

Background: Apolipoprotein C-II (apoC-II) is the cofactor for lipoprotein lipase (LPL), the central enzyme for plasma triglyceride metabolism.
Results: ApoC-II adopts two conformations at lipid surfaces that are favored at different surface pressures.
Conclusion: Conformational rearrangement of apoC-II at lipoprotein surfaces promotes interaction with LPL.
Significance: Our mechanism explains how apoC-II withstands increases in surface pressure during LPL-mediated lipoprotein metabolism.
Keywords: apolipoprotein, lipase, lipid droplet, lipoprotein metabolism, low-density lipoprotein (LDL), triglyceride
Abstract
Apolipoprotein C-II (apoC-II) is the co-factor for lipoprotein lipase (LPL) at the surface of triacylglycerol-rich lipoproteins. LPL hydrolyzes triacylglycerol, which increases local surface pressure as surface area decreases and amphipathic products transiently accumulate at the lipoprotein surface. To understand how apoC-II adapts to these pressure changes, we characterized the behavior of apoC-II at multiple lipid/water interfaces. ApoC-II adsorption to a triacylglycerol/water interface resulted in large increases in surface pressure. ApoC-II was exchangeable at this interface and desorbed on interfacial compressions. These compressions increase surface pressure and mimic the action of LPL. Analysis of gradual compressions showed that apoC-II undergoes a two-step desorption, which indicates that lipid-bound apoC-II can exhibit at least two conformations. We characterized apoC-II at phospholipid/triacylglycerol/water interfaces, which more closely mimic lipoprotein surfaces. ApoC-II had a large exclusion pressure, similar to that of apoC-I and apoC-III. However, apoC-II desorbed at retention pressures higher than those seen with the other apoCs. This suggests that it is unlikely that apoC-I and apoC-III inhibit LPL via displacement of apoC-II from the lipoprotein surface. Upon rapid compressions and re-expansions, re-adsorption of apoC-II increased pressure by lower amounts than its initial adsorption. This indicates that apoC-II removed phospholipid from the interface upon desorption. These results suggest that apoC-II regulates the activity of LPL in a pressure-dependent manner. ApoC-II is provided as a component of triacylglycerol-rich lipoproteins and is the co-factor for LPL as pressure increases. Above its retention pressure, apoC-II desorbs and removes phospholipid. This triggers release of LPL from lipoproteins.
Introduction
Human apolipoprotein C-II (apoC-II)2 is a 79-amino acid protein that is synthesized in the liver and intestine and circulates in plasma as a component of high-density lipoproteins (HDLs), very low-density lipoproteins (VLDLs), and chylomicrons (1–3). ApoC-II plays an essential role in the metabolism of plasma lipid as the cofactor for lipoprotein lipase (LPL) (4, 5). LPL hydrolyzes triacylglycerol (TG) and produces fatty acids and monoacylglycerol for uptake by adipose, muscle, and other tissues (6). LPL activity produces remnant particles from VLDL and chylomicrons, having ≤50% of the TG in substrate particles (7, 8). Defects in the structure or production of apoC-II and some other gene products share the phenotype of LPL deficiency in humans, hypertriglyceridemia and chylomicronemia (1, 9, 10).
Although the molecular details of the mechanism of LPL activation by apoC-II are largely unknown, significant progress has been made in elucidating the interactions between apoC-II, LPL, and lipid required for LPL activation. The N-terminal region of apoC-II is responsible for lipid binding, whereas the C-terminal one-third of the molecule contains the structures needed for LPL activation, as shown by in vitro assays using synthetic fragments of apoC-II, lipid substrates, and LPL (11–15). Lipid-free apoC-II has little α-helical content, as shown by far-UV circular dichroism (CD) (16). Binding of apoC-II to micelles of SDS or dodecylphosphocholine induces α-helical structure in 60% of the apoC-II sequence (17–20).
NMR structural analyses of apoC-II bound to SDS micelles showed helical structure in both the N- and C-terminal regions of the molecule (17, 19). Residues 14–38 form a class A amphipathic α-helix (Fig. 1), which is characterized by a large (30–50%) apolar face subtending less than 180°, positively charged residues at its polar/nonpolar interface, and negatively charged residues along its polar face (21–23). This type of helix is well suited for binding lipid. Residues 50–54 (central helix) and 63–76 (C-terminal helix) of apoC-II form class G amphipathic α-helices (Fig. 1), which are characterized by a random radial distribution of positively and negatively charged residues on the polar face (23). The C-terminal helix exhibited significantly higher levels of motion on the nanosecond time scale than the N-terminal helix (19). This suggests that the C-terminal helix can disengage from lipid surfaces, and this motion is supported by the less perfect helical structure of residues 58–63 (17, 19). The region spanning residues 40–65 is disordered in NMR structures of apoC-II·dodecylphosphocholine complexes (18), which suggests that helical structure formed in this region varies with different lipid-bound conformations of apoC-II molecules.
FIGURE 1.
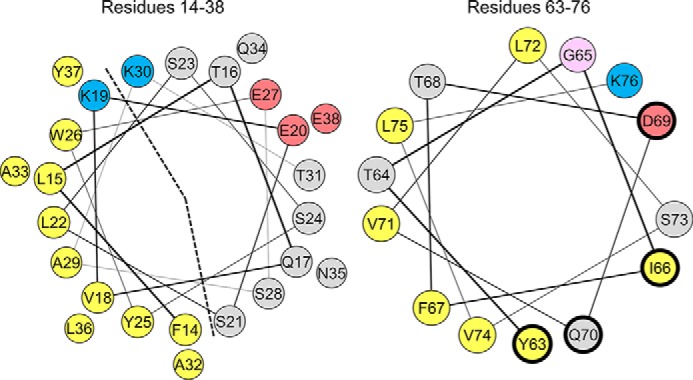
Helix wheel diagrams of the large N-terminal (left) and C-terminal (right) α-helices in lipid-bound apoC-II. The N-terminal helix (left) is a lipid-binding class A amphipathic helix, characterized by an apolar face (marked by a dashed line) of nine hydrophobic residues (yellow), basic residues (blue) at the polar/apolar lipid/water interface, and acidic residues (red) along the polar face. The C-terminal helix (right) is a class G helix, characterized by a random radial distribution of basic (blue) and acidic (red) residues. The four residues that interact with LPL (Tyr-63, Ile-66, Asp-69, and Gln-70) have a thicker, dark blue edge. Polar residues are gray, and glycine is pink.
Four residues (Tyr-63, Ile-66, Asp-69, and Gln-70) located on the same face of the C-terminal helix and exposed to solvent in NMR structures form a binding site for LPL (19, 24, 25). In vitro LPL activity assays showed that replacement of any of these four residues with alanine decreases the affinity of apoC-II for LPL and the catalytic activity of the LPL·apoC-II complex (24). Replacement of all four residues with alanine inhibits LPL activity. Disruption of the secondary structure of the C-terminal helix in apoC-II also impairs LPL activation (25). Together, these results indicate that the C-terminal helix forms an LPL binding site, whereas the N-terminal helix of apoC-II binds lipid. In vitro assays with LPL, apoC-II-deficient chylomicrons, and apoC-II peptides show that lipid binding of the N-terminal helix is required for the activation of LPL (26). In a recent study, a mimetic peptide that contained the C-terminal helix of apoC-II and a lipid-binding class A amphipathic helix was designed (27). This peptide enhanced cholesterol efflux from ABCA1-transfected cells and stimulated LPL-mediated lipolysis in several systems.
ApoC-II is not required for binding of LPL to lipid surfaces (14, 15, 28). Rather, apoC-II regulates LPL activity at lipid surfaces in ways that are highly dependent on surface pressure, as shown by surface chemistry studies (14, 15). Surface pressure (Π) correlates with the density of amphipathic molecules at lipid surfaces and is defined as the difference between the energies required to maintain various lipid/water interfaces (29, 30). As LPL hydrolyzes TG and produces amphiphilic fatty acids and monoacylglycerols, these transiently accumulate increasing surface lipid density and surface pressure (31). LPL is active at mixed TG/phospholipid and pure phospholipid monolayers of lower surface pressures but requires apoC-II for activity above a critical pressure (14, 15). These results and others (28) suggest that apoC-II interacts with LPL to induce correct alignment of the complex at the lipoprotein surface and facilitate substrate exposure to the catalytic site at higher pressures.
We hypothesize that the lipid-bound conformation of apoC-II strongly depends on surface pressure. As lipoprotein surface pressure increases, the surface area per lipid-bound apoC-II molecule decreases (32). Protein conformation probably changes, and mobility of the C-terminal helix may allow this region to desorb from lipid and interact with LPL. We speculate that the ability of apoC-II to remain bound to remnants of VLDL or chylomicrons as pressure increases during TG hydrolysis limits LPL activity. Above the retention pressure of apoC-II, protein molecules desorb from the remnants and transfer to HDL (33). Transfer between lipoproteins is negatively cooperative, such that apoC-II desorbs as steric crowding of protein and lipid molecules becomes too great on the lipid surface (34).
To probe the relation between apoC-II and lipoprotein surface pressure, we used oil-drop tensiometry (30, 35) to characterize the behavior of human apoC-II at triolein/water (TO/W) and 1-palmitoyl-2-oleoyl-phosphatidylcholine/triolein/water (POPC/TO/W) interfaces. TO is a common TG, and POPC is the most abundant phospholipid on lipoproteins. The TO/W interface provides a model for protein interactions with a TG core, a component of all TG-rich lipoproteins. POPC/TO/W interfaces more closely resemble the surface of nascent, cholesterol-poor lipoproteins (36). ApoC-II adsorption to TO/W and POPC/TO/W interfaces induces changes in surface pressure, reflective of the ability of apoC-II to remodel the lipoprotein surface (30–32, 37). Expansion and compression of these interfaces mimic the pressure changes induced by LPL activity (31, 32). Gradual compression of these interfaces reveals the pressures at which lipid-bound apoC-II molecules undergo conformational rearrangements (30–32, 37). These conformational rearrangements were probed by study of N- and C-terminal apoC-II fragments.
Experimental Procedures
Expression and Purification of Apolipoprotein
Human apoC-II cDNA was cloned into pET29a(+) followed by an N-terminal His6 tag (24). The pET-histag-hapoC-II plasmid was transformed into Epicurian coli BL21-condonPlus (DE3)-RIL competent cells (Stratagene) as described previously (24, 25, 38). Transformed bacteria were grown at 37 °C until A600 = 1.0 (24). Temperature was lowered to 16 °C, and after 1.5 h, expression of human apoC-II was induced overnight with 0.5 mm isopropyl 1-thio-β-d-galactopyranoside. Bacteria were harvested by centrifugation (3800 rpm (Beckman J-6B) for 20 min at 4 °C), and cell lysis was performed using denaturing conditions followed by purification by nickel-nitrilotriacetic acid-agarose as described previously (24, 38). Briefly, use of buffers containing 8 m urea, 0.1 m NaH2PO4, and 0.01 m Tris-Cl produced a discontinuous gradient from pH 8.0 to 4.5 (38). Fractions with high protein content (eluting at different pH) were pooled and dialyzed against 6 m urea, 10 mm Tris, pH 8.0, followed by DEAE-ion exchange chromatography using a continuous Tris gradient ranging from 10 to 115 mm in 6 m urea, pH 8.2. Fractions with the expected molecular weight of histag-hapoC-II, as judged by SDS-PAGE, were pooled and dialyzed for 24 h at 4 °C against ammonium bicarbonate and lyophilized.
Human apoC-III cDNA was cloned into pET23b followed by a C-terminal His6 tag (39). This plasmid was expressed, and apoC-III protein was purified as described previously (40). For both apoC-II and apoC-III, lyophilized protein was divided into small aliquots and frozen at −20 °C. The protein was over 95% in purity, as determined by SDS-polyacrylamide gels, MALDI-TOF mass spectrometry, and high performance liquid chromatography (HPLC).
Prior to interfacial studies, aliquots of apoC-II or apoC-III were thawed and solubilized with hexafluor-2-proponal (HFIP) at a concentration of 2.5 mg/ml. For interfacial studies, protein was injected into 7 ml of phosphate buffer at concentrations of 0.2–12.5 μg/ml. At these protein concentrations, HFIP represented 0.005–0.5% of the total volume. At protein concentrations at which most experiments were conducted (<8 μg/ml), HFIP had no effect on interfacial tension. At protein concentrations of >8 μg/ml, HFIP decreased interfacial tension by 1–2 mN/m. Protein concentration was determined by the Lowry protein assay and absorption at 280 nm. Far-UV circular dichroism showed that lipid-free apoC-II (at a concentration of 0.02 mg/ml in buffer with 0.8% HFIP) had 21 ± 5% α-helix, which is consistent with previous results (24, 41).
