

Background: Salt-inducible kinases (SIKs) are capable of suppressing gluconeogenic gene expression in hepatocytes when they are overexpressed.
Results: However, enhanced gluconeogenic programs are observed only in SIK3-defective hepatocytes.
Conclusion: SIK3 is the major kinase that down-regulates gluconeogenesis.
Significance: The present study proposes that SIK3 could be a new target of diabetic care.
Keywords: AMP-activated kinase (AMPK), cAMP response element-binding protein (CREB), gluconeogenesis, hepatocyte, liver kinase B1 (LKB1), CRTC, SIK
Abstract
Salt-inducible kinases (SIKs), members of the 5′-AMP-activated protein kinase (AMPK) family, are proposed to be important suppressors of gluconeogenic programs in the liver via the phosphorylation-dependent inactivation of the CREB-specific coactivator CRTC2. Although a dramatic phenotype for glucose metabolism has been found in SIK3-KO mice, additional complex phenotypes, dysregulation of bile acids, cholesterol, and fat homeostasis can render it difficult to discuss the hepatic functions of SIK3. The aim of this study was to examine the cell autonomous actions of SIK3 in hepatocytes. To eliminate systemic effects, we prepared primary hepatocytes and screened the small compounds suppressing SIK3 signaling cascades. SIK3-KO primary hepatocytes produced glucose more quickly after treatment with the cAMP agonist forskolin than the WT hepatocytes, which was accompanied by enhanced gluconeogenic gene expression and CRTC2 dephosphorylation. Reporter-based screening identified pterosin B as a SIK3 signaling-specific inhibitor. Pterosin B suppressed SIK3 downstream cascades by up-regulating the phosphorylation levels in the SIK3 C-terminal regulatory domain. When pterosin B promoted glucose production by up-regulating gluconeogenic gene expression in mouse hepatoma AML-12 cells, it decreased the glycogen content and stimulated an association between the glycogen phosphorylase kinase gamma subunit (PHKG2) and SIK3. PHKG2 phosphorylated the peptides with sequences of the C-terminal domain of SIK3. Here we found that the levels of active AMPK were higher both in the SIK3-KO hepatocytes and in pterosin B-treated AML-12 cells than in their controls. These results suggest that SIK3, rather than SIK1, SIK2, or AMPKs, acts as the predominant suppressor in gluconeogenic gene expression in the hepatocytes.
Introduction
Salt-inducible kinase (SIK)2 is a member of the 5′-AMP-activated protein kinase (AMPK)-related kinase family, and the SIK subfamily is composed of three isoforms (1). We isolated SIK1 from the adrenals of rats fed with a high Na+ or K+ diet (2) and identified SIK2 by its sequence similarity (3). SIK3 was characterized by another group in a wide range analysis (4). Although multiple SIK substrates have been identified based on information from the SIK phosphorylation motifs, LX(R/K)(S/T)XpSXXXL (5), only two types of substrates, CRTC (a coactivator for the cAMP response element (CRE)-binding protein (CREB)) and class IIa histone deacetylase (HDAC) (6), have been confirmed by independent research groups. Phosphorylation of these two substrates by SIKs results in a loss of their transcriptional regulatory activities by inducing nuclear export (5, 7).
The other important aspect of SIK is as a feedback regulator in PKA signaling (8). When PKA activates CREB by phosphorylation, it also counters SIK actions by phosphorylating the C-terminal regulatory domain of SIK, which in turn promotes the dephosphorylation rate of CRTC, enhances nuclear accumulation of this coactivator, and increases CREB-dependent gene expression (5, 7, 9). Conversely, when PKA activity wanes, the reactivated SIK terminates CREB activity by inactivating CRTC (5, 9). This is also the case for class IIa-HDAC (6). PKA-dependent inactivation SIK facilitates the activation of class IIa-HDAC. Because class IIa-HDAC is the major suppressor of myocyte enhancer factor 2 (MEF2) (10), the MEF2-dependent transcription is suppressed by PKA and reactivated by SIK (6).
The first physiological relevance of SIKs in vivo was found in the mouse liver as a suppression of gluconeogeneic programs (11). CREB is one of the key transcription factors that up-regulate gluconeogenic gene expression (12) by binding to their gene promoters, such as phosphoenolpyruvate carboxykinase (Pepck), glucose-6-phosphatase catalytic subunit (G6pc), and peroxisome proliferator-activated receptor gamma coactivator 1α (Pgc1a; formal gene name is Ppargc1a). The knockdown of SIK1 protein in the mouse liver by RNAi-adenovirus vectors results in an increase in blood glucose levels, which is accompanied by the dephospho-form of CRTC2 and enhanced levels of gluconenogenic gene expression (11). However, the effects of SIK1 knock-out (KO) on glucose metabolism have not been elucidated (13). SIK2 also may play an important role in glucose metabolism (3, 14–17).
Obvious abnormalities in glucose homeostasis have been found in SIK3-KO mice (18). SIK3-KO mice display a lean phenotype, resistance to a high fat diet, and excessive hypoglycemia. Although a response to insulin and lactate-induced gluconeogenesis in SIK3-KO mice appeared to be normal under fasting conditions, gluconeogenic gene expression was extremely higher in the SIK3-KO liver than in WT mice (18). In addition to the lean phenotype (small amount of energy storage), impaired bile acid, cholesterol, and retinoid homeostasis with skeletal abnormalities in SIK3-KO mice (19) can make it difficult to determine whether enhanced gluconeogenic programs in the SIK3-KO mice liver were the results of a cell autonomous action or systemic effects.
In addition to the issue of SIKs in gluconeogenesis, the role of AMPK in the regulation of CREB/CRTC-mediated gluconeogenic programs remains unclear (20–22). A recent excessive study using mice that simultaneously lost the Ampkα1/α2 and Sik2 genes in the liver and a kinase inhibitor that inhibited all SIKs suggested that a loss of activity of all SIKs resulted in enhanced gluconeogenic programs, whereas the triple loss of AMPKα1/α2 and SIK2 left flawlessly managed gluconeogenic programs (23). Here, using cultured hepatocytes and a small compound, we have tried to discuss the important or indispensable role of SIK3 in the regulation of gluconeogenic programs in the liver.
Experimental Procedures
Reagents and Mice
Forskolin (Fsk), dexamethasone, glucose oxidase, 4-aminoantipyrine, N-ethyl-N-(2-hydroxy-3-sulfopropyl)-3-methylaniline, sodium salt, and horseradish peroxidase were purchased from WAKO Pure Chemicals (Osaka, Japan). HG9-91-01 was obtained from MedChem Express (Monmouth Junction, NJ). Halo tag expression plasmids for phosphorylase kinase gamma 2 (PHKG2) and other kinases and phosphatases were purchased from Promega (Madison, WI).
The anti-AMPK, anti-phospho-AMPK, and anti-phospho antibodies were from Cell Signaling (Boston, MA), the anti-CREB antibody was from GenScript Corp. (Piscataway, NJ), the anti-GAPDH antibody was from WAKO (Tokyo, Japan), and anti-PHKG2 antibody was obtained from Santa Cruz Biotechnology, Inc. (Dallas, TX). Anti-pSIK3 (Thr(P)-163) was created against the keyhole limpet hemocyanin-conjugated peptide (CSNLFTPGQLLK(pT)W) in rabbits and purified with the same peptide. CRTC2 protein was separated on an 8% linear polyacrylamide gel, and other proteins were on 4–20% gradient gels. AML-12 (alpha mouse liver-12) cells from American Type Culture Collection were cultured in DMEM-Ham's F-12 medium supplemented with 10% FBS and an insulin, transferrin, and selenium supplement (Thermo Fisher, Carlsbad, CA).
Information about the KO mice, SIK1-KO, SIK2-KO, and SIK3-KO are described in (13, 15, 18), respectively. SIK1-KO mice were mated with C57BL/6J for six generations. The experimental mouse protocols were approved by the ethics committee of the National Institute of Biomedical Innovation (assigned nos. DS20–77, DS23–37, and DS25–54). The animals were maintained under standard conditions of light (on at 08:00 and off at 20:00) and temperature (23 °C, 60% humidity). The kinase inhibitor (∼90 compounds) libraries were from Enzo Life Science (Farmingdale, NY), and the unannotated compounds (∼2000) in the Molport libraries were purchased from Namiki Shouji (Tokyo, Japan). Flavonoids (∼50) and other natural compounds (∼150) are described in Refs. 24 and 25.
Pterosin B was extracted from Pteridium aquilinum (total 100 kg, wet) after soaking in 0.1% sodium bicarbonate at 70 °C overnight. The ingredients in the P. aquilinum chloroform/hexane (1:4) extract were separated by silica gel and charcoal column chromatography, and pterosin B was crystallized in chloroform by increasing the hexane content. Finally, we got 3 g of pterosin B whose purity was confirmed by nuclear magnetic resonance. Synthetic pterosin B (racemic) was obtained from Intelium Crop. (Tokyo, Japan).
