

Background: Tid1 is a human DnaJ protein and has a role in regulating cellular signaling pathways.
Results: Tid1 increases autophagic flux by interacting with the autophagy protein complex.
Conclusion: Tid1 is a novel mediator of macroautophagy.
Significance: As a new member of autophagy pathway, Tid1 may have a crucial role in participating in a broad range of biological aspects.
Keywords: 70 kilodalton heat shock protein (Hsp70), autophagy, Beclin-1 (BECN1), chaperone DnaJ (DnaJ), molecular chaperone, PI3KC3, Tid1
Abstract
One of the fundamental functions of molecular chaperone proteins is to selectively conjugate cellular proteins, targeting them directly to lysosome. Some of chaperones, such as the stress-induced Hsp70, also play important roles in autophagosome-forming macroautophagy under various stress conditions. However, the role of their co-chaperones in autophagy regulation has not been well defined. We here show that Tid1, a DnaJ co-chaperone for Hsp70 and the mammalian homologue of the Drosophila tumor suppressor Tid56, is a key mediator of macroautophagy pathway. Ectopic expression of Tid1 induces autophagy by forming LC3+ autophagosome foci, whereas silencing Tid1 leads to drastic impairment of autophagy as induced by nutrient deprivation or rapamycin. In contrast, Hsp70 is dispensable for a role in nutrient deprivation-induced autophagy. The murine Tid1 can be replaced with human Tid1 in murine fibroblast cells for induction of autophagy. We further show that Tid1 increases autophagy flux by interacting with the Beclin1-PI3 kinase class III protein complex in response to autophagy inducing signal and that Tid1 is an essential mediator that connects IκB kinases to the Beclin1-containing autophagy protein complex. Together, these results reveal a crucial role of Tid1 as an evolutionarily conserved and essential mediator of canonical macroautophagy.
Introduction
Tid1, the human counterpart of Drosophila tumor suppressor Tid56 encoded by the lethal tid (tumorous imaginal discs) gene, is a mammalian DnaJ protein that serves as a molecular co-chaperone for members of the heat shock protein 70 (Hsp70) chaperone family (1, 2). Tid1 has been identified as a cellular protein that binds to the viral oncoprotein E7 derived from human papilloma virus type 16 (2) and also serves as the intracellular target for other oncogenic viral proteins from human T cell leukemia virus type 1, Epstein-Bar virus, and human hepatitis B virus (3–5). These findings implicate a potential role of Tid1 in mediating viral oncogenesis. In addition to forming a molecular chaperone complex with Hsp70, it has been shown that Tid1 interacts with a variety of cellular signaling molecules including IκB kinase, Jak/Stat, Trk, RasGAP, ErbB-2, EGF receptor, Stat5b, agrin, and the tumor suppressors von Hippel-Lindau protein (pVHL) and p53 (6–16). The role of Tid1 in oncogenesis remains controversial. Tid1 may function as a mammalian tumor suppressor as overexpression of Tid1 induces cell senescence, promotes apoptosis of cancer cells, and represses tumor growth in mice (7, 17–20). In contrast, Tid1 facilitates c-Met-mediated tumorigenicity in the context of renal cell carcinoma (21).
Tid1 is an evolutionally conserved cellular protein and is ubiquitously expressed in human tissues. Both mammalian Tid1 proteins and Drosophila Tid56 comprise a well conserved, N-terminal signature J domain required for interaction with Hsp70 and herein are classified as DnaJ proteins or co-chaperones of the molecular chaperone superfamily (22). Typically, the ATPase activity of Hsp70 is necessary for its chaperone activity and is modulated by its co-chaperones (22). As molecular co-chaperones, DnaJ proteins are bound to Hsp70 proteins through their conserved J domains to form molecular chaperon complexes, enhancing the ATPase activity of Hsp70. Two spliced forms of human Tid1 have been identified that share almost identical amino acid sequence, differing only from their C termini (23). Tid1 associates with the stress-induced Hsp70, the constitutively expressed cytoplasmic Hsc70, and the mitochondrial Hsp70, mortalin (3, 24, 25), yet the co-chaperone function of Tid1 remains poorly understood. Analysis of the subcellular localization of Tid1 indicates that this DnaJ protein resides predominantly in the mitochondria (26). However, studies have also shown that Tid1 interacts with vast amounts of cytoplasmic and plasma membrane-bound cellular and viral proteins (3–16).
Molecular chaperones are the driving force for chaperone-mediated autophagy (27). It is known that Hsc70, one of the main chaperones, selectively conjugates cellular proteins, targeting them directly to lysosome for degradation. However, the molecular cross-talk between chaperone-mediated autophagy and autophagosome-forming macroautophagy is largely unclear. It has been recently shown that the stress-induced Hsp70 participates in macroautophagic process by interacting with Beclin1, a key component of the autophagy molecular complex containing PI3 kinase class III (PI3KC3) and Beclin1 (28). However, it remains to be determined whether or not the co-chaperone protein Tid1 is involved in this macroautophagic process. In the present study, we demonstrate that Tid1 is a key regulator of canonical macroautophagy, mediating autophagy independently of its co-chaperone function for Hsp70.
Experimental Procedures
Cell Lines, Antibodies, and Reagents
HeLa, U2OS, and HT1080 cell lines were described previously (7, 29), MT-1 was kindly provided by Drs. Atsushi Koito and Takeo Ohsugi, and NIH3T3 and HOS cells (human osteosarcoma cells) were obtained from the AIDS Reagent Program. These cell lines were cultured in RMPI1640 medium supplemented with 10% FBS plus antibiotics at 37 °C/5%CO2. Antibodies reacting to p62, BECN1, Tid1, Hsp70, GST, and HA were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-LC3B and LAMP1 were obtained from Cell Signaling Technology (Danvers, MA). Anti-β-actin antibody, protease, and phosphatase inhibitor cocktails were obtained from Sigma. Rapamycin was purchased from Selleck Chemicals (Houston, TX). VER-155008 (ATP-derivative inhibitor of HSP70) was from Sigma. EBSS2 and Opti-MEM were from Life Technologies, Inc. Glutathione-Sepharose 4B beads were purchased from GE Healthcare.
Plasmids, DNA Transfection, and Lentivirus Transduction
The expression plasmids for Tid1, Tax, Hsp70, IKKβKA, GFP-LC3, p40PX-GFP, Beclin1, PI3KC3, and UVRAG were described previously (3, 29). Tid1ΔJ, Tid1LΔN67, and Tid1LΔN100 were generated by PCR-based site-directed mutagenesis. PI3KC3 and Beclin1 shRNAs were purchased from Open Biosystems, and Atg5 and Tid1 shRNAs were described previously (7, 30). mTid1 shRNA was constructed in a modified pLL3.7 lentivirus vector in which the GFP fragment was replaced by puromycin-resistant gene. DNA transfection for HEK293 cells was performed with PolyFect reagent (Qiagen) and for HT1080 and HeLa cells with FuGENE HD (Promega, Madison, WI) following the manufacturer's recommended protocols. The methods for lentivirus production, purification, and transduction were reported previously (29).
Western Blot Analysis
Cells were collected and lysed in radioimmune precipitation assay buffer containing protease and phosphatase inhibitor mixtures at 4 °C for 30 min. Equal amounts of cellular proteins were analyzed by SDS-PAGE, followed by immunoblot. Anti-β-actin blot was used for the protein loading control. The heat shock procedure and GST pulldown were reported previously (29, 31). For starvation, NIH3T3 or U2OS cells were washed once with PBS and then incubated with EBSS for 30 min, 1 h, and 2 h. For rapamycin stimulation, cells were incubated with rapamycin or DMSO for 6 h or overnight.
