

Background: Down-regulation of junctophilin-2 is associated with a variety of cardiac diseases.
Results: Junctophilin-2 is cleaved by calpain at multiple sites, resulting in dysfunctional junctophilin-2 truncations.
Conclusion: Calpain-mediated proteolysis contributes to posttranslational down-regulation of junctophilin-2.
Significance: These data reveal, for the first time, the detailed molecular determinants responsible for calpain proteolysis of junctophilin-2.
Keywords: calcium, calpain, excitation-contraction coupling (E-C coupling), heart, proteolysis, junctophilin-2
Abstract
Junctophilin-2 (JP2), a membrane-binding protein that provides a structural bridge between the plasmalemma and sarcoplasmic reticulum, is essential for precise Ca2+-induced Ca2+ release during excitation-contraction coupling in cardiomyocytes. In animal and human failing hearts, expression of JP2 is decreased markedly, but the molecular mechanisms underlying JP2 down-regulation remain incompletely defined. In mouse hearts, ischemia/reperfusion injury resulted in acute JP2 down-regulation, which was attenuated by pretreatment with the calpain inhibitor MDL-28170 or by transgenic overexpression of calpastatin, an endogenous calpain inhibitor. Using a combination of computational analysis to predict calpain cleavage sites and in vitro calpain proteolysis reactions, we identified four putative calpain cleavage sites within JP2 with three N-terminal and one C-terminal cleavage sites. Mutagenesis defined the C-terminal region of JP2 as the predominant calpain cleavage site. Exogenous expression of putative JP2 cleavage fragments was not sufficient to rescue Ca2+ handling in JP2-deficient cardiomyocytes, indicating that cleaved JP2 is non-functional for normal Ca2+-induced Ca2+ release. These data provide new molecular insights into the posttranslational regulatory mechanisms of JP2 in cardiac diseases.
Introduction
Myocardial infarction, one of the most common causes of heart failure, is characterized by defects in cardiac excitation-contraction (E-C)2 coupling (1, 2). In a normal cardiomyocyte, E-C coupling depends on Ca2+-induced Ca2+ release, in which L-type Ca2+ channel-mediated Ca2+ influx triggers Ca2+ release from the sarcoplasmic reticulum (SR) via type 2 ryanodine receptors (RyR2) (3, 4). L-type Ca2+ channels and RyR2s are physically and functionally organized into a tightly regulated structure known as the Ca2+ release unit, which provides the structural basis for Ca2+-induced Ca2+ release (4, 5). The integrity of the Ca2+ release unit is maintained by the structural protein junctophilin-2 (JP2) that bridges the T-tubule membrane and the SR (6–8). Disruption of the fine architecture of the E-C coupling machinery impairs Ca2+-induced Ca2+ release, thereby leading to loss of contractility and heart failure (9).
JP2, the major junctophilin isoform expressed in the heart, contains eight N-terminal membrane occupation and recognition nexus (MORN) domains that mediate interactions with the plasmalemma, a space-spanning α helix, and a C-terminal transmembrane (TM) domain that anchors JP2 to the SR membrane (6). Consistent with a critical role for JP2 in E-C coupling, conditional silencing of JP2 in cardiomyocytes results in contractile dysfunction, abnormal Ca2+ handling, and acute heart failure (7, 10, 11). On the contrary, cardiac-specific overexpression of JP2 attenuated the development of heart failure induced by pressure overload (12). Moreover, mutations in the JP2 coding region have been discovered in patients with hypertrophic cardiomyopathy (13, 14). The pathological relevance of JP2 has been revealed by observations that dysregulation of JP2 protein is associated with pathological progression in multiple models of heart failure, including pressure overload-induced hypertrophy/heart failure and myocardial infarction (15–21). Toward understanding the mechanism of JP2 down-regulation, a recent report has demonstrated that JP2 is targeted by the microRNA miR-24, which may be responsible for the down-regulation of JP2 during long-term pressure overload-induced hypertrophy and heart failure (19, 22). Recently, a Ca2+-dependent mechanism of junctophilin proteolysis has been reported (23). More specifically, the activity of calpain, a family of Ca2+-dependent proteases has been found to be related to the degradation of JP2 (21). However, it remains unclear whether JP2 is a direct substrate of calpain and the specific molecular site for JP2 proteolysis.
In this study, our objective was to determine whether and how JP2 expression is regulated posttranslationally in response to cardiac injury. We first demonstrated that JP2 expression is down-regulated in failing hearts from patients with ischemic heart disease. Using a murine model of ischemia/reperfusion (I/R) injury, we identified the Ca2+-dependent protease calpain as a key mediator of JP2 down-regulation, including a comprehensive determination of the molecular sites for calpain cleavage. We also found that JP2 fragments corresponding to the primary cleavage site fail to rescue E-C coupling in JP2-deficient cardiomyocytes. These data provide important insights into the mechanism by which JP2 expression is lost in failing hearts.
Experimental Procedures
Human Heart Samples and Animal Studies
Samples from patients with ischemic heart disease were obtained from the University of Iowa Heart Failure Transplant Program. Rejected healthy donor hearts were obtained through the Donor Network. All human heart tissue samples were obtained under an organ research donation protocol that was approved by the Institutional Review Board of the University of Iowa. Animal experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals (National Institutes of Health publication no. 85-23, revised 1996) and were approved by the Institutional Animal Care and Use Committee at the University of Iowa.
Cardiac-specific JP2 Knockdown (JP2-KD) was achieved with the use of transgenic mice conditionally expressing a JP2 shRNA (shJP2). Specifically, conditional shJP2 mice (provided by Dr. Wehrens) (7) were crossed with mice carrying the cardiac-specific α-myosin heavy chain promoter upstream of Cre (αMHC-Cre, also FVB-Tg(Myh6-cre)2182Mds/J, The Jackson Laboratory). The shJP2 mice were maintained on a C57BL/6 background for more than 10 generations. The αMHC-Cre mice were maintained on an FVB background. PCR was used to genotype the offspring. The near complete depletion of JP2 protein in JP2-KD (αMHC-Cre X shJP2) hearts has been reported recently (11). JP2-KD mice were sacrificed at 2 months of age for isolation and culture of ventricular cardiomyocytes as described below.
