine residue in transition metal translocation. hZIP4 encodes six transmembrane histidine residues: 379 (TM2), 507 (TM4), 536, 540, and 550 (TM5), as well as 624 (TM8). We individually replaced each TM histidine residue with alanine.
Zinc transport kinetics were determined for each of the mutant transporters using our established uptake assay (3). Previously, we have demonstrated that the wild type hZIP4 has two distinct Km values: 76 ± 5 nm and 1.4 ± 0.3 μm (3). Alanine substitution at the TM histidine residues altered the nm Km for three mutant proteins: H379A, H507A, and H536A (Table 1). In addition, alanine replacement abolished the μm Km for one mutant protein (H379A). Equally, no zinc translocation was observed for the H624A mutant (data not shown). Representative data for one of these mutants (H540A) is shown in Fig. 7. Furthermore, to directly compare Vmax between the wild type and mutant constructs, oocytes expressing each construct were incubated with 3 μm 65ZnCl2. When normalizing the data to surface expression, the Vmax was significantly lower for the H379A, H507A, H536A, H540A, and H624Q mutants (but not H550A) when compared with the WT protein (Fig. 8). Analysis of transport data for some constructs revealed Hill coefficients with non-Michaelis-Menten behavior (Table 1). This could be due to one or more factors including a flip-flop model where a dimeric transporter has reciprocal conformations, a ping-pong mechanism, or random substrate binding if more than one zinc atom is transported per transport cycle (35, 36). It was also determined that zinc transport was roughly linear over the time of the assay (60 min) upon the addition of 21.75 μm 65ZnCl2 (Fig. 9). We have previously demonstrated that under these conditions, 65Zn2+ uptake is roughly linear for WT hZIP4 over 2 h (3).
TABLE 1.
Zinc transport kinetic parameters for WT and mutant hZIP4 transporters
Kinetic parameters were determined by measuring amount of zinc taken up with hZIP4-Strep mRNA as described under “Materials and Methods.” The data were fit to the Michaelis-Menten equation as described under “Materials and Methods.” Data originated from 5–11 oocytes, values are means ± S.E. Km values statistically different (p < 0.05) from WT hZIP4 are in bold. No statistical tests were performed on Vmax as assays were performed on separate days and surface expression was variable between days. ND, none detected.
| Vmax(1) | n1 | Km1 | Vmax(2) | n2 | Km2 | |
|---|---|---|---|---|---|---|
| pmol/oocyte/h | nm | pmol/oocyte/h | μm | |||
| WT | 1.4 ± 0.1 | 3.6 ± 0.6 | 76 ± 5 | 13 ± 1 | 1.2 ± 0.3 | 1.4 ± 0.3 |
| H379A | 0.8 ± 0.01 | 4.2 ± 0.7 | 370 ± 10 | ND | ND | ND |
| H507A | 0.6 ± 0.1 | 6.1 ± 2.9 | 220 ± 20 | 2.3 ± 0.2 | 0.9 ± 0.1 | 1.8 ± 0.4 |
| H536A | 0.7 ± 0.1 | 2.4 ± 1.1 | 210 ± 50 | 3.1 ± 0.3 | 1.3 ± 0.3 | 1.9 ± 0.5 |
| H540A | 0.9 ± 0.1 | 2.1 ± 0.8 | 70 ± 20 | 6.5 ± 0.9 | 1.2 ± 0.4 | 1.4 ± 0.6 |
| H550A | 0.4 ± 0.1 | 1.2 ± 0.7 | 60 ± 30 | 0.8 ± 0.01 | 1.8 ± 0.3 | 0.5 ± 0.1 |
| H624A | ND | ND | ND | ND | ND | ND |
| H624Q | 1.1 ± 0.1 | 1.8 ± 0.9 | 60 ± 17 | 3.3 ± 0.1 | 2.9 ± 0.9 | 0.8 ± 0.1 |
FIGURE 7.