Synthesis of ApoC-II Peptide Fragments
Peptide fragments representing the 25 residues of the N-terminal helix (residues 14–38; FLTQVKESLSSYWESAKTAAQNLYE) and 30 residues of the C-terminal region (residues 50–79; RDLYSKSTAAMSTYTGIFTDQVLSVLKGEE) of apoC-II (Fig. 1) were obtained commercially by solid state synthesis and purified by HPLC to >91% purity at 21st Century Biochemicals. The N termini of the peptides were acetylated. The C termini of the 25-residue fragments were amidated. The identity and purity of the proteins were confirmed by collision-induced dissociation tandem mass spectrometry. Prior to interfacial studies, aliquots of peptide were solubilized at a concentration of 2 mg/ml in solutions of HFIP and 5 mm sodium phosphate buffer at a 1:4 (v/v) ratio.
Lipids
Triolein (>99% pure) was purchased from NU-CHEK PREP, Inc. (Elysian, MN). Its interfacial tension was ∼32 mN/m at 25.0 ± 0.2 °C. POPC, dissolved in chloroform at 25.0 mg/ml, was purchased from Avanti Polar Lipids (Alabaster, Alabama) and stored at −20 °C. Purity of both lipids was checked using high-dose thin layer chromatography. POPC was dried under nitrogen and resuspended in 5 mm sodium phosphate buffer, pH 7.4, at 2.5 mg/ml. POPC in buffer was sonicated for 60 min with a pulsed duty cycle of 30% to form small unilamellar vesicles (SUVs) with diameter of ∼22 nm (42, 43).
Interfacial Tension (γ) and Surface Pressure (Π) Measurements
An oil-drop tensiometer designed by Teclis Instruments (Longessaigne, France) was used to measure the γ of lipid/water interfaces (30, 35). γ is the energy required to create one new cm2 of surface (i.e. γ = ergs/cm2 or mN/m) and is higher for apolar versus polar interfaces (29, 30). All tensiometry experiments were conducted at 25.0 ± 0.2 °C in a thermostated system and repeated at least three times.
To create TO/W interfaces, triolein drops of 16 μl were formed at the tip of a J-needle submerged in 7 ml of bulk buffer. The bulk buffer was 5 mm sodium phosphate buffer at a pH of 7.4 or 5.9. TO/W interfaces stabilized at γTO = 32.0 ± 1.0 mN/m, independent of pH. Adsorption of amphipathic molecules (i.e. phospholipid and apolipoprotein) to this interface shields TO from the aqueous phase and decreases γ. To create POPC/TO/W interfaces, triolein drops of 16 μl were formed in bulk buffer containing 0.75–1.25 mg of POPC. After POPC adsorbed to the triolein drop, the buffer was exchanged with 250 ml of POPC-free buffer, as described previously (42). This washout removed >99.9% of the original buffer and all POPC SUVs suspended in the bulk phase. For POPC/TO/W interfaces after a washout, γ = 23.0–25.5 mN/m.
Varied amounts of apoC-II were added to the bulk phase to obtain different protein concentrations (0.2–12.5 μg/ml). ApoC-II is monomeric at these concentrations over the time scales of our experiments (16, 41). ApoC-II adsorbed to TO/W and POPC/TO/W interfaces, and γ was monitored continuously as it fell to an equilibrium value (γeq) (Figs. 2A and 3A). Surface pressure (Π) was defined as the difference in γ between a pure TO/W interface (γTO = 32.0 mN/m) and the interface with bound POPC and/or protein (Π = γTO − γ). The initial pressures (Πi) of POPC/TO/W interfaces were calculated as the difference between γTO and γafter POPC adsorption, whereas Πi of a TO/W interface was 0 mN/m. After apoC-II adsorption, the equilibrium pressure of TO/W and POPC/TO/W interfaces was calculated as Πeq = γTO − γeq. The change in pressure (ΔΠ) of lipid/water interfaces due to apoC-II adsorption was ΔΠ = Πeq - Πi.
FIGURE 2.
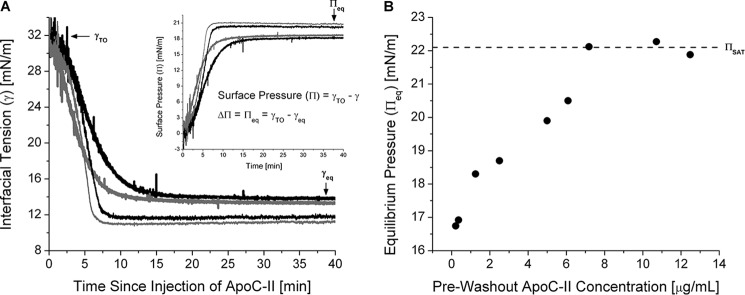
Adsorption of apoC-II leads to significant remodeling of TO/W interfaces. A, interfacial tension (γ) versus time curves for apoC-II at a TO/W interface. TO drops (16 μl) were formed in 7.0 ml of sodium phosphate buffer. ApoC-II was added at concentrations of 1.25 (black lines) or 2.5 μg/ml (gray lines) to a bulk phase of pH 7.4 (thick lines) or 5.9 (thin lines). γ was monitored continuously as it fell from γ of a clean TO/W interface (γTO = 32.0 ± 0.5 mN/m) to γeq. Inset, γ values were converted to surface pressures (Π) to determine the change in Π (ΔΠ) due to apoC-II adsorption. ΔΠ is equivalent to equilibrium pressure (Πeq) at a TO/W interface. B, Πeq increases to a saturation value (ΠSAT) as the apoC-II bulk phase concentration increases. ApoC-II was added at various concentrations to a bulk phase of pH 7.4, and γ was monitored as it fell to γeq. Values for γeq were converted to Πeq and plotted against the bulk phase concentration of apoC-II. Dashed line, ΠSAT.
FIGURE 3.
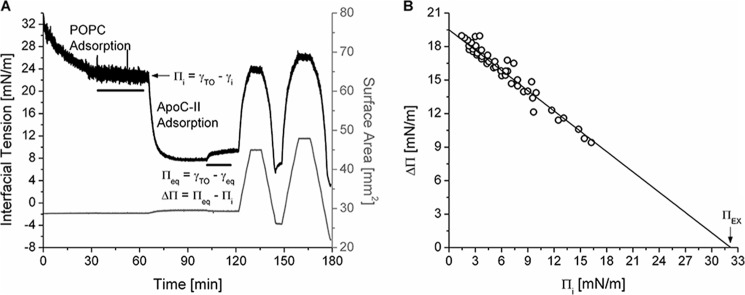
ApoC-II has a high ΠEX at POPC/TO/W interfaces. A, protocol to monitor apoC-II adsorption to and desorption from a POPC/TO/W interface. POPC adsorbed to a 16-μl TO drop and lowered γ (black line). POPC was removed from the bulk phase by a 250-ml washout (black bar, 8.35 ml/min for 30 min starting at 33.4 min). After the washout, γ was 22.5 mN/m (Πi = 9.5 mN/m). ApoC-II was added to the bulk phase at a concentration of 1.25 μg/ml, and γ was monitored as it fell to γeq. ApoC-II was removed from the bulk phase by a 150 ml washout (black bar, 8.35 ml/min for 18 min starting at 101.7 min). The drop volume underwent two sets of gradual expansions and compressions at a rate of 1.2 μl/min (shown as recordings of surface area in mm2, gray line). The volume reached upper limits of 30.1 and 33.0 μl (area = 44.9, 47.9 mm2) for expansions and lower limits of 13.5 and 9.0 μl (area = 26.1, 22.0 mm2) for compressions. B, apoC-II has a ΠEX of 32.2 mN/m. For a series of experiments similar to A over a range of Πi, ΔΠ was plotted against Πi. Linear regression of the data was significant (R2 = 0.95). The x intercept is ΠEX.
Exclusion Pressure (ΠEX) Measurement
ΠEX for apoC-II is the surface pressure above which the protein cannot bind and insert into POPC/TO/W interfaces (42, 44). With respect to tensiometry experiments, ΠEX is the pressure of a POPC/TO/W interface at which the addition of apoC-II to the bulk phase leads to no adsorption-induced change in surface pressure (ΔΠ = 0 mN/m).
After POPC adsorption and a 250-ml washout, the initial pressure of POPC/TO/W interfaces was Πi = 5.5–9.5 mN/m. To achieve a larger range of Πi values, TO drop volume was increased or decreased at a rate of 1.2 μl/min, decreasing or increasing Π to new Πi values (Fig. 3). ΔΠ values from apoC-II adsorption were plotted against Πi (Fig. 3B). Linear regression was applied to the data, and the x intercept represented ΠEX (i.e. the Πi at which ΔΠ = 0 mN/m). The surface concentration of POPC (ΓPOPC) was also calculated from the Πi of each POPC/TO/W interface, as described previously (42).
Compression and Expansion of Lipid/Water Interfaces
As an exchangeable apolipoprotein, bound apoC-II should desorb from lipid/water interfaces. Two protocols were used to characterize desorption of apoC-II from TO/W and POPC/TO/W interfaces.
The first protocol involved a series of rapid compressions and expansions of the lipid/water interfaces. After apoC-II adsorption lowered γ of either type of interface to γeq (increased Π to Πeq), the interface underwent a series of compressions and expansions as TO drop volume (16 μl initially) was rapidly decreased or increased by 1.3–8 μl (Figs. 4A and 9A). Decreases in volume, or compressions, correlated with decreases in surface area (A) and resulted in abrupt decreases in γ. After each compression, the TO drop was held at reduced volumes for >5 min. As apoC-II desorbed from the surface, γ rose toward new equilibrium values (observed as desorption curves). Increases in volume back to 16 μl upon interfacial expansions correlated with increases in surface area and resulted in abrupt increases in γ to values above the initial γeq. ApoC-II re-adsorbed from the bulk phase and γ fell toward the initial γeq (observed as readsorption curves).
FIGURE 4.
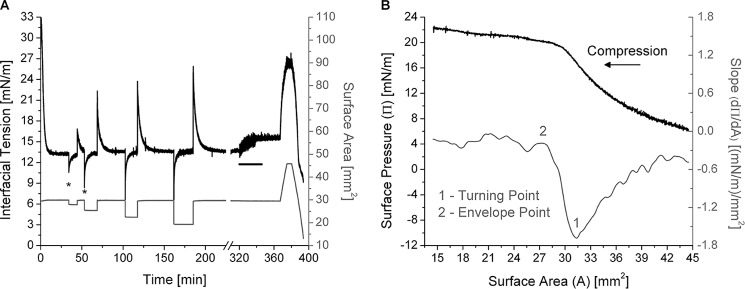
Desorption of apoC-II from a TO/W interface is a two-step process. A, example of rapid and gradual compressions of apoC-II/TO/W interfaces. ApoC-II was added to the aqueous phase at 2.5 μg/ml. After apoC-II adsorption to a TO drop (V = 16 μl) lowered γ to γeq = 13.3 mN/m, the drop volume was rapidly compressed and re-expanded in increments of ±1.5, ±3.3, ±5.3, and ±7.4 μl. Asterisks mark the minimum tension for the first two compressions. ApoC-II was then removed from the bulk phase by a 150-ml washout (black bar, 8.35 ml/min for 18 min starting at 320 min), and γ increased to 15.4 mN/m. From 367 to 374.8 min, drop volume was expanded at 1.2 μl/min to 30.3 μl (area = 45.8 mm2). From 380 to 393.8 min, drop volume was compressed at 1.2 μl/min to 5.5 μl (area = 14.0 mm2). B, the compression Π/A isotherm of apoC-II/TO/W interfaces shows two significant inflection points. From the experiment in A, γ values during the gradual, post-washout compression were converted to Π values and plotted against area (black line). The direction of compression is indicated by an arrow. A spline fit of smoothing parameter 0.5 was applied to this Π/A isotherm. The first derivative of the fit gave slope values (dΠ/dA), which were plotted against area (gray line). As area decreased, the slope profile featured a minimum at the turning point (1) and the start of a plateau at the envelope point (2), which both indicate a relaxation in Π due to partial or complete desorption of apoC-II molecules.
FIGURE 9.

Upon desorption, apoC-II removes POPC from PC/TO/W interfaces. A, example of rapid compressions and expansions of an apoC-II/PC/TO/W interface. POPC SUVs with 1–2% PNPC, a fluorescently labeled derivative of POPC, adsorbed to a 16-μl TO drop and lowered γ. POPC/PNPC was removed from the bulk phase by a 250-ml washout (black bar, 8.35 ml min for 30 min starting at 40 min). After the washout, γi = 24.7 mN/m (Πi = 7.3 mN/m). At 71.1 min, TO drop volume was increased by 3 μl to V = 19.0 μl. Πi decreased to 5.5 mN/m. ApoC-II was added to the aqueous phase at 1.25 μg/ml, and γ fell to γeq = 9.1 mN/m (dotted line). The TO drop was compressed and re-expanded in increments of ±1.6, ±2.7, ±3.7, ±4.7, and ±7.8 μl. Upon each re-expansion, γ fell to γeq > 9.2 mN/m. B, fluorescent signal from PNPC in the bulk phase increases as apoC-II removes PC lipid from the PC/TO/W interface. 3-ml aliquots were removed from the bulk phase at points 1–3 in A. These aliquots were placed in a FluoroMax-2, and the emission spectrum from 490 to 600 nm (λem = 533 nm for PNPC) was captured with λex = 474 nm. The emission spectra from aliquot 1 (solid line), before apoC-II was added, and from aliquot 3 (dashed line), after a series of rapid compressions and expansions, are shown. Dotted line, difference between the two spectra. Aliquot 2 had the same spectrum as aliquot 1. Inset, ApoC-I and apoC-III remove relatively little POPC. The net change in emission spectra of aliquots from the bulk phase between peptide injection and interfacial compressions and expansions was determined as in B for the other apoCs. Each apoC was added to the bulk phase at the same concentration (1.25 μg/ml), adsorbed to interfaces of similar ΓPOPC (34.5 + 1.3%), and ejected by the same number and amplitudes of compressions.
