

Background: NHE1 is a therapeutic drug target, yet knowledge of sites involved in cation translocation and drug binding remains incomplete.
Results: Mutagenesis analyses identified residues in transmembrane helix M9 and exofacial re-entrant loop EL5 that affect substrate affinities and/or drug sensitivity.
Conclusion: M9 and EL5 form part of the cation permeation pathway.
Significance: These findings provide new insight into the structure-function domains of NHE1.
Keywords: membrane protein, molecular biology, pH regulation, site-directed mutagenesis, sodium-proton exchange, transporter
Abstract
Mammalian Na+/H+ exchangers (NHEs) regulate numerous physiological processes and are involved in the pathogenesis of several diseases, including tissue ischemia and reperfusion injuries, cardiac hypertrophy and failure, and cancer progression. Hence, NHEs are being targeted for pharmaceutical-based clinical therapies, but pertinent information regarding the structural elements involved in cation translocation and drug binding remains incomplete. Molecular manipulations of the prototypical NHE1 isoform have implicated several predicted membrane-spanning (M) helices, most notably M4, M9, and M11, as important determinants of cation permeation and drug sensitivity. Here, we have used substituted-cysteine accessibility mutagenesis and thiol-modifying methanethiosulfonate (MTS) reagents to further probe the involvement of evolutionarily conserved sites within M9 (residues 342–363) and the adjacent exofacial re-entrant loop 5 between M9 and M10 (EL5; residues 364–415) of a cysteine-less variant of rat NHE1 on its kinetic and pharmacological properties. MTS treatment significantly reduced the activity of mutants containing substitutions within M9 (H353C, S355C, and G356C) and EL5 (G403C and S405C). In the absence of MTS, mutants S355C, G403C, and S405C showed modest to significant decreases in their apparent affinities for Na+o and/or H+i. In addition, mutations Y370C and E395C within EL5, whereas failing to confer sensitivity to MTS, nevertheless, reduced the affinity for Na+o, but not for H+i. The Y370C mutant also exhibited higher affinity for ethylisopropylamiloride, a competitive antagonist of Na+o transport. Collectively, these results further implicate helix M9 and EL5 of NHE1 as important elements involved in cation transport and inhibitor sensitivity, which may inform rational drug design.
Introduction
Electroneutral countertransport of alkali cations such as Na+, K+, and Li+ for H+ across membranes of mammalian cells are catalyzed by a heterogeneous family of at least 11 secondary active solute carriers (SLC9 gene family) generically termed Na+/H+ exchangers or antiporters (NHE/NHA)3 (1, 2). The activities of these transporters are tightly controlled and important for efficient execution of numerous physiological processes, ranging from cellular and systemic pH and volume homeostasis (3–5) to the regulation of cell shape (6), migration (7–10), and mitosis (11), among others (12–17). On the other hand, aberrant overactivation of NHE activity, notably the ubiquitous NHE1 isoform, occurs in several pathophysiological conditions and contributes to disease progression, including tissue injuries following ischemic and hemorrhagic stroke (18–20), acute myocardial infarction (21–23), cardiac hypertrophy and failure (24–27), and cancer metastasis and invasion (28–31); damages that can be mitigated in animal models by pharmacological inhibition of NHE1 activity. However, in the case of heart disease, attempts to translate some of these promising experimental findings into clinical therapies have thus far proven inconclusive due to modest efficacy and adverse side effects of the tested compounds (32–35). Despite these setbacks, an improved understanding of the molecular determinants that underlie the catalytic and pharmacological properties of NHE1 could assist in the development of more efficacious drugs and treatment regimens.
Information regarding the structural and functional properties of the mammalian NHEs has been derived mainly from analyses of the ubiquitous NHE1 and epithelial NHE3 isoforms. Early computational modeling and substituted-cysteine accessibility mutagenesis studies of human NHE1 (hNHE1) by Wakabayashi et al. (36) predicted a configuration of 12 membrane-spanning (M) helices at its N terminus (∼400–500 amino acids) involved in cation translocation and drug binding, and a cytoplasmically oriented segment at its C terminus (∼350 amino acids) that confers responsiveness to various regulatory stimuli (illustrated in Fig. 1A). Biochemical (37–44) analyses indicated that the exchanger most likely assembles as a homodimer, although higher ordered structures have also been proposed (45).
FIGURE 1.
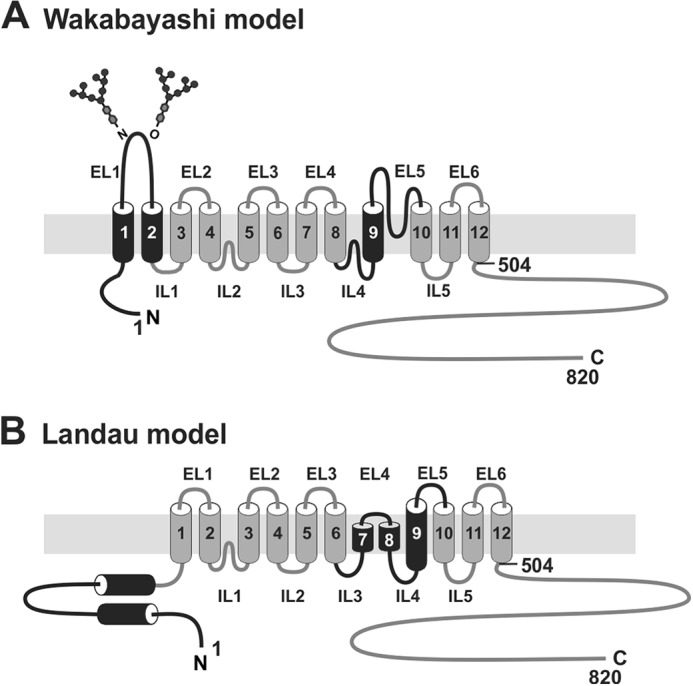
Schematic illustration of the predicted membrane topology of mammalian NHE1. Transmembrane organization of NHE1 according to the models proposed by (A) Wakabayashi et al. (36) and (B) Landau et al. (65). The black shading highlights regions that differ between the two models.
To identify sites involved in Na+ binding and translocation, initial studies (46–48) took advantage of pyrazine- (e.g. amiloride and ethylisopropylamiloride (EIPA)) or benzyol-guanidinium-based (e.g. HOE642 and HOE694) compounds that inhibit NHE activity by acting as competitive or mixed-type antagonists of Na+ influx, suggesting that they bind at or near sites involved in Na+ binding. Random or site-directed mutagenesis of NHE1 followed by functional selection for altered drug sensitivity identified several residues located in the predicted second exofacial loop (EL2) (49) and two membrane-spanning segments, M4 (50–54) and M9 (49, 55, 56), which are significant determinants of drug recognition and/or transport velocity. However, only mutations at Phe162 in M4 of hNHE1 were found to appreciably reduce Na+ affinity (52). Additional mutations of conserved residues within the fifth intracellular loop (IL5) (i.e. Arg440) and adjacent transmembrane helix M11 (Gly445, Gly446) of hNHE1 decreased and increased, respectively, its sensitivity to intracellular H+ (57, 58). Other mutagenesis analyses have confirmed the importance of M4 (59), M9 (60), and M11 (61), and further implicated residues in M6 (62) and M7 (63) as pore-lining elements.
Although there is general support for this model, the precise arrangement of transmembrane helices remains controversial (64–66). High-resolution crystal structures of the bacterial Escherichia coli Na+/H+ antiporter NhaA (67, 68) that is structurally (∼10% amino acid identity) and kinetically (i.e. electrogenic) distinct from NHE1 also indicated a 12-transmembrane structure that assembles as a dimer. Such parallels prompted Landau et al. (65) and Nygaard et al. (69) to use the atomic coordinates of NhaA as a template to generate a three-dimensional model of hNHE1. However, whereas the model proposed by Nygaard et al. (69) closely mirrors that of Wakabayashi et al. (36), Landau and colleagues (65) derived a novel membrane topology that challenges this view (Fig. 1B). Notably, in their new model, the original transmembrane helices M1 and M2 were repositioned intracellularly and the catalytically important M9 helix was reconfigured to form two short intramembrane helices relabeled as M7 and M8, whereas the adjoining exofacial loop 5 (EL5, amino acids 364–415) between M9 and M10 formed a new intracellular loop and a new transmembrane segment M9 that is largely inaccessible to external reagents. Although the computationally derived reorientation of M1 and M2 seems unlikely given that the intervening EL1 segment was shown previously to undergo N- and O-linked glycosylation and therefore places this segment extracellularly (70), the topology of the M9-EL5 region remains uncertain. This region is of particular interest because the putative EL5 loop was originally postulated to invaginate into the membrane (36) in a manner analogous to the pore-lining loops present in ion channels (71, 72), and therefore may contribute to cation permeation and drug sensitivity.
Based on discordances between these models, we have further probed the potential contributions of evolutionarily conserved as well as certain known drug-sensitive sites located in the originally designated M9 and adjacent re-entrant loop EL5 segments (as depicted in Fig. 1A) to cation translocation using the substituted-cysteine accessibility method (73). The data revealed additional amino acids in both M9 and EL5 that contribute to the affinities of the exchanger for substrates and pharmacological antagonists and hence are likely to line the cation permeation pathway. The relevance of these findings to the two different membrane topologies proposed for NHE1 is discussed.
Experimental Procedures
Materials
Carrier free 22NaCl (radioactivity, 5 mCi/ml) was obtained from PerkinElmer Life Sciences. Amiloride, ouabain, and nigericin were purchased from Sigma. α-Minimal essential medium, fetal bovine serum, G418®, and trypsin-EDTA were purchased from Invitrogen. Murine monoclonal HA antibody was purchased from BabCo (Richmond, CA), whereas horseradish peroxidase-conjugated goat α-mouse IgG was obtained from Jackson ImmunoResearch Laboratories Inc. (West Grove, PA). 2-Sulfonatoethyl methanethiosulfonate (MTSES) and 2-(trimethylammonium)ethyl methanethiosulfonate (MTSET) were purchased from Toronto Research Chemicals (North York, ON). All other chemicals and reagents used in these experiments were purchased from Fisher Scientific and were of the highest grade available.