Kinetics of H540A hZIP4 zinc transport. The transport parameters, Km and Vmax, of the H540A hZIP4 mutant were elucidated at low (A) and high (B) concentrations of zinc by measuring the amount of 65Zn2+ uptake into oocytes injected with hZIP4-Strep (with H540A mutant) mRNA over 1 h in assay buffer, which contained varying amounts of 65ZnCl2. The data were fit to the Michaelis-Menten equation described under “Materials and Methods.” Data originated from 4–7 oocytes; values are means ± S.E.
FIGURE 8.
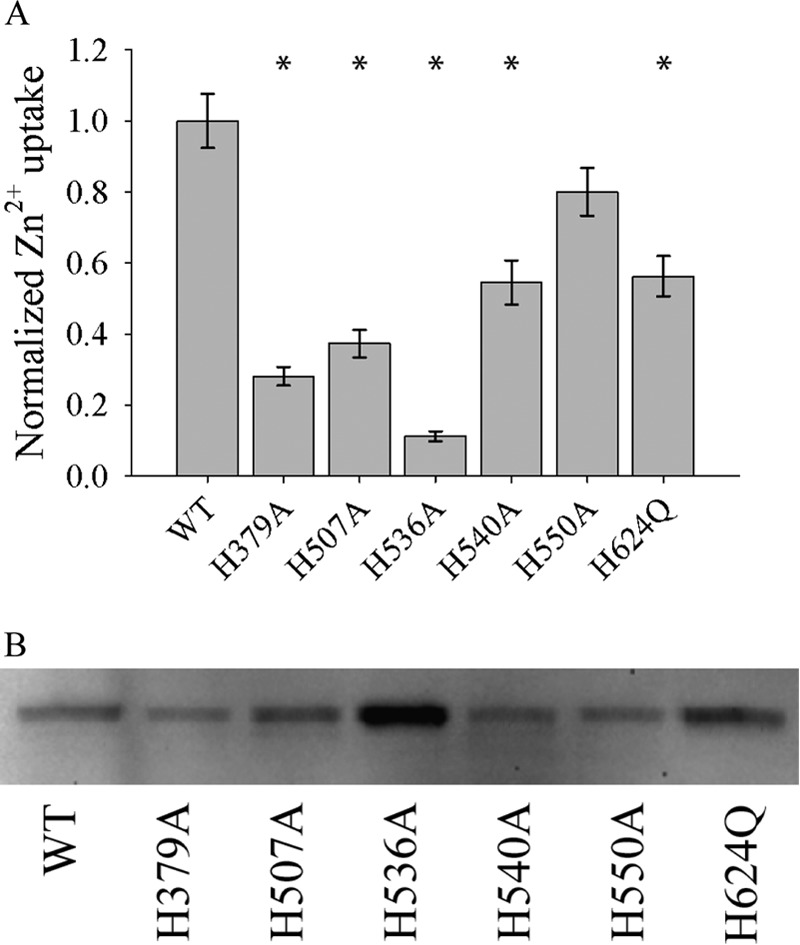
Relative velocity of WT hZIP4 and mutant transporters. A, oocytes were injected with 25 ng of WT hZIP4 or mutant hZIP4 mRNA. After 3 days of incubation at 16 °C, oocytes were subjected to radioisotope uptake experiment. Oocytes were incubated with 3 μm 65Zn2+, and zinc uptake was measured after 60 min. Transport data were normalized to surface expressed protein (B). * indicates a statistically significant difference in zinc uptake between mutant hZIP4 and WT after uptake was normalized to the level of protein surface expression (p < 0.05). Values are means ± S.E. B, Western blot of surface-expressed WT and mutant hZIP4 proteins isolated as described under “Materials and Methods.” The relative expression of hZIP4 was quantified using Quantity One software (Bio-Rad Laboratories).
FIGURE 9.

Time course of zinc uptake for mutant constructs. Oocytes, injected with 25 ng of hZIP4 mutant constructs, were incubated with 21.75 μm 65ZnCl2. The assay was quenched every 30 min up to 120 min, and data originated from 5–8 oocytes; values are means ± S.E. Fitting the data to the equation to a standard linear equation (y = mx + b) resulted in the following R2 values: H379A (0.9685), H507A (0.9683), H536A (0.9824), H540A (0.9816), H550A (0.9949), and H624Q (0.9493).