In Vitro and in Vivo I/R Protocols in Mouse Hearts
For in vitro I/R injury, hearts were excised rapidly from anesthetized mice and perfused with Krebs-Henseleit solution at 37 °C using a Langendorff apparatus at a constant pressure of 80 mm Hg with 95% O2, 5% CO2. After 20 min of equilibration, hearts were subjected to 35 min of global no-flow ischemia followed by 50 min of reperfusion. Some hearts were treated with MDL-28170 (10 μm) via perfusion 10 min before ischemia and after the onset of reperfusion. For in vivo I/R injury in mouse hearts, myocardial ischemia was produced by ligation of the left anterior descending coronary artery for 20 min, followed by reperfusion for 30 min (longer periods of ischemia/reperfusion lead to a high death rate under our conditions). Left ventricles were dissected for analysis by Western blotting.
Molecular Cloning, Mutagenesis, and Adenovirus Construction
Wild-type JP2 cDNA was provided by Dr. Takeshima (6). FLAG and HA epitope tags were added to 5′ and 3′ ends, respectively, in the same reading frame of the coding sequence of JP2 cDNA by PCR. The dual epitope-tagged cDNA was cloned into the pCMV-XL5 plasmid under the control of a CMV promoter. cDNAs of JP2 truncations were cloned using PCR. QuikChange II (Agilent) was used for site-directed mutagenesis. Adenoviruses containing full-length JP2 (Ad-JP2), truncated JP2(1–565) (Ad-JP2(1–565)), or JP2(566-end) (Ad-JP2(566-end)) or empty Ad5 virus (Ad-empty) were produced by the University of Iowa Gene Transfer Vector Core. In the adenovirus construct, the full-length JP2 and truncation JP2(1–565) were tagged with FLAG at the N terminus and HA at the C terminus. The truncation JP2(566-end) was tagged with HA at the C terminus.
Adult Cardiomyocyte Culture, Cell Transfection, and Adenovirus Infection
Genecarrior-1 (Epoch Lab) was used to transfect plasmids into HEK293T cells. Isolation and culture of cardiomyocytes were performed as described previously (24). Adenoviruses were applied at a multiplicity of infection of 100, which resulted in transfection of almost all cells, as confirmed by immunostaining of epitope tags. Ca2+ imaging experiments were performed 40 h after adenoviral infection.
In Vitro Calpain-mediated Proteolysis Reaction
Mouse and human heart tissues as well as HEK 293T cells (obtained from the ATCC and cultured according to the protocol of the manufacturer) overexpressing tagged JP2 were washed with PBS and homogenized in lysis buffer (20 mm Tris-HCl, 150 mm NaCl, 2 mm EDTA, and 1% Triton X-100), followed by sonication. Homogenates were centrifuged for 10 min at 14,000 rpm at 4 °C. Protein content in the supernatant was determined using the BCA assay (Thermo Scientific). Cell extracts were diluted in ice-cold calpain reaction buffer (135 mm NaCl, 5 mm KCl, 1 mm MgCl2, 10 mm glucose, 10 mm Hepes (pH 7.25), 2 μm lactacystin, and 10 mm 2-mercaptoethanol) to a concentration of 4 mg protein/ml. Just prior to starting the protease reaction, CaCl2 was added to achieve a final concentration of 3 mm free Ca2+ (in the presence of EDTA), as calculated by winmaxc32 (Stanford University). For some experiments, the free Ca2+ concentration was adjusted to 500 μm, as calculated by winmaxc32. Purified human calpain I (Calbiochem) was added at a concentration of 1 mg of total protein/1 μg of calpain I. Reactions without Ca2+ or without purified calpain I were used as controls. Reactions were incubated at 30 °C for the indicated time and stopped by adding EDTA to a final concentration of 10 mm and incubating for 5 min at 95 °C. The reaction products were subjected to Western blotting.
Western Blot Analysis and Immunostaining
Western blotting and immunostaining of cardiomyocytes were performed as described previously (24) using antibodies to JP2 (catalog no. sc-51313, Santa Cruz Biotechnology) and epitope tags (anti-FLAG, catalog no. A00187, Genescript; anti-HA, catalog no. sc-805, Santa Cruz Biotechnology). Immunofluorescence was imaged by using a confocal microscope (Carl Zeiss MicroImaging Inc.).
Confocal Ca2+ Imaging
Cells were loaded with Rhod-2 AM at 37 °C for 20 min, followed by washing with Tyrode's solution at room temperature for 15 min before Ca2+ imaging. Confocal images were acquired using a ×63, 1.3 numerical aperture oil immersion objective mounted on a Zeiss LSM 510 confocal microscope. Confocal line scanning was used to record Ca2+ signals. Steady-state Ca2+ transients were recorded in Tyrode's solution containing 1.8 mm Ca2+ under field stimulation of 1 Hz. At least five steady-state transients for each cell were analyzed and averaged to represent Ca2+ signals of the cell. Ca2+ imaging data were analyzed by using a home-complied software, CaTeasy, which is coded in MATLAB 2013a (Mathworks). Briefly, the program normalizes Ca2+ transient images on a column-by-column basis (512 columns/image). The characteristic parameters of normalized transients were detected automatically. The firing time of every scanning pixel was detected column by column on the basis of the maximum local variability of pixel intensity. The profile of the point-to-point firing time was indicated by a red line overlapping on the Ca2+ image. Index of dyssynchronization, defined by the mean absolute deviation of firing time of each scanning pixel (with every 8 pixels binned), was used to evaluate the dyssynchrony of Ca2+ transients. This automated software for calcium transient analysis is available upon request.
Statistics
Data are presented as mean ± S.E. Analysis of variance and Student's t test were applied when appropriate. A p value of <0.05 was considered statistically significant.