Construction of Na+/H+ Exchanger Mutants
The rat cDNA, engineered to contain a series of unique restriction endonuclease sites that provide convenient DNA cassettes for mutagenesis, was subcloned into a mammalian expression vector under the control of the enhancer/promoter region from the immediate early gene of human cytomegalovirus (pCMV), as previously described (56). A single copy of an influenza virus hemagglutinin (HA) peptide (YPYDVPDYA) was inserted at the C terminus of NHE1 using polymerase chain reaction (PCR) mutagenesis (the construct is referred to herein as NHE1HA). NHE1HAΔC was constructed by substituting the eight endogenous cysteine residues (amino acids 117, 137, 216, 425, 481, 542, 565, and 799) with the structurally conservative residue serine. The substitution of serine did not generate any obvious consensus motifs for phosphorylation by known serine/threonine kinases. Single cysteine residues were introduced into this cysteine-less background using the QuikChangeTM Site-directed Mutagenesis system purchased from Stratagene (La Jolla, CA). All mutant constructs were sequenced to verify their fidelity.
Stable Transfection and Expression of the Na+/H+ Exchanger cDNAs
Chinese hamster ovary cells (AP-1 cells), a chemically mutagenized cell-line devoid of endogenous plasmalemmal NHE activity (74), were transfected with mammalian expression plasmids containing either wild-type NHE1HA or NHE1HAΔC-based constructs using LipofectamineTM. Cells were maintained in complete α-minimal essential medium supplemented with 10% fetal bovine serum, 100 units/ml of penicillin, 100 μg/ml of streptomycin, and 25 mm NaHCO3, pH 7.4, and incubated in an humidified atmosphere of 95% air, 5% CO2 at 37 °C. Starting 48 h after transfection, the AP-1 cells were selected for survival in response to repeated (∼6 times over a 2-week period) acute NH4Cl-induced acid loads (i.e. H+-killing technique) (75, 76) to discriminate between NHE1 positive and negative transfectants. Six clonal isolates per mutant were routinely selected and the one exhibiting the highest amiloride-sensitive H+i-activated 22Na+o uptake was subjected to further analyses.
Measurement of Na+/H+ Exchanger Activity and Covalent Modification with Sulfhydryl-reactive Reagents
Clonal cells were grown to confluence in 24-well plates and then acidified (to ∼pHi 6.0) using the NH4Cl pre-pulse technique (75, 76). Briefly, the cell culture medium was aspirated and replaced by isotonic NH4Cl medium (50 mm NH4Cl, 70 mm choline chloride, 5 mm KCl, 1 mm MgCl2, 2 mm CaCl2, 5 mm glucose, 20 mm HEPES-Tris, pH 7.4). Cells were incubated in this medium for 30 min at 37 °C in a nominally CO2-free atmosphere. After acid loading, the monolayers were rapidly washed twice with isotonic choline chloride solution (125 mm choline chloride, 1 mm MgCl2, 2 mm CaCl2, 5 mm glucose, 20 mm HEPES-Tris, pH 7.4). 22Na+ influx assays were initiated by incubating the cells in isotonic choline chloride solution containing 1 μCi of 22NaCl/ml (∼120 nm 22NaCl, carrier-free). The assay medium was K+-free to prevent 22Na+ influx and efflux by the Na+-K+-Cl− cotransporter and the Na+/K+-ATPase, respectively. Influx of 22Na+ was terminated by rapidly washing the cell monolayers three times with 4 volumes of ice-cold isotonic saline solution (130 mm NaCl, 1 mm MgCl2, 2 mm CaCl2, 20 mm HEPES-Tris, pH 7.4). The washed cell monolayers were solubilized in 0.25 ml of 0.5 n NaOH and the wells were washed with 0.25 ml of 0.5 n HCl. Both the solubilized cell extract and wash solutions were added to vials and radioactivity was assayed using a liquid scintillation counter. Under the H+-loading conditions used in this study, uptake of 22Na+ was linear with time for 8 to 10 min (at low Na+ concentrations, 22 °C). Therefore, 22Na+ uptakes were measured after 5 min except when examining the kinetics of NHE activity as a function of the extracellular Na+ (Na+o) concentration.
When assessing the Km for [Na+o], a modified version of the aforementioned protocol was applied. Previous measurements indicated that when [Na+o] is increased to 100–125 mm, 22Na+ uptake is linear for several minutes. Therefore, when conducting this kinetic analysis in which the [Na+o] concentration ranges from 1.25 to 120 mm, 22Na+ uptake was terminated after 1 min. Measurements of 22Na+ influx specific to the Na+/H+ exchanger were determined as the difference between the initial rates of H+-activated 22Na+ influx in the absence and presence of either 2 mm amiloride or 100 μm EIPA (concentrations sufficient to inhibit NHE1 under these experimental conditions) and expressed as amiloride- or EIPA-inhibitable 22Na+ influx. To make quantitative comparisons of the intrinsic rates of transport of the various NHE constructs in stably transfected cells, the cellular rates of drug-sensitive H+-activated 22Na+ influx were measured under near maximal acid-load conditions and normalized per mg of total cellular protein per abundance of the fully glycosylated NHE1 protein present at the plasma membrane (assessed by Western blotting and densitometry), as described previously (49).
To examine NHE activity as a function of intracellular H+ concentration, pHi was set over the range of 5.4–7.4 using the K+-nigericin method as previously described (77, 78). Briefly, the confluent monolayers were washed with isotonic N-methyl-d-glucamine (NMG)-chloride solution (140 mm NMG-Cl, 1 mm MgCl2, 2 mm CaCl2, 5 mm glucose, 10 mm HEPES-Tris, pH 7.4), and then incubated for 4 min at room temperature in NMG-balanced salt solutions specific for each pHi (pHi-clamp solutions). All solutions contained 2 mm NaCl, 1 mm MgCl2, and 10 mm HEPES-Tris, pH 7.4, varying concentrations of K+ (1.4 to 140 mm KCl/K+-glutamate, adjusted as needed with NMG-glutamate to bring the final concentration to 140 mm), and the K+/H+ exchange ionophore, nigericin (10 μm). These pHi clamp solutions are designed to fix the pHi at a desired level by adjusting the extracellular K+ concentration as described (78). In essence, the desired pHi can be established according to the following equation: [K+i]/[K+o] = [H+i]/[H+o], assuming that the intracellular K+ concentration is 140 mm and the extracellular H+ concentration is set at 7.4. 22Na+ uptake was initiated in the same pHi clamp solutions supplemented with 1 μCi/ml of 22Na+ and 1 mm ouabain in the absence or presence of 2 mm amiloride. Uptake occurred for a period of 10 min and was terminated in the same fashion as described above. Protein content was assessed using the Bio-Rad DC protein assay kit as per the manufacturer's protocol. Rates were expressed as nanomole of Na+/min/mg of protein.
To evaluate the effect of the methanethiosulfonate derivatives (73) on NHE1 activity, cell monolayers were incubated in phosphate-buffered saline containing 1 mm MgCl2 and 0.1 mm CaCl2, pH 7.2, with or without 1 mm MTSET or 10 mm MTSES for 30 min at 37 °C. Cells were washed twice with isotonic choline chloride solution and then intracellular pH was clamped at pHi 5.6 using the K+-nigericin method. 22Na+ uptake was initiated using the same clamp solution supplemented with 1 μCi of 22Na+ and 1 mm ouabain in the absence or presence of 2 mm amiloride or 100 μm EIPA. 22Na+ uptake was terminated after 10 min using ice-cold isotonic saline solution in the same fashion as mentioned above. Residual NHE1 activity was determined as the ratio of amiloride or EIPA-inhibitable 22Na+ uptake in the presence and absence of MTSET or MTSES.
Immunoblotting
Stably transfected cells were grown to confluence in 10-cm plates and were lysed using 1% Triton X-100. Total cellular protein extracts (30 μg) were resolved by 10% SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto PVDF Hybond-PTM membranes (Amersham Biosciences). The blots were rinsed briefly with PBST (1 × PBS containing 0.1% Tween 20), blocked with 5% nonfat skim milk in PBST, and then incubated with a murine monoclonal anti-HA antibody (dilution 1:5,000). Following extensive washing with PBST, blots were incubated with goat anti-murine IgG secondary antibody conjugated with horseradish peroxidase (dilution 1:5,000). Immunoreactive bands were visualized using enhanced chemiluminescence (PerkinElmer Life Sciences) and recorded on x-ray film.
Statistics
The data were analyzed using Origin and Microsoft Excel. The data from the sodium affinity, drug resistance, and pH profile experiments was fitted with a Hill function, whereas the EIPA and pH profile data were fitted with a dose-response function. Unless otherwise stated, error bars represent the mean ± S.E. and statistical analysis was performed by using the one-way analysis of variance Tukey post hoc test, with a significance level of 0.05.
Results
Characterization of NHE1HAΔC and Single-substituted Cysteine Mutants
To further explore the structure-function domains of NHE1 in the M9-EL5 region (the nomenclature used will follow the Wakabayashi model illustrated in Fig. 1A), we used the substituted-cysteine accessibility method (73). This method relies on covalent interaction between the ionized sulfhydryl group of cysteines located in a water-accessible environment, such as the pore, with membrane-impermeant sulfhydryl-reactive reagents that have the potential to irreversibly impair transmembrane ion fluxes by either steric blockage or charge attraction/repulsion. Such covalent modifications might also hinder critical conformation changes that occur during cation translocation either at the cation pore itself or at a more distant site that nevertheless impacts cation translocation.