Histidine 624 Is Essential for Biometal Translocation
Hydropathy analysis, as well as our structural model, predicts that His-624 is located on the cytoplasmic end of TM8 and is not conserved among ZIP family proteins. Considering that ZIP member proteins that function as iron transporters (ZIP8 and ZIP14) encode glutamine at this position, we replaced histidine with glutamine at this position to further elucidate the role of this residue in transition metal permeation. The H624Q mutation retained zinc translocation function, while having a tighter μm Km (Table 1). To further explore the functional role of His-624 in biometal translocation, we performed a competition experiment where 200-fold excess of various divalent cations was added to our uptake assay in the presence of 3 μm 65Zn2+. Previously, we have shown that in the wild type transporter, zinc transport is inhibited by 200-fold excess Cu2+, Ni2+, and Zn2+ (3). In addition, Zn2+, Cu2+, and Ni2+ are transported by hZIP4. In contrast to the wild type transporter, 65Zn2+ transport was significantly inhibited by Cu2+, Ba2+, Co2+, Fe2+, Mn2+, Ni2+, and Zn2+ for the H624Q mutant transporter (Fig. 10).
FIGURE 10.

Competition of 65Zn2+ uptake with a series of divalent cations. To determine which divalent cations inhibited hZIP4-mediated 65Zn2+ uptake, oocytes expressing hZIP4 were preincubated in 600 μm cold ZnCl2, BaCl2, CdCl2, CoCl2, CuCl2, FeCl2, MgCl2, MnCl2, or NiCl2 in the uptake assay buffer. The uptake assay was initiated by adding 3.0 μm 65ZnCl2. The assay was quenched after 1 h as described under “Materials and Methods.” Data were normalized to the amount of 65Zn2+ uptake in the absence of competing cation. Data originated from 14–17 oocytes; values are means ± S.E. Data marked with * represent statistically significant difference from WT (p < 0.05).
Residues Contribute to Cation Selectivity of ZIP Transporters
To explore the role of the transmembrane histidine residues in cation specificity for hZIP4, we examined the relative velocity of Fe2+ and Zn2+ for WT and each mutant construct (Table 2). The rationale for this experiment was the observation that zinc transport was inhibited for the H624Q mutant construct by high levels of iron in our competition assay. Therefore, we hypothesized that the H624Q mutant hZIP4 transporter would translocate Fe2+. Analysis of our experiments demonstrated that the wild type hZIP4 transports Fe2+, but that the H624Q mutant transporter is a poor iron transporter. Equally, replacement of three histidine residues with alanine significantly altered the velocity ratio of Fe2+ and Zn2+ transport (H379A, H507A, and H536A) (Table 2).
TABLE 2.
Relative Specificity for Zn2+ and Fe2+ for WT and mutant hZIP4 transporters
A final concentration of 5 μm of indicated metal was applied to oocytes expressing WT or mutant hZIP4 protein. Data originated from 8–11 oocytes, values are means ± S.E. Zn2+/Fe2+ uptake values for mutant proteins statistically different (p > 0.05) from WT hZIP4 are in bold. Similar results were obtained upon application of 10 μm of indicated metal. ND = none detected.
| Zn2+ uptake | Fe2+ uptake | Zn2+/Fe2+ uptake | |
|---|---|---|---|
| pmol/oocyte/h | pmol/oocyte/h | ||
| WT | 2.2 ± 0.2 | 0.61 ± 0.07 | 3.6 ± 0.7 |
| H379A | 1.3 ± 0.3 | 0.64 ± 0.04 | 2.0 ± 0.6 |
| H507A | 2.7 ± 0.3 | 0.38 ± 0.07 | 7 ± 2 |
| H536A | 1.4 ± 0.2 | 2.1 ± 0.1 | 0.7 ± 0.1 |
| H540A | 3.5 ± 0.5 | 0.65 ± 0.04 | 5 ± 3 |
| H550A | 1.9 ± 0.2 | 0.46 ± 0.07 | 4.1 ± 0.8 |
| H624A | ND | 0.24 ± 0.05 | Undefined |
| H624Q | 2.4 ± 0.2 | 0.26 ± 0.06 | 9 ± 2 |
Structural Alignment of hZIP4 with Membrane Protein Structures
To examine whether the model of hZIP4 was similar in structure to more distal membrane proteins, we performed structural alignments between the model and all known crystal structures of membrane proteins, in the Protein Data Bank of Transmembrane Proteins), using TM-align (30, 31, 37). The hZIP4 dimeric model is closest in structure to MFS transporters (Fig. 11). The highest ranked MFS transporter is PiPT, a eukaryotic phosphate transporter (Fig. 12). The MFS and CDF proteins are separate families within the SLC group of proteins (38). PiPT encodes 12 TMs in a two-fold axis of symmetry where phosphate translocation occurs at the two-fold axis.