Fluorescence Microscopy
A Nikon Eclipse E800 Fluorescence microscope was used. Cells were plated on coverslips one night before being transfected with indicated constructs in Opti-MEM using FuGENE HD reagent. 24 h after treatment, slides were fixed using 4% paraformaldehyde/PBS at room temperature for 15 min and washed twice with PBS. For immunofluorescent analysis, slides were then washed twice with PBS, permeabilized by using PBS containing 0.25% Triton X-100, and blocked with 2% BSA in PBST for 30 min, and the first antibodies were added following Abcam immunofluorescence protocol. The autophagic process was analyzed by counting percentage of cells showing accumulation of GFP (RFP) LC3 puncta and the average number of GFP (RFP) LC3 puncta per cell. For the calculation of percentage of cells showing LC3 accumulation, a total of 300 cells were calculated each time for three duplicates. For the calculation of average number of puncta per cell, a total number of 100 cells were calculated each time for three duplicates. The representative fluorescent images were collected using SPOT Advanced software.
Results
Tid1 Induces Formation of LC3+ Autophagosome
To investigate the role of Tid1 in regulating autophagy, we first transfected FLAG-targeted long and short forms of Tid1 (Tid1L-FLAG and Tid1S-FLAG, respectively) into HT1080 cells. Tid1L or Tid1S, when ectopically expressed in cells, displayed two molecular weights. The full-length protein represents the unprocessed form of Tid1, whereas the cleaved protein is the processed form of Tid1 resulting from its N-terminal cleavage (Fig. 1A). We found that Tid1L, Tid1S, or the retroviral oncoprotein Tax, a known autophagy-inducing viral protein used for control (30), induced a drastic increase of LC3-II, a lipidated form of LC3 that is associated with autophagosome membrane, whereas vector did not do so (Fig. 1A). Correlating with increased LC3-II levels in Tid1- or Tax-transfected cells, the levels of p62, a key mediator of autophagy, were reduced (Fig. 1A). Because p62 is degraded following autophagy flux, this finding suggested that either Tid1L or Tid1S promoted autophagy flux.
FIGURE 1.
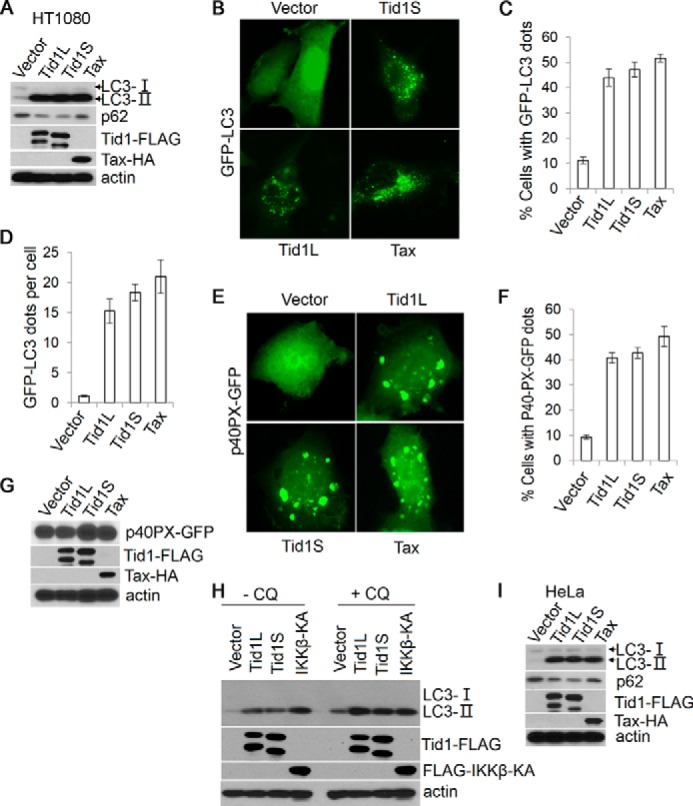
Tid1 induces macroautophagy. A, Tid1L-FLAG, Tid1S-FLAG, or Tax-HA was transiently transfected into HT1080 cells, and total protein lysates were collected 24 h post-transfection and were analyzed with anti-LC3, anti-p62, anti-FLAG (for detecting ectopically expressed Tid1-FLAG), and anti-HA (for detecting Tax-HA). β-Actin was used as protein loading control. B, GFP-LC3 was co-transfected with vector, Tid1L-FLAG, Tid1S-FLAG, or Tax-HA in HT1080 cells. C and D, the formation of cytoplasmic LC3+ foci was examined with fluorescence imaging, and the percentage of cells with LC3+ puncta (C) and average dots per cell (D) were statistically analyzed. p40PX-GFP was co-transfected with vector, Tid1L-FLAG, Tid1S-FLAG, or Tax-HA in HT1080 cells. E–G, the cytoplasmic p40PX-GFP aggregates, the percentage of cells with p40PX-GFP aggregates, and protein expression evaluation with immunoblot are shown in E, F, and G, respectively. H, HT1080 cells were transfected with vector, Tid1L-FLAG, Tid1S-FLAG, or FLAG-IKKβKA. 24 h following transfection, cells were treated with DMSO or with chloroquine (CQ, 50 μm) for 2 h, and total protein lysates were collected for immunoblot analysis with anti-LC3, anti-FLAG, and anti-HA blots. I, Tid1L-FLAG, Tid1S-FLAG, or Tax-HA were transiently transfected into HeLa cells, and total protein lysates were collected 24 h post-transfection for detecting LC3, p62, Tid1-FLAG, and Tax-HA.
Next, we applied fluorescence imaging technique to visualize LC3 puncta by co-transfecting Tid1 with GFP-LC3 in HT1080 cells. The cytoplasmic LC3+ puncta reminiscent of autophagosomes were observed in Tid1L-, Tid1S-, or Tax-transfected cells, whereas vector-transfected cells showed even distribution of the GFP-LC3 fluorescent signals (Fig. 1B). Quantitative analysis of the GFP-LC3 fluorescence showed that both forms of Tid1 increased the percentage of LC3+ puncta-containing cells and the number of GFP-LC3+ dots in a single cell (Fig. 1, C and D).
Further, we utilized p40PX-GFP to determine the activity of PI3KC3. Upon activation of PI3KC3, p40PX-GFP molecules bind to the phosphorylated lipids, forming p40PX cytoplasmic aggregates. Either Tid1L or Tid1S or Tax induced significant amounts of p40PX-GFP aggregates (Fig. 1, E–G). Chloroquine, a compound that inhibits fusion of autophagosome with lysosome, further enhanced Tid1-mediated augmentation of the LC3-II level (Fig. 1H), providing additional evidence supporting the notion that Tid1 promoted autophagy flux. The activity of Tid1 in inducing formation of LC3+ autophagosome appeared not to be unique in HT1080 cells, because this DnaJ protein also increased the levels of LC3-II in HeLa cells (Fig. 1I). The finding that Tid1 increased LC3-II levels and p40PX-GFP aggregates with simultaneous reduction of the p62 level strongly supported the role of Tid1 in promoting autophagy flux.