Primary Hepatocytes
Hepatocytes were isolated from mice as described previously (23). Briefly, under isoflurane anesthesia, the mouse livers were perfused with Hanks' balanced salt solution, which contained 0.5 mm EGTA, followed by perfusion with liver digest medium (Thermo Fisher). Isolated hepatocytes were cultured in DMEM supplemented with 10% FBS, 100 nm insulin, and 1 μm dexamethasone (1× hepatocyte medium). Before the treatments, the hepatocytes were incubated with DMEM supplemented with 1% FBS, 10 nm insulin, and 0.1 μm dexamethasone (0.1× hepatocyte medium) for 12 h.
DNA Constructs and Site-directed Mutagenesis
pM-MEF2C (26), pM-CRTC2 (27), GFP-CRTC2 (7), GFP-HDAC5 (26), pTarget-SIK3 (27), and pEBG-SIK3 (27) had been previously constructed. The SIK3 mutants (S493A, T411A, and the double Ala mutant (DA)) were constructed by site-directed mutagenesis using pTarget-hSIK3 plasmids and the following primers: for S493A (5′-CCCTTGGCCGGAGGGCTGCAGATGGAGGAGCCAAC/5′-GTTGGCTCCTCCATCTGCAGCCCTCCGGCCAAGGG), and for T411A (5′-TTGTCAATGAGGAGGCATGCCGTGGGTGTGGCTGACCCA/5′-TGGGTCAGCCACACCCACGGCATGCCTCCTCATTGACAA). The SIK3 DA mutant was constructed by using pTarget-hSIK3 S493A as the template with the primers for T411A.
To prepare an adenovirus vector for SIK3 (WT, DA), the SIK3 cDNA fragments were amplified by PCR with the attB primers. The amplified products were then constructed into pDONR221 vectors by using BP clonase enzyme mix (Thermo Fisher). The resultant cDNAs were finally cloned into pAd/DMV/V5-DEST Gateway vectors using LR clonase enzyme mix (Thermo Fisher).
To screen the SIK3 inhibitory compounds, we constructed the LexA reporter assay system. A DNA fragment containing 3× LexA elements was prepared by annealing the oligonucleotides (5′-GATCTACTGTATATATATACAGTAGAGTACTGTATATATATACAGTACACTACTGTATATATATACAGTA/5′-AATTTACTGTATATATATACAGTAGTGTACTGTATATATATACAGTACTCTACTGTATATATATACAGTA), and the fragment was ligated into the BglII/EcoRI site of the Renilla-Luc internal reporter vector (pRL-TK). To prepare the LEXA-CRTC2 expression vector, a DNA fragment for LEXA DNA-binding domain was amplified by PCR with primers (5′-GCAAAAAGCTAGATCATGAAAGCGTTAACGGCCAGGCAA/5′-ACCATAATGAGAGTCCAGCCAGTCGCCGTTGCGAA) and Escherichia coli genome DNA. An In-Fusion HD cloning kit (Clontech) was used to replace the DNA fragment for the GAL4 DNA-binding domain in the pM-CRTC2 vector with the LEXA fragments in pM-CRTC2. The HEK293 cells were placed into 96-well white-bottomed plates and transfected with the SIK3 expression vectors (WT or its empty vector; 5 ng), DNA-binding domain-linked expression vectors (pM-MEF2C and pMLexA-CRTC2; 5 ng), pTAL-GAL4 (20 ng), and pRL-LexA (20 ng) per well, using Lipofectamine 2000 (Thermo Fisher). To measure the reporter activity, we used the Dual-Luciferase reporter assay system (Promega). The cells were lysed with 10 μl of passive lysis buffer, and all of the lysate was used for the assay.
Quantitative Real Time PCR Analysis and Reporter Assay
The total RNA was extracted using an EZ1 RNA universal tissue kit (Qiagen), and the cDNA was synthesized using a ReverTra Ace qPCR RT Master Mix (TOYOBO, Kyoto, Japan). PCR amplification was performed using an EXPRESS SYBR GreenER (Thermo Fisher). Primers used in this study were Pgc1a (5′-GCGAACCTTAAGTGTGGAAC/5′-CACCACGGTCTTGCAAGAGG), Pepck (5′-AGAACAAGGAGTGGAGACCG/5′-GCTTCATAGACAAGGGGGAC), G6pc (5′-CGCAGCAGGTGTATACTATG/5′-CCCAGAATCCCAACCACAAG), and Tbp (5′-GAGCTCTGGAATTGTACCGC/5′-TGTGCACACCATTTTTCCAG).
Levels of Gluconenogenic mRNA Were Normalized by Tbp mRNA
HEK293 and AML-12 cells were transfected with the SIK3 expression vectors (pTarget hSIK3 WT, T411A, S493A, T411A/S493A (DA), or its empty vector; 50 ng), GAL4 DNA-binding domain-linked expression vectors (pM-MEF2C, pM-CRTC2, or its empty vector; 50 ng), pTAL-GAL4 (150 ng) (8), and the Renilla-Luc internal reporter (pRL-(Int-)TK, 50 ng) using Lipofectamine 2000 reagent (Thermo Fisher). To measure reporter activity, we used the Dual-Luciferase reporter assay system (Promega). The cells were lysed with 100 μl of passive lysis buffer, and 10 μl was used for the assay. The activities of firefly luciferase were normalized by those of Renilla luciferase. The reporter activity measurement was performed as described above. The cells were lysed with 100 μl of passive lysis buffer, and 10 μl was used for the assay. The activities of firefly luciferase were normalized by those of Renilla luciferase.
To construct vectors for miRNA(s) against PHKG2, double-stranded oligonucleotides (oligonucleotide 1: 5′-TGCTGAGAATGTGCATCTCTCGCCGTGTTTTGGCCACTGACTGACACGGCGAGATGCACATTCT/5′-CCTGAGAATGTGCATCTCGCCGTGTCAGTCAGTGGCCAAAACACGGCGAGAGATGCACATTCTC and oligonucleotide 2: 5′-TGCTGTCCAGTAGAGACCTCATGATGGTTTTGGCCACTGACTGACCATCATGATCTCTACTGGA/5′-CCTGTCCAGTAGAGATCATGATGGTCAGTCAGTGGCCAAAACCATCATGAGGTCTCTACTGGAC) were introduced into the pcDNA6.2-GW/EmGFP-miR vector (Life Technologies), and adenoviruses were prepared by the Gateway system. Knockdown of PHKG2 protein in AML-12 cells was achieved by infection/transformation two times with a 2-day interval.
Glucose Production and cAMP Measurement
Primary hepatocytes were seeded on a 24-well plate (1.0 × 106) and incubated in 1× hepatocyte medium for 3 h and then in 0.1× hepatocyte medium for 12 h. The AML-12 cells were plated in 24-well plate (1.0 × 106) and incubated in DMEM/F-12 medium for 72 h (with medium change every 24 h) to increase the cellular lipid droplets. After the medium replacement with 0.1× hepatocyte medium, the cells were further cultured for 12 h. The glucose production assay was performed as described (22). Briefly, the cells were washed with phosphate-buffered saline followed by adding 300 μl of assay medium (DMEM without glucose or glutamate containing 1 mm sodium pyruvate, 10 mm lactate, and 100 nm dexamethasone) containing the compounds (Fsk, etc.) per well. After ∼0–8 h of incubation, the assay medium was harvested and centrifuged at 12,000 rpm for 10 min. Then 20 μl of the supernatant was used for the assay. To measure the glucose levels, 180 μl of reaction buffer (50 mm sodium phosphate, pH 7.0, 1 units/ml glucose oxidase, 0.1 units/ml horse radish peroxidase, 50 μm 10-acetyl-3,7-dihydroxyphenoxazine) was added to each well of a 96-well black plate and incubated at room temperature for 30 min. The fluorescence at 590 nm was measured with excitation at 535 nm.
The AML-12 cells were placed into a 96-well plate at a density of 2.0 × 104. The pGloSensor-22F cAMP Plasmids (Promega) were transfected into the cells using Lipofectamine 2000. The cAMP levels were detected with chemiluminescence using GloSensor cAMP reagent (Promega).
Purification of SIK3 and in Vitro Kinase Activity
The pEBG-hSIK3 WT vector was transfected into the HEK293 cells. After 36 h of transfection, the cells were lysed with IP lysis buffer (50 mm Tris-HCl, pH 8.0, 5 mm EDTA, 5 mm EGTA, 2 mm DTT, 50 mm glycerol 3-phosphate, 50 mm NaF, 1 mm NaVO4, 0.5% Triton X-100, 1 mm phenylmethylsulfonyl fluoride, 10 μg/ml leupeptin, and 14 μg/ml aprotinin) and incubated with glutathione-Sepharose 4B (GE Healthcare) at 4 °C for 15 min. The protein complexes were washed with IP lysis buffer and eluted with 10 mm of glutathione. Aliquots of purified SIK3 were subjected to an in vitro kinase assay and Western blot analyses.
Adenoviruses of hSIK3 WT and DA was transfected to AML-12 cells. After the stimulation with pterosin B and Fsk, the cells were lysed with 1 ml of IP lysis buffer and incubated with protein G-Sepharose (GE Healthcare) containing anti-SIK3 antibody at 4 °C for 1 h. Purified SIK3 was eluted with 100 μl of 3× SDS and detected by Western blot analyses.