To this end, a cysteine-less HA epitope-tagged version of rat NHE1 (NHE1ΔCHA) was constructed by replacing the eight native cysteines in NHE1 (indicated in Fig. 2A) with serine, a conservative substitution that maintains amino acid polarity and side chain length and hence minimizes potential structural perturbations of the transporter. The NHE1ΔCHA was stably expressed in mutagenized Chinese hamster ovary AP-1 cells devoid of endogenous NHE1 as previously described (75). Levels of protein expression for NHE1HA wild-type (WT) and ΔC are displayed in the inset of Fig. 2B. Previous studies demonstrated that the slower migrating band at ∼100 kDa represents the fully glycosylated form of the protein present at the cell surface, whereas the faster migrating band at ∼75 kDa represents the partially processed or core-glycosylated form of the protein that is largely retained in the endoplasmic reticulum (79). Importantly, substitution of the cysteines residues with serine did not impair the biosynthesis and maturation of the transporter.
FIGURE 2.

Functional characterization of a cysteine-less variant of NHE1. A, schematic representation of the membrane topology of rat NHE1 according to Wakabayashi et al. (36) and sites of endogenous cysteines replaced with serine to form the cysteine-less (ΔC) NHE1. B-D, NHE1 wild-type (WT) and ΔC were stably expressed in AP-1 cells and assayed for their affinities for Na+ (B) and H+ (C) and their sensitivity to inhibition by ethylisopropylamiloride (EIPA) (D) according to protocols described under ”Experimental Procedures.“ Values represent the mean ± S.E. of 3–5 experiments, each performed in quadruplicate. Error bars smaller than the symbol are absent.
We next compared the kinetic properties of the WT and ΔC transporters. The initial rates of NHE1 activity as a function of the Na+o concentration were examined using solutions containing tracer amounts of the radioisotope 22Na+ added to Na+o concentrations ranging from 2.5 to 120 mm following an imposed NH4Cl-induced intracellular acidification. The data for both WT and ΔC were best fit to a Hill equation and revealed an apparent sigmoidal dependence on the Na+ concentration for both constructs, indicative of positive cooperative binding with comparable Hill coefficients of ∼1.5 and 1.7, respectively (Table 1). Replacement of the cysteine residues also did not alter its maximal velocity (Vmax, nmol/min/mg of protein: WT, 105.5 ± 1.0; ΔC, 105.0 ± 0.7), but did cause a reduction (∼36%) in its affinity for Na+o (KNa: WT, 29.5 ± 0.1; ΔC, 46.1 ± 0.9; p < 0.05) (Fig. 2B; Table 1). To determine their affinities for intracellular H+, the H+i concentration was adjusted by clamping pHi at specific levels within the range of 5.4–7.4 using the K+/H+ ionophore nigericin, and measuring the rate of 22Na+ influx. As shown in Fig. 2C, the WT and ΔC transporters exhibited similar affinities for H+i (KH pH: WT, 6.14 ± 0.05; ΔC, 6.26 ± 0.03; p > 0.05). On the other hand, ΔC showed an approximate 8-fold decrease in sensitivity to the pharmacological antagonist EIPA (IC50, nm: WT, 5.4 ± 1.2; ΔC, 40.7 ± 10.4; p < 0.05) (Fig. 2D). Based on these data, the endogenous cysteines moderately influenced, either directly or indirectly, the Na+ and drug affinities of the transporter, but they were not structurally essential. Hence, NHE1ΔCHA could serve as a suitable template for further cysteine-substitution mutagenesis and functional analyses.
TABLE 1.
Kinetic constants of wild-type and mutant NHE1
Values represent the mean ± S.E.
| NHE1 | KNa (n) | Na+, Hill coefficient | KH (n), pHi | Vmax |
|---|---|---|---|---|
| mm | nmol/min/mg protein | |||
| WT | 29.5 ± 0.1 (9) | 1.53 ± 0.01 | 6.14 ± 0.05 (6) | 105.5 ± 1.1 |
| ΔC | 46.1 ± 0.9a (9) | 1.73 ± 0.02 | 6.26 ± 0.03 (6) | 105.0 ± 0.7 |
| H353C | 43.6 ± 2.6 (9) | 1.13 ± 0.02b | 6.20 ± 0.07 (3) | 60.9 ± 3.1b |
| S355A | 50.8 ± 1.0† (9) | 1.13 ± 0.02a | 6.34 ± 0.03† (6) | 89.9 ± 1.0a |
| S355C | 70.7 ± 0.6b (9) | 1.72 ± 0.01 | 6.37 ± 0.11 (3) | 143.1 ± 3.0b |
| G356C | 40.1 ± 1.6 (9) | 1.62 ± 0.04 | 6.39 ± 0.13 (3) | 84.3 ± 3.1b |
| Y370C | 92.8 ± 2.0b (9) | 1.21 ± 0.01b | 6.19 ± 0.03 (5) | 19.6 ± 0.2b |
| N374C | NDc | ND | ND | |
| S376C | 58.2 ± 4.9 (9) | 1.12 ± 0.03b | ND | 31.6 ± 1.3b |
| T381C | 44.1 ± 3.8 (3) | 2.01 ± 0.20b | ND | 25.4 ± 3.9b |
| Y385C | 50.7 ± 1.9 (9) | 1.42 ± 0.03b | 6.13 ± 0.05 (6) | 30.5 ± 1.1b |
| S392C | 41.5 ± 0.5 (9) | 1.61 ± 0.02 | 6.30 ± 0.02 (3) | 53.2 ± 0.9b |
| E395C | 65.7 ± 3.6b (10) | 1.12 ± 0.02b | 6.19 ± 0.02 (4) | 19.8 ± 1.3b |
| L397C | ND | ND | ND | |
| G403C | 41.9 ± 1.2 (9) | 1.28 ± 0.02b | 5.82 ± 0.04b (12) | 25.6 ± 0.7b |
| S405C | 68.0 ± 3.4b (9) | 1.25 ± 0.03b | 6.00 ± 0.04b (8) | 89.0 ± 2.3b |
a Indicates a significant difference of the mean from NHE1 WT, where p < 0.05.
b Indicates a significant difference of the mean from NHE1ΔC, where p < 0.05.
c ND, not determined.
To rationalize potential targets for cysteine substitution, certain sites in M9 (Glu350, His353, and Gly356) (49, 55, 56) were selected based on their known involvement in conferring sensitivity to amiloride- or benzyolguanidinium-based compounds that act as either simple or mixed competitors of Na+o binding and therefore are likely in close proximity to the permeation pathway. Additional amino acids in M9 and EL5 were chosen based on their conservation among the plasmalemmal NHE isoforms (NHE1-NHE5) (Figs. 3A and 4A), their relative predicted position in a transmembrane α-helix (Fig. 3B), and their hydrophilic nature, which likely orients them in an aqueous environment and potentially accessible to the thiol-modifying reagents. In total, 20 single cysteine substitutions were generated in M9 (S342C, Y343C, Y346C, S348C, E350C, H353C, S355C, G356C, and A359C) and EL5 (Y370C, N374C, S376C, T381C, Y385C, S392C, E395C, L397C, G403C, S405C, and W415C). The resulting constructs were then stably expressed in AP-1 cells based on their ability to confer cell survival followed repeated intracellular acid loads that are lethal to AP-1 cells lacking a functional transporter.
FIGURE 3.

Functional analysis of single cysteine substitutions in membrane-spanning segment M9. A, alignment of M9 sequences from rat (r) NHE1 to NHE5. Amino acids that are highly conserved are shaded in black, whereas moderately conserved are shaded in gray. Asterisks indicate residues that were mutated to cysteine. B, helical wheel representation of M9 with hydrophobic and hydrophilic amino acids depicted by blue and red shading, respectively. C, Western blot analysis displaying the protein expression levels of the cysteine-less NHE1HA (ΔC) and the single cysteine-substituted mutations in M9. The slower migrating band represents the fully glycosylated (fg) form of the protein, whereas the faster migrating band represents the core glycosylated (cg) form of the protein. D, NHE1 activity, defined as rates of amiloride-inhibitable H+-activated 22Na+ influx, for WT, ΔC, and the single cysteine-substituted mutants in the presence of 10 mm MTSES or 1 mm MTSET. Results are expressed as a percentage of the uptake catalyzed by each mutant in the absence of the MTS compounds. Values represent the mean ± S.E. of at least 3 experiments, each performed in quadruplicate. Asterisks indicate statistical significance at the 0.05 level by Student's t test (p < 0.05).
FIGURE 4.

Functional analysis of single cysteine substitutions in the EL5 R-loop. A, alignment of the EL5 R-loop from rat (r) NHE1 to NHE5. Amino acids that are highly conserved are shaded in black, whereas moderately conserved are shaded in gray. Asterisks indicate residues that were mutated to cysteine. B, Western blot analysis displaying the protein expression levels of NHE1HAΔC (ΔC) and the single cysteine-substituted mutations in EL5. C, NHE1 activity, defined as rates of amiloride-inhibitable H+-activated 22Na+ influx, for WT, ΔC, and the single cysteine-substituted mutants in the presence of 10 mm MTSES or 1 mm MTSET. Results are expressed as a percentage of the uptake catalyzed by each mutant in the absence of the MTS compounds. Values represent the mean ± S.E. of at least 3 experiments, each performed in quadruplicate. Asterisks indicate statistical significance at the 0.05 level by Student's t test (p < 0.05).