Tid1 Mediates Autophagy Independent of Its Association with Hsp70
We next determined whether or not Tid1 and Hsp70 are mutually dependent in induction of autophagy. Similar to Tid1S or Tax, ectopic expression of Hsp70 led to accumulation of LC3-II (Fig. 2A). Unlike the wild type Tid1S that interacted with Hsp70, the Tid1SΔJ mutant failed to do so (Fig. 2B). Tid1SΔJ maintained its ability to induce LC3-II augmentation, although it had a lower activity than the wild type Tid1S (Fig. 2C). Similarly, Tid1LΔJ and Tid1SΔJ, the J domain deletion mutants of Tid1L and Tid1S, respectively, did not interact with Hsp70 (Fig. 2, B and D). Tid1SΔJ displayed a decreased ability to induce formation of LC3+ cytoplasmic puncta (Fig. 2, C and E). These experimental findings had two lines of implication. Tid1 might mediate autophagy in part in cooperation with Hsp70 through its conserved J domain, or Tid1 could execute its function for induction of autophagy in a manner independent of interacting with Hsp70 because the J domain of Tid1 was found to interact with cellular proteins other than Hsp70 (12).
FIGURE 2.
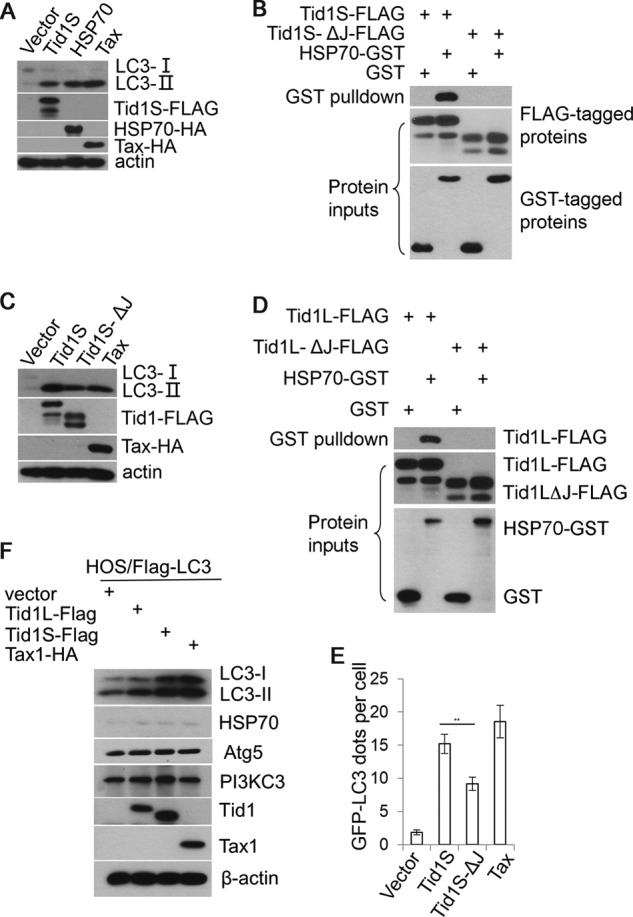
Tid1 mediates autophagy independently of its interaction with Hsp70. A, vector, Tid1S-FLAG, HSP70-HA, or Tax-HA was transiently transfected into HT1080 cells, and total protein lysates were collected 24 h post-transfection for detecting LC3, Tid1S-FLAG, HSP70-HA, or Tax-HA using immunoblots. B, Tid1S-FLAG or Tid1SΔJ-FLAG was transiently co-transfected with HSP70-GST. 24 h post-transfection, total protein extracts were prepared for GST pulldown assay. C, anti-LC3 blot in HT1080 cells transfected with vector, Tid1S-FLAG, Tid1SΔJ-FLAG, or Tax-HA. D, GST pulldown assay to evaluate the interaction of HSP70 with Tid1L-FLAG or with Tid1LΔJ-FLAG. E, the average numbers of GFP-LC3 puncta per cell in HT1080 cells transfected with Tid1S-FLAG, Tid1SΔJ-FLAG, or Tax-HA. **, p < 0.01. F, HOS/FLAG-LC3 cells were transduced with control lentivirus or the lentivirus expressing Tid1L-FLAG, Tid1S-FLAG, or Tax-HA. 4 days post-transduction, immunoblot analysis was performed to detect proteins indicated in the figure.
To test these possibilities, we utilized HOS cells in which the level of Hsp70 was found barely detectable. HOS cells, which were engineered to constitutively express FLAG-LC3 driven by human elongation factor promoter, were transduced with lentivirus expressing Tid1L, Tid1S, or Tax. As shown in Fig. 2F, ectopic expression of Tid1L, Tid1S, or Tax increased FLAG-LC3-II, whereas the endogenous Hsp70 protein was maintained at very low levels. The levels of Atg5 and PI3KC3, the essential mediators of autophagy, were not altered (Fig. 2F). It was noted that Tid1L and Tid1S were fully processed in HOS cells (Fig. 2F), indicating that the processed forms of Tid1 had the capacity to mediate autophagy.
Tid1 and Hsp70 Mediate Autophagy under Various Stresses
To determine whether Tid1 is required for Hsp70 induction of autophagy, we depleted endogenous Tid1, followed by transient transfection of Hsp70 into these cells. We found that regardless of the presence of Tid1, Hsp70 increased LC3-II at comparable levels (Fig. 3A). Depletion of Tid1 did not significantly affect the ability of Hsp70 in inducing formation of LC3+ foci (Fig. 3, B and C). To further investigate the role of Hsp70 in induction of autophagy under various stresses, we selected MT-1 T cell line in which Hsp70 was not expressed under normal culture condition. 4 h following hyperthermia stress, Hsp70 was detected, and the level of Hsp70 was further increased over time following heat shock (Fig. 3D). In correlation with increasing levels of Hsp70, the level of LC3-II was augmented in heat-stressed MT-1 cells (Fig. 3D). Heavy metal Cd2+ also induced Hsp70 expression, which correlated with increased levels of LC3-II (Fig. 3E). Under nutrient deprivation condition, Hsp70 remained undetectable (data not shown); however, increased LC3-II levels were still observed (Fig. 3F). DMSO increased the basal level of Hsp70, and heat stress further enhanced Hsp70 expression in MT-1 cells (Fig. 3G). Although LC3-II was very slightly increased in DMSO-treated MT-1 cells following heat stress, a reduction of LC3-I was noted, suggesting that heat shock still induced autophagic flux in DMSO-treated cells. We found that treatment of MT-1 cells with VER-155008, a chemical inhibitor for Hsp70 family proteins, caused p62 accumulation following heat stress regardless of the induction of LC3-II (Fig. 3G), suggesting that the autophagy process was blocked after generation of LC3-II. Together, these findings suggested that Hsp70 had no apparent role in nutrient deprivation-induced autophagy but played a key role in heat stress-induced macroautophagy.
FIGURE 3.