Results
Calpain Mediates Down-regulation of JP2 in Response to Cardiac Ischemia/Reperfusion Injury
A Western blotting assay showed that JP2 protein was reduced by 60% in left ventricular lysates from end-stage heart failure patients with ischemic heart disease (Fig. 1A), similar to previous reports on human failing hearts of different etiologies, such as dilated and hypertrophied cardiomyopathies (10, 25). To dissect the molecular mechanisms responsible for JP2 down-regulation in disease, we used a murine model of acute cardiac stress. Following global cardiac I/R injury in Langendorff-perfused WT mouse hearts, we observed a significant down-regulation of JP2 protein in the left ventricle (Fig. 1B). JP2 down-regulation was also observed in an in vivo I/R model induced by in situ left anterior descending coronary artery ligation/reperfusion in mouse hearts (Fig. 1D). These data suggest that JP2 down-regulation is not solely accomplished by miRNA-mediated gene silencing (19), given that loss of JP2 expression was evident within the short time period of I/R injury in our study.
FIGURE 1.

JP2 is down-regulated by calpain in response to in vitro and in vivo I/R injury. A, Western blot of JP2 expression in left ventricular lysates from patients with ischemic heart failure (HF) (n = 4) or rejected donor hearts (Control) (n = 5). B, representative Western blot and summary data of JP2 expression in WT murine left ventricles after in vitro I/R injury under Langendorff perfusion (at least four hearts for each group). MDL-28170 (10 μm) perfusion after the onset of reperfusion and 10 min before ischemia attenuated JP2 down-regulation. C, representative Western blot and summary data of JP2 expression in left ventricles from calpastatin (CAST) transgenic mice after in vitro I/R injury under Langendorff perfusion (at least three hearts for each group). D and E, JP2 protein level in left ventricles from WT mice (D) and calpastatin transgenic mice (E) after in vivo coronary artery occlusion-induced I/R injury (at least three hearts for each group). Data are normalized to GAPDH and expressed relative to the values of control or sham-operated hearts for each genotype. *, p < 0.05; **, p < 0.01; N.S., not significant.
JP2 down-regulation was attenuated by perfusion with the calpain inhibitor MDL-28170 before and after ischemia (Fig. 1B). These data are in line with the observed increase in calpain activity under a variety of pathological conditions, such as pressure overload, myocardial infarction, I/R injury, and isoproterenol-induced cardiac disease (26–37). In the post-ischemic heart, calpain degrades a myriad of structural and myofilament proteins, resulting in cardiac dysfunction (29, 38–41). We next performed studies in transgenic mice overexpressing the endogenous calpain inhibitor protein calpastatin (CAST) (42, 43). JP2 levels were statistically unchanged in hearts from CAST mice after both in vitro and in vivo I/R injury (Fig. 1, C and E), further suggesting that calpain contributes to the reduction in JP2 protein expression.
To determine whether calpain directly cleaves JP2, we next performed in vitro cleavage reactions by incubating protein lysates from WT mouse hearts with purified human calpain I. Blotting with a JP2 C-terminal antibody revealed a significant reduction in full-length JP2 upon addition of calpain I (Fig. 2A). The Ca2+ chelator EDTA or the calpain inhibitor Z-LLY-FMK inhibited JP2 degradation. In vitro calpain cleavage assays using human heart lysates also showed a time-dependent and Ca2+/calpain-dependent JP2 down-regulation, identifying JP2 as a direct substrate for calpain (Fig. 2B). These data provide direct evidence that, in cardiac tissue, JP2 is a substrate of the Ca2+-dependent protease calpain.
FIGURE 2.
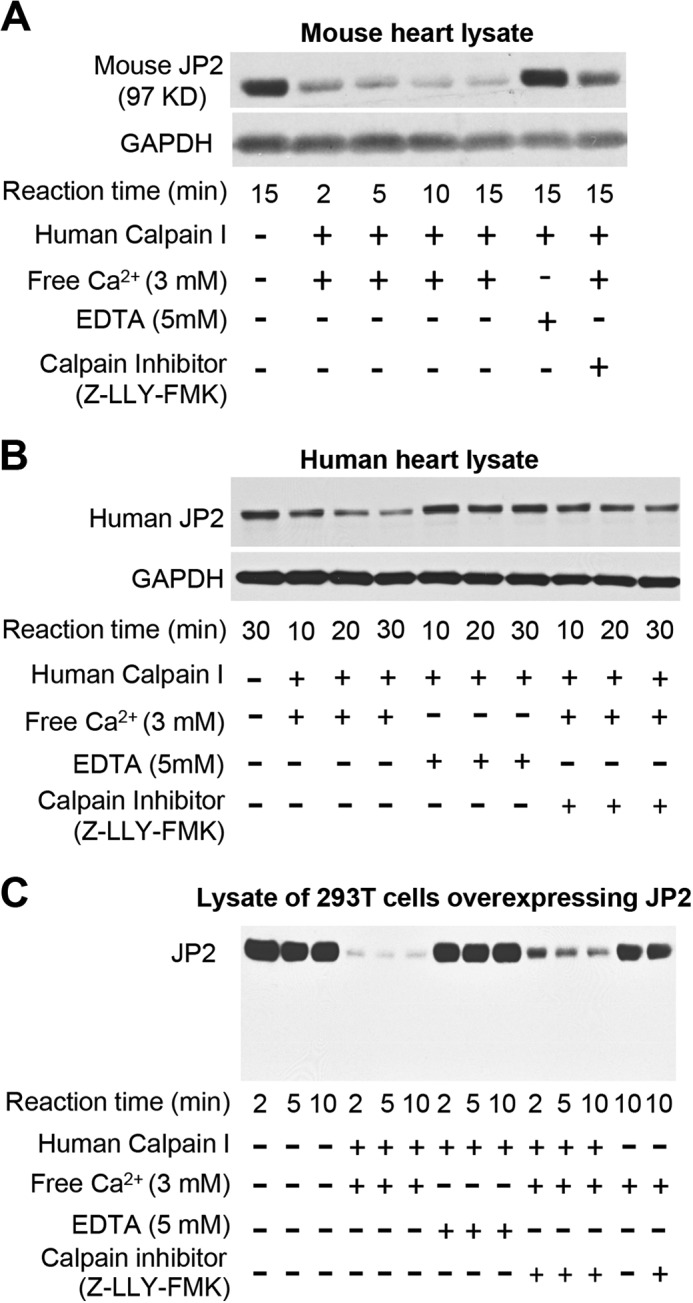
In vitro calpain cleavage reactions identify calpain as the JP2 protease in mouse and human heart. A and B, in vitro proteolysis of JP2 in mouse (A) and human (B) heart lysates (1 mg total protein) in the presence of human calpain I (1 μg of calpain I) and free Ca2+ (3 mm) with or without EDTA (5 mm) or the calpain inhibitor Z-LLY-FMK (20 μm). C, protein lysates from 293T cells overexpressing JP2 (untagged) were incubated with purified human calpain I in the presence or absence of Ca2+ or calpain inhibitor. Data are representative of at least three independent experiments.