To make initial estimates of the relative activities of parental and single cysteine-substituted mutants of NHE1HAΔC in stably transfected cells, the cellular rates of amiloride-sensitive H+i-activated 22Na+o influx were measured in the presence of nominal non-radioactive Na+o (∼120 nm 22NaCl, carrier-free; 5 min uptake) and then normalized for the level of fully glycosylated NHE1 protein at the cell surface as determined by immunoblotting and densitometry. Levels of protein expression for NHE1HAΔC and the single cysteine-substituted mutants are displayed in Figs. 3C and 4B. Within M9, the relative activity levels of the single cysteine-substituted mutants were generally higher (i.e. ∼2-fold) than the parental NHE1HAΔC (Fig. 3C), with the notable exception of S348C which, whereas not highly expressed, exhibited an apparent dramatic 24-fold increase in its rate of transport. However, because of its low level of expression, the calculated rate of activity per unit NHE1 protein is more subject to error and hence the determined value may overestimate its actual rate of transport. By comparison, the majority of the mutants in EL5 exhibited lower rates of transport with the exception of L397C, which displayed a 2.5-fold increase, although its expression level was also quite low, which made an accurate assessment of its relative rate of transport more difficult (Fig. 4B). These initial findings implicate both regions as important elements in cation translocation.
MTS Inhibition of the M9 and EL5 Single Cysteine Mutants
To further probe the involvement of these regions in cation permeation, the cysteine-substituted transporters were subjected to chemical modification using the membrane-impermeant sulfhydryl-active reagents MTSET and MTSES that are positively and negatively charged, respectively, and assessed for their effects on NHE1 activity. The activities of the majority of the M9 mutants were unaffected by the MTS derivatives, suggesting that the sites are either inaccessible to these reagents or alternatively that they did react with the compounds but that this did not lead to inhibition (Fig. 3D). However, three mutants containing neighboring substitutions, H353C, S355C, and G356C showed ∼40, 30, and 98% inhibition of activity, respectively, following treatment with MTSET (Fig. 3D). The negatively charged MTSES caused a similar reduction (∼30%) in the activity of the S355C mutant, but had a considerably lesser effect on H353C and G356C. Because both reagents are similar in size, the differing degrees of inhibition caused by the respective MTS reagents are likely due to charge effects rather than steric hindrance. Collectively, these data suggest that amino acids His353, Ser355, and Gly356 (equivalent to hNHE1 His349, Ser351, and Gly352) comprise an accessible segment that faces an aqueous environment and are potentially involved in cation translocation. Likewise, the majority of the EL5 mutants were unaffected by exposure to the MTS derivatives, with the exceptions of G403C and S405C, which showed significant reductions (∼30 and 70%, respectively) in 22Na+o influx in the presence of MTSET, and to a lesser extent in the presence of MTSES (Fig. 4C).
Kinetic Properties of Mutants Sensitive to Thiol Modification
Having identified sites where cysteine substitutions confer sensitivity to thiol-reactive compounds, as revealed by reductions in transport activity, we next investigated whether the mutations alone (i.e. without MTS treatment) influenced the kinetic properties of the exchanger.
For sites in M9 (i.e. H353C, S355C, and G356C), calculation of their maximal velocities as a function of the external Na+o concentration (Fig. 5, A–C) revealed a significant reduction in transport for H353C (∼42%) (Vmax, nmol/min/mg of protein: 60.9 ± 3.1; p < 0.05) and to a lesser extent for G356C (∼20%) (84.3 ± 3.1; p < 0.05), whereas the apparent Vmax for S355C increased by ∼36% (S355C, 143.1 ± 3.0; p < 0.05) compared with control NHE1ΔC (Vmax: WT, 105.0 ± 0.7). The curve obtained for H353C also displayed a reduction in cooperative Na+ binding (Hill coefficient (n): ΔC, 1.73 ± 0.02; H353C, 1.13 ± 0.02; p < 0.05), whereas those for S355C (n = 1.72 ± 0.01) and G356C (n = 1.62 ± 0.04) were unchanged (Table 1). However, calculation of the apparent Na+o affinity constants (KNa) for H353C and G356C yielded values similar to that determined for the parental ΔC construct (KNa, mm: ΔC, 46.1 ± 0.9; H353C, 43.6 ± 2.6; G356C, 40. 1 ± 1.6) (Table 1), suggesting that these amino acids, whereas important for maximal transport velocity, are not critical determinants of Na+o binding. By contrast, the transport velocity of the S355C mutant did not approach saturation within the Na+o concentration range tested and exhibited a significant reduction (∼53%) in Na+o affinity (KNa, 70.7 ± 0.6 mm, p < 0.05) (Table 1). This finding is particularly striking given the conservative nature of the S355C substitution, and therefore implicates Ser355 as an important determinant of Na+o transport. On the other hand, measurement of the intracellular H+i affinities of H353C, S355C, and G356C were equivalent to parental ΔC (KH, pHi: ΔC, 6.26 ± 0.03; H353C, 6.20 ± 0.07; S355C, 6.37 ± 0.11; G356C, 6.39 ± 0.13; p > 0.05) (Fig. 5, D–F; Table 1). Collectively, these findings suggest that cysteine substitutions at these sites have a preferential impact on Vmax, with certain sites also affecting the cooperativity (H353C) or affinity (S355C) for Na+o binding.
FIGURE 5.

Kinetic analyses of NHE1 MTS-responsive mutants in helix M9. NHE1 ΔC and mutants H353C, S355C, and G356C were stably expressed in AP-1 cells and assayed for their affinities for Na+ (A–C) and H+ (D--F) according to protocols described under ”Experimental Procedures.“ Values represent the mean ± S.E. of 3–9 experiments, each performed in quadruplicate. Error bars smaller than the symbol are absent.
To further examine the importance of Ser355 within the context of WT NHE1 (as opposed to the ΔC variant), Ser was replaced with Ala (S355A), a non-polar residue whose side chain length is comparable in size. This mutant was properly synthesized and processed at levels comparable with WT, ΔC, and S355C (Fig. 6A). However, unlike S355C, its maximal velocity was decreased slightly, albeit significantly, compared with WT (Vmax: WT, 105.5 ± 1.1; S355A, 89.9 ± 1.0; p < 0.05). Nevertheless, S355A, like S355C, showed a significant reduction (∼73%) in Na+o affinity (KNa, mm: WT, 29.5 ± 0.1; S355A, 50.8 ± 1.0, p < 0.05) as well as a decrease in cooperative Na+ binding (Hill coefficient (n): WT, 1.53 ± 0.01; S355A, 1.13 ± 0.02; p < 0.05) (Fig. 6B; Table 1). S355A was also more active at more alkaline values compared with WT, resulting in an apparent 58% increase in H+i affinity (KH, pHi: WT, 6.14 ± 0.05; S355A, 6.34 ± 0.03; p < 0.05) (Fig. 6C; Table 1). Thus, these findings corroborate a critical role for Ser355 in cation translocation.
FIGURE 6.
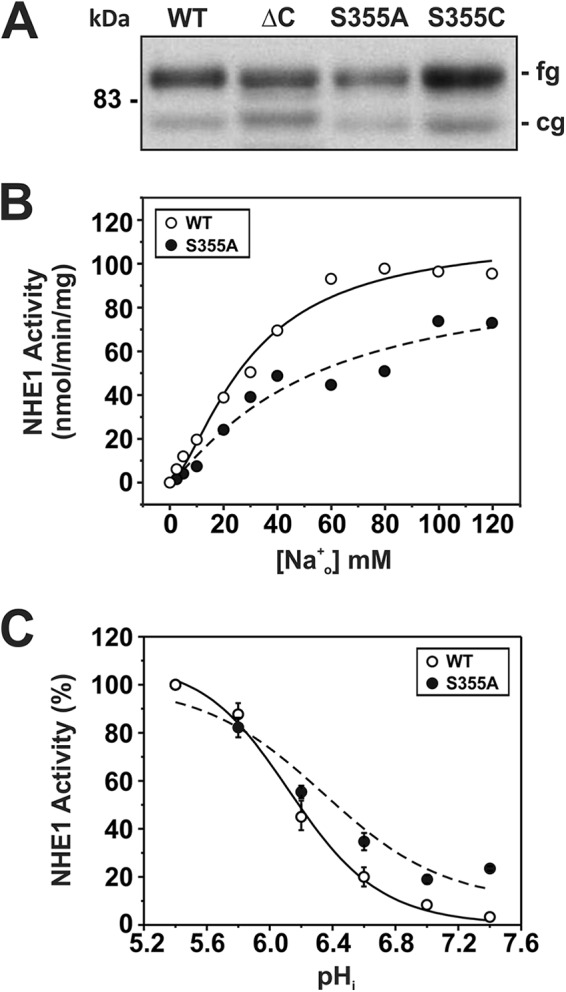
Kinetic analysis of NHE1 mutant S355A. A, NHE1 WT and S355A were stably expressed in AP-1 cells and their relative expression levels were compared with ΔC and S355C by Western blot analysis. NHE1 WT and S355A were assayed for their affinities for Na+ (B) and H+ (C) according to protocols described under ”Experimental Procedures.“ Values represent the mean ± S.E. of 6–9 experiments, each performed in quadruplicate. Error bars smaller than the symbol are absent.
Kinetic analyses of the two MTS-sensitive mutants in EL5, G403C and S405C, revealed decreases in their maximal velocities, especially for S403C (Vmax: ΔC, 105.0 ± 0.7; S403C, 25.6 ± 0.7; S405C, 89.0 ± 2.3, p < 0.05) (Fig. 7, A and B; Table 1). In the case of G403C, this was accompanied by a marked reduction in its affinity for H+i (i.e. acidic shift) (KH pHi: ΔC, 6.26 ± 0.03; G403C, 5.82 ± 0.04, p < 0.05) (Fig. 7E; Table 1), whereas its affinity for Na+o was apparently unaffected (KNa mm: ΔC = 46.1 ± 0.9; S403C = 41.9 ± 1.2, p > 0.05) (Fig. 7A; Table 1). By contrast, S405C exhibited reductions in affinities for both Na+o (KNa mm: ΔC, 46.1 ± 0.9; S405C, 68.0 ± 3.4, p < 0.05) and H+i (KH pHi: ΔC, 6.26 ± 0.03; S405C, 6.00 ± 0.04, p < 0.05 (Fig. 7, B and F; Table 1). Thus, Gly403 and Ser405 are also important for cation binding and permeation.