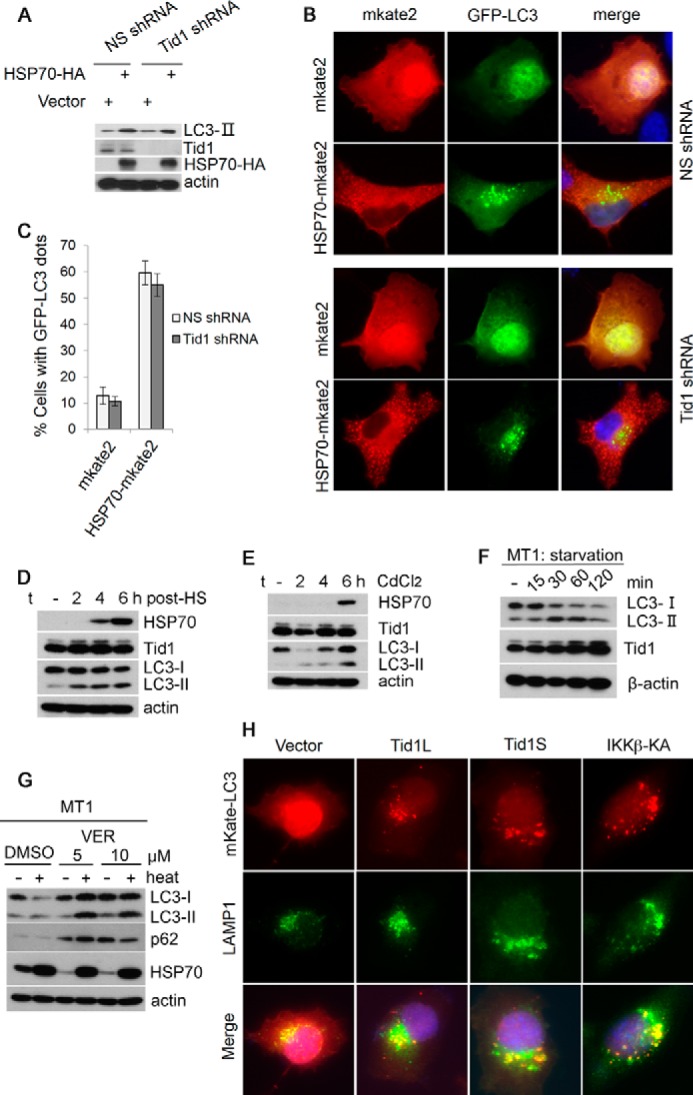
Tid1 and Hsp70 mediate autophagy under distinct stresses. A, HT1080 cells stably expressing NS shRNA or Tid1shRNA were transfected with vector or with HSP70. The levels of LC3-II were examined using anti-LC3 blot. B, HT1080 cells stably expressing NS shRNA or Tid1shRNA were transfected with mKate2 or with HSP70-mKate2. LC3+ puncta were examined with fluorescence imaging. C, the percentage of cells with LC3+ puncta from the experiments in B was shown. D, MT-1 cells under hyperthermia stress at various time points indicated in the figure were examined for the levels of LC3-II, HSP70, and Tid1 with immunoblots. E, MT-1 cells treated with CdCl2 for indicated time points were analyzed for the levels of LC3-II, HSP70, and Tid1 with immunoblots. F, MT-1 cells treated with EBSS (nutrient deprivation) for various amounts of time were examined with anti-LC3 and anti-Tid1 blots. G, MT-1 cells were pretreated with DMSO (control) or 5 or 10 μm of VER-155008 (VER) for 1 h. Then these cells were heat-shocked at 43 °C for 30 min. 4 h following the treatment, whole cell lysates were collected and immunoblotted with anti-LC3, anti-p62, or anti-Hsp70 antibody. β-Actin was used for protein loading control. H, mKate2-LC3 was co-transfected with vector, Tid1L-FLAG, Tid1S-FLAG, or FLAG-IKKβKA in HT1080 cells. The transfected cells were stained with anti-LAMP1 (green). Representative pictures are shown.
Fluorescence imaging was employed to validate the notion that Tid1 promotes autophagy flux to the stage of autolysosome formation. We co-transfected Tid1 with mKate2-LC3, followed by staining the transfected cells with anti-LAMP1 antibody. As shown in Fig. 3H, co-localization of mKate2-LC3 and LAMP1, a lysosome marker protein, was observed as evidenced by forming yellow puncta in cells transfected with Tid1 or IKKβKA, a kinase active form of IKKβ that was known to promote autophagy flux (29, 30). Together, these results suggested that Tid1 and Hsp70 were both positive regulators of macroautophagy pathway, and they appeared to mediate autophagy under various stresses.
Tid1 Is a Crucial Component of Canonical Autophagy Pathway
To evaluate the role of Tid1 in canonical macroautophagy, we generated Tid1-depleted cell lines using lentivirus transduction of Tid1-specific shRNA. The Tid1 knockdown efficiency was verified using anti-Tid1 blot, showing significant reduction of both long and short forms of the endogenous human Tid1 proteins (Fig. 4, A and B). Depletion of the endogenous Tid1 appeared not to be detrimental to cell growth (data not shown). We found that the Tid1-depleted cells displayed diminished LC3-II levels when treated with rapamycin or EBSS (Fig. 4, A and B). In contrast, the NS shRNA-transduced cells maintained their ability to respond to rapamycin or EBSS with increased LC3-II levels (Fig. 4, A and B).
FIGURE 4.

Depletion of Tid1 impairs autophagy induction. A, U2OS cells stably expressing NS shRNA or Tid1shRNA were treated with DMSO or with rapamycin at 0.125, 0.25, 0.5, and 1 μm rapamycin for 6 h. The levels of LC3-II were examined with anti-LC3 blot. B, U2OS cells stably expressing NS shRNA or Tid1shRNA were treated with EBSS at various time points indicated in the figure. The levels of LC3-II were examined with anti-LC3 blot. C–E, HT1080 cells stably expressing NS shRNA, Beclin1 shRNA (C), Atg5 shRNA (D), or PI3KC3 shRNA (E) were transfected with vector, Tid1L-FLAG, Tid1S-FLAG, or HSP70-HA. The levels of LC3-II and other related proteins as indicated in the each figure were examined with specific antibodies. F, U2OS cells stably expressing NS shRNA or Tid1 shRNA were treated with DMSO or 6.25, 12.5, 25, or 50 μm cisplatin for 24 h. The levels of LC3-II, Tid1, and actin were detected with immunoblot using relevant antibodies. U2OS cells stably expressing NS shRNA or mTid1 shRNA were treated with DMSO or 100 μm of cisplatin for 24 h. G, disrupted nucleus was examined with DAPI staining. H, the percentage of cells with disrupted nucleus was statistically analyzed. **, p < 0.01.
To determine whether Tid1 targets canonical macroautophagy pathway, we developed cell lines with depletion of key components of autophagy complexes including Beclin1, Atg5, and PI3KC3. We found that in Beclin1-, Atg5-, or PI3KC3-depleted cells, neither Tid1L nor Tid1S induced accumulation of LC3-II (Fig. 4, C–E). In contrast, the NS shRNA-transduced cells responded to ectopic expression of Tid1L, Tid1S, or Hsp70 as evidenced by increased levels of LC3-II (Fig. 4, C–E). The levels of p62 were reduced following transfection of Tid1 or Hsp70 in the NS shRNA-expressing cells but were accumulated in the Beclin1-depleted cells (Fig. 4C). These results validated the role of Tid1 in promoting autophagy flux, probably by targeting Beclin1-containing autophagy molecular complex. Chemotherapeutic agents frequently induce autophagy, which potentially contributes to chemoresistance. To determine whether silencing Tid1 sensitizes to cisplatin-mediated apoptosis, we deleted Tid1 in U2OS cells. As shown in Fig. 4F, cisplatin induced formation of LC3-II in NS shRNA-expressing cells but failed to do so in Tid1-depleted cells. Increased disrupted nucleus was detected in Tid1-depleted cells as compared with the NS shRNA-expressing cells (Fig. 4, G and H). This finding supported a role of Tid1-mediated autophagy for the survival of cancer cells under cisplatin treatment.
To investigate whether or not the function of Tid1 in induction of autophagy is conserved in mammalian cells of different species, we utilized NIH3T3 mouse fibroblast cells. In the NS shRNA-transduced NIH3T3 cells, significantly increased levels of LC3-II were seen following treatment of rapamycin or EBSS, whereas depletion of the endogenous mouse Tid1 (mTid1) drastically impaired LC3-II augmentation (Fig. 5, A and B). Reconstitution of human Tid1 (hTid1L or hTid1S) in mTid1-depleted NIH3T3 cells restored the ability of these cells in response to rapamycin as evidenced by increased LC3-II levels (Fig. 5, C and D). Together, these data strongly suggested that both human and mouse Tid1 proteins exhibited a conserved function in regulating macroautophagy and that both Tid1L and Tid1S displayed comparable activities in induction of autophagy.