The purified SIK3 enzyme was incubated with coumarin-labeled CRTC2 peptides and compounds in 40 μl of reaction buffer (5 mm Tris-HCl, pH 7.4, 1 mm ATP, 1 mm DTT, 5 mm MgCl2). The reactions were performed at 25 °C for 1 h and were stopped by the addition of 40 μl of 3× SDS sample buffer. The phosphorylated peptide was separated by electrophoresis on a 1.5% agarose gel in 50 mm Tris borate buffer (pH 8.5) and visualized by ultraviolet light (28).
The PHKG2 enzyme was also prepared by the same methods as for SIK3 and incubated with coumarin-labeled SIK3 T411 peptide (KKLSMRRHTVGVADP) or S493 peptide (KKPLGRRASDGGANI) in 40 μl of the reaction buffer (5 mm Tris-HCl, pH 6.8, 1 mm ATP, 1 mm DTT, 5 mm MgCl2). The phosphorylated peptide was separated on the agarose gel in 50 mm Tris acetate buffer (pH 3.5).
Glycogen Measurement
AML-12 cells were lysed in 0.1 m sodium citrate buffer (pH 4.2) supplemented with 60 mm NaF, and the supernatants were recovered by centrifugation at 14,000 × g for 5 min. The concentration of glycogen was measured using the EnzyChrom glycogen assay kit (BioAssay Systems, Hayward, CA). Reagents for periodic acid-Schiff stain were obtained from Mutokagaku Co. Ltd. (Tokyo, Japan).
Statistical Analyses
Data from each group were characterized by the means ± S.D. Student's t test was used to assess all experimental data in Microsoft Excel.
Results
Gluconeogenic Program Is Constitutively Up-regulated in SIK3-KO Mice Hepatocytes
In cultured cell systems, SIK1–3 regulates energy metabolism by apparently sharing the same transcriptional modulators, CRTCs (27), and class IIa HDACs (29). However, in vivo, we observed that only SIK3-KO mice had dramatic phenotypes for energy metabolism (18). To confirm the indispensability of SIK3 in glucose metabolism in vivo, all SIKs KO mice with the same genetic background (C57BL/6J) were subjected to a glucose challenge after 4 h of starvation (Fig. 1A). Although the blood glucose levels in both the WT and SIK3-KO mice were significantly elevated in response to the glucose treatment, the maximum level was significantly lower in the SIK3-KO than WT mice. However, no significant difference in glucose clearance between the SIK1-KO or SIK2-KO mice and their WT mice was observed. Next, we examined the gluconeogenic potency in these mice by performing a lactate (a source of gluconeogenesis) challenge under a fed condition in which the gluconeogenic programs had not been run in the WT liver because of insulin actions. The blood glucose levels in the SIK3-KO mice were quickly increased to the levels of the WT mice after lactate treatment, whereas there was no significant response in the glucose level to lactate in WT, SIK1-KO, or SIK2-KO mice (Fig. 1B), suggesting a constitutively activated gluconeogenic ability in the SIK3-KO mice.
FIGURE 1.

Glucose metabolism in SIK1–3 KO mice. A, WT and SIK1–3 KO mice (8-week-old females n = 6) were starved for 4 h (4 h after lights were on) and subjected to a glucose tolerance test (1.5 g/kg); the blood glucose level was measured. Means and S.D. are indicated. **, p < 0.01 compare with 0 time. B, a lactate tolerance test (1.5 g/kg) was performed under the fed condition (just after the light was turned on).
Although the gluconeogenic gene expression has been up-regulated in SIK3-KO mouse liver (18), the systemic effects, e.g. hypoglycemia, made it difficult to discuss the cell autonomous actions in the SIK3-KO mouse liver. To eliminate the systemic effects, we prepared hepatocytes from SIK1–3 KO mice, and gluconeogenic programs were induced by the cAMP agonist Fsk. As shown in Fig. 2A, the SIK3-KO hepatocytes produced glucose more quickly than the WT hepatocytes (at 2 h post-Fsk treatment), despite a lower maximum glucose production (at 8 h). No significant difference in glucose production was observed between SIK1-KO or SIK2-KO and their WT hepatocytes. Gene expression analyses (Fig. 2B, genes related to gluconenogenesis: Pgc1a, Pepck, and G6pc) suggested that SIK3-KO hepatocytes possessed a high gluconeogenic potency even in unstimulated cells with almost fully enhanced gluconeogenic gene expression levels. Briefly, starvation (glucose/glutamate-free) did not alter gluconeogenic gene expression levels in both WT and SIK3-KO mice hepatocytes. The stimulation of WT hepatocytes with Fsk up-regulated the levels of gluconenogenic gene expression up to (Pgc1a and Pepck) or beyond (G6pc) those increased levels in the SIK3-KO hepatocytes. Again, we observed no significant difference in the gluconeogenic gene expression levels between in the SIK1-KO and SIK2-KO and WT mice hepatocytes.
FIGURE 2.

Enhanced gluconenogenic programs in SIK3-KO hepatocytes. A, hepatocytes were prepared from 8-week-old female mice (n = 3 or 4) and cultured in a low serum (1%) medium for 12 h. Then cells were pretreated with a starvation (Starv.) medium (serum- and glucose/glutamate-free) for 1 h. Glucose production was induced by 10 mm lactate, 20 μm Fsk, and 0.1 μm dexamethasone. Means and S.D. are indicated. *, p < 0.05; **, p < 0.01. B, the hepatocytes were preincubated with a low serum (1%) medium for 12 h and incubated in a serum-free medium, a starvation medium alone, or supplemented with 20 μm Fsk for 3 h. Total RNA was extracted for quantitative PCR. (n = 3). C, the same treatment was used for Western blot analyses.
Western blot analyses (Fig. 2C) detected a constitutively dephosphorylated CRTC2 in the SIK3-KO hepatocytes, but not in others. Curiously, Fsk-induced phosphorylation of CREB was not observed in the SIK3-KO hepatocytes. AMPK had already been phosphorylated in unstimulated SIK3-KO hepatocytes, suggesting an energy-depleted environment in SIK3-KO hepatocytes; however, the starved condition might not be one of the major factors for the enhanced gluconeogenic programs in SIK3-KO mice hepatocytes.
Pterosin B Inhibited SIK3 Downstream Cascades
To precisely characterize the SIK3 actions in defined culture systems, we chose the mouse hepatoma AML-12 cells. Despite a successful knockdown of SIK1 and SIK2 protein in AML-12 cells, we could not knock down the SIK3 protein. Moreover, an in vitro screening for SIK3 inhibitors from our chemical library (∼2,500 compounds) resulted in only an identification of nonspecific inhibitors, such as the flavonoid fisetin (24). To modulate the SIK3-specific signal, we performed reporter-based screening using transcription regulators (class IIa-HDAC and CRTC2) that were suppressed by SIKs. Fig. 3A indicates the principles of the reporter system. Because SIK3 inhibited the suppressive action of class IIa-HDAC on the transcription factor MEF2, the firefly luciferase activity under the regulation of the GAL4 fusion MEF2 is up-regulated by SIK3 and down-regulated by the SIK3 inhibitory compounds, such as the nonspecific kinase inhibitor staurosprine (STS) (27) (Fig. 3B). CRTC2 is also inhibited by SIK3; thus, the Renilla luciferase activity directly regulated by LEXA fusion CRTC2 was suppressed by SIK3, and STS up-regulated the LEXA-Renilla luciferase activity. The combination of these two reporters in the same cells produced ∼25 times the S/N ratio, when SIK3 was inhibited by STS.
FIGURE 3.

Pterosin B inhibits SIK3 signaling. A, a principle of reporter-based chemical screening. B, a model experiment of reporter assays using the nonspecific kinase inhibitor STS (10 nm). The fold difference in un-normalized luciferase activities between SIK3 overexpression alone and SIK3 + STS is indicated (n = 3). C, the result of a representative plate containing pterosin B is indicated. Fsk (20 μm) and okadaic acid (OA, 1 μm) are positive controls for SIK3 inhibition. Compounds were classified into kinase inhibitors, uncharacterized compounds, and natural compounds and were treated for 36 h at 10 μm, 10 μg/ml, and 50 μg/ml, respectively. Some compounds showed high ratios caused by errors, such as cell toxicity. D, MEF2 and CRTC2 activity were measured in the same GAL4-based firefly luciferase system in the AML-12 cells. E, structural activity relationship of pterosin B and its derivatives. HEK293 cells were transformed with the MEF2 or CRTC2 reporter together with the SIK3 expression vector. Compounds (300 μm) were treated for 36 h. n = 2–3. Cont, control.
Using these reporters and HEK293 cells, we identified pterosin B, an ingredient in P. aquilinum, as a candidate for SIK3 signaling inhibition (Fig. 3C). The action of pterosin B on MEF2 and CRTC2 were also confirmed in separately assayed firefly luciferase reporter systems in the AML-12 cells (Fig. 3D). The structural-activity relationship assay suggested that only pterosin S failed to inhibit SIK3 signaling (Fig. 3E). Synthetic pterosin B (racemic) also inhibited SIK3 signaling, indicating that pterosin B, not non-pterosin contaminants, inhibited SIK3 signaling.
To visually evaluate the inhibitory actions of pterosin B on SIK3 signaling, GFP fusion HDAC5 (a class IIa-HDAC) and CRTC2 were expressed together with SIK3. Both HDAC5 (Fig. 4A) and CRTC2 (Fig. 4B) were localized in the cytoplasm in the SIK3-overexpressing cells, and pterosin B inhibited this cytoplasmic localization, suggesting that pterosin B was able to inhibit downstream cascades of SIK3 signaling.