FIGURE 7.

Kinetic analyses of NHE1 mutants in re-entrant loop EL5. NHE1ΔC and mutants Y370C, E395C, G403C, and S405C were stably expressed in AP-1 cells and assayed for their affinities for Na+ (A–D) and H+ (E–H) according to protocols described under ”Experimental Procedures.“ Values represent the mean ± S.E. of 3–9 experiments, each performed in quadruplicate. Error bars smaller than the symbol are absent.
The majority of the other Cys substitutions in the predicted re-entrant loop EL5 did not confer sensitivity to the MTS compounds. Nevertheless, we explored whether some of these sites (i.e. Y370C, N374C, S376C, T381C, Y385C, S392C, E395C, and L397C) could contribute to substrate affinities and in some cases to their sensitivity to inhibition by EIPA. The majority of the mutations caused marked reductions in Vmax and cooperativity of Na+ binding (Table 1). We were unable to determine the kinetic properties of the N374C and L397C mutants because whereas their velocities increased as a function of the Na+ concentration, the values were erratic and did not display saturable binding kinetics (data not shown). Among these mutations, only Y370C and E395C exhibited marked reductions in Na+o affinity (KNa mm: ΔC, 46.1 ± 0.9; Y370C, 92.8 ± 2.0, p < 0.05; E395C, 65.7 ± 3.6, p < 0.05) (Fig. 7, C and D; Table 1), but no significant shift in H+ affinity (Fig. 7, G and H; Table 1). When analyzed for sensitivity to inhibition by EIPA, Y370C exhibited a significant 4-fold increase (K0.5 nm: ΔC, 40.7 ± 9.1, n = 11; Y370C, 9.6 ± 3.2, n = 8; p < 0.05) (Fig. 8), whereas E395C was not significantly altered (K0.5 nm: 67.6 ± 17.3, n = 8; p > 0.05). This suggests that whereas the Y370C and E395C mutants were unaffected by the MTS derivatives, the sites nevertheless contributed to cation permeation and partly to drug sensitivity.
FIGURE 8.
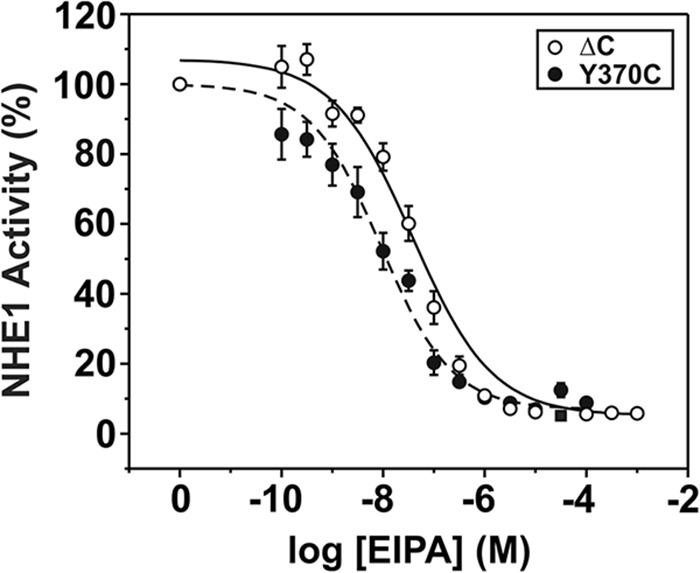
Effect of Y370C on the drug sensitivity of NHE1. AP-1 cells stably expressing NHE1ΔC or Y370C were grown to confluence in 24-well plates and their activities were measured in the presence of increasing concentrations of the NHE antagonist EIPA. Transport activity was measured as described under ”Experimental Procedures.“ Values represent the mean ± S.E. of 8 experiments, each performed in quadruplicate.
Discussion
In this study, the substituted-cysteine accessibility method (73) was applied to rNHE1 to identify candidate amino acids in two segments of the transporter, the predicted transmembrane helix M9 (rNHE1-(342–363); equivalent to hNHE1-(338–359)) and the adjoining exofacial re-entrant loop between M9 and M10 (EL5; rNHE1-(364–415) or hNHE1-(360–411)), which had previously been implicated in conferring sensitivity to competitive antagonists of Na+o binding and therefore may potentially line the cation permeation pathway of the transporter (49, 56, 60). The data show that of the 20 cysteine substitutions made at a select number of known drug-sensitive or evolutionarily conserved sites, only a limited number of these in M9 (i.e. rat H353C, S355C, G356C; human H349C, S351C, and G352C) and EL5 (i.e. rat G403C and S405C; human G399C and S401C) caused marked reductions in NHE1 activity upon modification with MTS sulfhydryl reagents. Significantly, kinetic measurements of unmodified rNHE1 S355C, G403C, and S405C as well as two other substitutions in EL5 (rat Y370C and E395C; human Y366C and E391C) showed reduced affinities for Na+o and/or H+i. Interestingly, the Y370C mutant, whereas possessing a 2-fold decrease in Na+ affinity, exhibited a 4-fold increase in affinity for the competitive antagonist EIPA. This Tyr residue is highly conserved in the mammalian NHEs and therefore it is tempting to speculate that it may serve as a common binding site for Na+o and side chain substituents of amiloride derivatives. Other cysteine substitutions within EL5 also significantly impaired the maximal activity of the transporter, although they did not alter the apparent affinities for Na+o or H+i, suggesting that they may fulfill other structural roles in ion translocation. Collectively, these data implicate these amino acids as important elements involved in cation permeation and drug sensitivity.
Among the three MTS-sensitive residues within M9, the G356C mutation was the most reactive. The activity of G356C was almost completely inhibited (∼98%) in the presence of positively charged MTSET. Likewise, transport activity was also significantly reduced by negatively charged MTSES, although to a lesser extent (∼40%). Mechanistically, thiol modification of G356C may block cation transport by sterically hindering critical conformation changes that occur during cation transport, either at the cation pore itself or at a more remote site that nevertheless influences cation translocation. However, the observed sizeable differences in the degree of inhibition elicited by the two oppositely charged, but similarly sized, MTS reagents also implicates the involvement of electrostatic forces on the flow of cations through the protein, suggesting that Gly356 is more likely in close proximity to the cation translocation pathway. Thus, aside from possible steric effects, the positive charge of MTSET could further impede the flow of Na+ by electrostatic repulsion, whereas the negatively charged MTSES reagent could act as an attractant and retard passage of Na+ ions through the pore, but with less efficiency than the repelling effects of MTSET. We have also reported (49) that this residue is an important determinant of the high sensitivity of NHE1 to inhibition by both amiloride- and benzyolguanidinium-based compounds, further supporting an important structural and functional role for Gly356.
Aside from G356C, cysteine substitution of the two adjacent N-terminal residues, H353C and S355C, also rendered the transporter moderately sensitive to inhibition by the MTS reagents. The reactivity of S355C agrees with an earlier study showing that the equivalent mutation in a cysteine-less variant of hNHE1, S351C, is also sensitive to MTS (60). The lesser reactivity of these two residues to MTS reagents might be expected if they lie deeper within the membrane, as predicted by the Wakabayashi model (see Fig. 9A) and their thiol side chains would be directed more toward the lipid bilayer, assuming 3.6 amino acids per turn of an α-helix relative to position Gly356 and thereby making them less available for modification by the MTS reagents. This might also explain why MTS reagents did not have a detectable effect on rNHE1 mutants containing cysteine substitutions N-terminal to His353, although one previous study (60) reported that E350C (equivalent to hE346C) was sensitive to MTSET. The basis for the variance is unclear.
FIGURE 9.
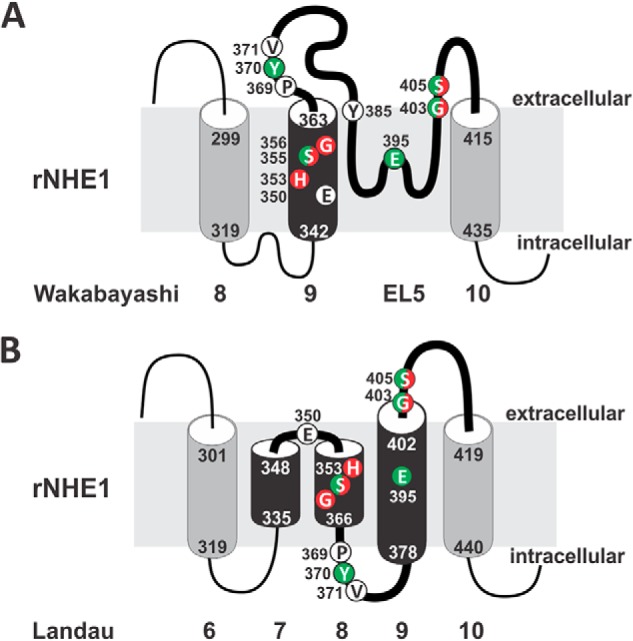
Comparison of different models of the predicted transmembrane arrangement of the helix M9 and EL5 region of NHE1. Schematic representation of the transmembrane organization of NHE1 in the M9-EL5 region according to the models proposed by (A) Wakabayashi et al. (36) and (B) Landau et al. (65) that highlight functionally relevant amino acids identified in the present study. The red or green shading identifies sites that conferred sensitivity to MTS reagents or affected substrate affinities (Na+ and/or H+), respectively, when replaced with cysteine. Mutations of some sites affected both parameters and are dual labeled.