The second protocol involved the gradual expansion and compression of TO/W or POPC/TO/W interfaces. For interfaces of various Πi, apoC-II was added to the bulk phase, and protein adsorption to the interface lowered γ to γeq. Buffer was exchanged with 150 ml of protein-free buffer, removing apoC-II from the bulk phase. Drop volume was increased at a rate of 1.2 μl/min to V > 30 μl (Figs. 3–5). Volume was held constant for >2 min and decreased at 1.2 μl/min to various terminal volumes. Pressure-area (Π-A) isotherms were calculated from the γ and surface area (A) profiles of each compression and expansion, as described previously (42).
FIGURE 5.
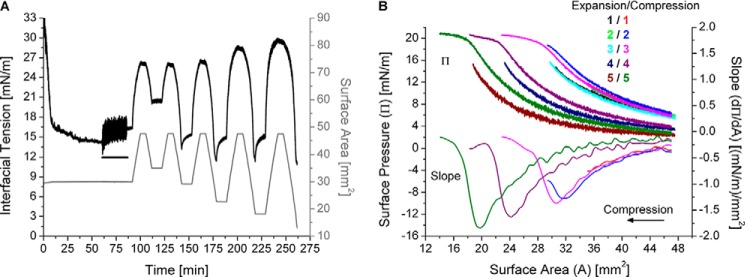
ApoC-II molecules only leave a TO/W interface above the retention pressure (ΠENV). A, example of gradual, post-washout expansions and compressions of apoC-II/TO/W interfaces. ApoC-II was added to the aqueous phase at 1.25 μg/ml. After apoC-II adsorption to a TO drop (V = 16 μl) lowered γ to γeq = 14.4 mN/m, apoC-II was removed from the bulk phase by a 150-ml washout (black bar, 6.26 ml/min for 25 min starting at 60.8 min). Drop volume underwent a series of five gradual expansions and compressions at a rate of 1.2 μl/min. Each expansion ended at V = 31.8 μl, A = 47.6 mm2. Each compression ended at a smaller volume (i.e. area) than the previous compression(s), ending at areas of 35.0, 29.2, 22.7, 18.2, and 13.1 mm2. B, apoC-II molecules partially desorb before completely desorbing from a TO/W interface. From the experiment in (A), γ values during the expansions and compressions were converted to Π values and plotted against area (top set of curves). The expansions and compressions are labeled by color and number, starting at 1 for the first post-washout expansion and compression. A spline fit of smoothing parameter 0.5 was applied to the compression Π/A isotherms. The first derivative of the fits gave slope values (dΠ/dA), which were plotted against area (bottom five curves).
Measurement of Retention Pressures (ΠENV)
The retention pressure of apoC-II at a lipid/water interface is the maximum surface pressure apoC-II molecules withstand before they desorb from the interface. Our laboratory has used envelope pressure (ΠENV) to refer to retention pressure (i.e. the highest pressure at which a peptide can be retained on an interface before being ejected) (31, 45).
To determine ΠENV values of apoC-II at TO/W and POPC/TO/W interfaces, the Π-A isotherms from gradual interfacial expansions and compressions were analyzed (Figs. 4–6). A spline fit (i.e. a piecewise polynomial) with a smoothing parameter between 0.25 and 0.6 was applied to each isotherm (Figs. 4B, 5B, and 6A). The slope of the isotherm was determined by calculating the first derivative of the spline fit, using Matlab's Curve Fitting toolbox. The slope (dΠ/dA) of compression isotherms to areas smaller than the retention area (AENV) followed this trend. 1) The slope (dΠ/dA) decreased as the change in pressure (dΠ) became more negative for incremental decreases in area (dA). This occurred because vacant binding sites present on the expanded interface were eliminated, and the density of surface-active amphipathic molecules increased. 2) The slope reached a minimum. As area continued to decrease, dΠ/dA increased as dΠ became less negative for each dA. This minimum represented a turning point (AP, ΠP), above which bound apoC-II molecules at least partially desorbed to relax the surface and reduce the dΠ for each dA. 3) The slope reached a plateau near dΠ/dA = 0. This point represented the envelope point (AENV, ΠENV), above which bound apoC-II molecules completely desorbed from the surface. Increases in pressure were minimized (dΠ ≈ 0 mN/m) as area decreased further, indicating that peptide was leaving the surface phase and partitioning to the bulk phase. All of the tensiometry protocols described above were repeated for human apoC-III for the sake of comparison.
FIGURE 6.
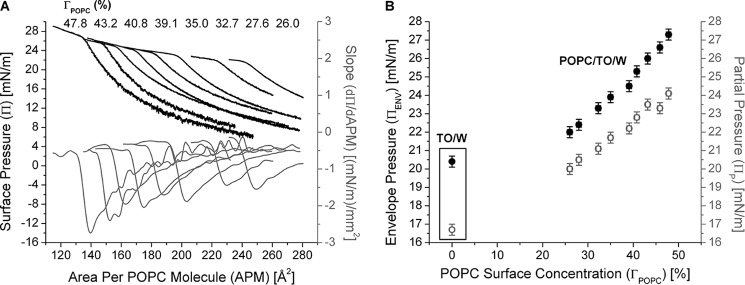
ApoC-II molecules desorb from POPC/TO/W interfaces at higher pressures with greater POPC/ApoC-II ratios. A, the retention areas of apoC-II decrease as ΓPOPC increases. Experiments similar to those in Fig. 3A were conducted over a range of Πi values. Πi was converted to ΓPOPC, as described previously (42). γ values during compression were converted to Π values and plotted against the area per POPC molecule (APM), which was calculated as described previously (42). The ΓPOPC for each compression isotherm, from left to right, is listed at the top of the graph. B, the pressures at which apoC-II molecules partially (ΠP) and completely (ΠENV) desorb from POPC/TO/W interfaces increase as ΓPOPC increases. For each compression isotherm in Fig. 6A, ΠP and ΠENV values were calculated as described in the legend to Fig. 4B. ΠP (gray, open circles) and ΠENV (black, filled circles) were plotted as a function of ΓPOPC. For comparison, data for Πp and Πenv for apoC-II are shown within the frame for a TO/W interface. x and y error bars, S.D. from n = 2–5 experiments.
Results
ApoC-II Binds to Lipid/Water Interfaces with High Affinity Resulting in Large Surface Pressure Modifications
This study characterized the effects of apoC-II adsorption on the surface pressure of lipoprotein-like lipid/water interfaces, which reflect the ability of apoC-II to bind and insert into the interfaces. Fig. 2A shows a set of interfacial tension-time curves for apoC-II at a TO/W interface. Tension values were converted to surface pressures (Fig. 2A, inset). For bulk phase concentrations of 1.25 and 2.5 μg/ml, apoC-II adsorption increased surface pressure by ΔΠ = 18.2–19.0 mN/m.
The ability of apoC-II to modify a TO/W interface increased with lower bulk phase pH (Fig. 2A) or with greater bulk phase concentration (Fig. 2B). If the pH of the bulk phase was decreased to approach the isoelectric point of apoC-II (pI = 5.6), apoC-II adsorption to a TO/W interface resulted in greater ΔΠ. At an apoC-II bulk phase concentration of 1.25 and 2.5 μg/ml, ΔΠ increased to 20.1–20.7 mN/m (Fig. 2A, inset). The change in pH had no effect on γ of a clean TO/W interface.
As the concentration of apoC-II was increased in the bulk phase, Πeq increased up to a saturation pressure of ΠSAT = 22.1 mN/m at concentrations of ≥7.2 μg/ml (Fig. 2B). This result suggests that lipid-free and lipid-bound forms of apoC-II are in a concentration-dependent equilibrium that influences Πeq (32, 46). As the concentration of lipid-free apoC-II increases, the concentration of lipid-bound apoC-II increases until, at saturation, no binding sites are available at the lipid surface. A pH of 7.4, reflective of the pH of plasma, was used for the remainder of experiments unless otherwise noted.
The ability of apoC-II to remodel POPC/TO/W interfaces was determined for several ΓPOPC values. After POPC adsorption to a TO drop and a washout to remove POPC in the bulk phase, the Πi could be changed by increasing or decreasing drop volume (Fig. 3A). Πi was used to determine ΓPOPC in units of μmol/m2 or as a percentage of TO drop coverage by POPC, as described previously (42). ΔΠ values were recorded for apoC-II adsorption to interfaces of various ΓPOPC and plotted as a function of Πi (Fig. 3B). A linear fit was applied to the data, and the x intercept represents the ΠEX. At Π ≥ ΠEX, lipid-free apoC-II cannot insert into POPC/TO/W interfaces, and ΔΠ = 0 mN/m. The ΠEX of apoC-II was 32.2 mN/m, which is similar to ΠEX of apoC-II at an egg PC/air interface (∼30 mN/m) (15).
ApoC-II Desorbs from Lipid/Water Interfaces at High Surface Pressures
This study also characterized the effect of increases in surface pressure on the ability of apoC-II to remain bound to lipoprotein-like lipid/water interfaces, which mimics the response of apoC-II to LPL activity at the lipoprotein surface (15, 31). Our results indicate that apoC-II dissociates from a TO/W interface upon instantaneous increases in Π. Fig. 4A shows γ and area changes during rapid compressions and expansions of an apoC-II/TO/W interface. Rapid compressions in volume correlated with decreases in surface area and resulted in sharp decreases in surface tension (marked by asterisks for the first two compressions in Fig. 4A). In the 10–15 min after each compression, γ gradually returned to the initial equilibrium, γeq. This suggests that apoC-II molecules desorb from compressed interfaces.
Re-expansions from compressed volumes correlated with increases in surface area. This resulted in sharp increases in surface tension to values higher than γeq, which indicates that vacant binding sites are present on the re-expanded surface. As apoC-II from the bulk phase adsorbed to these binding sites, γ fell to the initial γeq. The γ profile was markedly different for rapid compressions and expansions of apoC-II/TO/W interfaces after apoC-II was removed from the bulk phase by a washout (data not shown). After each compression, γ rapidly decreased and then slowly increased as apoC-II desorbed from the surface. After each expansion, γ rapidly increased above γeq and remained at elevated levels because no apoC-II was present in the aqueous phase to bind to the newly exposed binding sites. Altogether, these results suggest that apoC-II molecules desorb from lipid/water interfaces as Π increases above the retention pressure of the protein.
The retention or envelope pressure of apoC-II at a lipid/water interface is the maximum surface pressure that apoC-II molecules can withstand before they desorb from the interface. Because the rate of increases in Π correlates with the rate of interfacial compression, retention pressure is most directly observed from gradual compressions. The right-hand side of Fig. 4A shows γ and area changes during the gradual, post-washout expansion and compression of an apoC-II/TO/W interface. After a 150-ml washout, γ gradually increased (Π decreased) as the TO drop volume was expanded at a rate of 1.2 μl/min from 16 to 30.3 μl. This indicates that more TG is exposed at the surface as the drop is expanded, increasing the energy cost (γ) to maintain the TO/W interface.
In contrast, γ gradually decreased (Π increased) as the TO drop volume was compressed at a rate of 1.2 μl/min from 30.3 to 5.5 μl. The γ values during compression were converted to Π values and plotted against surface area (Fig. 4B). The slope (dΠ/dA) of the compression Π/A isotherm was determined and plotted against area (Fig. 4B). As area decreased, dΠ/dA decreased to a minimum value of −1.7 (mN/m)/mm2 at A = 31.7 ± 0.3 mm2 (point 1 in Fig. 4B) and then increased as area decreased to 27.0 mm2. For A ≤ 27.0 ± 0.3 mm2 (point 2 in Fig. 4B), dΠ/dA approached an asymptote of dΠ/dA = 0 (mN/m)/mm2. Surface areas of 31.7 ± 0.3 and 27.0 ± 0.3 mm2 correspond to pressures of 16.7 ± 0.2 and 20.3 ± 0.3 mN/m on the Π/A isotherm. This experiment was repeated five or more times at different pre-washout concentrations of apoC-II, and the values for these inflection points were the same.