FIGURE 11.

Structural homology of hZIP4 dimer to other membrane proteins. The red filled circles are MFS proteins. EmrE and YiiP are the efflux multidrug transporter and Znt zinc exporter, respectively.
FIGURE 12.

A, similarity of the Rosetta model of hZIP4 with the structure of the MFS transporter (PDB: 4j05). Loop regions and helices 1 and 6 of hZIP4 are not shown for clarity. TM-score = 0.66 (3.53 root mean square deviation over aligned region). B, alignment of transmembrane helices in sequential space. The matching colors of hZIP4 and MFS indicate structural alignment. The gray regions did not align and were not shown in A.
Oligomeric State of hZIP4
TM7s from each monomeric unit are adjacent helices in each of the top ranked models (Fig. 6). Therefore, to elucidate whether the oligomeric state of hZIP4 could be disrupted using our model as a template, we replaced residues within TM7 with tryptophan. Oocytes were injected with WT or mutant hZIP4 mRNA. Total membrane proteins were isolated after 72 h. Following Western blot analysis, it was observed that WT hZIP4 is composed of both monomeric and dimeric complexes (Fig. 13). Replacement of some, but not all, residues along TM7 with tryptophan resulted in mutant proteins, which favors a monomeric state. Notably, the L602W mutation resulted in nearly all of hZIP4 in the monomeric state, but this protein was not glycosylated. Equally, the D604W mutation greatly diminished the population of dimer; however, glycosylation is not affected for this mutant protein. Finally, the M605W mutation did not affect the relative distribution of hZIP4 oligomeric states.
FIGURE 13.
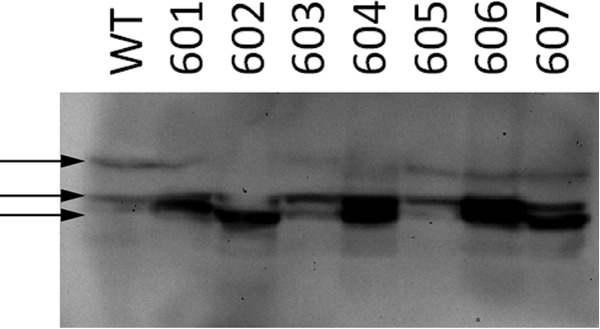
Western blot analysis of tryptophan-scanning mutagenesis studies along TM7. Residues replaced with tryptophan are indicated above blot. Arrows represent (from top to bottom) dimer, glycosylated monomer, and nonglycosylated monomer.
Discussion
We have combined GREMLIN contact prediction using the pseudo-likelihood approach with the Rosetta membrane protein structure prediction method to model the TMs of hZIP4. The 327-residue N-terminal domain was excluded because there were not enough homologous sequences to perform co-evolution analysis. The 76-residue cytosolic domain between TM3 and TM4 was included in the structural modeling, but was removed during clustering and analysis because it did not converge to a low number of structures. The eight transmembrane domains encode a core structure composed of TMs 2, 4, 5, and 7 with additional transmembrane domains (TMs 1, 3, 6, and 8) peripheral to this core structure. Multiple oligomeric structures could satisfy the predicted contacts and retain membrane-like topologies (Fig. 6). However, the interface for each oligomeric structure is composed of TMs 3, 4, 7, and 8. It is important to note that although our approach can define residue pairs that are co-evolving, co-evolution between residues does not always mean contact for all conformational states of the protein. Some contacts may only be made in one conformational state, whereas other contacts may comprise higher order oligomeric assemblies. Therefore, the top four potential models are shown here. Furthermore, models with a z-score greater than or equal to two were selected for the clustering analysis (Fig. 3). Published experimental support for the dimeric model is derived from two sets of experiments. First, direct analysis of size exclusion HPLC suggested that the oligomeric state of ZIPB in detergent micelles was consistent with a dimer (39). More recently, it was observed that hZIP13 is dimeric following chemical cross-linking (40). Analysis of our tryptophan-scanning mutagenesis experiments suggests that dimer formation can be disrupted upon mutagenesis along TM7.