FIGURE 4.

Pterosin B inhibits cytoplasmic localization of HDAC5 and CRTC2. GFP fusion HDAC5 (A) and CRTC2 (B) were overexpressed in HEK293 cells with or without SIK3 overexpression. Pterosin B (300 μm) was treated for 36 h. The localization of GFP signals in 100 positive cells was classified into three categories: cytoplasmic (C), nucleus (N), and both (C/N) and shown as %.
Pterosin B Does Not Affect SIK3-KO Hepatocytes
To examine whether pterosin B inhibits SIK3 signaling and mimics the glucose metabolism observed in SIK3-KO hepatocytes (Fig. 2), both WT and SIK3-KO hepatocytes were treated with pterosin B, and glucose production was monitored (Fig. 5A). Pterosin B quickly increased the medium glucose levels in WT hepatocytes, which was accompanied by enhanced expression of gluconeogenic genes (Fig. 5B). However, no further enhancement of glucose production or gluconeogenic gene expression by pterosin B was observed in the SIK3-KO hepatocytes, suggesting that pterosin B-induced gluconeogenesis may be mediated by SIK3. Prolonged treatment with pterosin B was found to decrease glucose levels in the WT hepatocyte medium, and G6pc gene expression in SIK3-KO hepatocytes was down-regulated by pterosin B.
FIGURE 5.
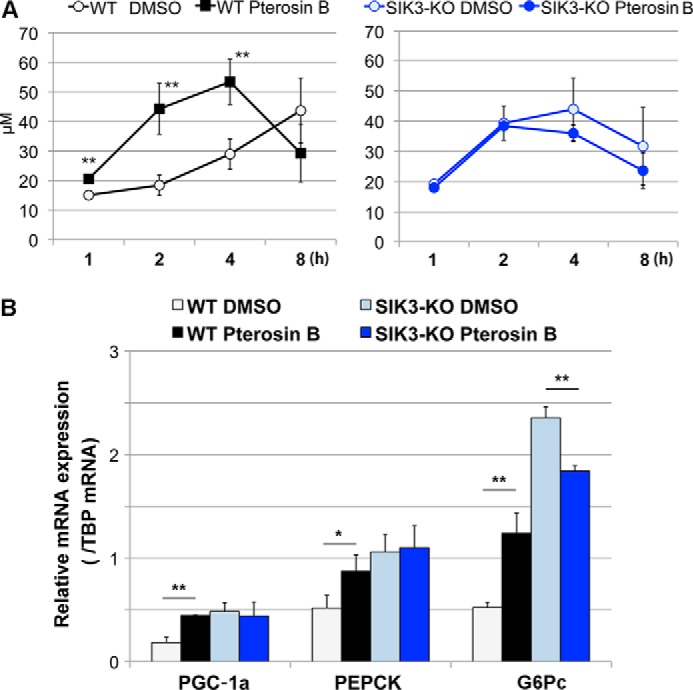
Pterosin B up-regulates glucose production and gluconeogenic gene expression in WT hepatocytes, but not in SIK3-KO hepatocytes. A, hepatocytes were prepared from 12-week-old female WT (left panel) or SIK3-KO (right panel) mice (n = 3) and cultured in a low serum (1%) medium for 12 h. The cells were then pretreated with a starvation medium (serum- and glucose/glutamate-free) for 1 h. Glucose production was induced by 10 mm lactate and 0.1 μm dexamethasone in the presence or absence of pterosin B (300 μm). The means and S.D. are shown. **, p < 0.01. B, the hepatocytes were preincubated with a low serum (1%) medium for 12 h and incubated in a serum-free medium, a starvation medium alone, or supplemented with pterosin B (300 μm). Total RNA was extracted for quantitative PCR. (n = 3).
Pterosin B Represses SIK3 Signaling via the C-terminal Regulatory Region
To test the specificity of pterosin B toward SIKs, SIK1–3 was examined in the same reporter assay in the HEK293 cells (Fig. 6A). The up-regulation of MEF2 and down-regulation of CRTC2 activities by SIK1 or SIK2 were not affected by pterosin B, indicating that pterosin B specifically inhibited SIK3 signaling. On the other hand, pterosin B did not inhibit SIK3 kinase activity up to 1 mm, whereas the strong pan-SIK inhibitor HG9-91-01 (30) completely inhibited SIK3 kinase activity even at 1 μm (Fig. 6B).
FIGURE 6.

Pterosin B represses SIK3 signaling via the C-terminal regulatory region. A, HEK293 cells were transformed with the MEF2 or CRTC2 reporter together with the SIK1–3 expression vectors and treated with pterosin B (300 μm) for 36 h. The fold differences in the reporter activities by the pterosin B treatment are indicated (n = 3, means and S.D.). B, in vitro kinase assay. The GST-SIK3 enzyme was expressed in HEK293 cells, purified with a glutathione resin, and incubated with compounds, the coumarin-labeled CRTC2 peptide, and 1 mm ATP for 1 h. The phosphorylated and nonphosphorylated peptides were separated by electrophoresis. C, HEK293 cells overexpressing GST-SIK3 (WT and T163A mutant) were treated with pterosin B (300 μm) for 36 h, and then the GST-SIK3 were purified and subjected to the in vitro kinase assay and Western blot analysis. D, the upper diagram shows phosphorylation sites in SIK3. HEK293 cells were transformed with reporters together with the SIK3 expression vectors as A. E, the same experiments with SIK3-phoshorylation site mutants were performed.
Because SIK3 was activated by phosphorylation at Thr-163 by the upstream kinase LKB1 (4), we examined the phosphorylation status at this site on the overexpressed SIK3 in HEK293 cells (Fig. 6C). Western blot analyses with anti-Thr(P)-163 revealed that pterosin B did not alter the phosphorylation level at this site. The alternative candidate sites for inhibition of the SIK3 actions were in the C-terminal regulatory domains of Thr-411 and Ser-493 (Fig. 6D) (31). The phosphorylation of these sites suppressed the CRTC inhibitory activity of SIK3. To confirm the importance of the C-terminal domain, we performed reporter assays with various SIK3 mutants. The truncation of the C-terminal domain (kinase domain only) converted SIK3 into a pterosin B-resistant mutant (Fig. 6D). The actions of double mutant SIK3 (DA: T411A and S493A) were also not affected by pterosin B (Fig. 6E), despite a small difference between the MEF2 and CRTC2 systems (Thr-411 may be more critical for CRTC2 regulation than Ser-493).
Indeed, the phosphorylation levels at Thr-411 and Ser-493 were up-regulated in mouse hepatoma AML-12 cells treated with pterosin B, which was also found in Fsk-treated cells (Fig. 7A). Ser-493 may be phosphorylated by PKA (1), and overexpression of PKA in HEK293 cells mimicked the pterosin actions on MEF2 and CRTC2 (Fig. 7B). However, the PKA inhibitor H89 did not block pterosin B-dependent suppression of SIK3 signaling (Fig. 7C). Although Thr-411 is probably phosphorylated by PKA or CaMKs, the overexpression of CaMK I/II or the treatment with the CaMK inhibitor KN62 disproved the involvement of these kinases in the pterosin B-dependent suppression of SIK3 signaling (Fig. 7, B and C). In addition, the cAMP-responsible luciferase system showed that pterosin B reduced the intracellular cAMP levels induced by Fsk (Fig. 7D). These results suggested that unknown kinases, rather than PKA or CaMK I/II, might phosphorylate SIK3 in Thr-411 and Ser-491, which inhibited SIK3 signaling.
FIGURE 7.

PKA or CaMKs is not responsible kinase for the pterosin B-mediated suppression of SIK3 signaling. A, AML-12 cells were transfected with SIK3 (WT and DA mutant) adenoviruses and were treated with pterosin B (300 μm) or Fsk (20 μm). Three hours later, SIK3 protein was purified with anti-SIK3 antibodies and subjected to Western blot analyses. B, HEK293 cells were transformed with the MEF2 or CRTC2 reporter together with the SIK3 and PKA or CaMK I/II expression vector. Luciferase activities were measured after 36 h. n = 3. C, effects of various inhibitors on pterosin B-mediated suppression of SIK3-dependent MEF2C activity. H89 (20 μm, PKA inhibitor), KN62 (10 μm, CaMK inhibitor), rapamycin (1 μm, PI3K/Akt/mTOR signal inhibitor), LY364947 (10 μm, TGF-β receptor inhibitor), BAY 11–7082 (10 μm, IKK inhibitor), SB-203580 (10 μm, p38 inhibitor), SP600125 (10 μm, JNK inhibitor), PD98059 (10 μm, MEK inhibitor), and STO609 (30 μm, CaMKK inhibitor) were used. D, AML-12 cells that had been transformed with the cAMP sensor plasmid GloSensorTM were treated with Fsk (20 μm) together with or without pterosin B (300 μm). The cellular cAMP level was monitored as luciferase activities.