These results suggest that, as area decreases to 31.7 mm2, TG partitions from the surface to the core, and Π increases as γ decreases. As a result, the absolute value of dΠ/dA increases. At A = 31.7 mm2, Π returns to the post-washout Π of the apoC-II interface (see below), which suggests that all vacant binding sites created by expansion have been eliminated. As area decreases to 27.0 mm2, apoC-II molecules partially or completely desorb from the interface to relax the surface. As a result, the absolute value of dΠ/dA decreases. As area decreases further, apoC-II molecules completely desorb from the interface such that dΠ/dA is approximately equal to 0 (mN/m)/mm2.
To probe the nature of apoC-II desorption between A = 31.7 and 27.0 mm2, experiments similar to that in Fig. 4A were conducted but with multiple sets of gradual expansions to V = 31.8 μl and compressions to various, decreasing terminal volumes (Fig. 5). Terminal volumes for the compressions in Fig. 5A correlated with surface areas outside and within the region of interest: 27.0 mm2 ≤ A ≤ 31.7 mm2. The first compression (red) ends at A = 35.0 mm2, and the second compression (blue) ends at A = 29.2 mm2 (Fig. 5B). The third expansion (cyan) overlays with the first expansion (black), which indicates that none of the apoC-II molecules bound to the interface had desorbed from the interface during the first or second compressions. The third compression (magenta) ends at A = 22.7 mm2 and overlays with the first compression (Fig. 5B). The Π/A isotherms and their slopes closely overlay for the first three compressions. These results strongly indicate that part of the lipid-bound apoC-II molecule desorbs between pressures of 16.7 mN/m (ΠP) and 20.3 mN/m (ΠENV), such that Π/A isotherms are reversible to Π < 20.3 mN/m (i.e. the region of the apoC-II molecule that is off of the surface can snap back on during re-expansion).
As Π exceeds 20.3 mN/m, apoC-II molecules desorb from a TO/W interface. With less apoC-II at the lipid/water interface, subsequent expansion and compression Π/A isotherms are markedly shifted to the left (i.e. lower areas). This was observed for the fourth and fifth sets of expansions and compressions in Fig. 5B. Similar results were observed upon repetition of this experiment (n = 4). In sum, these results indicate that part of the apoC-II molecule begins to desorb from a TO/W interface at Π ≥ 16.7 mN/m. The apoC-II molecule completely desorbs at Π ≥ 20.3 mN/m. These pressures and their corresponding areas were identified as partial points (AP, ΠP) and envelope points (AENV, ΠENV) upon compression Π/A isotherms for apoC-II at a TO/W interface in Figs. 4B and 5B. At A ≤ AP, Π ≥ ΠP, apoC-II molecules begin to partially detach from a lipid surface, whereas at A ≤ AENV, Π ≥ ΠENV, apoC-II molecules begin to completely desorb from the surface. The retention pressure (ΠENV) of apoC-II at a TO/W interface is 20.3 ± 0.2 mN/m.
A two-step protein desorption was also evident in the Π/A compression isotherms of apoC-II/POPC/TO/W interfaces (Fig. 6A). Experiments similar to those in Fig. 3A were conducted over a range of Πi and ΓPOPC values. In addition to converting values of γ during compression to Π, area values were converted to area per POPC molecule as described previously (42). This conversion standardized the compression isotherms to the amount of POPC present at POPC/TO/W interfaces. Π/A compression isotherms were plotted for apoC-II/POPC/TO/W interfaces of various ΓPOPC (Fig. 6A). As ΓPOPC increased, Π/A isotherms shifted to the left. This indicates that apoC-II molecules desorb at smaller areas for greater POPC/apoC-II ratios at the lipid surface. The pressures at which apoC-II molecules partially (ΠP) and completely (ΠENV) desorb from POPC/TO/W interfaces were plotted as a function of ΓPOPC (Fig. 6B). Both ΠP and ΠENV increased as ΓPOPC increased. This suggests that POPC increases the affinity of apoC-II, in any conformational state, for the lipid/water interface.
The N Terminus of ApoC-II Mediates Binding to Lipid Surfaces
Compression isotherms (Figs. 4–6) indicate that that lipid-bound apoC-II exhibits various conformations differing in the area occupied at the lipid surface. To test the hypothesis that the N-terminal helix anchors apoC-II to the lipid surface, whereas the C-terminal helix exhibits multiple conformational states, we characterized the adsorption and desorption of peptide fragments at the TO/W interface (Fig. 7).
FIGURE 7.

The N-terminal domain of apoC-II mediates adsorption and desorption at the TO/W interface. A, the C terminus of apoC-II is unable to displace the N terminus. The N-terminal peptide was added to the aqueous phase at 2.85 μg/ml. Adsorption to a TO drop (V = 16 μl) lowered γ to γeq = 18.3 mN/m. The C-terminal peptide was added to the aqueous phase at 25.8 and 49.8 min (marked by arrows) to final concentrations of 2.85 and 5.70 μg/ml. There were no significant changes in γ. Drop volume was rapidly compressed and re-expanded in increments of ±2.1, ±4.0, ±6.0, and ±8.0 μl. In each case, re-adsorption decreased γ to γeq = 18.2 ± 0.1 mN/m. B, the N terminus of apoC-II displaces the C terminus. The C-terminal peptide was added to the aqueous phase at 2.85 μg/ml. Adsorption to the TO/W interface lowered γ to γeq = 23.1 mN/m. The N-terminal peptide was added to the aqueous phase at 31.1 min (marked by an arrow) to a concentration of 2.85 μg/ml. γ decreased to γeq = 18.5 mN/m. All peptide was removed from the bulk phase by a washout (black bar, 6.26 ml/min for 25 min starting at 65.5 min). Drop volume underwent two sets of gradual expansions and compressions at a rate of 1.2 μl/min. C, the N terminus adsorbs faster than the C terminus. The N- and C-terminal peptides were added in equal weight ratios to the aqueous phase at 4.6 μg/ml. Adsorption to the TO/W interface lowered γ to γeq = 18.6 mN/m. The drop volume was rapidly compressed and re-expanded in increments of ±3.8, ±5.7, ±8.0, and ±8.7 μl. In each case, re-adsorption decreased γ to γeq = 18.6 mN/m. D, the N terminus desorbs at higher pressures than the C terminus. From experiments similar to those in B, Π/A compression isotherms were generated for the N and C termini at the TO/W interface. These were plotted with an isotherm for full-length apoC-II. A spline fit of smoothing parameter 0.38 was applied to the isotherms. The first derivative of the fits gave slope values (dΠ/dA), which were plotted against area.
Two peptides were synthesized to model the N- and C-terminal helices of apoC-II, as described under “Experimental Procedures.” Each peptide was added to the aqueous phase at 2.85 μg/ml and adsorbed to a TO drop (Fig. 7). The N-terminal peptide induced ΔΠ = 13.7 mN/m (Fig. 7A), whereas ΔΠ was significantly lower (8.9 mN/m) for the C-terminal peptide (Fig. 7B). These ΔΠ were smaller than that of full-length apoC-II at a similar concentration (ΔΠ = 18.7 mN/m), which indicates that under these conditions, both helices coordinate the lipid binding of apoC-II.
Two sets of experiments were conducted to see if the N-terminal peptide could displace the C-terminal peptide from lipid surfaces. In one set, the C-terminal peptide was added to the aqueous phase after the N-terminal peptide had adsorbed to the TO/W interface and decreased γ to γeq = 18.3 mN/m (Fig. 7A). Tension did not change, which indicates that the C-terminal helix cannot insert into the interface and displace the N-terminal helix. In the second set of experiments, the N-terminal peptide was added to the aqueous phase after adsorption of the C-terminal peptide decreased γ to γeq = 23.1 ± 0.1 mN/m (Fig. 7B). Tension decreased to γeq = 18.5 mN/m, which corresponds to a net ΔΠ = 13.5 mN/m, which mirrors that of N-terminal adsorption to the TO/W interface in the absence of the C-terminal helix. These trends were also observed at POPC/TO/W interfaces (data not shown). These results indicate that the N-terminal helix remodels lipid surfaces with higher affinity, whereas the C-terminal helix is more easily displaced.
To probe adsorption kinetics, the two peptides were added to the aqueous phase simultaneously at the beginning of an experiment (Fig. 7C). Tension of the TO/W interface decreased to γeq = 18.6 mN/m, and the rate of adsorption mirrored that of the N-terminal helix alone. In another experiment (Fig. 7A), the C-terminal peptide was added to the aqueous phase after the N-terminal peptide had adsorbed to the TO/W interface. The final concentration of the C-terminal peptide was 2-fold higher than that of the N-terminal peptide. This interface was rapidly compressed and expanded by various amplitudes to induce desorption of the N-terminal helix (Fig. 7A). On re-expansion, γ decreased to γeq = 18.2 ± 0.1 mN/m with similar kinetics as the initial adsorption of the N-terminal helix. These data indicate that the N-terminal helix adsorbs to lipid surfaces at a faster rate and prevents binding of the C-terminal helix.
To probe the desorption behavior of these peptides, Π/A isotherms were generated from the post-washout, gradual expansion, and compression of peptide/TO/W interfaces. Π/A isotherms for compressions to A<AENV, along with their slope profile, were plotted for each peptide fragment and full-length apoC-II (Fig. 7D). Retention areas and pressures were calculated using both Π/A isotherms and dΠ/dA curves, as described in Figs. 5 and 6. The short peptides desorbed from the TO/W interface at retention areas and pressures smaller than corresponding values for full-length apoC-II. This indicates that the N- and C-terminal helices occupy smaller surface areas than full-length apoC-II at lipid surfaces. The N-terminal peptide desorbed at higher pressures (ΠENV = 11.9 ± 0.3 mN/m) than the C-terminal peptide (ΠENV = 8.4 ± 0.3 mN/m). The slope profile of isotherms for both peptides showed poorly defined minima at dΠ/dA ≈ −0.6 (mN/m)/mm2, 3-fold lower than the corresponding value for full-length apoC-II. Together, these data indicate that the N-terminal helix anchors apoC-II to the lipid surface, and the Π-induced conformational rearrangement of apoC-II requires both helices.
In addition, Π/A isotherms from experiments in which N-terminal peptide was added after C-terminal peptide adsorbed to TO/W or POPC/TO/W interfaces overlaid with isotherms from experiments in which N-terminal peptide adsorbed to peptide-free interfaces (data not shown). These data confirm that the N terminus of apoC-II can displace the C terminus from lipid surfaces.
ApoC-II Exhibits Multiple Conformations at Lipid/Water Interfaces
We further examined the conformational adaptability of apoC-II by analyzing the Π(t) profiles of apoC-II/TO/W interfaces during a washout. After apoC-II adsorption to TO/W or POPC/TO/W interfaces lowered γ to γeq, apoC-II could be removed from the bulk phase by a washout (see “Experimental Procedures”). During washouts, γ of apoC-II/TO/W and apoC-II/POPC/TO/W interfaces increased (Figs. 3–5 and 8), which suggests that apoC-II molecules desorb as the lipid-free concentration of apoC-II is reduced to zero. The γ(t) profiles of apoC-II/TO/W interfaces at various, pre-washout γeq values were recorded during a 150-ml washout and converted to Π(t) profiles (Fig. 8). γeq values were converted to equilibrium pressures (Πeq). Whereas Πeq increased with the pre-washout concentration of lipid-free apoC-II (Fig. 2B), the pressure after a washout (ΠWO) was independent of the pre-washout apoC-II bulk phase concentration or Πeq (Fig. 8 and Table 1). The difference between Πeq and ΠWO increased as Πeq increased, but ΠWO remained constant at 16.6 ± 0.2 mN/m (Table 1).
FIGURE 8.
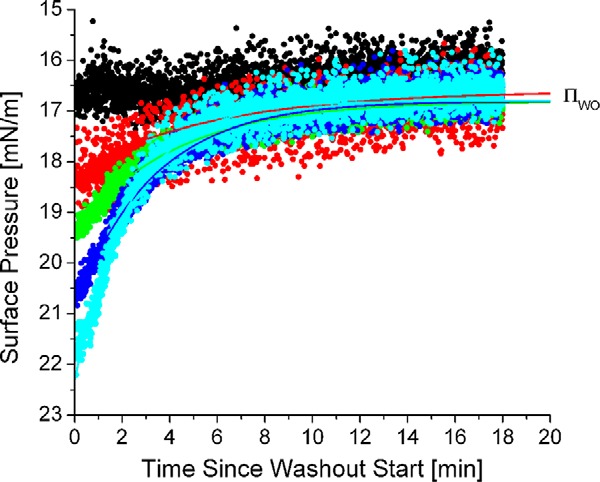
The surface pressure (Π) of apoC-II/TO/W interfaces after a washout (ΠWO) is independent of the pressures before a washout (Πeq). Π(t) profiles during washouts were plotted for five apoC-II/TO/W interfaces of different pre-washout equilibrium pressures (Πeq). Πeq varied from 16.7 mN/m (black) to 21.8 mN/m (cyan). To acquire each Π(t) profile, γ was monitored continuously during a 150-ml washout. The washout rate was 8.35 ml/min. γ values were converted to Π values and plotted against washout time. ΠWO = 16.6 ± 0.2 mN/m, independent of Πeq or washout rate. For Πeq ≥ 18.2, each Π(t) profile was fit with an exponential decay curve of the form Π(t) = (Πeq − ΠWO) × e−kd × t + ΠWO. The exponential decay curves of these Π(t) profiles of apoC-II/TO/W interfaces during a washout are shown in Table 1. R2 values for the curves are listed in Table 1.