Analysis of TM scoring demonstrates that the hZIP4 dimer model retains the same fold as MFS proteins. The closest MFS homolog is the 12-transmembrane monomeric eukaryotic phosphate transporter, PiPT (38). Recently, it has been proposed that MFS transporters evolved either from three TM unit rearrangements or from a common ancestor without three TM unit rearrangements (41–44). Although analysis of our data provides further insight into the evolutionary relationship between SLC proteins, it is important to note that there are caveats to this analysis. First, analysis of our modeling experiments identified four possible dimeric interfaces. Some of the lower scoring models have slightly lower TM-scores when compared with the PiPT transporter. If the lower scoring models prove to be more accurate, then the comparison between the PiPT structure and the hZIP4 model should be reanalyzed. Second, the transport pathway for PiPT is along the homologous dimer interface of the hZIP4 model. This could suggest a different mechanism of transport for these two families of transporters. Alternatively, it could also be true that the proposed three TM unit rearrangements described above resulted in proteins with new functionalities, which are exhibited in hZIP4.
Mechanistically, the hZIP4 model encodes a central transition metal coordination site similar to the YiiP crystal structure (Fig. 14). Mutagenesis of residues within the YiiP metal coordination site alters the transition metal specificity of YiiP (45). Equally, alanine replacement of histidine residues within the hZIP4 model coordination site alters the Km, Vmax, and metal specificity of hZIP4. Among ZIP member proteins, residues that comprise this putative metal coordination site are variable, and amino acid differences within this coordination site likely contribute to differing cation specificity among ZIP transporters (2).
FIGURE 14.

A, expanded view of putative YiiP and hZIP4 transition metal coordination sites. B, WebLogo showing the conservation at each position.
It is plausible that overexpression of hZIP4 may result in protein misfolding, which may lead to distinct protein populations with altered kinetics or metal specificity. Although Km or normalized Vmax values between differing expression systems may vary, it is likely that trends, such as mutations that result in higher/lower affinities or changes in normalized Vmax, will be consistent between overexpression systems. Furthermore, considering that both micromolar and nanomolar Km values have been observed upon heterologous expression in mammalian cells as well as X. laevis oocytes, this suggests that either both affinities are present in vivo or that overexpression in both cell types gives equivalent artifacts (3, 4, 46). Therefore, analysis of hZIP4 transport experiments performed in X. laevis oocytes is a viable and valuable method to examine transport properties for this class of proteins. By quantifying surface expression through the biotinylation and isolation of all surface expressed proteins, we have gained valuable insight into how mutations of histidine residues within the transmembrane domains influence Vmax. Equally, the non-Michaelis-Menten Hill coefficients could be due to one or more factors including a flip-flop model where a dimeric transporter has reciprocal conformations, a ping-pong mechanism, or random substrate binding if more than one zinc molecule is transported per transport cycle.
In summary, by employing Rosetta with co-evolution and membrane terms in tandem with functional experiments, we have developed the first molecular model for ZIP proteins. This model can be used to examine the location of residues that contribute to specificity (His-379, His-507, His-536, and His-624) as well as the velocity of metal transport (His-379, His-607, His-536, His-540, and His-624). We expect that this model will open new avenues and experimental approaches to identify more nuanced mechanistic questions such as residues that contribute to transition metal docking within the ZIP family of proteins.
Supplementary Material
Acknowledgments
We thank Elizabeth Bafaro and Ryan Richards for invaluable discussion. We thank Vladimir Yarov-Yarovoy and Per Jr. Greisen for their comments and helpful suggestions, and Rosetta@home participants for donating their computer time.