Identification of Calpain Cleavage Sites on JP2
We overexpressed murine JP2 in 293T cells and subjected lysates to a calpain proteolysis assay. Using an antibody against the JP2 C-terminal epitope, we detected a dramatic decrease in the amount of full-length JP2 but could not detect the faster-migrating species (Fig. 2C), indicating that calpain cleavage may have destroyed the antibody epitope.
To determine the extent of JP2 proteolysis by calpain, we next generated a JP2 construct with an N-terminal FLAG tag and a C-terminal HA tag (Fig. 3A). After cotransfection of the dually tagged JP2 and human calpain I into 293T cells, we used the Ca2+ ionophore ionomycin to induce Ca2+-dependent cleavage of JP2. Blotting with an antibody against the N-terminal FLAG tag demonstrated the calpain-dependent appearance of four N-terminal cleavage products, denoted N-I, II, III, and IV (Fig. 3B, a). The antibody against the HA tag detected one C-terminal cleavage product (denoted C-I, Fig. 3B, b). Treatment with the calpain inhibitor MDL-28170 resulted in a substantial reduction in the presence of both the N- and C-terminal cleavage products and restored JP2 full-length levels to those observed in untreated cells (Fig. 3B, a and b). Cleavage products of a similar molecular weight were also observed in the in vitro calpain cleavage assays at 3 mm Ca2+ (Fig. 3C) and 500 μm Ca2+ (Fig. 3D). Removing Ca2+ from the reaction or inhibition of calpain with Z-LLY-FMK resulted in a substantial reduction in cleavage products and preservation of full-length JP2 (Fig. 3, C and D). These data suggest that JP2 contains at least four calpain cleavage sites.
FIGURE 3.

Calpain cleaves JP2 at multiple sites in the N- and C-terminal regions. A, schematic of the N- and C-terminal epitope-tagged full-length JP2 cDNA. B, after cotransfection of 293T cells with calpain I and full-length JP2 containing an N-terminal FLAG tag and a C-terminal HA tag, cells were exposed to 5 μm ionomycin and 4 mm extracellular Ca2+ for 1 h in the absence or presence of 10 μm MDL-28170. JP2 expression and degradation were assessed by Western blotting with anti-FLAG (a) or anti-HA (b) to detect N- and C-terminal cleavage fragments, respectively. N-I, II, III, and IV denote the four N-terminal cleavage products detected with anti-FLAG. C-I denotes the major C-terminal cleavage product detected by anti-HA. C, in vitro calpain-I mediated cleavage assay as in Fig. 2 using lysates from 293T cells overexpressing JP2 with N-terminal FLAG and C-terminal HA epitopes. Cleavage products were detected with anti-FLAG (a) or anti-HA (b). D, in vitro calpain proteolysis of tagged JP2 at lower concentrations of Ca2+ (500 μm). Data are representative of at least three independent experiments.
Using the computational tools CaMPDB (44) and GPS-CCD 1.0 (45), we identified several putative calpain cleavage sites in JP2. We first generated a panel of tagged JP2 truncations on the basis of the predicted N- and C-terminal calpain cleavage sites (Fig. 4A). When expressed exogenously in 293T cells, JP2(1–155), JP2(1–201), and JP2(1–565) have a similar molecular weight as the cleavage products N-I, N-II, and N-IV, respectively (Fig. 4B, a). JP2(1–155), which corresponds to N-I, was only detectable in 293T cells treated with the proteasomal inhibitor lactacystin (Fig. 4B, a), suggesting that this cleavage product is not stable and undergoes further degradation by the proteasome (Fig. 4B, a). Note that JP2(566-end), which is the C-terminal counterpart of JP2(1–565), has a similar molecular weight as C-terminal cleavage product C-I (Fig. 4B, b), indicating N-IV and C-I may be generated by a single cleavage of JP2 at Arg-565/Thr-566. We also examined the susceptibility of JP2(1–565) to calpain-mediated proteolysis and found that this fragment can be further processed by calpain to generate N-I, N-II, and N-III fragments (Fig. 4C). These data provide evidence that deletion of the extreme C-terminal tail of JP2 does not attenuate the calpain recognition of other cleavage sites.
FIGURE 4.

C-terminal JP2 proteolysis at R565T is the primary site for calpain-mediated cleavage. A, schematic of the N- or C-terminal epitope-tagged truncated JP2 constructs on the basis of putative calpain cleavage sites. These cleavage sites were predicted by using GPS-CCD 1.0 and CaMPDB. Arrows indicate these predicted cleavage sites relative to the MORN and TM domains. B, expression of JP2 truncations in 293T cells. Where indicated, cells were pretreated with lactacystin (10 μm) after transfection. C, Western blot with anti-FLAG (N-terminal tag) following in vitro cleavage reaction with JP2(1–565) truncation. D, six to eight Amino acid deletions surrounding the predicted cleavage sites (V155R, Δ(153–158); L201L, Δ(198–205); R565T, Δ(563–568)) were introduced into the full-length JP2 construct with N-terminal FLAG and C-terminal HA epitope tags. Following expression of mutants in 293T cells, lysates were subjected to in vitro cleavage reactions. WT JP2 with tags was used as a control.