TABLE 1.
Dissociation rate of apoC-II from TO/W interfaces during washout
Dissociation rate (kd) of apoC-II from TO/W interfaces during a washout depends on initial surface concentration of apoC-II. A single exponential decay fit was applied to the Π(t) profile of various apoC-II/TO/W interfaces during a 150-ml washout with a rate of 8.35 ml/min. The exponential curve had the form Π(t) = (Πeq − ΠWO)*e−kd*t + ΠWO, where Πeq is the pre-washout equilibrium pressure, ΠWO is the post-washout pressure, and kd is the dissociation rate constant. Experimental values of Πeq and ΠWO (shown on the left) are compared with those predicted from the exponential fit (shown on the right). The data plotted in Fig. 8 are shown in italic type. NA, not applicable.
| ApoC-II pre-washout concentration | Experimental parameters |
Parameters from exponential fit |
|||||
|---|---|---|---|---|---|---|---|
| Πeq | ΠWO | Πeq − ΠWO | ΠWO | Πeq − ΠWO | kd | R2 | |
| μg/ml | mN/m | mN/m | mN/m | mN/m | mN/m | min−1 | |
| 0.2 | 16.7 | 16.2 | 0.5 | NA | NA | NA | NA |
| 1.25 | 18.2 | 16.5 | 1.7 | 16.6 | 1.57 | 0.166 | 0.74 |
| 2.5 | 19.1 | 16.5 | 2.6 | 16.8 | 2.44 | 0.187 | 0.90 |
| 5.0 | 19.3 | 16.8 | 2.5 | 16.8 | 2.57 | 0.237 | 0.93 |
| 6.1 | 20.4 | 16.6 | 3.8 | 16.8 | 4.07 | 0.305 | 0.97 |
| 11.0 | 21.7 | 16.3 | 5.4 | 16.5 | 5.76 | 0.370 | 0.99 |
| 12.5 | 21.8 | 16.7 | 5.1 | 16.8 | 5.51 | 0.433 | 0.97 |
| Averages | 16.6 ± 0.2 | 16.7 ± 0.1 | |||||
The dependence of Πeq of apoC-II/TO/W interfaces on the pre-washout bulk phase concentration of the protein demonstrates that one population of lipid-bound apoC-II is in equilibrium with lipid-free apoC-II (32). The independence of ΠWO from the pre-washout concentration of lipid-free apoC-II demonstrates that a second population of lipid-bound apoC-II is not in equilibrium with lipid-free apoC-II. These two populations may differ in conformation and are in a surface pressure-dependent equilibrium with each other. As the concentration of lipid-free apoC-II approaches zero during a washout, lipid-bound apoC-II molecules in a less exchangeable population remain bound, whereas apoC-II molecules in a more exchangeable population desorb (32). A direct corollary of this conclusion is that lipid-bound apoC-II molecules of the less exchangeable population occupy a larger surface area than molecules of the more exchangeable population. These results support the results of Figs. 4–6, which indicate that lipid-bound apoC-II exhibits multiple conformations. The area and pressure of the lipid surface dictate which conformation apoC-II molecules adapt.
To determine the dissociation rate (kd) of apoC-II from a TO/W interface during a washout, a single exponential was fit to the Π(t) profile of apoC-II/TO/W interfaces during a washout (Fig. 8 and Table 1). The goodness of the exponential fit (R2) increased as the difference between Πeq and ΠWO increased (Table 1). R2 ≥ 0.90 if Πeq − ΠWO ≥ 2.5 mN/m, but R2 = 0.74 for Πeq − ΠWO = 1.7 mN/m. This indicates that a single exponential fits the data better when a greater amount of apoC-II desorbs from the TO/W interface. Consistent with this, the kd increased from 0.17 to 0.40 min−1 as the difference between Πeq and ΠWO increased.
Desorption of ApoC-II Removes Phosphatidylcholines from Lipid/Water Interfaces
We hypothesized that apoC-II removes POPC molecules from POPC/TO/W interfaces as protein molecules desorb from the interface. A previous study at the PC/air/water interface showed that apoC-II, when injected into the subphase, decreased the surface radioactivity of monolayers of egg phosphatidyl[14C]choline (47).
Consistent with these data, apoC-II removed POPC from POPC/TO/W interfaces (Fig. 9). Rapid compression and re-expansion of apoC-II/POPC/TO/W interfaces with apoC-II in the bulk phase resulted in desorption of lipid-bound apoC-II molecules and re-adsorption of lipid-free apoC-II molecules. However, the γeq value after re-adsorption of protein to re-expanded POPC/TO/W interfaces increased for each subsequent compression and re-expansion (Fig. 9A). This suggests that apoC-II removes POPC on desorption from the interface, such that replacement of lipid-bound apoC-II with lipid-free apoC-II after re-expansion results in a net decrease in PC molecules at the interface and net increase in surface tension.
To verify that apoC-II removes POPC, PNPC (a derivative of POPC with an NBD fluorescent label at carbon 12 of its sn-2 acyl chain) was used. PNPC was incorporated at 1–2 weight % of POPC SUVs. These SUVs were used in experiments similar to that shown in Fig. 9A. Aliquots were taken from the bulk phase after phospholipid was removed from the bulk phase (1), after apoC-II adsorbed to the interface (2), and after a cycle of rapid interfacial compressions and expansions (3). Aliquots were analyzed on a fluorimeter for fluorescence of PNPC (Fig. 9B). The first aliquot exhibited low fluorescence, which indicates that most of the phospholipid in the bulk phase was removed by the washout (Fig. 9B, solid line). The second aliquot also exhibited low fluorescence (not shown), which indicates that apoC-II adsorption does not displace POPC from the interface. The third aliquot exhibited a 7–8-fold increase in fluorescence (Fig. 9B, dashed and dotted lines), which indicates that apoC-II desorption removes PC lipids from the interface. In similar experiments, apoC-III removed little to no PC, whereas apoC-I removed <50% of the PC lipid removed by apoC-II (Fig. 9B, inset).
ApoC-III Adsorption and Desorption at Lipid/Water Interfaces Provide Comparisons for ApoC-II Data
We used oil-drop tensiometry to characterize the affinity of apoC-III for TO/W and POPC/TO/W interfaces in order to complement our previous studies on apoC-I (31, 37) and provide comparisons between all three apoCs. Some data for apoC-III are shown in Figs. 10 and 11 and Table 2.
FIGURE 10.

ApoC-III adsorption and desorption from TO/W (A and B) and POPC/TO/W (C and D) interfaces. A, a tension (γ) versus time curve for apoC-III adsorption and desorption at a TO/W interface. A TO drop (16 μl) was formed in 7.0 ml of sodium phosphate buffer, pH 7.4. ApoC-III was added to the bulk phase at a concentration of 1.4 μg/ml. γ was monitored continuously as it fell to γeq due to apoC-III adsorption. ApoC-III was removed from the bulk phase by a 150-ml washout (black bar, 6.26 ml/min for 25 min starting at 45 min), and γ increased to 14.5 mN/m. The drop volume underwent two sets of gradual expansions and compressions at a rate of 1.2 μl/min (shown as recordings of surface area in mm2, gray line). The area reached upper limits of 45.6 and 48.6 mm2 for the two expansions and lower limits of 26.5 and 9.5 mm2 for the compressions. B, Πeq increases to ΠSAT = 22.0 mN/m as the apoC-III bulk phase concentration increases. ApoC-III was added at various concentrations to the bulk phase, and γ of a TO/W interface was monitored as it fell to γeq, as in A. Values for γeq were converted to Πeq and plotted against the bulk phase concentration of apoC-III. ΠSAT is marked by a dashed line. C, a tension (γ) versus time curve for apoC-III adsorption and desorption at a POPC/TO/W interface. POPC adsorbed to a 16-μl TO drop and lowered γ. POPC was removed from the bulk phase by a 250-ml washout (black bar, 8.35 ml/min for 30 min starting at 71.7 min). After the washout, Πi = 5.8 mN/m. ApoC-III was added to the bulk phase at a concentration of 1.3 μg/ml, and γ was monitored as it fell to γeq. ApoC-III was removed from the bulk phase by a 150-ml washout (black bar, 6.26 ml/min for 25 min starting at 170.5 min). The drop volume underwent four sets of gradual expansions and compressions at a rate of 1.2 μl/min (shown as recordings of surface area in mm2; gray line). Each expansion ended at A = 45.8 mm2. Each compression ended at a smaller areas than the previous compression(s), ending at A = 33.3, 28.3, 22.9, and 16.4 mm2. D, apoC-III has a ΠEX of 31.5 mN/m. From a series of experiments similar to C over a range of Πi, ΔΠ was plotted against Πi. Linear regression of the data was significant (R2 = 0.96). To characterize apoC-III desorption, compression Π/A isotherms were generated from experiments similar to those in A and C. Retention pressures (ΠENV) for apoC-III at TO/W and POPC/TO/W interfaces were determined from these isotherms, as described for apoC-II. These ΠENV values are listed in Table 2 and were plotted against Πi in Fig. 11.
FIGURE 11.
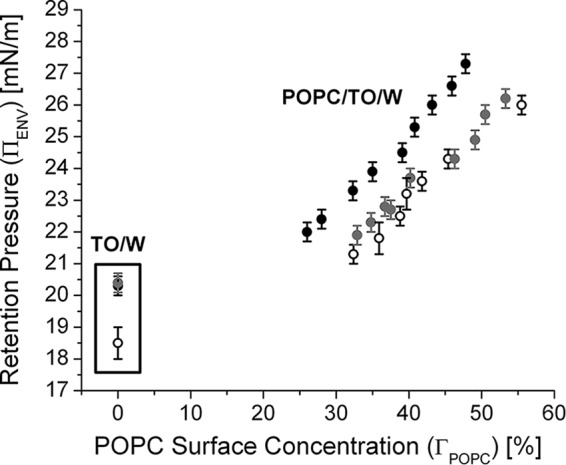
ApoC-II is retained at POPC/TO/W interfaces to higher pressures than the other apoCs. Retention pressures (ΠENV) for apoC-II (black circles), apoC-III (gray circles), and apoC-I (open black circles) were plotted against the ΓPOPC of various interfaces (ΓPOPC = 0% for a TO/W interface). Values for apoC-I were obtained from Ref. 31, whereas values for apoC-III were acquired from experiments similar to those in Fig. 10, A and C. Error bars, S.D.
TABLE 2.
Properties of exchangeable apolipoproteins at TO/W and POPC/TO/W interfaces
Shown is a comparison of the affinities of some exchangeable apolipoproteins (apoCs, full-length apoA-I, and the C terminus (C46) of apoA-I) for lipid/water interfaces. Parameters ΔΠ, ΠSAT, and ΠEX correlate with the abilities of the proteins to bind to and remodel TO/W and POPC/TO/W interfaces. The parameter ΠENV correlates with the abilities of the proteins to remain bound to lipid/water interfaces of various POPC surface concentrations (Γ). Values of these parameters for apoC-I, apoA-I, and C46 were obtained from Refs. 31, 42, 45, 46, and 48), whereas values for apoC-III are from Fig. 10.
| Protein | ΔΠa (TO/W) | ΠSAT (TO/W) | ΠEX (POPC/TO/W) | Retention pressure (ΠENV) |
||||
|---|---|---|---|---|---|---|---|---|
| TO/W | Γ = 32% | Γ = 35% | Γ = 40% | Γ = 46% | ||||
| mN/m | mN/m | mN/m | mN/m | |||||
| ApoC-II | 18.5 ± 0.5 | 22.1 | 32.2 | 20.3 ± 0.3 | 23.3 ± 0.3 | 23.9 ± 0.3 | 25.3 ± 0.3 | 26.6 ± 0.3 |
| ApoC-III | 19.5 ± 0.2 | 22.0 | 31.5 | 20.0 ± 0.5 | 21.9 ± 0.3 | 22.3 ± 0.3 | 23.7 ± 0.3 | 24.3 ± 0.3 |
| ApoC-I | 18.0 ± 0.2 | 21.5 | 32.3 | 18.5 ± 0.5 | 21.3 ± 0.3 | 21.8 ± 0.5 | 23.2 ± 0.3 | 24.3 ± 0.3 |
| ApoA-I | 14.0 ± 0.5 | 16.2 | 22.6 | 18.0 ± 0.4 | 21.2 ± 0.5 | 22.0 ± 0.5 | ||
| C46 | 15.3 ± 0.3 | 15.0 | 25.8 | 16.2 ± 0.4 | 17.0 ± 0.4 | 18.0 ± 0.5 | ||
a ΔΠ values at a TO/W interface were recorded for an aqueous phase protein concentration of 1.25 or 2.5 μg/ml.