Note Added in Proof
The pdb file generated from a computational model of hZIP4 was missing from the version of the article published as a Paper in Press on May 13, 2015. The model is now available as supplemental data.
The research described within this manuscript was supported by the Worcester Polytechnic Institute Research Foundation and National Institutes of Health Grants GM105964 (to R. E. D.) and GM092802 (to D. B.). The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental file hzip4_model_v1.pdb.
- ZIP
- Zrt and Irt protein family
- hZIP4
- human ZIP4
- TM
- transmembrane domain
- CDF
- cation diffusion facilitator
- SLC
- solute carrier
- MFS
- major facilitator superfamily
- ORI
- oocyte Ringer solution.
References
- 1. Eide D. J. (2004) The SLC39 family of metal ion transporters. Pflugers Arch. 447, 796–800 [DOI] [PubMed] [Google Scholar]
- 2. Dempski R. E. (2012) The cation selectivity of the ZIP transporters. Curr. Top. Membr. 69, 221–245 [DOI] [PubMed] [Google Scholar]
- 3. Antala S., Dempski R. E. (2012) The human ZIP4 transporter has two distinct binding affinities and mediates transport of multiple transition metals. Biochemistry 51, 963–973 [DOI] [PubMed] [Google Scholar]
- 4. Pinilla-Tenas J. J., Sparkman B. K., Shawki A., Illing A. C., Mitchell C. J., Zhao N., Liuzzi J. P., Cousins R. J., Knutson M. D., Mackenzie B. (2011) Zip14 is a complex broad-scope metal-ion transporter whose functional properties support roles in the cellular uptake of zinc and nontransferrin-bound iron. Am. J. Physiol. Cell Physiol. 301, C862–C871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gaither L. A., Eide D. J. (2001) Eukaryotic zinc transporters and their regulation. Biometals 14, 251–270 [DOI] [PubMed] [Google Scholar]
- 6. Dufner-Beattie J., Langmade S. J., Wang F., Eide D., Andrews G. K. (2003) Structure, function, and regulation of a subfamily of mouse zinc transporter genes. J. Biol. Chem. 278, 50142–50150 [DOI] [PubMed] [Google Scholar]
- 7. Wang K., Zhou B., Kuo Y. M., Zemansky J., Gitschier J. (2002) A novel member of a zinc transporter family is defective in acrodermatitis enteropathica. Am. J. Hum. Genet. 71, 66–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kury S., Dreno B., Bezieau S., Giraudet S., Kharfi M., Kamoun R., Moison J. P. (2002) Identification of SLC39A4, a gene involved in acrodermatitis enteropathica. Nat. Genet. 31, 239–240 [DOI] [PubMed] [Google Scholar]
- 9. Dufner-Beattie J., Wang F., Kuo Y. M., Gitschier J., Eide D., Andrews G. K. (2003) The acrodermatitis enteropathica gene ZIP4 encodes a tissue-specific, zinc regulated zinc transporter in mice. J. Biol. Chem. 278, 33474–33481 [DOI] [PubMed] [Google Scholar]
- 10. Kambe T. (2012) Molecular architecture and function of ZnT transporters. in Metal Transporters (Lutsenko S., Arguello J. M., eds), pp. 199–220, Academic Press, San Diego, CA: [DOI] [PubMed] [Google Scholar]
- 11. Coudray N., Valvo S., Hu M., Lasala R., Kim C., Vink M., Zhou M., Provasi D., Filizola M., Tao J., Fang J., Penczek P. A., Ubarretxena-Belandia I., Stokes D. L. (2013) Inward-facing conformation of the zinc transporter YiiP revealed by cryoelectron microscopy. Proc. Natl. Acad. Sci. U.S.A. 110, 2140–2145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hoch E., Lin W., Chai J., Hershfinkel M., Fu D., Sekler I. (2012) Histidine pairing at the metal transport site of mammalian ZnT transporters controls Zn2+ over Cd2+ selectivity. Proc. Natl. Acad. Sci. U.S.A. 109, 7202–7207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lu M., Fu D. (2007) Structure of the zinc transporter YiiP. Science 317, 1746–1748 [DOI] [PubMed] [Google Scholar]
- 14. Paulsen I. T., Saier M. H., Jr. (1997) A novel family of ubiquitous heavy metal ion transport proteins. J. Membr. Biol. 156, 99–103 [DOI] [PubMed] [Google Scholar]