We next verified the predicted calpain cleavage at Val-155/Arg-156, Leu-201/Leu-202, and Arg-565/Thr-566 by creating site-directed deletions in the surrounding area. A six-residue deletion around the putative cleavage site Val-155/Arg-156 (Δ(153–158)) completely abolished the generation of N-I in the in vitro calpain cleavage assay (Fig. 4D, a), whereas cleavage at the extreme C terminus remained largely intact compared with WT JP2 (Fig. 4D, a and b). Next, an eight residue deletion around the putative cleavage site Leu-201/Leu-202 (Δ(198–205)) resulted in complete loss of the JP2 cleavage product N-II but had no effect on the generation of N-I (Fig. 4D, c), suggesting that cleavage at Val-155/Arg-156 is not dependent on prior cleavage at Leu-201/Leu-202. Finally, a six-amino acid deletion around the putative cleavage site Arg-565/Thr-566 (Δ(563–568)) led to complete loss of N-IV (Fig. 4D, d) and a severe reduction in C-I (Fig. 4D, e). Several C-terminal cleavage products (arrows, Fig. 4D, e), which may be counterparts of N-terminal cleavage products detected by anti-FLAG antibody, were detected by the anti-HA antibody in the in vitro calpain cleavage reaction of Δ(563–568) but not WT JP2 (Fig. 4D, e). Unlike loss of the N-terminal cleavage sites, loss of the C-terminal Arg-565/Thr-566 cleavage site preserved full-length JP2 (Fig. 4D, d and e), suggesting that the C-terminal Arg-565/Thr-566 cleavage site is more sensitive to calpain than the N-terminal cleavage sites. Taken together, these data identify Val-155/Arg-156, Leu-201/Leu-202, and Arg-565/Thr-566 as calpain cleavage sites that result in the generation of the cleavage products N-I, N-II and N-IV, and C-I, respectively.
JP2 Fragments Corresponding to the Primary Cleavage Site (Arg-565/Thr-566) Have No Effect on Cardiomyocyte Ca2+ Handling
Cleavage at the primary calpain proteolysis site (Arg-565/Thr-566) splits apart the C-terminal SR-binding TM domain and N-terminal plasma membrane-binding MORN domains of JP2. It is believed that intact JP2 is required for its normal function to tether the plasma membrane and SR together, but it is unknown whether truncated JP2 fragments exert any additional functions on myocyte Ca2+ handling. To test this, we expressed epitope-tagged (see “Experimental Procedures”) full-length JP2 and JP2 truncations corresponding to the primary calpain cleavage site in cardiomyocytes via adenoviruses. Immunostaining showed the striated pattern of these three versions of JP2 in infected mouse ventricular cardiomyocytes (Fig. 5A), indicating that the two truncations can still be localized to the T-tubule/SR junction via either MORN domains or TM domains, respectively. Coimmunoprecipitation showed that full-length JP2 and JP2(1–565), but not JP2(566-end), interact with RyR2 and Cav1.2 (Fig. 5B), demonstrating that JP2 interacts with complexes of E-C coupling channels via its N terminus.
FIGURE 5.

JP2 truncations are nonfunctional in regulating Ca2+ transients. A, an antibody against the HA tag was used to reveal the localization of adenoviral expression of tagged full-length JP2 and truncations, as indicated, in adult wild-type cardiomyocytes. All the three version of JP2 can be localized in the striated pattern. B, full-length JP2 and JP2(1–565) forms complexes with RyR2 and Cav1.2 in vivo. An antibody against HA was used for immunoprecipitation (IP). The RyR2 and Cav1.2 that were pulled down were detected by Western blot analysis. Note that full-length JP2 and JP2(1–565) pulled down both RyR2 and Cav1.2, although JP(566-end) does not form complexes with RyR2 or Cav1.2. IB, immunoblot. C, adenovirus-mediated expression of full-length JP2, FLAG-JP2(1–565)-HA, and JP2(566-end)-HA in JP2-KD) cardiomyocytes. D, representative steady-state Ca2+ transients under 1-Hz field stimulation. The fluorescence intensity (F) of Ca2+ imaging was normalized to the baseline (F0). The red lines overlapping on Ca2+ imaging show the profile of the moment of Ca2+ transient firing on the scanning line in a point-by-point way. A straighter line means better synchronization of the Ca2+ transients. Note that expression of full-length JP2, but not JP2 truncations, improves the amplitude and synchronization of Ca2+ transients. E–G, summary of Ca2+ transient amplitude, index of dyssynchronization (mean absolute deviation of firing time), and duration of 50% decay (T50) (n = 68, 70, 70, and 52 for Ad-Empty, Ad-JP2, Ad-JP2(1–565), and Ad-JP2(566-end), respectively). Only full-length JP2 improves the amplitude, synchronization, and decay of Ca2+ transients. H, summary of SR Ca2+ content, which was assessed by caffeine-induced SR Ca2+ release (n = 17, 17, 17, and 12 for Ad-Empty, Ad-JP2, Ad-JP2(1–565), and Ad-JP2(566-end), respectively). **, p < 0.01 versus indicated groups; N.S., not significant.
Next we evaluated the potential role of JP2 truncates on Ca2+ handling in cardiomyocytes. We expressed epitope-tagged full-length JP2, N-terminal truncation JP2(1–565) and C-terminal truncation JP2(566-end) back into ventricular cardiomyocytes from mice with silenced endogenous JP2 (JP2-KD) (Fig. 5C). Expression of full-length JP2 in JP2-KD cardiomyocytes resulted in a significantly increased amplitude (by ∼50%) (Fig. 5, D and E), increased synchronization (quantified by the index of dyssyncronization, Fig. 5, D and F), as well as a shortened decay phase (T50) of Ca2+ transients (Fig. 5G). In contrast, expression of JP2(1–565) or JP2(566-end) failed to restore Ca2+ transients in JP2-KD cardiomyocytes (Fig. 5, E–G). SR Ca2+ content of JP2-KD cardiomyocytes was not influenced by expression of full-length JP2 or JP2 truncations (Fig. 5H).
Because JP2(1–565) and JP2(566-end) can still be localized to the striated T-tubule/SR junction, and because JP2(1–565) even interacts with RyR2 and Cav1.2, we next tested whether these truncations have a dominant negative effect on Ca2+ handling in WT cardiomyocytes. Our data showed that overexpression of full-length JP2 or truncations from either terminus did not alter the kinetics of steady-state Ca2+ transients under 1-Hz field stimulation and SR Ca2+ loading in WT cardiomyocytes (Fig. 6). These data indicate that proteolysis of JP2 at the C terminus results in E-C coupling dysfunction.
FIGURE 6.