Although His353 and Ser355 are hydrophilic residues, it is perhaps initially unexpected to find that a hydrophobic residue such as Gly356 would also directly face the aqueous pore. However, because Gly contains a hydrogen atom as its side chain group, it is only moderately nonpolar and confers flexibility to the peptide backbone structure that may be important for conformational changes that ensue during cation translocation. Moreover, previous studies have demonstrated that hydrophobic residues often found to line the translocation pores of transporters and channels where they are postulated to provide an inert surface that facilitates ion diffusion (80–82). Kinetic analyses, however, did not reveal significant changes in the cation affinities for H353C and/or G356C, but did show a marked reduction in Na+ affinity for S355C, which agrees with a previous observation (60). This finding was further corroborated in native NHE1, where substitution of Ser355 with Ala also reduced Na+ affinity by ∼2-fold. Taken together, these data are consistent with the notion that M9, and these three residues in particular, constitute an integral part of the cation translocation pore.
The adjacent putative EL5 R-loop between M9 and M10 was also found to contribute significantly to transport activity and drug sensitivity. Indeed, manipulation of sites in EL5 often had more profound effects on transporter function than sites in M9. The R-loop shares structural similarities to P-loops that are known to invaginate into the membrane bilayer and facilitate ion flow through channels and pumps (83–85). These structural elements are responsible for mediating antagonist binding, ion selectivity, and conductance. Regarding NHE1, the vast majority of single cysteine substitutions in EL5 significantly decreased maximal transport velocity. In most cases (i.e. Y370C, S376C, Y385C, E395C, G403C, and S405C), these changes were associated with a reduction in positive cooperativity of Na+ binding, as defined by the Hill coefficient. Previous kinetic analyses of the Na+ dependence of the NHEs have provided mixed results with some studies reporting simple Michaelis-Menten kinetics (86, 87), whereas others describe cooperative activating effects of Na+ (37, 39, 88, 89). Although the basis for these differences is not fully understood, part of these differences may relate to assay conditions and/or cell type. In a detailed study by Fuster et al. (89), they observed that the Hill coefficient for extracellular Na+ dependence was dependent on cytoplasmic pH. Specifically, the extracellular Na+ concentration dependence was sigmoidal at a cytoplasmic pH of 7.2 with a Hill coefficient of ∼1.8, whereas this cooperativity was diminished at more acidic values. The extracellular Na+ dependences of NHE1 were explained equally by either a parallel or serial model of dimer coupling with a 2Na+/2H+ stoichiometry of the monomer, with cooperative Na+o binding dependent on the H+i concentration. Our mutagenesis data suggest that EL5 may play an important role in the cooperative activating effects of Na+o.
In the case of four mutants in EL5, Y370C, E395C, G403C, and S405C, the changes in activity also correlated with marked reductions in Na+ affinity (Y370C, E395C, and S405C) and/or H+ affinity (G403C and S405C). Both the G403C and S405C mutants were also sensitive to modification by MTS reagents, suggesting that they were accessible from the extracellular medium. Furthermore, the Y370C mutation exhibited increased sensitivity to inhibition by EIPA. Although other mutations in this region did not appear to affect ion or drug binding, the impairment in Vmax suggests that these sites may nevertheless be important for the ensuing conformational changes required for optimal transport activity.
Presently, two different transmembrane topologies have been postulated for NHE1. The first model developed by Wakabayashi et al. (36) using the substituted-cysteine accessibility mutagenesis approach predicted 12 membrane-spanning helices with M9 encompassing amino acids 342–363 of rat NHE1 (human 338–359) followed by a re-entrant loop structure extending from amino acids 364 to 415 (human 360–411), as illustrated in Figs. 1A and 9A. Subsequently, Landau et al. (65) proposed a different helical arrangement (Fig. 1B and 9B) based on comparisons of evolutionary conserved sequences and secondary structure predictions, and then mapped the optimized alignment to the crystal structure of the E. coli NhaA antiporter. In this alternate model, the N-terminal segment encompassing the original helices M1 and M2 (residues ∼1–150) was located instead in the cytoplasm and postulated to serve as part of a truncated signal sequence, with the first transmembrane segment commencing at the former M3 helix. However, this seems unlikely given that the predicted EL1 segment between M1 and M2 is known to undergo N- and O-linked glycosylation (70) as it transits through the endoplasmic reticulum and Golgi compartments and therefore should be oriented extracellularly when the transporter is inserted into the plasma membrane. The M9 helix was also rearranged to form two short helices renamed M7 (amino acids 335–348) and M8 (amino acids 353–366), whereas the neighboring extracellular re-entrant loop 5 (EL5) was predicted to form a new intracellular loop (amino acids 367–377) and new M9 helix (amino acids 378–402) that was predicted to be largely inaccessible to external reagents, followed by residues that again were accessible to external reagents (amino acids 403–418).
The results from our analyses are partially consistent with both models, but overall are better accounted for by the Wakabayashi model. The susceptibility of both G403C and S405C to modification by MTS reagents places these residues in an environment that is accessible to the extracellular milieu, in agreement with both models. The MTS sensitivity of H353C, S355C, and G356C, with the latter residue being the most reactive, also positions these amino acids extracellularly and possibly facing the external funnel of the ion permeation pathway. Based on the hierarchy of their MTS reactivity, these amino acids could be positioned according to the Wakabayashi model, but seem at odds with the Landau model, which places Gly356 deep within the membrane. We further found that mutations to Ser355 affected substrate affinities, especially for Na+, and maximal transport velocity, consistent with an earlier finding (60). This suggests that this residue faces the ion pore and is catalytically important. In the Landau et al. (65) model, the side chain of Ser355 was oriented into the lipid bilayer which, in principle, would make it less accessible. However, Landau et al. (65) postulated that this helical segment could potentially rotate 180° around its axis to allow this residue to participate in cation permeation. Notwithstanding, the new proposed orientation of this segment also placed Tyr370 within an intracellular loop. Such a position would make it more difficult to account for our observation that Tyr370 is involved in external Na+o binding and drug sensitivity. These data are explained more simply by positioning this residue in the extracellular milieu, but which is unaffected by MTS reagents for reasons that remain obscure. Because Gly356 is also known to confer sensitivity to NHE antagonists (49), these residues may be in close proximity to each other. More recently, Reddy et al. (60) determined the structure of a peptide representing amino acids 342–369 (human 338–365) by high resolution nuclear magnetic resonance (NMR) in detergent micelles. The structure contained two helical regions (amino acids Met344–Ser348 and Ile357–Ser363) separated by a sharp, potentially flexible, segment (amino acids Ala349–Gly356) that bends immediately N-terminal to Ser355, resulting in a kinked “L”-shaped structure. If such an arrangement exists in the native transporter, then it has the potential to position Tyr370 as well as neighboring amino acids in an intramembranous, albeit externally facing, environment that constitutes part of the pore funnel involved in ion coordination.
In summary, residues in M9 and EL5 comprise important structural elements for cation translocation in NHE1. As a crystal structure has yet to be achieved, additional biochemical, biophysical, and structural analyses using approaches that combine substituted-cysteine accessibility mutagenesis with electron paramagnetic resonance spectroscopy and NMR may help to shed further structural and mechanistic insight into this catalytically important region of the transporter. Such information should prove valuable in the design and development of more efficacious NHE1-specific drugs to prevent or lessen tissue damage arising from certain cardio- and cerebrovascular diseases and cancer progression.
Author Contributions
J. O. conceived and coordinated the study and wrote the paper. T. J. designed, performed, and analyzed the experiments shown in Figs. 2D, 4B, 5, A-C, and 6–8. C. B. J. designed, performed, and analyzed the experiments shown in Figs. 2, A-C, 3, and 5, D-F. A. B. provided technical assistance. J. O., T. J., and C. B. J. contributed to the preparation of the figures. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgment
We also acknowledge the services provided by the Genome Quebec Innovation Centre.
This work was supported by Canadian Institutes for Health Research Grants MOP-11221 and MOP-111191 (to J. O.). The authors declare that they have no conflicts of interest with the contents of this article.
- NHE/NHA
- Na+/H+ exchanger or antiporter
- EIPA
- ethylisopropylamiloride
- AP-1
- a chemically mutagenized Chinese hamster ovary cell line devoid of plasma membrane Na+/H+ exchange activity
- MTSES
- 2-sulfonatoethyl methanethiosulfonate
- MTSET
- 2-(trimethylammonium)ethyl methanethiosulfonate
- NMG
- N-methyl-d-glucamine.