Pterosin B-resistant SIK3 Decreases Glucose Production and CRTC2 Dephosphorylation in AML-12 Cells
To specifically examine the signaling in pterosin B-induced gluconeogenesis and the relevance of the importance of the C-terminal domain of SIK3, we examined glucose production in AML-12 cells after pterosin B treatment. Pterosin B increased glucose production as quickly as Fsk, whereas HG9-91-01 increased it gradually (Fig. 8A). At 8 h post-stimulation, however, the glucose level in pterosin B-treated cells was returned to the basal level. Because the higher potential of pterosin B on glucose production than HG9-91-01 suggested the presence of SIK-independent pathways in pterosin B actions, we examined pterosin B-induced glucose production in the AML-12 cells that had been transfected with SIK3-adenoviruses. Overexpression of SIK3 lowered the pterosin B-induced glucose production, which was more evident in cells that expressed pterosin B-resistant SIK3 DA mutant than in pterosin B-sensitive SIK3 (WT) (Fig. 8B).
FIGURE 8.

Pterosin B-resistant SIK3 suppresses pterosin B-induced gluconeogenic programs. A, AML-12 cells that had been incubated in 1% serum medium for 12 h were further preincubated with the starvation medium (serum- and glucose/glutamate-free) for 1 h. Means and S.D. are indicated (n = 4). *, p < 0.05; **, p < 0.01 (compared with the control group, DMSO). Glucose production was induced by pterosin B (300 μm), Fsk (20 μm), or HG9-91-01 (1 μm) in the presence of 10 mm lactate and 0.1 μm dexamethasone. B, AML-12 cells that had been transfected with the SIK3 (WT and DA mutant) adenoviruses were subjected to glucose production assays (at 3 h). *, p < 0.05 (compared with the control pterosin B group, Vector) C, gluconeogenic mRNA were examined in the AML-12 cells. Cells that were incubated with a serum-free medium for 12 h and treated with compounds (same as A) in the starvation medium for 3 h (n = 3). D, the pterosin B-induced gene expression was analyzed in the SIK3-overexpressing AML-12 cells. E, Western blot analyses of AML-12 cells that were treated as in C. F, the pterosin B treatment was for 3 h. MOI, multiplicity of infection; Ptero.B, pterosin B.
Next, we examined the levels of gluconeogenic gene expression in the pterosin B-treated AML-12 cells. Cells were pretreated with serum-free medium for 1 h and then incubated with pterosin B or other compounds in the absence of glucose for 3 h. Pterosin B up-regulated Pgc1a, Pepck, and G6pc mRNA levels with different efficacies, which was also represented by Fsk and HG9-91-01 with different target specificities (Fig. 8C). Again, we confirmed whether SIK3 overexpression was able to suppress pterosin B-induced gluconeogenic gene expressions. All expressions were suppressed by SIK3 overexpression, and SIK3 DA was more efficient than SIK WT (Fig. 8D).
We then examined the protein status involved in gluconeogenesis. Pterosin B quickly induced the dephosphorylation of CRTC2 in AML-12 cells (Fig. 8E), which was also observed in Fsk- and HG9-91-01-treated cells. CREB phosphorylation at Ser-133 was up-regulated with starvation time in the control (DMSO) cells. However, the treatment with pterosin B or HG9-91-01 inhibited the CREB phosphorylation. An increase in AMPK phosphorylation levels at Thr-172 was further up-regulated in pterosin B-treated cells and down-regulated in Fsk-treated cells. HG9-91-01 apparently showed no effect on the AMPK phosphorylation levels. Similarly to other assays, SIK3 overexpression returned the levels of pterosin B-induced dephosphorylation of CRTC2 and phosphorylation of CREB (Fig. 8F), which was, again, more evidently with SIK3 DA than SIK3 WT. These results suggested that dephosphorylation of CRTC2 by pterosin B might induce gluconeogenic programs, and the inactivation of SIK3 caused by enhanced phosphorylation of its C-terminal domain might be a mechanism of these programs.
Involvement of PHKG2 in Pterosin B-mediated Inactivation of SIK3
Finally, we tried to identify the molecules that mediate pterosin B action on SIK3, which could be a kinase or a phosphatase. Some candidates were overexpressed in the MEF2 and CRTC2 reporter systems in HEK293 cells, and SIK3-dependent activation and repression were evaluated in the presence or absence of pterosin B (Fig. 9A). We previously reported that the phosphatases PP1, PP2A, and calcineurin modulate CRTC2 activity (32). However, these phosphatases modulate reporter activities of either MEF2 or CRTC2. Pyruvate dehydrogenase kinase and pyruvate dehydrogenase phosphatase are key regulators in mitochondria and regulate acetyl-CoA production by modulating pyruvate dehydrogenase activity (33). Pyruvate dehydrogenase activity is inextricably linked with pyruvate carboxylase, producing oxaloacetic acid, which is the initial metabolite of gluconeogenesis. However, pyruvate dehydrogenase kinase(s) and pyruvate dehydrogenase phosphatase(s) did not modulate SIK3-dependent MEF2 or CRTC2 activities.
FIGURE 9.

PHKG2 inactivates SIK3 in response to pterosin B. A, a reporter assay was performed in HEK293 cells. Some kinases and phosphatases were overexpressed together with GAL4-MEF2 or GAL4-CRTC2 reporters (see Fig. 2B). The bars indicate fold activation (MEF2) or repression (CRTC2) by SIK3 overexpression (n = 3). In SIK3 without transcriptional regulatory activity, the fold change (activation or repression) approaches 1 (no change). Pterosin B was added at 300 μm. B, AML-12 cells that had been cultured for 72 h with daily medium change were incubated with various concentrations of pterosin B (0–300 μm) for 3 h. The glycogen concentrations were then measured (left panel). Intracellular accumulation of glycogen was observed after periodic acid-Schiff staining (right panel). C, the GST fusion PHKG2 enzyme was overexpressed in COS-7 cells and purified using a glutathione column. Fluoro-peptides corresponding to SIK3 Thr-411 and Ser-493 were incubated with GST-PHKG2 in the presence of ATP. Phosphorylated peptides were separated by electrophoresis on agarose gel. D, GST-SIK3 were overexpressed in AML-12 cells in the presence of Halo-tagged PHKG1/2 and pulldown by glutathione-Sepharose after 3 h of pretreatment with pterosin B (300 μm). PHKG1/2 and phospho-SIK3 (pS) were detected by anti-Halo tag antibody and anti-pS493, respectively. E, GST-SIK3 WT and T411A/S493A mutant (DA) were used. F, GST SIK1–3 were used. G, PHKG2 protein was knocked down in AML-12 cells by transformation with miRNA plasmid vectors. H, the same sequences of miRNA were transferred into an adenovirus vector and knocked down PHKG2 protein in AML-12 cells to monitor pterosin B-induced gluconeogenic gene expression (300 μm, 2 h). n = 3. **, p < 0.01 (compared with the control group, LacZ-pterosin B). I, hypothetical model of SIK3 signaling.
When gluconeogenesis is activated in hepatocytes, glycogenolysis could also be activated. The key molecules regulating glycogenolysis are phosphorylases and are activated by phosphorylase kinase catalytic gamma 2 (PHKG2). Indeed, PHKG2, but not the muscle type PHKG1, inhibited the SIK3-dependent regulation of both MEF2 and CRTC2, which is accelerated by pterosin B (when SIK3 is inhibited, the fold change approaches 1). In addition, 3 h of treatment with pterosin B decreased the glycogen content in AML-12 cells in a dose-dependent manner (Fig. 9B), suggesting that pterosin B stimulates glycogenolysis.
An in vitro kinase assay of PHKG2 and SIK3 peptide corresponding to the regions of Thr-411 and Ser-493 indicates that PHKG2 can phosphorylate SIK3 (Fig. 9C). GST pulldown in AML-12 cells suggests that PHKG2 binds to SIK3 in response to pterosin B (Fig. 9D) in a Thr-411 or Ser-493 phosphorylation-independent manner (Fig. 9E). The association with PHKG2 was only observed when SIK3 was used as bait (Fig. 9F).
On the other hand, the overexpression of PHKG2 in AML-12 cells had less of an effect on the SIK3 cascades downstream, probably because of the limited amount of SIK3 protein. To show the indispensable role of PHKG2 in pterosin B-mediated SIK3 inactivation, knockdown experiments were performed in AML-12 cells. Two different miRNA vectors for mouse PHKG2 decreased the protein levels and lowered the phosphorylation levels of SIK3 Thr(P)-411 and Ser(P)-493 (Fig. 9G), in contrast to CRTC2 phosphorylation levels. Pterosin B-induced gluconeogenic gene expression was also suppressed by PHKG2 knockdown (Fig. 9H), suggesting that PHKG2 is a new regulator of SIK3 as elicited by pterosin B.
Discussion
The first clue about the involvement of SIK in the regulation of gluconeogenesis in the liver was based on the fact that SIK1 suppressed the CREB activity via inactivation of CRTC2 (11). During the proof of this suppression, the AMPK activator 5-aminoimidazole-4-carboxamide-1-s-d-ribofuranoside (AICAR)also represented the similar CRTC2 inactivation activity (11), which might deceive us into discussing the role of AMPK in gluconeogenic programs.