FIGURE 5.
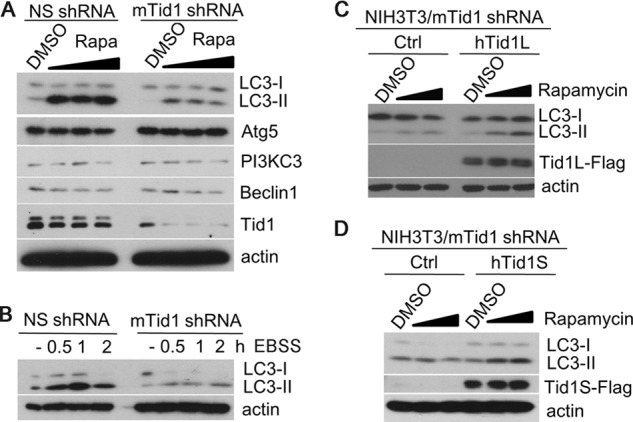
Reconstitution of human Tid1 in mTid1-depleted NIH3T3 cells restores sensitivity of cells to rapamycin or EBSS-induced autophagy. A, NIH3T3 cells stably expressing NS shRNA or mTid1 shRNA were treated with 0.01, 0.1, or 1 μm rapamycin (Rapa) overnight. The levels of LC3-II and other related proteins as indicated were detected with specific antibodies. B, NIH3T3 cells stably expressing NS shRNA or mTid1 shRNA were incubated with EBSS for indicated time points, and the LC3-II levels were examined. C and D, mTid1-depleted NIH3T3 cells were reconstituted with human Tid1L (C), with human Tid1S (D), or control (Ctrl), and these cells were treated with rapamycin. The levels of LC3-II, Tid1L, or Tid1S were examined with anti-LC3 or anti-Tid1 antibodies.
Tid1 Interacts with Autophagy Molecular Complex
To understand the mechanistic nature of Tid1 in mediating autophagy, we evaluated physical and functional interaction of Tid1 with the autophagy molecular complex containing Beclin1, PI3KC3, and UVRAG. Tid1L was found to co-precipitate with Beclin1 and PI3KC3, but not with UVRAG (Fig. 6A). The interaction of either Tid1L or Tid1S with Beclin1 appeared to be more potent than the interaction between Hsp70 and Beclin1 (Fig. 6B). However, the interaction of Tid1 with PI3KC3 seemed to be less efficient than the binding of Tax to PI3KC3 (Fig. 6C), suggesting that the interaction between Tid1 and PI3KC3 might be indirect. To validate this assumption, we evaluated the interaction between Tid1 and Beclin1 in PI3KC3-depleted cells. We found that removal of PI3KC3 did not impair the interaction of Tid1 with Beclin1 (Fig. 6, D and E), whereas depletion of Beclin1 led to drastic impairment of the co-precipitation between Tid1 and PI3KC3 (Fig. 6F). By analyzing various Tid1 mutants, we found that Tid1ΔN100 failed to interact with Beclin1, correlating with its diminished activity in inducing accumulation of LC3-II (Fig. 6, G and H). Hsp70 was apparently bound to Beclin1 independently of Tid1, because depletion of Tid1 did not affect the interaction between Hsp70 and Beclin1 (Fig. 6I). Further, we observed that rapamycin facilitated a physical interaction between Tid1 and Beclin1 using endogenously expressed proteins (Fig. 6J). Together, our data indicated that Tid1 targets the autophagy complex by interacting with Beclin1 for in response to autophagy stimulation.
FIGURE 6.

Tid1 interacts with Beclin1. A, transient co-transfection using various combinations of expression plasmids as indicated in the figure was performed in HEK293 cells, followed by GST pulldown assay. B, GST pulldown assay of HEK293 cells co-transfected with FLAG-BECN1 and GST or GST-tagged Tid1L, Tid1S, HSP70, and Tax. C, GST pulldown assay of HEK293 cells co-transfected with PI3KC3-FLAG and GST or GST-tagged protein expression plasmids. D, PI3KC3 knockdown efficiency in HEK293 cells was verified with anti-PI3KC3 blot. E, HEK293 cells stably expressing NS shRNA or PI3KC3 shRNA were transfected with GST or GST-tagged Tid1. F, co-precipitation of Tid1 and PI3KC3 in NS shRNA- or BECN1shRNA-expressing HEK293 cells using transient co-transfection method. G, GST pulldown assay of HEK293 cells co-transfected with FLAG-BECN1 and GST, GST-tagged Tid1L, or its mutants. H, HT1080 cells were transfected with vector, Tid1-FLAG, or its mutants, and the levels of LC3-II were determined with anti-LC3 blot. I, HEK293 cells stably expressing NS shRNA or Tid1 shRNA were co-transfected with Flag-Beclin1 and GST or HSP70-GST. J, co-precipitation of Tid1 with Beclin1 in DMSO or rapamycin (μm/ml, 1 h) HEK293 cells using endogenously expressed proteins. IP, immunoprecipitation.
Tid1 is a downstream molecule of IκB kinases
The IκB kinase complex plays an essential role in rapamycin or nutrient deprivation-induced autophagy (32), whereas Tid1 was shown to interact with IκB kinases (7). To determine whether Tid1 involves in IκB kinase-mediated autophagy, we transfected the retroviral Tax protein Tax1 or Tax2 or IKKβKA into Tid1-silenced cells as Tax was shown previously to increase autophagic flux through IκB kinases (29). In Fig. 7A, Tax1, Tax2, or IKKβKA induced formation of LC3-II in NS shRNA-expressing cells but failed to do so in Tid1-depleted cells. Typical cytoplasmic GFP-LC3 puncta were seen in cells co-transfected with Tax or IKKβKA in NS shRNA-expressing cells, but not in Tid1-depleted cells (Fig. 7B), which was statistically significant (Fig. 7C). In Tax-expressing HTLV-1-transformed T cell lines, SLB-1 and MT-2, silencing Tid1 reduced LC3-II formation (Fig. 7D). Furthermore, removal of the endogenous Tid1 led to impaired interaction of IκB kinases with Beclin1, and the interaction of Tax1 with Beclin1 was only slightly affected by Tid1 silencing (Fig. 7E). Thus, these results verified a mechanistic model that Tid1 connects IκB kinases to Beclin1-containing autophagy complex, leading to the induction of autophagy (Fig. 8).
FIGURE 7.