Fig. 10, A and C, shows interfacial tension-time curves for apoC-III at TO/W (A) and POPC/TO/W (C) interfaces. The ability of apoC-III to remodel these interfaces was recorded as ΔΠ due to protein adsorption. At a TO/W interface, ΔΠ = Πeq, and these values were measured over a range of apoC-III bulk phase concentrations in experiments similar to those in Fig. 10A. The results were plotted in Fig. 10B. As the concentration of apoC-III was increased, Πeq increased up to ΠSAT = 22.0 mN/m at concentrations of ≥7.1 μg/ml (Fig. 10B). At POPC/TO/W interfaces, ΔΠ values were measured over a range of Πi values in experiments similar to those in Fig. 10C. The results were plotted in Fig. 10D. A linear fit was applied to the data, and the x intercept (i.e. ΠEX of apoC-III) was 31.5 mN/m.
The ability of apoC-III to remain bound to TO/W and POPC/TO/W interfaces was recorded as ΠENV on protein desorption. Π-A isotherms were generated from gradual, post-washout compressions in experiments similar to those in Fig. 10, A and C. ApoC-III exhibited two-step desorption, and ΠENV values were determined using the isotherm slopes, as for apoC-II in Fig. 6. ΠENV was plotted against ΓPOPC for all apoCs in Fig. 11.
Discussion
Because surface pressure increases significantly at the site of LPL activity on lipoprotein surfaces, we hypothesized that the enzyme's cofactor apoC-II must have a high affinity for lipids. To test this hypothesis, we used oil-drop tensiometry to characterize the ability of apoC-II to remodel lipoprotein-like TO/W and POPC/TO/W interfaces. We characterized the ability of apoC-II to be retained as these interfaces were compressed and surface pressure increased, which mimics the actions of LPL. We can compare these results with those from similar experiments with other exchangeable apolipoproteins and peptides, such as apoC-I (31, 37), apoC-III (Figs. 10 and 11), apoA-I (45, 48), and the N- and C-terminal domains of apoA-I (32, 42, 46). This comparison reveals the ability of apoC-II, relative to these proteins, to bind to and travel with TG-rich lipoproteins during metabolism. This comparison provides novel insights into the mechanisms by which the apoCs regulate lipoprotein metabolism as apoC-II promotes but apoC-I and apoC-III inhibit the LPL-mediated catabolism of chylomicrons and VLDL (1, 40).
The apoCs have little secondary structure in solution (<30%) (17, 18, 31, 40, 49) and no tertiary structure, which influences lipid binding (50). Class A ampipathic α-helices are induced in all of these exchangeable proteins on lipid binding (17, 18, 31, 42, 49). The C-terminal region of apoA-I (C46) is predicted to be the most lipophilic region of apoA-I and has a higher lipid affinity than the N-terminal region (42, 46).
The results in Table 2 and Fig. 11 show that apoC-II has a higher lipid affinity than all other exchangeable, helical peptides we have studied. The ability of apoC-II to bind to and remodel TO/W and POPC/TO/W interfaces is markedly greater than that of full-length apoA-I and C46, as marked by greater pressure changes (ΔΠ), saturation pressures (ΠSAT), and ΠEX on adsorption (Table 2). By comparison, apoC-I and apoC-III remodeled TO/W and POPC/TO/W interfaces to a similar extent as apoC-II (Table 2). Similar ΔΠ, ΠSAT, and ΠEX values suggest that the apoCs, perhaps due to their similar sizes, structures, and hydrophobic moments (49, 51), remodel lipid surface to a similar extent and saturate the surfaces at similar pressures.
ApoC-II has the highest retention pressures (ΠENV) at lipid/water interfaces of the exchangeable peptides we have studied (Table 2 and Fig. 11). The rank order of ΠENV for exchangeable proteins at POPC/TO/W interfaces was apoC-II > apoC-III ≥ apoC-I > apoA-I > C46. This indicates that the apoCs are retained up to higher surface pressures than many other exchangeable apolipoproteins at the surface of VLDL and chylomicron remnants. ApoC-II has markedly higher ΠENV values than apoC-I and apoC-III at POPC/TO/W interfaces (Fig. 11). These results indicate that protein-phospholipid interactions are stronger between apoC-II and PC lipids than between the other apoCs and PC lipids. The heightened affinity of apoC-II for PC lipids is reflected in the ability of apoC-II to remove significantly more PC lipid than other apoCs, upon desorption from lipid/water interfaces (Fig. 9B, inset).
Our results indicate that, in normolipidemic subjects, lipid-bound apoC-II can activate LPL as the TG in VLDL or chylomicrons is consumed and surface pressure increases. Lipolysis of TG by LPL increases the concentration of phospholipid at the lipoprotein surface. Strong interactions between apoC-II and phospholipid allow apoC-II to remain bound and activate LPL above pressures at which LPL alone would be inactive (15). This occurs even as other exchangeable apolipoproteins with weaker affinity for phospholipid, such as apoC-I and apoC-III, desorb from the surface.
ApoC-III inhibits LPL (52–55), lipoprotein assembly and secretion (56), and lipoprotein remnant uptake (1, 57) and is a strong independent predictor of cardiovascular disease (58). The mechanism of LPL inhibition by apoC-III (and apoC-I) is unknown, but some of us speculated that apoC-I and apoC-III have a higher affinity that allows them to displace apoC-II from lipoproteins and inhibit LPL (31, 49). Our results (Table 2 and Fig. 11) invalidate this hypothesis; apoC-II has the greatest affinity for lipoproteins, as shown by retention pressures. Rather, the plasma concentrations of the apoCs must determine their relative ability to bind to or displace each other from the surface of TG-rich lipoproteins.
In a typical individual, plasma concentrations of the apoCs are 80–100 μg/ml (apoC-III), 60 μg/ml (apoC-I), and 30–40 μg/ml (apoC-II) (1, 55). At these concentrations, the apoCs have no inhibitory effect on LPL in normolipidemic subjects (55, 59). In hypertriglyceridemic patients, plasma apoC-III levels can increase to >300 μg/ml, whereas apoC-II levels increase to 50–60 μg/ml (60, 61). The percentage of total apoC-III on TG-rich lipoproteins increases from 20 to 50% in normo- versus hypertriglyceridemic patients (62). LPL inhibitory activity of the lipoprotein of plasma from hypertriglyceridemic subjects was strongly correlated with elevated levels of apoC-III (55).
At concentrations 5- or 6-fold higher than apoC-II, apoC-III probably outcompetes apoC-II or LPL through mass action for binding at the surface of VLDLs or chylomicrons. Consistent with this, some of us have shown that apoC-III and apoC-I compete with LPL for binding to intralipid emulsions in a concentration-dependent manner (40) and that more apoC-I or apoC-III is needed to expel LPL from the surface in the presence of apoC-II. In incubation conditions of 10:1 (w/w) apoC-III/apoC-II, only 30% of aqueous LPL bound to lipid emulsion particles, and there was a 40% reduction in LPL activity compared with incubation conditions without apoC-III (40). Other assays showed that LPL activity was abolished at a 20:1 apoC-III/apoC-II molar ratio upon co-incubation of apoC-III with apoC-II, LPL, and phospholipid/TG (1:20 molar ratio) emulsion particles (33).
We also hypothesized that lipid-bound apoC-II must be conformationally flexible in order to regulate LPL. To test this hypothesis, we used a recently developed protocol (32, 63) to analyze the surface pressure response of apoC-II at TO/W and POPC/TO/W interfaces to washout and compression. Whereas the pre-washout Πeq varied with the bulk phase concentration of apoC-II, the post-washout pressure of apoC-II/TO/W interfaces was a constant (ΠWO = 16.6 ± 0.2 mN/m) (Table 1 and Fig. 8). These results indicate that there are at least two populations of lipid-bound apoC-II: one that is in a concentration-dependent equilibrium with lipid-free apoC-II and one that is not (32).
Analysis of Π/A isotherms of apoC-II/lipid/water interfaces during compression (Figs. 4–6) indicates that the lipid-bound populations of apoC-II differ in conformation. As surface pressure increased, apoC-II molecules partially desorbed from lipid/water interfaces at areas A ≤ AP and Π ≥ ΠP, where AP, ΠP is a turning point in the isotherms. ΠP = ΠWO, such that apoC-II molecules are in a conformation that maximizes available surface area at lower pressures. ApoC-II molecules irreversibly desorb from lipid surfaces at A ≤ AENV and Π ≥ ΠENV, where AENV and ΠENV are the retention areas and pressures. ΠP and ΠENV differ by 2–3.5 mN/m at all interfaces (Fig. 6B). In this pressure range, the surface area per apoC-II molecule is decreased, and apoC-II adopts a second conformation to remain bound to the lipid surface. The helices of apoC-II exhibit cooperative binding to lipid surfaces, such that N- and C-terminal fragments of apoC-II were unable to adopt multiple conformations upon changes in Π (Fig. 7D).
Because the conformation that is favored at higher surface pressures occupies less surface area, and it is at higher pressures that apoC-II interaction with LPL is critical for LPL activity (14, 15), we speculate that the C-terminal helix of apoC-II desorbs from the lipid-water interface to interact with LPL and keep it in an active conformation. The C-terminal helix of apoC-II in apoC-II·SDS complexes exhibits high levels of motion (19) and has a significantly lower hydrophobic moment than the N-terminal helix (27). Consistent with our hypothesis, we showed that the N-terminal helix of apoC-II has a higher lipid affinity than the C-terminal helix and mediates protein binding to and desorption from lipid surfaces (Fig. 7). In contrast, the C-terminal helix can be easily displaced from the interface by increases in Π, such as those induced by adsorption of other protein (Fig. 7B) or TG hydrolysis.
We propose the following model for the regulation of LPL by apoC-II (Fig. 12). ApoC-II binds to nascent VLDL and chylomicrons and increases the local surface pressure. This pressure is low, and LPL is active independent of apoC-II (14, 15). ApoC-II adopts multiple conformations with a different amount of residues bound (Fig. 11). At lower pressures, apoC-II maximizes available surface area such that the N- and C-terminal helices are bound to lipid. ApoC-II may or may not interact with LPL in this state.
FIGURE 12.
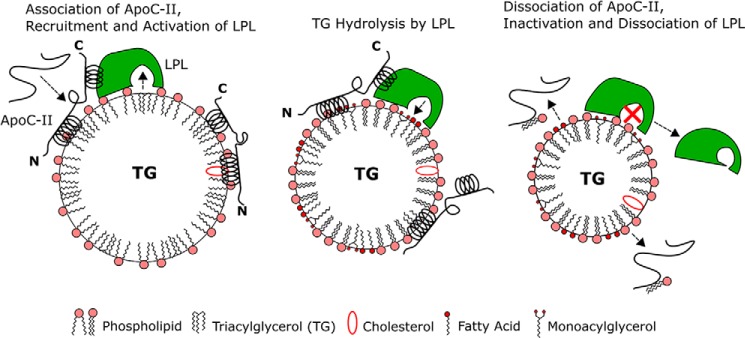
The regulation of LPL by apoC-II strongly depends on lipoprotein surface pressure. Left, newly secreted chylomicrons and VLDL have a relatively low surface pressure, allowing soluble apolipoproteins to bind as they interact in blood plasma. ApoC-II is largely unfolded in solution, but binding to nascent TG-rich lipoproteins induces ∼60% helical content in apoC-II, resulting in large N- and C-terminal helices. ApoC-II adsorption increases local lipoprotein surface pressure. At low pressures, bound apoC-II can maximize available surface area, and both helices are on the lipid surface. Middle, as LPL hydrolyzes TG, the lipoprotein core shrinks, and the local surface density of amphipathic lipids and proteins increases. Transient accumulation of fatty acids and monoacylglycerols at the surface, coupled with loss of large amounts of TG, results in high local surface pressure. This increase in pressure induces desorption of the C-terminal helix of apoC-II as less surface area is available to each protein molecule. The C-terminal helix interacts with LPL to promote TG hydrolysis. Right, apoC-II desorbs from the lipoprotein surface above its retention (or envelope) pressure and removes some PC lipids with it. At high pressures, LPL is inactive without its cofactor (marked by a red x). LPL dissociates from the VLDL or chylomicron remnant.
We speculate that apoC-II has stronger affinity for lipid near the site of LPL activity, due to a decrease in local pH. Our results indicate that the lipid affinity of apoC-II increases at lower pH (Fig. 2A). During LPL activity, fatty acids transiently accumulate at the surface. At a plasma pH (7.4), long-chain fatty acids are one-half ionized (i.e. they form an acid soap) (64, 65). Thus, some fatty acid is deprotonated (i.e. loses a hydrogen ion) as it is enzymatically released from the reaction site to the interface. This release of hydrogen ions lowers the local interfacial pH and could allow apoC-II to bind more strongly.