AMPK generally acts as energy-saving factors, whereas gluconeogenesis consumes energy (21, 34, 35). In this context, AMPK-mediated suppression of gluconeogenic programs is apparently reasonable. However, overexpression of AMPK in HEK293 cells fails to suppress the CRTC2 transactivating activity despite its ability as the CRTC2 kinase in vitro (27). If a kinase directly phosphorylated their putative substrate, the kinase would never choose a cell type, hepatocytes, or model cells (HEK293), and regulate the cellular downstream cascades of the substrate in the same manner, which may deny that AMPK is the CRTC2 kinase in hepatocytes. However, it does not deny the contribution of AMPK to gluconeogenesis in hepatocytes (36), although we have to mention here that other AMPK family kinases, MARKs (37, 38), may be capable of phosphorylating CRTC in cells (32, 39).
Other aspects about the upstream regulator of AMPK and SIK confused the discussion. LKB1 commonly phosphorylates AMPK and SIK and activates them (4). The loss of LKB1 in the liver results in hyperglycemia caused by enhanced gluconeogenesis (20, 40). The AMPK-CRTC2 cascade was first proposed to be the responsible cascade for gluconeogenic programs caused by LKB1 deficiency (20). However, some reports have provided negative evidence about this issue (22, 23). The liver-specific double knock-out mice with defective genes for Ampkα1/α2 have been found to have less dysregulation in glucose metabolism in vivo (22). The basal level of CRTC2 phosphorylation in the AMPKα1/α2-deficient hepatocytes is as high as that in WT hepatocytes. A recent extensive study using triple knock-out mice (AMPKα1/α2 and SIK2) showed an intact gluconeogenic regulation in the liver of this mouse (23). In contrast, the pan-SIK inhibitor HG9-91-01 that failed to inhibit AMPK induced the gluconeogenic programs followed by glucose production. Because the HG9-91-01-induced gluconeogenic programs are suppressed by overexpression of any drug-resistant isoforms of SIK1–3 (23), the presence of at least one SIK isoform may be sufficient to manage the gluconeogenic programs in hepatocytes.
Does AMPK really phosphorylate CRTC2 in hepatocytes? The phosphorylation level of CRTC2 in the cAMP-treated hepatocytes is increased by AMPK-activating compounds, metformin (41), and AICAR (42), which is not observed in AMPKα1/α2-deficient hepatocytes, suggesting unknown mechanisms by which AMPK can regulate CRTC phosphorylation (22, 23). It could be speculated that AMPK may inhibit the dephosphorylation steps of CRTC rather than the phosphorylation steps.
The next question that arose was whether all SIK isoforms equally suppressed gluconeogenic programs. SIK3-KO mice liver expressed gluconeogenic genes at a high level (18) and constitutively activated (the levels were comparable to those stimulated by Fsk) gluconeogenic programs in the SIK3-KO hepatocytes have been found in the present study, which may mimic LKB1-KO hepatocytes (20), but not AMPKα1/α2- and SIK2-triple KO hepatocytes (23). These results suggest that SIK3 may be the major regulator of gluconeogenic programs under the control of LKB1 in hepatocytes.
A number of reports succeeded to performing SIK3 knockdowns, and some showed impaired cholesterol transport in SIK3 knockdown cells (43), which explained the phenotype of cholesterol accumulation in the SIK3-KO liver (18). However, some phenotypes caused by SIK3 knockdown were observed in HeLa cells in which SIK3 could not possess kinase activity because of LKB1 deficiency (27). Although it was difficult to conclude whether the results in knockdown experiments were from on or off target effects, it was also true that we failed to knock down SIK3 even in HEK293 cells. Thus, we tried to identify SIK3-specific inhibitors.
Pterosin B suppressed SIK3 downstream cascades, including CRTC2 phosphorylation, despite not being capable of inhibiting SIK3 kinase activity. Pterosin B may modulate an unknown kinase or phosphatase signals and increase the self-inhibitory function of the C-terminal regulatory domain of SIK3. This mechanism was specific to SIK3, not SIK1 or SIK2; thus, we thought that the loss of SIK3 or its signaling resulted in an enhancement of gluconeogenic programs in hepatocytes via CRTC2 dephosphorylation. However, it was also true that pterosin B and Fsk induced higher levels of glucose production than that by the pan-SIK inhibitor HG9-91-01 (23). Because glucagon and Fsk increased cellular cAMP levels and activated PKA followed by CREB phosphorylation, the synergistic actions between dephosho-CRTC2 and phospho-CREB might result in higher potency of the gluconeogenic programs than the solo action of dephosho-CRTC2 produced by HG9-91-01 (23). On the other hand, pterosin B suppressed CREB phosphorylation levels in common with HG9-91-01. Pterosin B may activate an unidentified cascade up-regulating gluconenogenic programs in which SIK3 was fatefully inactivated as by PKA signaling.
Re-evaluation of metabolic regulators whose activities were modulated along with gluconeogenesis suggests that PHKG2 is a candidate for SIK3-inactivating kinase. PHKG2 belongs to the CaMK family (44) and shares phosphorylation motifs with CaMKs and PKA (45). A major role of PHKG2 is the initiation of glycogen breakdown in response to glucagon-cAMP-PKA or Ca2+ signaling by phosphorylating phosphorylase (46). Mutations in the PHKG2 gene cause type IXc glycogen storage disease (liver glycogenosis), which is characterized by hypoglycemia, lactic acidosis, and cirrhosis (47). Although glucagon response was found in type IX glycogen storage disease patients (48), some patients with PHKG2 gene mutations were reported to not respond or to only weakly respond to glucagon (49). In addition, type Ia glycogen storage disease, G6Pc deficiency (50), and PEPCK deficiency (liver isoform PCK1) (51) commonly cause lactic acidosis, suggesting the dysregulation of gluconeogenesis in PHKG2-mutated patients. A decrease in the glycogen content in pterosin B-treated AML-12 cells, capability of SIK3 phosphorylation by PHKG2 in vitro, specific association of PHKG2 with SIK3, and weakened action of pterosin B in PHKG2 knocked down AML-12 suggest that pterosin B inactivates SIK3 via the activation of PHKG2.
Pterosin B may activate both glycogenolysis and gluconeogenesis (Fig. 9I) and is therefore capable of quickly producing glucose (∼4 h post-treatment). Once the glycogen is depleted (SIK3-KO hepatocytes are also under an energy deficiency state), pterosin B may be unable to promote glucose production, which decreases the rate of glucose production later on (8 h). In addition, unexplained weaker expression of G6pc by pterosin B (compared with by Fsk or HG9-91-01) may result in insufficient glycogenolysis and gluconeogenesis, which is also observed in SIK3-KO hepatocytes, suggesting that secondary effects, such as modulation of AMPK signaling via glycogen metabolism (52), may affect G6pc expression, probably via a CRTC2-independent mechanism (e.g. through forkhead transcription factor FKHR) (53).
Pterosin A, a derivative of pterosin B, was reported to lower, not increase, blood glucose levels in diabetic animals (stroptozotocine-treated, high fat-fed, and db/db mice) (54). Pterosin A simultaneously activated conflicting signals in energy metabolism, AKT, and AMPK in the skeletal muscle and livers. In the liver, pterosin A increased glycogen content and decreased cAMP/dexamethasone-induced Pepck gene expression, which was a mechanism of pterosin A-mediated blood lowering effects. However, pterosin B lowered the glycogen content and initiated gluconeogenic programs, followed by glucose production. A number of assay conditions were different between ours and the pterosin A experiments (e.g. the absence or presence of serum and pterosin alone or with gluconenogenic inducers); we noted here that the most critical difference might be from acute or chronic effects. It was true that pterosin B resulted in a lowered glucose production at later phase (after 8 h), and pterosin A experiments for mRNA analyses were performed after 24 h, suggesting that pterosin(s) may act as bilateral effectors in glucose metabolism. A reduction in glucose production (content in the culture medium) at the late phase was also observed in SIK3-KO mouse hepatocytes, and hypoglycemia was also the representative phenotype of these mice. These results suggested that SIK3-KO hepatocytes and pterosin B-treated AML-12 cells may share similar mechanisms that may be linked to glycogen breakdown. Further studies on SIK3-mediated regulation of gluconeogenesis, glycogenolysis, and an uncharacterized crosstalk between AMPK and SIK3 signaling are needed.
Author Contributions
Y. I., M. S., H. F., Y. Y., A. K., M. K., and D. T. performed the experiments. J. D., M. O., N. T., and N. K. analyzed the data. H. T. designed the experiments and wrote the manuscript.
Acknowledgments
We thank Prof. Alejandro M. Bertorello (Karolinska Institutet) for the gift of SIK1-KO mice and for his dedication to research, including SIK.
This study was supported by Grants-in-Aid for Scientific Research from the A-STEP (Adaptable & Seamless Technology Transfer Program through Target-driven R&D); a grant from the Ministry of Health, Labor, and Welfare (2013–2017 and 2014–2016); funds from Scientific Research on Innovative Areas, a MEXT Grant-in-Aid Project 2012–2013; and a grant from the Uehara Memorial Foundation. The authors declare that they have no conflicts of interest with the contents of this article.