References
- 1. Brett C. L., Donowitz M., Rao R. (2005) Evolutionary origins of eukaryotic sodium/proton exchangers. Am. J. Physiol. Cell Physiol. 288, C223-C239 [DOI] [PubMed] [Google Scholar]
- 2. Orlowski J., Grinstein S. (2011) Na+/H+ exchangers. Compr. Physiol. 1, 2083–2100 [DOI] [PubMed] [Google Scholar]
- 3. Roos A., Boron W. F. (1981) Intracellular pH. Physiol. Rev. 61, 296–434 [DOI] [PubMed] [Google Scholar]
- 4. Hoffmann E. K., Lambert I. H., Pedersen S. F. (2009) Physiology of cell volume regulation in vertebrates. Physiol. Rev. 89, 193–277 [DOI] [PubMed] [Google Scholar]
- 5. Casey J. R., Grinstein S., Orlowski J. (2010) Sensors and regulators of intracellular pH. Nat. Rev. Mol. Cell Biol. 11, 50–61 [DOI] [PubMed] [Google Scholar]
- 6. Denker S. P., Huang D. C., Orlowski J., Furthmayr H., Barber D. L. (2000) Direct binding of the Na-H exchanger NHE1 to ERM proteins regulates the cortical cytoskeleton and cell shape independently of H+ translocation. Mol. Cell 6, 1425–1436 [DOI] [PubMed] [Google Scholar]
- 7. Denker S. P., Barber D. L. (2002) Cell migration requires both ion translocation and cytoskeletal anchoring by the Na-H exchanger NHE1. J. Cell Biol. 159, 1087–1096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hayashi H., Aharonovitz O., Alexander R. T., Touret N., Furuya W., Orlowski J., Grinstein S. (2008) Na+/H+ exchange and pH regulation in the control of neutrophil chemokinesis and chemotaxis. Am. J. Physiol. Cell Physiol. 294, C526-C534 [DOI] [PubMed] [Google Scholar]
- 9. Schneider L., Stock C. M., Dieterich P., Jensen B. H., Pedersen L. B., Satir P., Schwab A., Christensen S. T., Pedersen S. F. (2009) The Na+/H+ exchanger NHE1 is required for directional migration stimulated via PDGFR-α in the primary cilium. J. Cell Biol. 185, 163–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Clement D. L., Mally S., Stock C., Lethan M., Satir P., Schwab A., Pedersen S. F., Christensen S. T. (2013) PDGFRα signaling in the primary cilium regulates NHE1-dependent fibroblast migration via coordinated differential activity of MEK1/2-ERK1/2-p90RSK and AKT signaling pathways. J. Cell Sci. 126, 953–965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Putney L. K., Barber D. L. (2003) Na-H exchange-dependent increase in intracellular pH times G2/M entry and transition. J. Biol. Chem. 278, 44645–44649 [DOI] [PubMed] [Google Scholar]
- 12. Gekle M., Freudinger R., Mildenberger S. (2001) Inhibition of Na+-H+ exchanger-3 interferes with apical receptor-mediated endocytosis via vesicle fusion. J. Physiol. 531, 619–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Xinhan L., Matsushita M., Numaza M., Taguchi A., Mitsui K., Kanazawa H. (2011) Na+/H+ exchanger isoform 6 (NHE6/SLC9A6) is involved in clathrin-dependent endocytosis of transferrin. Am. J. Physiol. Cell Physiol. 301, C1431-C1444 [DOI] [PubMed] [Google Scholar]
- 14. Deisl C., Simonin A., Anderegg M., Albano G., Kovacs G., Ackermann D., Moch H., Dolci W., Thorens B., A. Hediger M., Fuster D. G. (2013) Sodium/hydrogen exchanger NHA2 is critical for insulin secretion in β-cells. Proc. Natl. Acad. Sci. U.S.A. 110, 10004–10009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Park K., Evans R. L., Watson G. E., Nehrke K., Richardson L., Bell S. M., Schultheis P. J., Hand A. R., Shull G. E., Melvin J. E. (2001) Defective fluid secretion and NaCl absorption in the parotid glands of Na+/H+ exchanger-deficient mice. J. Biol. Chem. 276, 27042–27050 [DOI] [PubMed] [Google Scholar]
- 16. Schultheis P. J., Clarke L. L., Meneton P., Harline M., Boivin G. P., Stemmermann G., Duffy J. J., Doetschman T., Miller M. L., Shull G. E. (1998) Targeted disruption of the murine Na+/H+ exchanger isoform 2 gene causes reduced viability of gastric parietal cells and loss of net acid secretion. J. Clin. Invest. 101, 1243–1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gawenis L. R., Greeb J. M., Prasad V., Grisham C., Sanford L. P., Doetschman T., Andringa A., Miller M. L., Shull G. E. (2005) Impaired gastric acid secretion in mice with a targeted disruption of the NHE4 Na+/H+ exchanger. J. Biol. Chem. 280, 12781–12789 [DOI] [PubMed] [Google Scholar]
- 18. Horikawa N., Nishioka M., Itoh N., Kuribayashi Y., Matsui K., Ohashi N. (2001) The Na+/H+ exchanger SM-20220 attenuates ischemic injury in in vitro and in vivo models. Pharmacology 63, 76–81 [DOI] [PubMed] [Google Scholar]
- 19. Cengiz P., Kleman N., Uluc K., Kendigelen P., Hagemann T., Akture E., Messing A., Ferrazzano P., Sun D. (2011) Inhibition of Na+/H+ exchanger isoform 1 is neuroprotective in neonatal hypoxic ischemic brain injury. Antioxid. Redox Signal. 14, 1803–1813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wu D., Russano K., Kouz I., Abraham W. M. (2013) NHE1 inhibition improves tissue perfusion and resuscitation outcome after severe hemorrhage. J. Surg. Res. 181, e75-e81 [DOI] [PubMed] [Google Scholar]
- 21. Karmazyn M. (1996) The sodium-hydrogen exchange system in the heart: its role in ischemic and reperfusion injury and therapeutic implications. Can. J. Cardiol. 12, 1074–1082 [PubMed] [Google Scholar]
- 22. Ayoub I. M., Kolarova J., Yi Z., Trevedi A., Deshmukh H., Lubell D. L., Franz M. R., Maldonado F. A., Gazmuri R. J. (2003) Sodium-hydrogen exchange inhibition during ventricular fibrillation: beneficial effects on ischemic contracture, action potential duration, reperfusion arrhythmias, myocardial function, and resuscitability. Circulation 107, 1804–1809 [DOI] [PubMed] [Google Scholar]
- 23. Park J. W., Roh H. Y., Jung I. S., Yun Y. P., Yi K. Y., Yoo S. E., Kwon S. H., Chung H. J., Shin H. S. (2005) Effects of [5-(2-methoxy-5-fluorophenyl)furan-2-ylcarbonyl]guanidine (KR-32560), a novel sodium/hydrogen exchanger-1 inhibitor, on myocardial infarct size and ventricular arrhythmias in a rat model of ischemia/reperfusion heart injury. J. Pharmacol. Sci. 98, 439–449 [DOI] [PubMed] [Google Scholar]
- 24. Karmazyn M. (2001) Role of sodium-hydrogen exchange in cardiac hypertrophy and heart failure: a novel and promising therapeutic target. Basic Res. Cardiol. 96, 325–328 [DOI] [PubMed] [Google Scholar]
- 25. Chahine M., Bkaily G., Nader M., Al-Khoury J., Jacques D., Beier N., Scholz W. (2005) NHE-1-dependent intracellular sodium overload in hypertrophic hereditary cardiomyopathy: prevention by NHE-1 inhibitor. J. Mol. Cell Cardiol. 38, 571–582 [DOI] [PubMed] [Google Scholar]
- 26. Kevelaitis E., Qureshi A. A., Mouas C., Marotte F., Kevelaitiene S., Avkiran M., Menasché P. (2005) Na+/H+ exchange inhibition in hypertrophied myocardium subjected to cardioplegic arrest: an effective cardioprotective approach. Eur. J. Cardiothorac. Surg. 27, 111–116 [DOI] [PubMed] [Google Scholar]
- 27. Karmazyn M., Kilić A., Javadov S. (2008) The role of NHE-1 in myocardial hypertrophy and remodelling. J. Mol. Cell Cardiol. 44, 647–653 [DOI] [PubMed] [Google Scholar]
- 28. Stock C., Schwab A. (2009) Protons make tumor cells move like clockwork. Pflugers Arch. 458, 981–992 [DOI] [PubMed] [Google Scholar]
- 29. Loo S. Y., Chang M. K., Chua C. S., Kumar A. P., Pervaiz S., Clement M. V. (2012) NHE-1: a promising target for novel anti-cancer therapeutics. Curr. Pharm. Des. 18, 1372–1382 [DOI] [PubMed] [Google Scholar]
- 30. Amith S. R., Fliegel L. (2013) Regulation of the Na+/H+ exchanger (NHE1) in breast cancer metastasis. Cancer Res. 73, 1259–1264 [DOI] [PubMed] [Google Scholar]
- 31. Reshkin S. J., Greco M. R., Cardone R. A. (2014) Role of pHi, and proton transporters in oncogene-driven neoplastic transformation. Philos. Trans. R. Soc. Lond. B Biol. Sci. 369, 20130100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chaitman B. R. (2003) A review of the GUARDIAN trial results: clinical implications and the significance of elevated perioperative CK-MB on 6-month survival. J. Card. Surg. 18, 13–20 [DOI] [PubMed] [Google Scholar]
- 33. Mentzer R. M., Jr., Bartels C., Bolli R., Boyce S., Buckberg G. D., Chaitman B., Haverich A., Knight J., Menasché P., Myers M. L., Nicolau J., Simoons M., Thulin L., Weisel R. D., and EXPEDITION Study Investigators (2008) Sodium-hydrogen exchange inhibition by cariporide to reduce the risk of ischemic cardiac events in patients undergoing coronary artery bypass grafting: results of the EXPEDITION study. Ann. Thorac. Surg. 85, 1261–1270 [DOI] [PubMed] [Google Scholar]
- 34. Zeymer U., Suryapranata H., Monassier J. P., Opolski G., Davies J., Rasmanis G., Linssen G., Tebbe U., Schröder R., Tiemann R., Machnig T., Neuhaus K. L., and ESCAMI Investigators (2001) The Na+/H+ exchange inhibitor eniporide as an adjunct to early reperfusion therapy for acute myocardial infarction. Results of the evaluation of the safety and cardioprotective effects of eniporide in acute myocardial infarction (ESCAMI) trial. J. Am. Coll. Cardiol. 38, 1644–1650 [DOI] [PubMed] [Google Scholar]