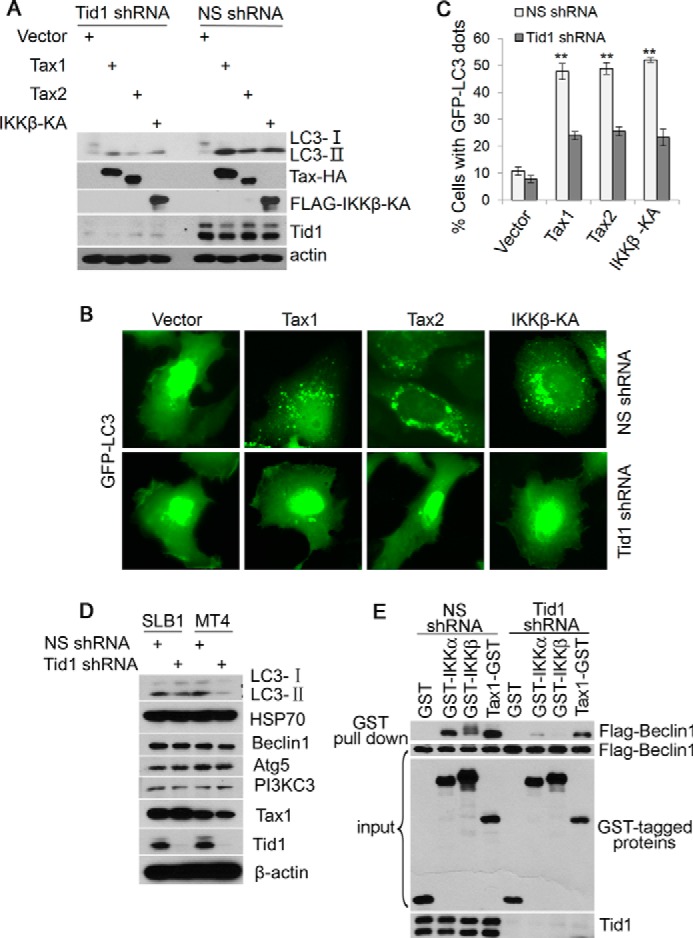
Tid1 is a downstream molecule of the IκB kinase complex. A, HT1080 cells stably expressing NS shRNA or Tid1 shRNA were transfected with vector, Tax1 (HTLV-1 Tax), Tax2 (HTLV-2 Tax), or IKKβKA, Total protein lysates were analyzed using antibodies indicated in the figure. B, in HT1080 cells stably expressing NS shRNA or Tid1 shRNA, GFP-LC3 was co-transfected with vector, Tax1, Tax2, or IKKβKA, The cytoplasmic GFP-LC3 puncta was visualized with fluorescence imaging. C, statistical analysis of the experiment in B. D, Tax-expressing HTLV-1-tranformed T cell lines SLB-1 and MT-4 were transduced with lentivirus expressing NS shRNA or Tid1shRNA; total protein lysates were examined using immunoblot with antibodies indicated in the figure. E, in NS shRNA or Tid1shRNA-expressing HEK293 cells, FLAG-Beclin1 was co-transfected with GST or GST-tagged IKKα, IKKβ, and Tax1. GST pulldown assay was performed to determine the interaction of IκB kinase and Beclin1.
FIGURE 8.
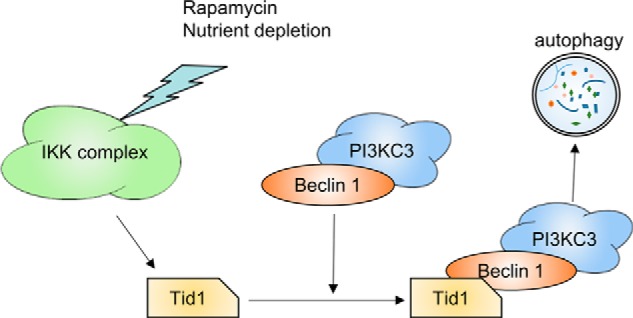
A hypothetical model of Tid1 participation in autophagy pathway. Rapamycin, nutrient deprivation, or the viral Tax protein activates IκB kinases, which in turn promotes their Tid1-dependent interaction of IκB kinases with the Beclin1-containing autophagy protein complex. This action induces activation of PI3KC3, facilitating autophagic flux.
Discussion
In the present study, we identify a novel function of the DnaJ protein Tid1 for its role in the regulation of macroautophagy. Our data have shown that Tid1 plays an essential role in the canonical autophagy pathway by facilitating assembly of the autophagy molecular complex containing Beclin1-PI3KC3 at the stage of vesicular nucleation. We find that the phosphorylation of p76S6K, the substrate of mTOR, is not inhibited by ectopic expression of Tid1 (data not shown), which suggests that Tid1-mediated autophagy is not related to the inhibition of the mTOR complex.
Tid1-mediated autophagy is apparently independent of its association with Hsp70 because the Tid1 mutants with complete deletion of the J domain retain their ability to increase LC3-II level. The DnaJ proteins of the molecular chaperone superfamily are evolutionarily conserved proteins for maintaining cellular homeostasis. Indeed, knock-out of tid1 was found to be embryotic lethal in mice, suggesting its life-supporting role during embryotic development (33). In addition to its co-chaperone function, vast amounts of evidence have shown that Tid1 regulates a variety of cellular signaling molecules, and these activities are apparently unrelated to the co-chaperone function of Tid1. We find that Tid1 possesses two domains that are important for induction of autophagy: an N-terminal domain adjacent to the J domain that mediates the binding of Tid1 to Beclin1 and the J domain that executes the full capacity of Tid1 in inducing autophagy. The observation that the Tid1ΔJ mutants partially lose their ability for induction of autophagy suggests that the J domain is one of the functional domains. Our data show that Tid1 induces canonical macroautophagy independent of Hsp70, because Tid1 is capable of mediating autophagy in the absence of Hsp70. Moreover, Hsp70 induces autophagy in Tid1-depleted cells, suggesting that Tid1 and Hsp70 are mutually independent in promoting the autophagic process. Furthermore, Tid1 and Hsp70 appear to mediate autophagy under various stresses. Tid1 plays an essential role in nutrient deprivation or rapamycin-induced canonical autophagy, whereas Hsp70 appears to be important for hyperthermia- or heavy metal-induced autophagy.
Our data demonstrate that the ability of Tid1 to induce autophagy is not exclusively related to its mitochondria localization. Tid1 possesses a consensus mitochondrial signal peptide at its N terminus, which presumably targets it into the mitochondria for processing into cleaved mature forms (23). Although Tid1 is predominantly localized in the mitochondria to maintain mitochondrial membrane integrity (34), a handful of studies have demonstrated that Tid1 exhibits cytoplasmic functions by interacting with the cellular and viral proteins that are not localized in the mitochondria. In the present study, the N-terminal mitochondria targeting sequence of Tid1 is dispensable for induction of autophagy, which suggests that at least the cytoplasmic portion of Tid1 is sufficient for such function.
Our study also shows that Tid1L and its spliced variant, Tid1S, function similarly in mediating autophagy. These two spliced variants may function differently in some biological aspects. It has been shown that Tid1L and Tid1S exhibit opposing activities in regulating apoptosis (23), although the underlying mechanism remains unclear. In addition, Tid1L, but not Tid1S, promotes ubiquitination-dependent degradation of EGF receptor, which may contribute to its tumor suppressive activity (12). However, other studies indicate that both isoforms of Tid1 display similar functions. For instance, both Tid1L and Tid1S inhibit IFNα signaling and induce cell senescence (18, 24). In this study, we examined two forms of Tid1 for their activities in the regulation of autophagy by depleting endogenous mTid1 in NIH3T3 cells, followed by reconstituting human Tid1L or Tid1S into these cells. We found that either hTid1L or hTid1S interacts with Beclin1-containing autophagy molecular complex and restores the capacity of mTid1-depleted NIH3T3 cells in mediating autophagy, indicating that they are functionally redundant in this biological aspect.