- SIK
- salt-inducible kinase
- AMPK
- 5′-AMP-activated protein kinase
- CREB
- cAMP-responsive element binding protein
- CRTC
- CREB-regulated transcription coactivator
- HDAC
- histone deacetylase
- MEF2
- myocyte enhancer factor 2
- Pepck
- phosphoenolpyruvate carboxykinase
- G6pc
- glucose-6-phosphatase catalytic subunit
- Pgc1a
- peroxisome proliferator-activated receptor gamma coactivator 1α
- AML-12
- alpha mouse liver-12
- Fsk
- forskolin
- STS
- staurosprine
- AICAR
- 5-aminoimidazole-4-carboxamide-1-s-d-ribofuranoside
- LKB1
- liver kinase B1
- CaMK
- Ca2+/calmodulin-dependent protein kinase
- PHKG
- phosphorylase kinase gamma.
References
- 1. Takemori H., Okamoto M. (2008) Regulation of CREB-mediated gene expression by salt inducible kinase. J. Steroid Biochem. Mol. Biol. 108, 287–291 [DOI] [PubMed] [Google Scholar]
- 2. Wang Z., Takemori H., Halder S. K., Nonaka Y., Okamoto M. (1999) Cloning of a novel kinase (SIK) of the SNF1/AMPK family from high salt diet-treated rat adrenal. FEBS Lett. 453, 135–139 [DOI] [PubMed] [Google Scholar]
- 3. Horike N., Takemori H., Katoh Y., Doi J., Min L., Asano T., Sun X. J., Yamamoto H., Kasayama S., Muraoka M., Nonaka Y., Okamoto M. (2003) Adipose-specific expression, phosphorylation of Ser794 in insulin receptor substrate-1, and activation in diabetic animals of salt-inducible kinase-2. J. Biol. Chem. 278, 18440–18447 [DOI] [PubMed] [Google Scholar]
- 4. Lizcano J. M., Göransson O., Toth R., Deak M., Morrice N. A., Boudeau J., Hawley S. A., Udd L., Mäkelä T. P., Hardie D. G., Alessi D. R. (2004) LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J. 23, 833–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Screaton R. A., Conkright M. D., Katoh Y., Best J. L., Canettieri G., Jeffries S., Guzman E., Niessen S., Yates J. R., 3rd, Takemori H., Okamoto M., Montminy M. (2004) The CREB coactivator TORC2 functions as a calcium- and cAMP-sensitive coincidence detector. Cell 119, 61–74 [DOI] [PubMed] [Google Scholar]
- 6. Berdeaux R., Goebel N., Banaszynski L., Takemori H., Wandless T., Shelton G. D., Montminy M. (2007) SIK1 is a class II HDAC kinase that promotes survival of skeletal myocytes. Nat. Med. 13, 597–603 [DOI] [PubMed] [Google Scholar]
- 7. Katoh Y., Takemori H., Min L., Muraoka M., Doi J., Horike N., Okamoto M. (2004) Salt-inducible kinase-1 represses cAMP response element-binding protein activity both in the nucleus and in the cytoplasm. Eur. J. Biochem. 271, 4307–4319 [DOI] [PubMed] [Google Scholar]
- 8. Doi J., Takemori H., Lin X.-Z., Horike N., Katoh Y., Okamoto M. (2002) Salt-inducible kinase represses PKA-mediated activation of human cholesterol side chain cleavage cytochrome promoter through the CREB basic leucine zipper domain. J. Biol. Chem. 277, 15629–15637 [DOI] [PubMed] [Google Scholar]
- 9. Takemori H., Katoh Y., Horike N., Doi J., Okamoto M. (2002) ACTH-induced nucleocytoplasmic translocation of salt-inducible kinase. Implication in the protein kinase A-activated gene transcription in mouse adrenocortical tumor cells. J. Biol. Chem. 277, 42334–42343 [DOI] [PubMed] [Google Scholar]
- 10. McKinsey T. A., Zhang C. L., Lu J., Olson E. N. (2000) Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature 408, 106–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Koo S. H., Flechner L., Qi L., Zhang X., Screaton R. A., Jeffries S., Hedrick S., Xu W., Boussouar F., Brindle P., Takemori H., Montminy M. (2005) The CREB coactivator TORC2 is a key regulator of fasting glucose metabolism. Nature 437, 1109–1111 [DOI] [PubMed] [Google Scholar]
- 12. Imai E., Miner J. N., Mitchell J. A., Yamamoto K. R., Granner D. K. (1993) Glucocorticoid receptor-cAMP response element-binding protein interaction and the response of the phosphoenolpyruvate carboxykinase gene to glucocorticoids. J. Biol. Chem. 268, 5353–5356 [PubMed] [Google Scholar]
- 13. Bertorello A. M., Pires N. M., Igreja B., Pinho M. J., Vorkapic E., Wagsater D., Wikstrom J., Behrendt M., Hamsten A., Eriksson P., Soares-da-Silva P., Brion L. (2015) Increased arterial blood pressure and vascular remodeling in mice lacking salt-inducible kinase 1 (SIK1). Circ. Res. 116, 642–652 [DOI] [PubMed] [Google Scholar]
- 14. Muraoka M., Fukushima A., Viengchareun S., Lombès M., Kishi F., Miyauchi A., Kanematsu M., Doi J., Kajimura J., Nakai R., Uebi T., Okamoto M., Takemori H. (2009) Involvement of SIK2/TORC2 signaling cascade in the regulation of insulin-induced PGC-1alpha and UCP-1 gene expression in brown adipocytes. Am. J. Physiol. Endocrinol. Metab. 296, E1430–E1439 [DOI] [PubMed] [Google Scholar]
- 15. Horike N., Kumagai A., Shimono Y., Onishi T., Itoh Y., Sasaki T., Kitagawa K., Hatano O., Takagi H., Susumu T., Teraoka H., Kusano K., Nagaoka Y., Kawahara H., Takemori H. (2010) Downregulation of SIK2 expression promotes the melanogenic program in mice. Pigment Cell Melanoma Res. 23, 809–819 [DOI] [PubMed] [Google Scholar]
- 16. Park J., Yoon Y. S., Han H. S., Kim Y. H., Ogawa Y., Park K. G., Lee C. H., Kim S. T., Koo S. H. (2014) SIK2 is critical in the regulation of lipid homeostasis and adipogenesis in vivo. Diabetes 63, 3659–3673 [DOI] [PubMed] [Google Scholar]
- 17. Sakamaki J., Fu A., Reeks C., Baird S., Depatie C., Al Azzabi M., Bardeesy N., Gingras A. C., Yee S. P., Screaton R. A. (2014) Role of the SIK2-p35-PJA2 complex in pancreatic beta-cell functional compensation. Nat. Cell Biol. 16, 234–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Uebi T., Itoh Y., Hatano O., Kumagai A., Sanosaka M., Sasaki T., Sasagawa S., Doi J., Tatsumi K., Mitamura K., Morii E., Aozasa K., Kawamura T., Okumura M., Nakae J., Takikawa H., Fukusato T., Koura M., Nish M., Hamsten A., Silveira A., Bertorello A. M., Kitagawa K., Nagaoka Y., Kawahara H., Tomonaga T., Naka T., Ikegawa S., Tsumaki N., Matsuda J., Takemori H. (2012) Involvement of SIK3 in glucose and lipid homeostasis in mice. PLoS One 7, e37803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sasagawa S., Takemori H., Uebi T., Ikegami D., Hiramatsu K., Ikegawa S., Yoshikawa H., Tsumaki N. (2012) SIK3 is essential for chondrocyte hypertrophy during skeletal development in mice. Development 139, 1153–1163 [DOI] [PubMed] [Google Scholar]
- 20. Shaw R. J., Lamia K. A., Vasquez D., Koo S. H., Bardeesy N., Depinho R. A., Montminy M., Cantley L. C. (2005) The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science 310, 1642–1646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Viollet B., Athea Y., Mounier R., Guigas B., Zarrinpashneh E., Horman S., Lantier L., Hebrard S., Devin-Leclerc J., Beauloye C., Foretz M., Andreelli F., Ventura-Clapier R., Bertrand L. (2009) AMPK: Lessons from transgenic and knockout animals. Front. Biosci. (Landmark Ed) 14, 19–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Foretz M., Hébrard S., Leclerc J., Zarrinpashneh E., Soty M., Mithieux G., Sakamoto K., Andreelli F., Viollet B. (2010) Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J. Clin. Invest. 120, 2355–2369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Patel K., Foretz M., Marion A., Campbell D. G., Gourlay R., Boudaba N., Tournier E., Titchenell P., Peggie M., Deak M., Wan M., Kaestner K. H., Göransson O., Viollet B., Gray N. S., Birnbaum M. J., Sutherland C., Sakamoto K. (2014) The LKB1-salt-inducible kinase pathway functions as a key gluconeogenic suppressor in the liver. Nat. Commun. 5, 4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kumagai A., Horike N., Satoh Y., Uebi T., Sasaki T., Itoh Y., Hirata Y., Uchio-Yamada K., Kitagawa K., Uesato S., Kawahara H., Takemori H., Nagaoka Y. (2011) A potent inhibitor of SIK2, 3,3′,7-trihydroxy-4′-methoxyflavon (4′-O-methylfisetin), promotes melanogenesis in B16F10 melanoma cells. PLoS One 6, e26148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ratnoglik S. L., Aoki C., Sudarmono P., Komoto M., Deng L., Shoji I., Fuchino H., Kawahara N., Hotta H. (2014) Antiviral activity of extracts from Morinda citrifolia leaves and chlorophyll catabolites, pheophorbide a and pyropheophorbide a, against hepatitis C virus. Microbiol. Immunol. 58, 188–194 [DOI] [PubMed] [Google Scholar]