JP2 truncations do not have a dominant negative effect on Ca2+ transients in wild-type cardiomyocytes. A, representative steady-state Ca2+ transients under 1-Hz field stimulation. See the legend of Fig. 5 for the definition of the red lines overlapping on Ca2+ imaging. B–D, summary of Ca2+ transient amplitude, index of dyssynchronization, and duration of 50% decay (T50) (n = 51, 52, 51, and 61 for Ad-Empty, Ad-JP2, Ad-JP2(1–565), and Ad-JP2(566-end), respectively). E, summary of SR Ca2+ content, which was assessed by caffeine-induced SR Ca2+ release (n = 24, 24, 29, and 33 for Ad-Empty, Ad-JP2, Ad-JP2(1–565), and Ad-JP2(566-end), respectively).
Discussion
These data reveal, for the first time, the molecular determinants responsible for posttranslational calpain proteolysis of JP2, providing a molecular mechanism of JP2 down-regulation following cardiac injury. Sequence prediction of calpain cleavage sites identified several putative sites, three of which we confirmed by mutagenesis. Our data suggest that C-terminal calpain cleavage of JP2 may be a prerequisite for proteolysis at N-terminal sites. We also found that neither N-terminal nor C-terminal truncations of JP2 are sufficient to restore Ca2+ transients in cardiomyocytes in which endogenous JP2 has been knocked down. These data provide important insights into JP2 regulation in cardiomyocyte E-C coupling and may explain, in part, the significant cardiac phenotype associated with loss of JP2 in vivo and the protective effect of calpain inhibition (32, 38).
We and others have reported that JP2 is required for maintaining T-tubule structural integrity in adult hearts (7, 18). More recently, we identified JP2 as an essential factor in T-tubule and E-C coupling maturation during development (17). In another study, we found that overexpression of JP2 attenuates the transition from hypertrophy to heart failure, suggesting its significance as a potential therapeutic target (12). JP2 protein down-regulation has been observed in failing human hearts as well as multiple animal models of cardiac disease (10, 15, 17–21, 23, 25, 46). This loss of JP2 is associated with alterations in T-tubule ultrastructure and E-C coupling dysfunction, thereby contributing to the development of heart failure (7, 10, 11, 19). To date, miR-24-mediated translational repression and Ca2+-dependent proteolysis have been implicated mechanistically in JP2 down-regulation (19, 22, 23, 46). Our data extend these latter findings by identifying calpain as the JP2 protease. Moreover, we define the molecular determinants of JP2 proteolysis and provide evidence for this mechanism in an animal model of cardiac stress.
Previous reports have suggested two mechanisms by which JP2 regulates E-C coupling. First, JP2 acts as a physical bridge between the T-tubule and SR membranes to maintain normal Ca2+ handling in cardiomyocytes (7, 11). Second, JP2 has been shown to interact with RyR2 and Cav1.2 and regulate their gating function.(7, 47) Our data suggest that down-regulation of JP2 results in E-C coupling dysfunction primarily through disrupting the junctional complex. Specifically, we found that the N-terminal truncation fragment of JP2 interacts with RyR2 and Cav1.2 but is not sufficient to restore Ca2+ transients in JP2-KD cardiomyocytes. These data provide compelling evidence that full-length JP2 is required for normal E-C coupling function. Taken together, this study provides novel molecular insights into the structure and function of JP2 in cardiomyocytes. Identification of the precise calpain cleavage sites on JP2 may lead to the development of new targeted therapies that maintain JP2 expression in the setting of cardiac stress.
Author Contributions
A. G. and L. S. S. conceived and designed the research. A. G., D. H., C. Z., and W. K. performed research and analyzed data. J. D. M. and F. J. L. provided human heart samples. T. P. provided reagents. C. E. G. and R. Z. L. supervised the experiments, data analysis, and interpretations. A. G. and L. S. S. wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Dr. Hiroshi Takeshima for JP2 cDNA and Dr. Xander Wehrens for providing shJP2 transgenic mice.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 HL090905 (to L. S. S.). This work was also supported by American Heart Association Postdoctoral Fellowship 13POST14630077 (to A. G.). The authors declare that they have no conflicts of interest with the contents of this article.
- E-C
- excitation-contraction
- SR
- sarcoplasmic reticulum
- RyR2
- type 2 ryanodine receptor
- MORN
- membrane occupation and recognition nexus
- MORN
- membrane occupation and recognition nexus
- KD
- knockdown
- TM
- transmembrane
- I/R
- ischemia/reperfusion
- T-tubule
- transverse tubule
- Z-LLY-FMK
- benzyloxycarbonyl-LLY-fluoromethyl ketone
- HF
- heart failure
- CAST
- calpastatin
- JP2
- junctophilin-2.