The ability of Tid1 in induction of autophagy may be connected to its tumor suppressive activity. Autophagy is crucial for maintaining chromosomal stability and is implicated in playing a key role in tumor suppression. Reduced expression of autophagic molecules such as Beclin1 or Bif1 in mice renders mice prone to the development of lymphoma (35). Heterozygous loss of beclin1 in human breast cancer is linked to oncogenesis, whereas overexpression of Beclin1 suppresses growth of cancer in mice (36), defining its role as a tumor suppressor. In opposition to the tumor suppressive role of autophagy, studies have also shown that the fundamental cytoprotective role of autophagy may contribute to chemotherapy resistance (37–39). Combination therapy employing chemotherapeutics and autophagy inhibitor displays a better therapeutic efficacy than chemotherapy alone (40–42). Furthermore, in the context of HTLV-1-mediated transformation of T lymphocytes, expression of a functional autophagy molecule such as Beclin1 or PI3KC3 is crucial for supporting leukemia cell survival and proliferation (30). In the case of Tid1, aberrant expression of Tid1 has been found in some types of human cancer. In lung cancer, Tid1 is expressed at lower levels than normal counterpart cells, whereas Tid1 is overexpressed in colon cancer (12, 43). In mouse models, overexpression of Tid1 suppresses growth of human head and neck cancer cells and melanoma cells (7, 19). In contrast, Tid1 assists c-Met-mediated oncogenesis in renal cell carcinoma (21). Collectively, like other autophagy molecules, the involvement of Tid1 in cancer development may be cell context- and/or cancer stage-dependent. The autophagy-inducing function of Tid1 in correlation with its role in oncogenesis needs to be further investigated.
Author Contributions
G. N. performed and analyzed experiments; H. Z., D. L., and L. C. performed parts of the experiments; C. B. and H.-G. W. analyzed data; and H. C. designed the experiments, analyzed the data, and wrote the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Karl Munger for pCMV/Tid1S plasmid and anti-Tid1 antibody, Atsushi Koito and Takeo Ohsugi for MT-1 cell line, and Thomas D. Martin and Vineet N. KewalRamani for NIH3T3 cell line.
This work was supported, in whole or in part, by National Institutes of Health Grant R01AI090113 (to H. C.). The authors declare that they have no conflicts of interest with the contents of this article.
- EBSS
- Earle's balanced salt solution
- NS
- non-specific.
References
- 1. Kurzik-Dumke U., Gundacker D., Renthrop M., Gateff E. (1995) Tumor suppression in Drosophila is causally related to the function of the lethal(2) tumorous imaginal discs gene, a dnaJ homolog. Dev. Genet. 16, 64–76 [DOI] [PubMed] [Google Scholar]
- 2. Schilling B., De-Medina T., Syken J., Vidal M., Münger K. (1998) A novel human DnaJ protein, hTid-1, a homolog of the Drosophila tumor suppressor protein Tid56, can interact with the human papillomavirus type 16 E7 oncoprotein. Virology 247, 74–85 [DOI] [PubMed] [Google Scholar]
- 3. Cheng H., Cenciarelli C., Shao Z., Vidal M., Parks W. P., Pagano M., Cheng-Mayer C. (2001) Human T cell leukemia virus type 1 Tax associates with a molecular chaperone complex containing hTid-1 and Hsp70. Curr. Biol. 11, 1771–1775 [DOI] [PubMed] [Google Scholar]
- 4. Sohn S. Y., Kim S. B., Kim J., Ahn B. Y. (2006) Negative regulation of hepatitis B virus replication by cellular Hsp40/DnaJ proteins through destabilization of viral core and X proteins. J. Gen. Virol. 87, 1883–1891 [DOI] [PubMed] [Google Scholar]
- 5. Wang L., Tam J. P., Liu D. X. (2006) Biochemical and functional characterization of Epstein-Barr virus-encoded BARF1 protein: interaction with human hTid1 protein facilitates its maturation and secretion. Oncogene 25, 4320–4331 [DOI] [PubMed] [Google Scholar]
- 6. Cheng H., Cenciarelli C., Tao M., Parks W. P., Cheng-Mayer C. (2002) HTLV-1 Tax-associated hTid-1, a human DnaJ protein, is a repressor of IκB kinase beta subunit. J. Biol. Chem. 277, 20605–20610 [DOI] [PubMed] [Google Scholar]
- 7. Cheng H., Cenciarelli C., Nelkin G., Tsan R., Fan D., Cheng-Mayer C., Fidler I. J. (2005) Molecular mechanism of hTid-1, the human homolog of Drosophila tumor suppressor l(2)Tid, in the regulation of NF-κB activity and suppression of tumor growth. Mol. Cell. Biol. 25, 44–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Liu H. Y., MacDonald J. I., Hryciw T., Li C., Meakin S. O. (2005) Human tumorous imaginal disc 1 (TID1) associates with Trk receptor tyrosine kinases and regulates neurite outgrowth in nnr5-TrkA cells. J. Biol. Chem. 280, 19461–19471 [DOI] [PubMed] [Google Scholar]
- 9. Trentin G. A., Yin X., Tahir S., Lhotak S., Farhang-Fallah J., Li Y., Rozakis-Adcock M. (2001) A mouse homologue of the Drosophila tumor suppressor l(2)tid gene defines a novel Ras GTPase-activating protein (RasGAP)-binding protein. J. Biol. Chem. 276, 13087–13095 [DOI] [PubMed] [Google Scholar]
- 10. Schaaf C. P., Benzing J., Schmitt T., Erz D. H., Tewes M., Bartram C. R., Janssen J. W. (2005) Novel interaction partners of the TPR/MET tyrosine kinase. FASEB J. 19, 267–269 [DOI] [PubMed] [Google Scholar]
- 11. Kim S. W., Chao T. H., Xiang R., Lo J. F., Campbell M. J., Fearns C., Lee J. D. (2004) Tid1, the human homologue of a Drosophila tumor suppressor, reduces the malignant activity of ErbB-2 in carcinoma cells. Cancer Res. 64, 7732–7739 [DOI] [PubMed] [Google Scholar]
- 12. Chen C. Y., Jan C. I., Lo J. F., Yang S. C., Chang Y. L., Pan S. H., Wang W. L., Hong T. M., Yang P. C. (2013) Tid1-L inhibits EGFR signaling in lung adenocarcinoma by enhancing EGFR Ubiquitinylation and degradation. Cancer Res. 73, 4009–4019 [DOI] [PubMed] [Google Scholar]
- 13. Dhennin-Duthille I., Nyga R., Yahiaoui S., Gouilleux-Gruart V., Régnier A., Lassoued K., Gouilleux F. (2011) The tumor suppressor hTid1 inhibits STAT5b activity via functional interaction. J. Biol. Chem. 286, 5034–5042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Linnoila J., Wang Y., Yao Y., Wang Z. Z. (2008) A mammalian homolog of Drosophila tumorous imaginal discs, Tid1, mediates agrin signaling at the neuromuscular junction. Neuron 60, 625–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bae M. K., Jeong J. W., Kim S. H., Kim S. Y., Kang H. J., Kim D. M., Bae S. K., Yun I., Trentin G. A., Rozakis-Adcock M., Kim K. W. (2005) Tid-1 interacts with the von Hippel-Lindau protein and modulates angiogenesis by destabilization of HIF-1α. Cancer Res. 65, 2520–2525 [DOI] [PubMed] [Google Scholar]
- 16. Ahn B. Y., Trinh D. L., Zajchowski L. D., Lee B., Elwi A. N., Kim S. W. (2010) Tid1 is a new regulator of p53 mitochondrial translocation and apoptosis in cancer. Oncogene 29, 1155–1166 [DOI] [PubMed] [Google Scholar]