- 26. Takemori H., Katoh Hashimoto Y., Nakae J., Olson E. N., Okamoto M. (2009) Inactivation of HDAC5 by SIK1 in AICAR-treated C2C12 myoblasts. Endocr. J. 56, 121–130 [DOI] [PubMed] [Google Scholar]
- 27. Katoh Y., Takemori H., Lin X. Z., Tamura M., Muraoka M., Satoh T., Tsuchiya Y., Min L., Doi J., Miyauchi A., Witters L. A., Nakamura H., Okamoto M. (2006) Silencing the constitutive active transcription factor CREB by the LKB1-SIK signaling cascade. FEBS J. 273, 2730–2748 [DOI] [PubMed] [Google Scholar]
- 28. Hashimoto Y. K., Satoh T., Okamoto M., Takemori H. (2008) Importance of autophosphorylation at Ser186 in the A-loop of salt inducible kinase 1 for its sustained kinase activity. J. Cell Biochem. 104, 1724–1739 [DOI] [PubMed] [Google Scholar]
- 29. Walkinshaw D. R., Weist R., Kim G. W., You L., Xiao L., Nie J., Li C. S., Zhao S., Xu M., Yang X. J. (2013) The tumor suppressor kinase LKB1 activates the downstream kinases SIK2 and SIK3 to stimulate nuclear export of class IIa histone deacetylases. J. Biol. Chem. 288, 9345–9362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Clark K., MacKenzie K. F., Petkevicius K., Kristariyanto Y., Zhang J., Choi H. G., Peggie M., Plater L., Pedrioli P. G., McIver E., Gray N. S., Arthur J. S., Cohen P. (2012) Phosphorylation of CRTC3 by the salt-inducible kinases controls the interconversion of classically activated and regulatory macrophages. Proc. Natl. Acad. Sci. U.S.A. 109, 16986–16991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sasaki T., Takemori H., Yagita Y., Terasaki Y., Uebi T., Horike N., Takagi H., Susumu T., Teraoka H., Kusano K., Hatano O., Oyama N., Sugiyama Y., Sakoda S., Kitagawa K. (2011) SIK2 is a key regulator for neuronal survival after ischemia via TORC1-CREB. Neuron 69, 106–119 [DOI] [PubMed] [Google Scholar]
- 32. Uebi T., Tamura M., Horike N., Hashimoto Y. K., Takemori H. (2010) Phosphorylation of the CREB-specific coactivator TORC2 at Ser(307) regulates its intracellular localization in COS-7 cells and in the mouse liver. Am. J. Physiol. Endocrinol. Metab. 299, E413–E425 [DOI] [PubMed] [Google Scholar]
- 33. Zhang S., Hulver M. W., McMillan R. P., Cline M. A., Gilbert E. R. (2014) The pivotal role of pyruvate dehydrogenase kinases in metabolic flexibility. Nutr. Metab. (Lond.) 11, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hardie D. G. (2014) AMPK-sensing energy while talking to other signaling pathways. Cell Metab. 20, 939–952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gan R. Y., Li H. B. (2014) Recent progress on liver kinase B1 (LKB1): expression, regulation, downstream signaling and cancer suppressive function. Int. J. Mol. Sci. 15, 16698–16718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Guigas B., Bertrand L., Taleux N., Foretz M., Wiernsperger N., Vertommen D., Andreelli F., Viollet B., Hue L. (2006) 5-Aminoimidazole-4-carboxamide-1-beta-d-ribofuranoside and metformin inhibit hepatic glucose phosphorylation by an AMP-activated protein kinase-independent effect on glucokinase translocation. Diabetes 55, 865–874 [DOI] [PubMed] [Google Scholar]
- 37. Hurov J. B., Huang M., White L. S., Lennerz J., Choi C. S., Cho Y. R., Kim H. J., Prior J. L., Piwnica-Worms D., Cantley L. C., Kim J. K., Shulman G. I., Piwnica-Worms H. (2007) Loss of the Par-1b/MARK2 polarity kinase leads to increased metabolic rate, decreased adiposity, and insulin hypersensitivity in vivo. Proc. Natl. Acad. Sci. U.S.A. 104, 5680–5685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sun C., Tian L., Nie J., Zhang H., Han X., Shi Y. (2012) Inactivation of MARK4, an AMP-activated protein kinase (AMPK)-related kinase, leads to insulin hypersensitivity and resistance to diet-induced obesity. J. Biol. Chem. 287, 38305–38315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jansson D., Ng A. C., Fu A., Depatie C., Al Azzabi M., Screaton R. A. (2008) Glucose controls CREB activity in islet cells via regulated phosphorylation of TORC2. Proc. Natl. Acad. Sci. U.S.A. 105, 10161–10166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Miller R. A., Chu Q., Le Lay J., Scherer P. E., Ahima R. S., Kaestner K. H., Foretz M., Viollet B., Birnbaum M. J. (2011) Adiponectin suppresses gluconeogenic gene expression in mouse hepatocytes independent of LKB1-AMPK signaling. J. Clin. Invest. 121, 2518–2528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhou G., Myers R., Li Y., Chen Y., Shen X., Fenyk-Melody J., Wu M., Ventre J., Doebber T., Fujii N., Musi N., Hirshman M. F., Goodyear L. J., Moller D. E. (2001) Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Invest. 108, 1167–1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sullivan J. E., Brocklehurst K. J., Marley A. E., Carey F., Carling D., Beri R. K. (1994) Inhibition of lipolysis and lipogenesis in isolated rat adipocytes with AICAR, a cell-permeable activator of AMP-activated protein kinase. FEBS Lett. 353, 33–36 [DOI] [PubMed] [Google Scholar]
- 43. Blattmann P., Schuberth C., Pepperkok R., Runz H. (2013) RNAi-based functional profiling of loci from blood lipid genome-wide association studies identifies genes with cholesterol-regulatory function. PLoS Genet. 9, e1003338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hanks S. K. (1987) Homology probing: identification of cDNA clones encoding members of the protein-serine kinase family. Proc. Natl. Acad. Sci. U.S.A. 84, 388–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chan K. F., Hurst M. O., Graves D. J. (1982) Phosphorylase kinase specificity. A comparative study with cAMP-dependent protein kinase on synthetic peptides and peptide analogs of glycogen synthase and phosphorylase. J. Biol. Chem. 257, 3655–3659 [PubMed] [Google Scholar]
- 46. Chrisman T. D., Exton J. H. (1980) Activation of endogenous phosphorylase kinase in liver glycogen pellet by cAMP-dependent protein kinase. J. Biol. Chem. 255, 3270–3273 [PubMed] [Google Scholar]
- 47. Maichele A. J., Burwinkel B., Maire I., Søvik O., Kilimann M. W. (1996) Mutations in the testis/liver isoform of the phosphorylase kinase gamma subunit (PHKG2) cause autosomal liver glycogenosis in the gsd rat and in humans. Nat. Genet. 14, 337–340 [DOI] [PubMed] [Google Scholar]
- 48. Dunger D. B., Leonard J. V. (1982) Value of the glucagon test in screening for hepatic glycogen storage disease. Arch. Dis. Child. 57, 384–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Burwinkel B., Rootwelt T., Kvittingen E. A., Chakraborty P. K., Kilimann M. W. (2003) Severe phenotype of phosphorylase kinase-deficient liver glycogenosis with mutations in the PHKG2 gene. Pediatr. Res. 54, 834–839 [DOI] [PubMed] [Google Scholar]
- 50. Oei T. L. (1962) Hexose monophosphate, pyruvate and lactate in the peripheral blood in glycogen-storage disease type I. Clin Chim. Acta 7, 193–198 [DOI] [PubMed] [Google Scholar]
- 51. Haworth J. C., Robinson B. H., Perry T. L. (1981) Lactic acidosis due to pyruvate carboxylase deficiency. J. Inherit. Metab. Dis. 4, 57–58 [DOI] [PubMed] [Google Scholar]
- 52. Milan D., Jeon J. T., Looft C., Amarger V., Robic A., Thelander M., Rogel-Gaillard C., Paul S., Iannuccelli N., Rask L., Ronne H., Lundström K., Reinsch N., Gellin J., Kalm E., Roy P. L., Chardon P., Andersson L. (2000) A mutation in PRKAG3 associated with excess glycogen content in pig skeletal muscle. Science 288, 1248–1251 [DOI] [PubMed] [Google Scholar]
- 53. Ota S., Horigome K., Ishii T., Nakai M., Hayashi K., Kawamura T., Kishino A., Taiji M., Kimura T. (2009) Metformin suppresses glucose-6-phosphatase expression by a complex I inhibition and AMPK activation-independent mechanism. Biochem. Biophys. Res. Commun. 388, 311–316 [DOI] [PubMed] [Google Scholar]
- 54. Hsu F. L., Huang C. F., Chen Y. W., Yen Y. P., Wu C. T., Uang B. J., Yang R. S., Liu S. H. (2013) Antidiabetic effects of pterosin A, a small-molecular-weight natural product, on diabetic mouse models. Diabetes 62, 628–638 [DOI] [PMC free article] [PubMed] [Google Scholar]