- 15. Hediger M. A., Romero M. F., Peng J. B., Rolfs A., Takanaga H., Bruford E. A. (2004) The ABCs of solute carriers: physiological, pathological and therapeutic implications of human membrane transport proteinsIntroduction. Pflugers Arch. 447, 465–468 [DOI] [PubMed] [Google Scholar]
- 16. Pao S. S., Paulsen I. T., Saier M. H. (1998) Major facilitator superfamily. Microbiol. Mol. Biol. Rev. 62, 1–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Simons K. T., Ruczinski I., Kooperberg C., Fox B. A., Bystroff C., Baker D. (1999) Improved recognition of native-like protein structures using a combination of sequence-dependent and sequence-independent features of proteins. Proteins 34, 82–95 [DOI] [PubMed] [Google Scholar]
- 18. Kamisetty H., Ovchinnikov S., Baker D. (2013) Assessing the utility of coevolution-based residue-residue contact predictions in a sequence- and structure-rich era. Proc. Natl. Acad. Sci. U.S.A. 110, 15674–15679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Söding J. (2005) Protein homology detection by HMM-HMM comparison. Bioinformatics 21, 951–960 [DOI] [PubMed] [Google Scholar]
- 20. Barth P., Schonbrun J., Baker D. (2007) Toward high-resolution prediction and design of transmembrane helical protein structures. Proc. Natl. Acad. Sci. U.S.A. 104, 15682–15687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yarov-Yarovoy V., DeCaen P. G., Westenbroek R. E., Pan C. Y., Scheuer T., Baker D., Catterall W. A. (2012) Structural basis for gating charge movement in the voltage sensor of a sodium channel. Proc. Natl. Acad. Sci. U.S.A. 109, E93–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cong Q., Grishin N. V. (2012) MESSA: MEta-Server for protein Sequence Analysis. BMC Biol. 10, 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Raman S., Vernon R., Thompson J., Tyka M., Sadreyev R., Pei J., Kim D., Kellogg E., DiMaio F., Lange O., Kinch L., Sheffler W., Kim B. H., Das R., Grishin N. V., Baker D. (2009) Structure prediction for CASP8 with all-atom refinement using Rosetta. Proteins 77, Suppl. 9, 89–99, 10.1002/prot.22540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lange O. F., Rossi P., Sgourakis N. G., Song Y., Lee H. W., Aramini J. M., Ertekin A., Xiao R., Acton T. B., Montelione G. T., Baker D. (2012) Determination of solution structures of proteins up to 40 kDa using CS-Rosetta with sparse NMR data from deuterated samples. Proc. Natl. Acad. Sci. U.S.A. 109, 10873–10878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang Y., Skolnick J. (2004) Scoring function for automated assessment of protein structure template quality. Proteins 57, 702–710 [DOI] [PubMed] [Google Scholar]
- 26. Bafaro E., Antala S., Nguyen T. V., Dzul S. P., Doyon B., Stemmler T. L., Dempski R. E. (2015) The large intracellular loop of hZIP4 is an intrinsically disordered zinc binding domain. Metallomics, 10.1039/C5MT00066A [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. DiMaio F., Leaver-Fay A., Bradley P., Baker D., André I. (2011) Modeling symmetric macromolecular structures in Rosetta3. PLoS One 6, e20450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Richards R., Dempski R. E. (2011) Examining the conformational dynamics of membrane proteins in situ with site-directed fluorescence labeling. J. Vis. Exp. 10.3791/2627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dürr K. L., Tavraz N. N., Dempski R. E., Bamberg E., Friedrich T. (2009) Functional significance of E2 state stabilization by specific α/β-subunit interactions of Na,K- and H,K-ATPase. J. Biol. Chem. 284, 3842–3854 [DOI] [PubMed] [Google Scholar]