- 17. Kim S. W., Hayashi M., Lo J. F., Fearns C., Xiang R., Lazennec G., Yang Y., Lee J. D. (2005) Tid1 negatively regulates the migratory potential of cancer cells by inhibiting the production of interleukin-8. Cancer Res. 65, 8784–8791 [DOI] [PubMed] [Google Scholar]
- 18. Tarunina M., Alger L., Chu G., Munger K., Gudkov A., Jat P. S. (2004) Functional genetic screen for genes involved in senescence: role of Tid1, a homologue of the Drosophila tumor suppressor l(2)tid, in senescence and cell survival. Mol. Cell. Biol. 24, 10792–10801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen C. Y., Chiou S. H., Huang C. Y., Jan C. I., Lin S. C., Hu W. Y., Chou S. H., Liu C. J., Lo J. F. (2009) Tid1 functions as a tumour suppressor in head and neck squamous cell carcinoma. J. Pathol. 219, 347–355 [DOI] [PubMed] [Google Scholar]
- 20. Qian J., Perchiniak E. M., Sun K., Groden J. (2010) The mitochondrial protein hTID-1 partners with the caspase-cleaved adenomatous polyposis cell tumor suppressor to facilitate apoptosis. Gastroenterology 138, 1418–1428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Copeland E., Balgobin S., Lee C. M., Rozakis-Adcock M. (2011) hTID-1 defines a novel regulator of c-Met receptor signaling in renal cell carcinomas. Oncogene 30, 2252–2263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sterrenberg J. N., Blatch G. L., Edkins A. L. (2011) Human DNAJ in cancer and stem cells. Cancer Lett. 312, 129–142 [DOI] [PubMed] [Google Scholar]
- 23. Syken J., De-Medina T., Münger K. (1999) TID1, a human homolog of the Drosophila tumor suppressor l(2)tid, encodes two mitochondrial modulators of apoptosis with opposing functions. Proc. Natl. Acad. Sci. U.S.A. 96, 8499–8504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sarkar S., Pollack B. P., Lin K. T., Kotenko S. V., Cook J. R., Lewis A., Pestka S. (2001) hTid-1, a human DnaJ protein, modulates the interferon signaling pathway. J. Biol. Chem. 276, 49034–49042 [DOI] [PubMed] [Google Scholar]
- 25. Iosefson O., Sharon S., Goloubinoff P., Azem A. (2012) Reactivation of protein aggregates by mortalin and Tid1: the human mitochondrial Hsp70 chaperone system. Cell Stress Chaperones 17, 57–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lu B., Garrido N., Spelbrink J. N., Suzuki C. K. (2006) Tid1 isoforms are mitochondrial DnaJ-like chaperones with unique carboxyl termini that determine cytosolic fate. J. Biol. Chem. 281, 13150–13158 [DOI] [PubMed] [Google Scholar]
- 27. Massey A. C., Zhang C., Cuervo A. M. (2006) Chaperone-mediated autophagy in aging and disease. Curr. Topics Dev. Biol. 73, 205–235 [DOI] [PubMed] [Google Scholar]
- 28. Yang Y., Fiskus W., Yong B., Atadja P., Takahashi Y., Pandita T. K., Wang H. G., Bhalla K. N. (2013) Acetylated hsp70 and KAP1-mediated Vps34 SUMOylation is required for autophagosome creation in autophagy. Proc. Natl. Acad. Sci. U.S.A. 110, 6841–6846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ren T., Dong W., Takahashi Y., Xiang D., Yuan Y., Liu X., Loughran T. P., Jr., Sun S. C., Wang H. G., Cheng H. (2012) HTLV-2 Tax immortalizes human CD4+ memory T lymphocytes by oncogenic activation and dysregulation of autophagy. J. Biol. Chem. 287, 34683–34693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ren T., Takahashi Y., Liu X., Loughran T. P., Sun S. C., Wang H. G., Cheng H. (2015) HTLV-1 Tax deregulates autophagy by recruiting autophagic molecules into lipid raft microdomains. Oncogene 34, 334–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yan G., Huang J., Jarbadan N. R., Jiang Y., Cheng H. (2008) Sequestration of NF-κB signaling complexes in lipid rafts contributes to repression of NF-κB in T lymphocytes under hyperthermia stress. J. Biol. Chem. 283, 12489–12500 [DOI] [PubMed] [Google Scholar]
- 32. Criollo A., Senovilla L., Authier H., Maiuri M. C., Morselli E., Vitale I., Kepp O., Tasdemir E., Galluzzi L., Shen S., Tailler M., Delahaye N., Tesniere A., De Stefano D., Younes A. B., Harper F., Pierron G., Lavandero S., Zitvogel L., Israel A., Baud V., Kroemer G. (2010) The IKK complex contributes to the induction of autophagy. EMBO J. 29, 619–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lo J. F., Hayashi M., Woo-Kim S., Tian B., Huang J. F., Fearns C., Takayama S., Zapata J. M., Yang Y., Lee J. D. (2004) Tid1, a cochaperone of the heat shock 70 protein and the mammalian counterpart of the Drosophila tumor suppressor l(2)tid, is critical for early embryonic development and cell survival. Mol. Cell. Biol. 24, 2226–2236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ng A. C., Baird S. D., Screaton R. A. (2014) Essential role of TID1 in maintaining mitochondrial membrane potential homogeneity and mitochondrial DNA integrity. Mol. Cell. Biol. 34, 1427–1437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Takahashi Y., Coppola D., Matsushita N., Cualing H. D., Sun M., Sato Y., Liang C., Jung J. U., Cheng J. Q., Mulé J. J., Pledger W. J., Wang H. G. (2007) Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat. Cell Biol. 9, 1142–1151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shin J. Y., Hong S. H., Kang B., Minai-Tehrani A., Cho M. H. (2013) Overexpression of beclin1 induced autophagy and apoptosis in lungs of K-rasLA1 mice. Lung Cancer 81, 362–370 [DOI] [PubMed] [Google Scholar]
- 37. Holohan C., Van Schaeybroeck S., Longley D. B., Johnston P. G. (2013) Cancer drug resistance: an evolving paradigm. Nat. Rev. Cancer 13, 714–726 [DOI] [PubMed] [Google Scholar]
- 38. Cui J., Gong Z., Shen H. M. (2013) The role of autophagy in liver cancer: molecular mechanisms and potential therapeutic targets. Biochim. Biophys. Acta 1836, 15–26 [DOI] [PubMed] [Google Scholar]
- 39. Wei Y., Zou Z., Becker N., Anderson M., Sumpter R., Xiao G., Kinch L., Koduru P., Christudass C. S., Veltri R. W., Grishin N. V., Peyton M., Minna J., Bhagat G., Levine B. (2013) EGFR-mediated Beclin 1 phosphorylation in autophagy suppression, tumor progression, and tumor chemoresistance. Cell 154, 1269–1284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kimura T., Takabatake Y., Takahashi A., Isaka Y. (2013) Chloroquine in cancer therapy: a double-edged sword of autophagy. Cancer Res. 73, 3–7 [DOI] [PubMed] [Google Scholar]
- 41. Janku F., McConkey D. J., Hong D. S., Kurzrock R. (2011) Autophagy as a target for anticancer therapy. Nat. Rev. Clin. Oncol. 8, 528–539 [DOI] [PubMed] [Google Scholar]
- 42. Guo X. L., Li D., Hu F., Song J. R., Zhang S. S., Deng W. J., Sun K., Zhao Q. D., Xie X. Q., Song Y. J., Wu M. C., Wei L. X. (2012) Targeting autophagy potentiates chemotherapy-induced apoptosis and proliferation inhibition in hepatocarcinoma cells. Cancer Lett. 320, 171–179 [DOI] [PubMed] [Google Scholar]
- 43. Kurzik-Dumke U., Hörner M., Czaja J., Nicotra M. R., Simiantonaki N., Koslowski M., Natali P. G. (2008) Progression of colorectal cancers correlates with overexpression and loss of polarization of expression of the htid-1 tumor suppressor. Int. J. Mol. Med. 21, 19–31 [PubMed] [Google Scholar]