Once LPL activity increases surface pressure above the ΠP of apoC-II, a region of apoC-II desorbs from the lipoprotein surface. This conformational rearrangement promotes interactions between apoC-II and LPL that keep the enzyme in a lipid-bound, active state above its critical pressure (Fig. 12) (14, 15). As pressure increases above the retention pressure (ΠENV) of apoC-II, apoC-II molecules desorb from the lipoprotein surface. Upon desorption, apoC-II molecules remove PC phospholipids (Fig. 12) and transfer to HDLs. In support of this, HDL significantly accelerated desorption of apoC-II·PC lipid complexes from phospholipid/water monolayers (47). In vitro assays also showed that apoC-II can transfer from VLDL to HDL during lipolysis (66). We speculate that apoC-II molecules remove PC phospholipids in order to relax the surface pressure to a greater degree than desorption of apoC-II alone. As apoC-II molecules desorb from VLDL and chylomicron remnants, the local Π is too high for unaided LPL to hydrolyze TG (14, 15), and LPL activity ceases.
The complex formed by apoC-II and LPL has not been well characterized, because the lipid-bound forms of the proteins strongly interact, whereas the lipid-free forms may interact weakly but not in a productive way (24). It is unknown whether apoC-II and LPL desorb together or independently from the lipoprotein surface. Studies that compared the association of apoC-II and LPL in the absence or presence of triolein globules showed that the apoC-II·LPL dissociation constant is >100-fold smaller on lipid than in solution, as shown by changes in the fluorescence of dansylated apoC-II and in the steady-state velocity of lipid hydrolysis (67). This suggests that the apoC-II·LPL complex could have a higher retention pressure than apoC-II and that, after dissociation of apoC-II·LPL complexes from VLDL and chylomicron remnants, apoC-II and LPL rapidly dissociate.
In contrast, several results suggest that LPL molecules remain bound to lipoproteins as apoC-II molecules desorb. LPL has a higher exclusion pressure than apoC-II at a phospholipid/water interface (15), but it is not known whether this is true at a phospholipid/TG/water interface. Binding assays that monitored tryptophan fluorescence showed that LPL has a dissociation constant from vesicles of non-hydrolyzable phospholipid (1,2-ditetradecyl-rac-glycerol-3-phosphocholine) that is >100-fold smaller than that of apoC-II (68). In experiments using lipid emulsion particles, apoC-II, apoC-III, and LPL, we noted that the main inhibitory effect of apoC-III on LPL activity was due to decreased binding of the enzyme to the lipid particles and that the presence of apoC-II rescued LPL from being expelled (40).
In this study, we used oil-drop tensiometry (30, 35) to characterize interactions between apoC-II and lipid. This approach has several unique strengths. This technique allows for the precise control of lipid surface composition and density at lipid/water interfaces (42, 43) and for measurement of the resulting surface pressure, which are not possible for free floating lipid droplets or lipoproteins in other systems. For a known surface concentration of amphipathic lipid, the surface area of TG-rich drops occupied by peptide is easily determined (42). The oil-drop tensiometer is unique in its real-time capability to measure surface pressure of lipid drops. The tensiometer overcomes many limitations of the Pockels-Langmuir surface balance, which is well suited to studies at the non-physiological air/water interface, but studies at oil/water interfaces are extremely difficult and suffer from non-uniform deformations and leakage of surface-active material upon interfacial compressions (35, 69). Our technique provides physiologically relevant biophysical characteristics, such as ΔΠ, ΠEX, and ΠENV, for peptides at various lipid surfaces and reveals the exchangeability of peptides (30). In contrast to helical peptides at lipid surfaces, we previously showed that the β-strand-rich regions of apoB were non-exchangeable and elastic and had a higher affinity for TG than phospholipid (30, 63, 70).
Characterization of protein-lipid interactions with the tensiometer has several limitations. The lipid surface is a simplified surface from the complex lipid and protein environment of the lipoprotein surface (8). The TG/W interface can be refined by adding amphipathic lipids, such as phospholipid, cholesterol, and fatty acid, to better mimic the lipoprotein surface (37), but a rigorous set of experiments is required to determine their interfacial concentrations. These experiments have been done for POPC (42) and egg yolk PC (43) but are more difficult for cholesterol, which partitions between the core and surface of lipid drops. Interpretation of tensiometry data relies on known protein structures (here, NMR structures of apoC-II in complex with SDS or dodecylphosphocholine (17–19)). Independent experiments to confirm these interpretations are very difficult, due both to the design of the tensiometer (i.e. extracting lipid drops at select Π is very difficult) and the limitations of external experiments (i.e. Π of freely suspended drops has not been quantified). Despite these limitations, oil-drop tensiometry is the best way to artificially mimic lipoprotein surfaces and is superior to techniques used previously in studies similar to ours (51, 71).
In summary, we have shown that the apoCs have a higher affinity for lipid than other exchangeable apolipoproteins, such as apoA-I. Of the apoCs, apoC-II has the highest retention pressure at lipid/water interfaces, which allows the protein to remain bound to triacylglycerol-rich lipoproteins up to higher pressures as LPL activity increases local surface pressure. Our data strongly suggest that apoC-II exhibits at least two conformations at the lipoprotein surface. This may allow the protein to regulate LPL in a variety of ways that are dependent on surface pressure.
Author Contributions
All four authors conceived the study. M. L. expressed and purified the proteins used in the study, under the supervision of G. O. N. L. M. conducted all of the tensiometry experiments and analyzed the data with D. M. S. M. L. wrote the part of “Experimental Protocols” discussing protein expression. N. L. M. wrote all other parts of the manuscript, although it underwent significant revisions from the input of all four authors. All authors reviewed the results and approved the final version of the manuscript.
This work was supported by American Heart Association Grant 12PRE12060584 and Swedish Science Council Grant 12203. The authors declare that they have no conflicts of interest with the contents of this article.
- apo
- apolipoprotein
- TG
- triacylglycerol
- LPL
- lipoprotein lipase
- TO
- triolein
- POPC
- 1-palmitoyl-2-oleoylphosphatidylcholine
- SUV
- small unilamellar vesicle
- TO/W
- triolein/water
- POPC/TO/W
- palmitoyloleoylphosphatidylcholine/triolein/water
- mN
- millinewtons
- HFIP
- hexafluor-2-proponal
- PNPC
- 1-palmitoyl-2-{12-[7-nitro-2-1,3-benzoxadiazol-4-yl)amino]dodecanoyl}-sn-glycero-3-phosphocholine.
References
- 1. Jong M. C., Hofker M. H., Havekes L. M. (1999) Role of ApoCs in lipoprotein metabolism: functional differences between ApoC1, ApoC2, and ApoC3. Arterioscler. Thromb. Vasc. Biol. 19, 472–484 [DOI] [PubMed] [Google Scholar]
- 2. Kei A. A., Filippatos T. D., Tsimihodimos V., Elisaf M. S. (2012) A review of the role of apolipoprotein C-II in lipoprotein metabolism and cardiovascular disease. Metabolism 61, 906–921 [DOI] [PubMed] [Google Scholar]
- 3. Wang C. S. (1991) Structure and functional properties of apolipoprotein C-II. Prog. Lipid Res. 30, 253–258 [DOI] [PubMed] [Google Scholar]
- 4. Havel R. J., Fielding C. J., Olivecrona T., Shore V. G., Fielding P. E., Egelrud T. (1973) Cofactor activity of protein components of human very low density lipoproteins in the hydrolysis of triglycerides by lipoproteins lipase from different sources. Biochemistry 12, 1828–1833 [DOI] [PubMed] [Google Scholar]
- 5. LaRosa J. C., Levy R. I., Herbert P., Lux S. E., Fredrickson D. S. (1970) A specific apoprotein activator for lipoprotein lipase. Biochem. Biophys. Res. Commun. 41, 57–62 [DOI] [PubMed] [Google Scholar]
- 6. Eckel R. H. (1989) Lipoprotein lipase. A multifunctional enzyme relevant to common metabolic diseases. N. Engl. J. Med. 320, 1060–1068 [DOI] [PubMed] [Google Scholar]
- 7. Bergman E. N., Havel R. J., Wolfe B. M., Bohmer T. (1971) Quantitative studies of the metabolism of chylomicron triglycerides and cholesterol by liver and extrahepatic tissues of sheep and dogs. J. Clin. Invest. 50, 1831–1839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cohen D. E., Fisher E. A. (2013) Lipoprotein metabolism, dyslipidemia, and nonalcoholic fatty liver disease. Semin. Liver Dis. 33, 380–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Breckenridge W. C., Little J. A., Steiner G., Chow A., Poapst M. (1978) Hypertriglyceridemia associated with deficiency of apolipoprotein C-II. N. Engl. J. Med. 298, 1265–1273 [DOI] [PubMed] [Google Scholar]
- 10. Brahm A. J., Hegele R. A. (2015) Chylomicronaemia-current diagnosis and future therapies. Nat. Rev. Endocrinol. 11, 352–362 [DOI] [PubMed] [Google Scholar]
- 11. Kinnunen P. K., Jackson R. L., Smith L. C., Gotto A. M., Jr., Sparrow J. T. (1977) Activation of lipoprotein lipase by native and synthetic fragments of human plasma apolipoprotein C-II. Proc. Natl. Acad. Sci. U.S.A. 74, 4848–4851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. MacPhee C. E., Howlett G. J., Sawyer W. H. (1999) Mass spectrometry to characterize the binding of a peptide to a lipid surface. Anal. Biochem. 275, 22–29 [DOI] [PubMed] [Google Scholar]
- 13. Sparrow J. T., Gotto A. M., Jr. (1980) Phospholipid binding studies with synthetic apolipoprotein fragments. Ann. N.Y. Acad. Sci. 348, 187–211 [DOI] [PubMed] [Google Scholar]
- 14. Vainio P., Virtanen J. A., Kinnunen P. K., Gotto A. M., Jr., Sparrow J. T., Pattus F., Bougis P., Verger R. (1983) Action of lipoprotein lipase on mixed triacylglycerol/phosphatidylcholine monolayers. Activation by apolipoprotein C-II. J. Biol. Chem. 258, 5477–5482 [PubMed] [Google Scholar]
- 15. Vainio P., Virtanen J. A., Kinnunen P. K., Voyta J. C., Smith L. C., Gotto A. M., Jr., Sparrow J. T., Pattus F., Verger R. (1983) Action of lipoprotein lipase on phospholipid monolayers. Activation by apolipoprotein C-II. Biochemistry 22, 2270–2275 [DOI] [PubMed] [Google Scholar]
- 16. Hatters D. M., Howlett G. J. (2002) The structural basis for amyloid formation by plasma apolipoproteins: a review. Eur. Biophys. J. 31, 2–8 [DOI] [PubMed] [Google Scholar]
- 17. MacRaild C. A., Hatters D. M., Howlett G. J., Gooley P. R. (2001) NMR structure of human apolipoprotein C-II in the presence of sodium dodecyl sulfate. Biochemistry 40, 5414–5421 [DOI] [PubMed] [Google Scholar]
- 18. MacRaild C. A., Howlett G. J., Gooley P. R. (2004) The structure and interactions of human apolipoprotein C-II in dodecyl phosphocholine. Biochemistry 43, 8084–8093 [DOI] [PubMed] [Google Scholar]
- 19. Zdunek J., Martinez G. V., Schleucher J., Lycksell P. O., Yin Y., Nilsson S., Shen Y., Olivecrona G., Wijmenga S. (2003) Global structure and dynamics of human apolipoprotein CII in complex with micelles: evidence for increased mobility of the helix involved in the activation of lipoprotein lipase. Biochemistry 42, 1872–1889 [DOI] [PubMed] [Google Scholar]
- 20. Storjohann R., Rozek A., Sparrow J. T., Cushley R. J. (2000) Structure of a biologically active fragment of human serum apolipoprotein C-II in the presence of sodium dodecyl sulfate and dodecylphosphocholine. Biochim. Biophys. Acta 1486, 253–264 [DOI] [PubMed] [Google Scholar]
- 21. Segrest J. P., De Loof H., Dohlman J. G., Brouillette C. G., Anantharamaiah G. M. (1990) Amphipathic helix motif: classes and properties. Proteins 8, 103–117 [DOI] [PubMed] [Google Scholar]
- 22. Segrest J. P., Garber D. W., Brouillette C. G., Harvey S. C., Anantharamaiah G. M. (1994) The amphipathic α helix: a multifunctional structural motif in plasma apolipoproteins. Adv. Protein Chem. 45, 303–369 [DOI] [PubMed] [Google Scholar]
- 23. Segrest J. P., Jones M. K., De Loof H., Brouillette C. G., Venkatachalapathi Y. V., Anantharamaiah G. M. (1992) The amphipathic helix in the exchangeable apolipoproteins: a review of secondary structure and function. J. Lipid Res. 33, 141–166 [PubMed] [Google Scholar]