- 30. Xu J., Zhang Y. (2010) How significant is a protein structure similarity with TM-score = 0.5? Bioinformatics 26, 889–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang Y., Skolnick J. (2005) TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 33, 2302–2309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hopf T. A., Colwell L. J., Sheridan R., Rost B., Sander C., Marks D. S. (2012) Three-dimensional structures of membrane proteins from genomic sequencing. Cell 149, 1607–1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nugent T., Jones D. T. (2012) Accurate de novo structure prediction of large transmembrane protein domains using fragment-assembly and correlated mutation analysis. Proc. Natl. Acad. Sci. U.S.A. 109, E1540–E1547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wu C. H., Apweiler R., Bairoch A., Natale D. A., Barker W. C., Boeckmann B., Ferro S., Gasteiger E., Huang H., Lopez R., Magrane M., Martin M. J., Mazumder R., O'Donovan C., Redaschi N., Suzek B. (2006) The Universal Protein Resource (UniProt): an expanding universe of protein information. Nucleic Acids Res. 34, D187–D191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lazdunski M., Petitclerc C., Chappelet D., Lazdunski C. (1971) Flip-flop mechanisms in enzymology. A model: the alkaline phosphatase of Escherichia coli. Eur. J. Biochem. 20, 124–139 [DOI] [PubMed] [Google Scholar]
- 36. Sanders D. (1986) Generalized kinetic analysis of ion-driven cotransport systems: II. Random ligand binding as a simple explanation for non-michaelian kinetics. J. Membr. Biol. 90, 67–87 [DOI] [PubMed] [Google Scholar]
- 37. Kozma D., Simon I., Tusnády G. E. (2013) PDBTM: Protein Data Bank of Transmembrane Proteins after 8 years. Nucleic Acids Res. 41, D524–529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pedersen B. P., Kumar H., Waight A. B., Risenmay A. J., Roe-Zurz Z., Chau B. H., Schlessinger A., Bonomi M., Harries W., Sali A., Johri A. K., Stroud R. M. (2013) Crystal structure of a eukaryotic phosphate transporter. Nature 496, 533–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lin W., Chai J., Love J., Fu D. (2010) Selective electrodiffusion of zinc ions in a Zrt-, Irt-like protein, ZIPB. J. Biol. Chem. 285, 39013–39020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bin B. H., Fukada T., Hosaka T., Yamasaki S., Ohashi W., Hojyo S., Miyai T., Nishida K., Yokoyama S., Hirano T. (2011) Biochemical characterization of human ZIP13 protein: a homo-dimerized zinc transporter involved in the spondylocheiro dysplastic Ehlers-Danlos syndrome. J. Biol. Chem. 286, 40255–40265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Madej M. G., Dang S.., Yan N., Kaback H. R. (2013) Evolutionary mix-and-match with MFS transporters. Proc. Natl. Acad. Sci. U.S.A. 110, 5870–5874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Madej M. G., Kaback H. R. (2013) Evolutionary mix-and-match with MFS transporters II. Proc. Natl. Acad. Sci. U.S.A. 110, E4831–E4838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Västermark A., Lunt B., Saier M. (2014) Major facilitator superfamily porters, LacY, FucP and XylE of Escherichia coli appear to have evolved positionally dissimilar catalytic residues without rearrangement of 3-TMS repeat units. J. Mol. Microbiol. Biotechnol. 24, 82–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Västermark A., Saier M. H. (2014) Major Facilitator Superfamily (MFS) evolved without 3-transmembrane segment unit rearrangements. Proc. Natl. Acad. Sci. U.S.A. 111, E1162–1163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wei Y., Fu D. (2006) Binding and transport of metal ions at the dimer interface of the Escherichia coli metal transporter YiiP. J. Biol. Chem. 281, 23492–23502 [DOI] [PubMed] [Google Scholar]
- 46. Franz M. C., Simonin A., Graeter S., Hediger M. A., Kovacs G. (2014) Development of the first fluorescence screening assay for the SLC39A2 zinc transporter. J. Biomol. Screen 19, 909–916 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.