References
- 1. Gómez A. M., Guatimosim S., Dilly K. W., Vassort G., Lederer W. J. (2001) Heart failure after myocardial infarction: altered excitation-contraction coupling. Circulation 104, 688–693 [DOI] [PubMed] [Google Scholar]
- 2. Litwin S. E., Zhang D., Bridge J. H. (2000) Dyssynchronous Ca2+ sparks in myocytes from infarcted hearts. Circ. Res. 87, 1040–1047 [DOI] [PubMed] [Google Scholar]
- 3. Bers D. M. (2002) Cardiac excitation-contraction coupling. Nature 415, 198–205 [DOI] [PubMed] [Google Scholar]
- 4. Wang S. Q., Song L. S., Lakatta E. G., Cheng H. (2001) Ca2+ signalling between single L-type Ca2+ channels and ryanodine receptors in heart cells. Nature 410, 592–596 [DOI] [PubMed] [Google Scholar]
- 5. Franzini-Armstrong C., Protasi F., Ramesh V. (1999) Shape, size, and distribution of Ca2+ release units and couplons in skeletal and cardiac muscles. Biophys. J. 77, 1528–1539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Takeshima H., Komazaki S., Nishi M., Iino M., Kangawa K. (2000) Junctophilins: a novel family of junctional membrane complex proteins. Mol. Cell 6, 11–22 [DOI] [PubMed] [Google Scholar]
- 7. van Oort R. J., Garbino A., Wang W., Dixit S. S., Landstrom A. P., Gaur N., De Almeida A. C., Skapura D. G., Rudy Y., Burns A. R., Ackerman M. J., Wehrens X. H. (2011) Disrupted junctional membrane complexes and hyperactive ryanodine receptors after acute junctophilin knockdown in mice. Circulation 123, 979–988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jayasinghe I. D., Baddeley D., Kong C. H., Wehrens X. H., Cannell M. B., Soeller C. (2012) Nanoscale organization of Junctophilin-2 and Ryanodine receptors within peripheral couplings of rat ventricular cardiomyocytes. Biophys. J. 102, L19–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Song L. S., Sobie E. A., McCulle S., Lederer W. J., Balke C. W., Cheng H. (2006) Orphaned ryanodine receptors in the failing heart. Proc. Natl. Acad. Sci. U.S.A. 103, 4305–4310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Landstrom A. P., Kellen C. A., Dixit S. S., van Oort R. J., Garbino A., Weisleder N., Ma J., Wehrens X. H., Ackerman M. J. (2011) Junctophilin-2 expression silencing causes cardiocyte hypertrophy and abnormal intracellular calcium-handling. Circ. Heart Fail. 4, 214–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen B., Guo A., Zhang C., Chen R., Zhu Y., Hong J., Kutschke W., Zimmerman K., Weiss R. M., Zingman L., Anderson M. E., Wehrens X. H., Song L. S. (2013) Critical roles of junctophilin-2 in T-tubule and excitation-contraction coupling maturation during postnatal development. Cardiovasc. Res. 100, 54–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Guo A., Zhang X., Iyer V. R., Chen B., Zhang C., Kutschke W. J., Weiss R. M., Franzini-Armstrong C., Song L. S. (2014) Overexpression of junctophilin-2 does not enhance baseline function but attenuates heart failure development after cardiac stress. Proc. Natl. Acad. Sci. U.S.A. 111, 12240–12245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Landstrom A. P., Weisleder N., Batalden K. B., Bos J. M., Tester D. J., Ommen S. R., Wehrens X. H., Claycomb W. C., Ko J. K., Hwang M., Pan Z., Ma J., Ackerman M. J. (2007) Mutations in JPH2-encoded junctophilin-2 associated with hypertrophic cardiomyopathy in humans. J. Mol. Cell Cardiol. 42, 1026–1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Takeshima H., Hoshijima M., Song L. S. (2015) Ca microdomains organized by junctophilins. Cell Calcium 10.1016/j.ceca.2015.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chen B., Li Y., Jiang S., Xie Y. P., Guo A., Kutschke W., Zimmerman K., Weiss R. M., Miller F. J., Anderson M. E., Song L. S. (2012) β-Adrenergic receptor antagonists ameliorate myocyte T-tubule remodeling following myocardial infarction. FASEB J. 26, 2531–2537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Guo A., Zhang C., Wei S., Chen B., Song L. S. (2013) Emerging mechanisms of T-tubule remodelling in heart failure. Cardiovasc. Res. 98, 204–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Minamisawa S., Oshikawa J., Takeshima H., Hoshijima M., Wang Y., Chien K. R., Ishikawa Y., Matsuoka R. (2004) Junctophilin type 2 is associated with caveolin-3 and is down-regulated in the hypertrophic and dilated cardiomyopathies. Biochem. Biophys. Res. Commun. 325, 852–856 [DOI] [PubMed] [Google Scholar]
- 18. Wei S., Guo A., Chen B., Kutschke W., Xie Y. P., Zimmerman K., Weiss R. M., Anderson M. E., Cheng H., Song L. S. (2010) T-tubule remodeling during transition from hypertrophy to heart failure. Circ. Res. 107, 520–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Xu M., Wu H. D., Li R. C., Zhang H. B., Wang M., Tao J., Feng X. H., Guo Y. B., Li S. F., Lai S. T., Zhou P., Li L. L., Yang H. Q., Luo G. Z., Bai Y., Xi J. J., Gao W., Han Q. D., Zhang Y. Y., Wang X. J., Meng X., Wang S. Q. (2012) Mir-24 regulates junctophilin-2 expression in cardiomyocytes. Circ. Res. 111, 837–841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xu M., Zhou P., Xu S. M., Liu Y., Feng X., Bai S. H., Bai Y., Hao X. M., Han Q., Zhang Y., Wang S. Q. (2007) Intermolecular failure of L-type Ca2+ channel and ryanodine receptor signaling in hypertrophy. PLoS Biol. 5, e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wu C. Y., Chen B., Jiang Y. P., Jia Z., Martin D. W., Liu S., Entcheva E., Song L. S., Lin R. Z. (2014) Calpain-dependent cleavage of junctophilin-2 and T-tubule remodeling in a mouse model of reversible heart failure. J. Am. Heart Assoc. 3, e000527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Song L. S., Guo A., Lin R. Z. (2012) MicroRNA: a toolkit fine-tuning the dyadic “fuzzy space”? Circ. Res. 111, 816–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Murphy R. M., Dutka T. L., Horvath D., Bell J. R., Delbridge L. M., Lamb G. D. (2013) Ca2+-dependent proteolysis of junctophilin-1 and junctophilin-2 in skeletal and cardiac muscle. J. Physiol. 591, 719–729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Guo A., Cala S. E., Song L. S. (2012) Calsequestrin accumulation in rough endoplasmic reticulum promotes perinuclear Ca2+ release. J. Biol. Chem. 287, 16670–16680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang H. B., Li R. C., Xu M., Xu S. M., Lai Y. S., Wu H. D., Xie X. J., Gao W., Ye H., Zhang Y. Y., Meng X., Wang S. Q. (2013) Ultrastructural uncoupling between T-tubules and sarcoplasmic reticulum in human heart failure. Cardiovasc. Res. 98, 269–276 [DOI] [PubMed] [Google Scholar]