- 24. Shen Y., Lookene A., Nilsson S., Olivecrona G. (2002) Functional analyses of human apolipoprotein CII by site-directed mutagenesis: identification of residues important for activation of lipoprotein lipase. J. Biol. Chem. 277, 4334–4342 [DOI] [PubMed] [Google Scholar]
- 25. Shen Y., Lookene A., Zhang L., Olivecrona G. (2010) Site-directed mutagenesis of apolipoprotein CII to probe the role of its secondary structure for activation of lipoprotein lipase. J. Biol. Chem. 285, 7484–7492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Olivecrona G., Beisiegel U. (1997) Lipid binding of apolipoprotein CII is required for stimulation of lipoprotein lipase activity against apolipoprotein CII-deficient chylomicrons. Arterioscler. Thromb. Vasc. Biol. 17, 1545–1549 [DOI] [PubMed] [Google Scholar]
- 27. Amar M. J. A., Sakurai T., Sakurai-Ikuta A., Sviridov D., Freeman L., Ahsan L., Remaley A. T. (2015) A novel apolipoprotein C-II mimetic peptide that activates lipoprotein lipase and decreases serum triglycerides in apolipoprotein E-knockout mice. J. Pharmacol. Exp. Ther. 352, 227–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bengtsson G., Olivecrona T. (1980) Lipoprotein lipase: some effects of activator proteins. Eur. J. Biochem. 106, 549–555 [DOI] [PubMed] [Google Scholar]
- 29. Lee A. G. (1987) A big fat book. Nature 10.1038/328486b0 [DOI] [Google Scholar]
- 30. Small D. M., Wang L., Mitsche M. A. (2009) The adsorption of biological peptides and proteins at the oil/water interface: a potentially important but largely unexplored field. J. Lipid Res. 50, S329–S334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Meyers N. L., Wang L., Gursky O., Small D. M. (2013) Changes in helical content or net charge of apolipoprotein C-I alter its affinity for lipid/water interfaces. J. Lipid Res. 54, 1927–1938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mitsche M. A., Small D. M. (2013) Surface pressure-dependent conformation change of apolipoprotein-derived amphipathic α-helices. J. Lipid Res. 54, 1578–1588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jackson R. L., Tajima S., Yamamura T., Yokoyama S., Yamamoto A. (1986) Comparison of apolipoprotein C-II-deficient triacylglycerol-rich lipoproteins and trioleoylglycerol/phosphatidylcholine-stabilized particles as substrates for lipoprotein lipase. Biochim. Biophys. Acta 875, 211–219 [DOI] [PubMed] [Google Scholar]
- 34. McKeone B. J., Massey J. B., Knapp R. D., Pownall H. J. (1988) Apolipoproteins C-I, C-II, and C-III: kinetics of association with model membranes and intermembrane transfer. Biochemistry 27, 4500–4505 [DOI] [PubMed] [Google Scholar]
- 35. Labourdenne S., Cagna A., Delorme B., Esposito G., Verger R., Rivière C. (1997) Oil-drop tensiometer: applications for studying the kinetics of lipase action. Methods Enzymol. 286, 306–326 [DOI] [PubMed] [Google Scholar]
- 36. Miller K. W., Small D. M. (1983) Surface-to-core and interparticle equilibrium distributions of triglyceride-rich lipoprotein lipids. J. Biol. Chem. 258, 13772–13784 [PubMed] [Google Scholar]
- 37. Meyers N. L., Wang L., Small D. M. (2012) Apolipoprotein C-I binds more strongly to phospholipid/triolein/water than triolein/water interfaces: a possible model for inhibiting cholesterol ester transfer protein activity and triacylglycerol-rich lipoprotein uptake. Biochemistry 51, 1238–1248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Andersson Y., Lookene A., Shen Y., Nilsson S., Thelander L., Olivecrona G. (1997) Guinea pig apolipoprotein C-II: expression in E. coli, functional studies of recombinant wild-type and mutated variants, and distribution on plasma lipoproteins. J. Lipid Res. 38, 2111–2124 [PubMed] [Google Scholar]
- 39. Liu H., Talmud P. J., Lins L., Brasseur R., Olivecrona G., Peelman F., Vandekerckhove J., Rosseneu M., Labeur C. (2000) Characterization of recombinant wild type and site-directed mutations of apolipoprotein C-III: lipid binding, displacement of ApoE, and inhibition of lipoprotein lipase. Biochemistry 39, 9201–9212 [DOI] [PubMed] [Google Scholar]
- 40. Larsson M., Vorrsjö E., Talmud P., Lookene A., Olivecrona G. (2013) Apolipoproteins C-I and C-III inhibit lipoprotein lipase activity by displacement of the enzyme from lipid droplets. J. Biol. Chem. 288, 33997–34008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hatters D. M., MacPhee C. E., Lawrence L. J., Sawyer W. H., Howlett G. J. (2000) Human apolipoprotein C-II forms twisted amyloid ribbons and closed loops. Biochemistry 39, 8276–8283 [DOI] [PubMed] [Google Scholar]
- 42. Mitsche M. A., Small D. M. (2011) C-terminus of apolipoprotein A-I removes phospholipids from a triolein/phospholipids/water interface, but the N-terminus does not: a possible mechanism for nascent HDL assembly. Biophys. J. 101, 353–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mitsche M. A., Wang L., Small D. M. (2010) Adsorption of egg phosphatidylcholine to an air/water and triolein/water bubble interface: use of the 2-dimensional phase rule to estimate the surface composition of a phospholipid/triolein/water surface as a function of surface pressure. J. Phys. Chem. B 114, 3276–3284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Weinberg R. B., Anderson R. A., Cook V. R., Emmanuel F., Denèfle P., Tall A. R., Steinmetz A. (2002) Interfacial exclusion pressure determines the ability of apolipoprotein A-IV truncation mutants to activate cholesterol ester transfer protein. J. Biol. Chem. 277, 21549–21553 [DOI] [PubMed] [Google Scholar]
- 45. Wang L., Mei X., Atkinson D., Small D. M. (2014) Surface behavior of apolipoprotein A-I and its deletion mutants at model lipoprotein interfaces. J. Lipid Res. 55, 478–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wang L., Hua N., Atkinson D., Small D. M. (2007) The N-terminal (1–44) and C-terminal (198–243) peptides of apolipoprotein A-I behave differently at the triolein/water interface. Biochemistry 46, 12140–12151 [DOI] [PubMed] [Google Scholar]
- 47. Jackson R. L., Pattus F., Demel R. A. (1979) Interaction of plasma apolipoproteins with lipid monolayers. Biochim. Biophys. Acta 556, 369–387 [DOI] [PubMed] [Google Scholar]
- 48. Wang L., Atkinson D., Small D. M. (2005) The interfacial properties of ApoA-I and an amphipathic α-helix consensus peptide of exchangeable apolipoproteins at the triolein/water interface. J. Biol. Chem. 280, 4154–4165 [DOI] [PubMed] [Google Scholar]
- 49. Gangabadage C. S., Zdunek J., Tessari M., Nilsson S., Olivecrona G., Wijmenga S. S. (2008) Structure and dynamics of human apolipoprotein CIII. J. Biol. Chem. 283, 17416–17427 [DOI] [PubMed] [Google Scholar]
- 50. Weers P. M., Narayanaswami V., Choy N., Luty R., Hicks L., Kay C. M., Ryan R. O. (2003) Lipid binding ability of human apolipoprotein E N-terminal domain isoforms: correlation with protein stability? Biophys. Chem. 100, 481–492 [DOI] [PubMed] [Google Scholar]
- 51. Bolanos-Garcia V. M., Renault A., Beaufils S. (2008) Surface rheology and adsorption kinetics reveal the relative amphiphilicity, interfacial activity, and stability of human exchangeable apolipoproteins. Biophys. J. 94, 1735–1745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Brown W. V., Baginsky M. L. (1972) Inhibition of lipoprotein lipase by an apoprotein of human very low density lipoprotein. Biochem. Biophys. Res. Commun. 46, 375–382 [DOI] [PubMed] [Google Scholar]
- 53. Ekman R., Nilsson-Ehle P. (1975) Effects of apolipoproteins on lipoprotein lipase activity of human adipose tissue. Clin. Chim. Acta 63, 29–35 [DOI] [PubMed] [Google Scholar]
- 54. Havel R. J., Kane J. P., Kashyap M. L. (1973) Interchange of apolipoproteins between chylomicrons and high density lipoproteins during alimentary lipemia in man. J. Clin. Invest. 52, 32–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wang C. S., McConathy W. J., Kloer H. U., Alaupovic P. (1985) Modulation of lipoprotein lipase activity by apolipoproteins. Effect of apolipoprotein C-III. J. Clin. Invest. 75, 384–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sundaram M., Zhong S., Bou Khalil M., Links P. H., Zhao Y., Iqbal J., Hussain M. M., Parks R. J., Wang Y., Yao Z. (2010) Expression of apolipoprotein C-III in McA-RH7777 cells enhances VLDL assembly and secretion under lipid-rich conditions. J. Lipid Res. 51, 150–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sehayek E., Eisenberg S. (1991) Mechanisms of inhibition by apolipoprotein C of apolipoprotein E-dependent cellular metabolism of human triglyceride-rich lipoproteins through the low density lipoprotein receptor pathway. J. Biol. Chem. 266, 18259–18267 [PubMed] [Google Scholar]
- 58. Ooi E. M., Barrett P. H., Chan D. C., Watts G. F. (2008) Apolipoprotein C-III: understanding an emerging cardiovascular risk factor. Clin. Sci. 114, 611–624 [DOI] [PubMed] [Google Scholar]
- 59. Wang C. S., Bass H. B., Downs D., Whitmer R. K. (1981) Modified heparin-Sepharose procedure for determination of plasma lipolytic activities of normolipidemic and hyperlipidemic subjects after injection of heparin. Clin. Chem. 27, 663–668 [PubMed] [Google Scholar]
- 60. Cohn J. S., Patterson B. W., Uffelman K. D., Davignon J., Steiner G. (2004) Rate of production of plasma and very-low-density lipoprotein (VLDL) apolipoprotein C-III is strongly related to the concentration and level of production of VLDL triglyceride in male subjects with different body weights and levels of insulin sensitivity. J. Clin. Endocrinol. Metab. 89, 3949–3955 [DOI] [PubMed] [Google Scholar]
- 61. Huff M. W., Breckenridge W. C., Strong W. L., Wolfe B. M. (1988) Metabolism of apolipoproteins C-II, C-III, and B in hypertriglyceridemic men. Changes after heparin-induced lipolysis. Arteriosclerosis 8, 471–479 [DOI] [PubMed] [Google Scholar]
- 62. Fredenrich A., Giroux L. M., Tremblay M., Krimbou L., Davignon J., Cohn J. S. (1997) Plasma lipoprotein distribution of apoC-III in normolipidemic and hypertriglyceridemic subjects: comparison of the apoC-III to apoE ratio in different lipoprotein fractions. J. Lipid Res. 38, 1421–1432 [PubMed] [Google Scholar]
- 63. Mitsche M. A., Packer L. E., Brown J. W., Jiang Z. G., Small D. M., McKnight C. J. (2014) Surface tensiometry of apolipoprotein B domains at lipid interfaces suggests a new model for the initial steps in triglyceride-rich lipoprotein assembly. J. Biol. Chem. 289, 9000–9012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Cistola D. P., Atkinson D., Hamilton J. A., Small D. M. (1986) Phase behavior and bilayer properties of fatty acids: hydrated 1:1 acid-soaps. Biochemistry 25, 2804–2812 [DOI] [PubMed] [Google Scholar]
- 65. Cistola D. P., Hamilton J. A., Jackson D., Small D. M. (1988) Ionization and phase behavior of fatty acids in water: application of the Gibbs phase rule. Biochemistry 27, 1881–1888 [DOI] [PubMed] [Google Scholar]
- 66. Murdoch S. J., Breckenridge W. C. (1996) Effect of lipid transfer proteins on lipoprotein lipase induced transformation of VLDL and HDL. Biochim. Biophys. Acta 1303, 222–232 [DOI] [PubMed] [Google Scholar]
- 67. Clarke A. R., Holbrook J. J. (1985) The mechanism of activation of lipoprotein lipase by apolipoprotein C-II: the formation of a protein-protein complex in free solution and at a triacylglycerol/water interface. Biochim. Biophys. Acta 827, 358–368 [DOI] [PubMed] [Google Scholar]
- 68. McLean L. R., Jackson R. L. (1985) Interaction of lipoprotein lipase and apolipoprotein C-II with sonicated vesicles of 1,2-ditetradecylphosphatidylcholine: comparison of binding constants. Biochemistry 24, 4196–4201 [DOI] [PubMed] [Google Scholar]
- 69. Benjamins J., Cagna A., Lucassen-Reynders E. H. (1996) Viscoelastic properties of triacylglycerol/water interfaces covered by proteins. Colloids Surf. A 114, 245–254 [Google Scholar]
- 70. Mitsche M. A., Wang L., Jiang Z. G., McKnight C. J., Small D. M. (2009) Interfacial properties of a complex multi-domain 490 amino acid peptide derived from apolipoprotein B (residues 292–782). Langmuir 25, 2322–2330 [DOI] [PubMed] [Google Scholar]
- 71. Krebs K. E., Phillips M. C., Sparks C. E. (1983) A comparison of the surface activities of rat plasma apolipoproteins C-II, C-III-0, C-III-3. Biochim. Biophys. Acta 751, 470–473 [DOI] [PubMed] [Google Scholar]