- 26. Chen M., Won D. J., Krajewski S., Gottlieb R. A. (2002) Calpain and mitochondria in ischemia/reperfusion injury. J. Biol. Chem. 277, 29181–29186 [DOI] [PubMed] [Google Scholar]
- 27. French J. P., Quindry J. C., Falk D. J., Staib J. L., Lee Y., Wang K. K., Powers S. K. (2006) Ischemia-reperfusion-induced calpain activation and SERCA2a degradation are attenuated by exercise training and calpain inhibition. Am. J. Physiol. Heart Circ. Physiol. 290, H128–136 [DOI] [PubMed] [Google Scholar]
- 28. Inserte J., Hernando V., Garcia-Dorado D. (2012) Contribution of calpains to myocardial ischaemia/reperfusion injury. Cardiovasc. Res. 96, 23–31 [DOI] [PubMed] [Google Scholar]
- 29. Yoshida K., Inui M., Harada K., Saido T. C., Sorimachi Y., Ishihara T., Kawashima S., Sobue K. (1995) Reperfusion of rat heart after brief ischemia induces proteolysis of calspectin (nonerythroid spectrin or fodrin) by calpain. Circ. Res. 77, 603–610 [DOI] [PubMed] [Google Scholar]
- 30. Greyson C. R., Schwartz G. G., Lu L., Ye S., Helmke S., Xu Y., Ahmad H. (2008) Calpain inhibition attenuates right ventricular contractile dysfunction after acute pressure overload. J. Mol. Cell Cardiol. 44, 59–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hall S. R., Wang L., Milne B., Hong M. (2005) Left ventricular dysfunction after acute intracranial hypertension is associated with increased hydroxyl free radical production, cardiac ryanodine hyperphosphorylation, and troponin I degradation. J. Heart Lung Transplant. 24, 1639–1649 [DOI] [PubMed] [Google Scholar]
- 32. Li Y., Ma J., Zhu H., Singh M., Hill D., Greer P. A., Arnold J. M., Abel E. D., Peng T. (2011) Targeted inhibition of calpain reduces myocardial hypertrophy and fibrosis in mouse models of type 1 diabetes. Diabetes 60, 2985–2994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sandmann S., Yu M., Unger T. (2001) Transcriptional and translational regulation of calpain in the rat heart after myocardial infarction–effects of AT(1) and AT(2) receptor antagonists and ACE inhibitor. Br. J. Pharmacol. 132, 767–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Arthur G. D., Belcastro A. N. (1997) A calcium stimulated cysteine protease involved in isoproterenol induced cardiac hypertrophy. Mol. Cell Biochem. 176, 241–248 [PubMed] [Google Scholar]
- 35. Patterson C., Portbury A. L., Schisler J. C., Willis M. S. (2011) Tear me down: role of calpain in the development of cardiac ventricular hypertrophy. Circ. Res. 109, 453–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Heidrich F. M., Ehrlich B. E. (2009) Calcium, calpains, and cardiac hypertrophy: a new link. Circ. Res. 104, e19–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Letavernier E., Perez J., Bellocq A., Mesnard L., de Castro Keller A., Haymann J. P., Baud L. (2008) Targeting the calpain/calpastatin system as a new strategy to prevent cardiovascular remodeling in angiotensin II-induced hypertension. Circ. Res. 102, 720–728 [DOI] [PubMed] [Google Scholar]
- 38. Chen M., He H., Zhan S., Krajewski S., Reed J. C., Gottlieb R. A. (2001) Bid is cleaved by calpain to an active fragment in vitro and during myocardial ischemia/reperfusion. J. Biol. Chem. 276, 30724–30728 [DOI] [PubMed] [Google Scholar]
- 39. Singh R. B., Chohan P. K., Dhalla N. S., Netticadan T. (2004) The sarcoplasmic reticulum proteins are targets for calpain action in the ischemic-reperfused heart. J. Mol. Cell Cardiol. 37, 101–110 [DOI] [PubMed] [Google Scholar]
- 40. Maekawa A., Lee J. K., Nagaya T., Kamiya K., Yasui K., Horiba M., Miwa K., Uzzaman M., Maki M., Ueda Y., Kodama I. (2003) Overexpression of calpastatin by gene transfer prevents troponin I degradation and ameliorates contractile dysfunction in rat hearts subjected to ischemia/reperfusion. J. Mol. Cell Cardiol. 35, 1277–1284 [DOI] [PubMed] [Google Scholar]
- 41. Kashef F., Li J., Wright P., Snyder J., Suliman F., Kilic A., Higgins R. S., Anderson M. E., Binkley P. F., Hund T. J., Mohler P. J. (2012) Ankyrin-B protein in heart failure: identification of a new component of metazoan cardioprotection. J. Biol. Chem. 287, 30268–30281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li X., Li Y., Shan L., Shen E., Chen R., Peng T. (2009) Over-expression of calpastatin inhibits calpain activation and attenuates myocardial dysfunction during endotoxaemia. Cardiovasc. Res. 83, 72–79 [DOI] [PubMed] [Google Scholar]
- 43. Peltier J., Bellocq A., Perez J., Doublier S., Dubois Y. C., Haymann J. P., Camussi G., Baud L. (2006) Calpain activation and secretion promote glomerular injury in experimental glomerulonephritis: evidence from calpastatin-transgenic mice. J. Am. Soc. Nephrol. 17, 3415–3423 [DOI] [PubMed] [Google Scholar]
- 44. DuVerle D. A., Ono Y., Sorimachi H., Mamitsuka H. (2011) Calpain cleavage prediction using multiple kernel learning. PLoS ONE 6, e19035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Liu Z., Cao J., Gao X., Ma Q., Ren J., Xue Y. (2011) GPS-CCD: a novel computational program for the prediction of calpain cleavage sites. PLoS ONE 6, e19001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Li R. C., Tao J., Guo Y. B., Wu H. D., Liu R. F., Bai Y., Lv Z. Z., Luo G. Z., Li L. L., Wang M., Yang H. Q., Gao W., Han Q. D., Zhang Y. Y., Wang X. J., Xu M., Wang S. Q. (2013) In vivo suppression of microRNA-24 prevents the transition toward decompensated hypertrophy in aortic-constricted mice. Circ. Res. 112, 601–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Golini L., Chouabe C., Berthier C., Cusimano V., Fornaro M., Bonvallet R., Formoso L., Giacomello E., Jacquemond V., Sorrentino V. (2011) Junctophilin 1 and 2 proteins interact with the L-type Ca2+ channel dihydropyridine receptors (DHPRs) in skeletal muscle. J. Biol. Chem. 286, 43717–43725 [DOI] [PMC free article] [PubMed] [Google Scholar]