- 35. Karmazyn M. (2013) NHE-1: still a viable therapeutic target. J. Mol. Cell Cardiol. 61, 77–82 [DOI] [PubMed] [Google Scholar]
- 36. Wakabayashi S., Pang T., Su X., Shigekawa M. (2000) A novel topology model of the human Na+/H+ exchanger isoform 1. J. Biol. Chem. 275, 7942–7949 [DOI] [PubMed] [Google Scholar]
- 37. Otsu K., Kinsella J., Sacktor B., Froehlich J. P. (1989) Transient state kinetic evidence for an oligomer in the mechanism of Na+-H+ exchange. Proc. Natl. Acad. Sci. U.S.A. 86, 4818–4822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Otsu K., Kinsella J. L., Koh E., Froehlich J. P. (1992) Proton dependence of the partial reactions of the sodium-proton exchanger in renal brush border membranes. J. Biol. Chem. 267, 8089–8096 [PubMed] [Google Scholar]
- 39. Otsu K., Kinsella J. L., Heller P., Froehlich J. P. (1993) Sodium dependence of the Na+-H+ exchanger in the pre-steady state. Implications for the exchange mechanism. J. Biol. Chem. 268, 3184–3193 [PubMed] [Google Scholar]
- 40. Lacroix J., Poët M., Maehrel C., Counillon L. (2004) A mechanism for the activation of the Na/H exchanger NHE-1 by cytoplasmic acidification and mitogens. EMBO Rep. 5, 91–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fafournoux P., Noël J., Pouysségur J. (1994) Evidence that Na+/H+ exchanger isoforms NHE1 and NHE3 exist as stable dimers in membranes with a high degree of specificity for homodimers. J. Biol. Chem. 269, 2589–2596 [PubMed] [Google Scholar]
- 42. Hisamitsu T., Pang T., Shigekawa M., Wakabayashi S. (2004) Dimeric interaction between the cytoplasmic domains of the Na+/H+ exchanger NHE1 revealed by symmetrical intermolecular cross-linking and selective co-immunoprecipitation. Biochemistry 43, 11135–11143 [DOI] [PubMed] [Google Scholar]
- 43. Hisamitsu T., Ben Ammar Y., Nakamura T. Y., Wakabayashi S. (2006) Dimerization Is crucial for the function of the Na+/H+ exchanger NHE1. Biochemistry 45, 13346–13355 [DOI] [PubMed] [Google Scholar]
- 44. Moncoq K., Kemp G., Li X., Fliegel L., Young H. S. (2008) Dimeric structure of human Na+/H+ exchanger isoform 1 overproduced in Saccharomyces cerevisiae. J. Biol. Chem. 283, 4145–4154 [DOI] [PubMed] [Google Scholar]
- 45. Béliveau R., Demeule M., Potier M. (1988) Molecular size of the Na+-H+ antiport in renal brush border membranes, as estimated by radiation inactivation. Biochem. Biophys. Res. Commun. 152, 484–489 [DOI] [PubMed] [Google Scholar]
- 46. Kinsella J. L., Aronson P. S. (1981) Amiloride inhibition of the Na+-H+ exchanger in renal microvillus membrane vesicles. Am. J. Physiol. 241, F374-F379 [DOI] [PubMed] [Google Scholar]
- 47. Ives H. E., Yee V. J., Warnock D. G. (1983) Mixed type inhibition of the renal Na+/H+ antiporter by Li+ and amiloride. Evidence for a modifier site. J. Biol. Chem. 258, 9710–9716 [PubMed] [Google Scholar]
- 48. Masereel B., Pochet L., Laeckmann D. (2003) An overview of inhibitors of Na+/H+ exchanger. Eur. J. Med. Chem. 38, 547–554 [DOI] [PubMed] [Google Scholar]
- 49. Khadilkar A., Iannuzzi P., Orlowski J. (2001) Identification of sites in the second exomembrane loop and ninth transmembrane helix of the mammalian Na+/H+ exchanger important for drug recognition and cation translocation. J. Biol. Chem. 276, 43792–43800 [DOI] [PubMed] [Google Scholar]
- 50. Counillon L., Franchi A., Pouysségur J. (1993) A point mutation of the Na+/H+ exchanger gene (NHE1) and amplification of the mutated allele confer amiloride resistance upon chronic acidosis. Proc. Natl. Acad. Sci. U.S.A. 90, 4508–4512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Counillon L., Noël J., Reithmeier R. A., Pouysségur J. (1997) Random mutagenesis reveals a novel site involved in inhibitor interaction within the fourth transmembrane segment of the Na+/H+ exchanger-1. Biochemistry 36, 2951–2959 [DOI] [PubMed] [Google Scholar]
- 52. Touret N., Poujeol P., Counillon L. (2001) Second-site revertants of a low-sodium-affinity mutant of the Na+/H+ exchanger reveal the participation of TM4 into a highly constrained sodium-binding site. Biochemistry 40, 5095–5101 [DOI] [PubMed] [Google Scholar]
- 53. Slepkov E. R., Chow S., Lemieux M. J., Fliegel L. (2004) Proline residues in transmembrane segment IV are critical for activity, expression and targeting of the Na+/H+ exchanger isoform 1. Biochem. J. 379, 31–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Slepkov E., Ding J., Han J., Fliegel L. (2007) Mutational analysis of potential pore-lining amino acids in TM IV of the Na+/H+ exchanger. Biochim. Biophys. Acta 1768, 2882–2889 [DOI] [PubMed] [Google Scholar]
- 55. Wang D., Balkovetz D. F., Warnock D. G. (1995) Mutational analysis of transmembrane histidines in the amiloride-sensitive Na+/H+ exchanger. Am. J. Physiol. 269, C392-C402 [DOI] [PubMed] [Google Scholar]
- 56. Orlowski J., Kandasamy R. A. (1996) Delineation of transmembrane domains of the Na+/H+ exchanger that confer sensitivity to pharmacological antagonists. J. Biol. Chem. 271, 19922–19927 [DOI] [PubMed] [Google Scholar]
- 57. Wakabayashi S., Hisamitsu T., Pang T., Shigekawa M. (2003) Mutations of Arg440 and Gly455/Gly456 oppositely change pH sensing of Na+/H+ exchanger 1. J. Biol. Chem. 278, 11828–11835 [DOI] [PubMed] [Google Scholar]
- 58. Wakabayashi S., Hisamitsu T., Pang T., Shigekawa M. (2003) Kinetic dissection of two distinct proton binding sites in Na+/H+ exchangers by measurement of reverse mode reaction. J. Biol. Chem. 278, 43580–43585 [DOI] [PubMed] [Google Scholar]
- 59. Slepkov E. R., Rainey J. K., Li X., Liu Y., Cheng F. J., Lindhout D. A., Sykes B. D., Fliegel L. (2005) Structural and functional characterization of transmembrane segment IV of the NHE1 isoform of the Na+/H+ exchanger. J. Biol. Chem. 280, 17863–17872 [DOI] [PubMed] [Google Scholar]
- 60. Reddy T., Ding J., Li X., Sykes B. D., Rainey J. K., Fliegel L. (2008) Structural and functional characterization of transmembrane segment IX of the NHE1 isoform of the Na+/H+ exchanger. J. Biol. Chem. 283, 22018–22030 [DOI] [PubMed] [Google Scholar]
- 61. Lee B. L., Li X., Liu Y., Sykes B. D., Fliegel L. (2009) Structural and functional analysis of transmembrane XI of the NHE1 isoform of the Na+/H+ exchanger. J. Biol. Chem. 284, 11546–11556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Tzeng J., Lee B. L., Sykes B. D., Fliegel L. (2010) Structural and functional analysis of transmembrane VI of the NHE1 isoform of the Na+/H+ exchanger. J. Biol. Chem. 285, 36656–36665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ding J., Rainey J. K., Xu C., Sykes B. D., Fliegel L. (2006) Structural and functional characterization of transmembrane segment VII of the Na+/H+ exchanger isoform 1. J. Biol. Chem. 281, 29817–29829 [DOI] [PubMed] [Google Scholar]
- 64. Slepkov E. R., Rainey J. K., Sykes B. D., Fliegel L. (2007) Structural and functional analysis of the Na+/H+ exchanger. Biochem. J. 401, 623–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Landau M., Herz K., Padan E., Ben-Tal N. (2007) Model structure of the Na+/H+ exchanger 1 (NHE1): functional and clinical implications. J. Biol. Chem. 282, 37854–37863 [DOI] [PubMed] [Google Scholar]
- 66. Schushan M., Landau M., Padan E., Ben-Tal N. (2011) Two conflicting NHE1 model structures: compatibility with experimental data and implications for the transport mechanism. J. Biol. Chem. 286, le9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Williams K. A. (2000) Three-dimensional structure of the ion-coupled transport protein NhaA. Nature 403, 112–115 [DOI] [PubMed] [Google Scholar]
- 68. Hunte C., Screpanti E., Venturi M., Rimon A., Padan E., Michel H. (2005) Structure of a Na+/H+ antiporter and insights into mechanism of action and regulation by pH. Nature 435, 1197–1202 [DOI] [PubMed] [Google Scholar]
- 69. Nygaard E. B., Lagerstedt J. O., Bjerre G., Shi B., Budamagunta M., Poulsen K. A., Meinild S., Rigor R. R., Voss J. C., Cala P. M., Pedersen S. F. (2011) Structural modeling and electron paramagnetic resonance spectroscopy of the human Na+/H+ exchanger isoform 1, NHE1. J. Biol. Chem. 286, 634–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Counillon L., Pouysségur J., Reithmeier R. A. (1994) The Na+/H+ exchanger NHE-1 possesses N- and O-linked glycosylation restricted to the first N-terminal extracellular domain. Biochemistry 33, 10463–10469 [DOI] [PubMed] [Google Scholar]
- 71. Sun Z. P., Akabas M. H., Goulding E. H., Karlin A., Siegelbaum S. A. (1996) Exposure of residues in the cyclic nucleotide-gated channel pore: P region structure and function in gating. Neuron 16, 141–149 [DOI] [PubMed] [Google Scholar]
- 72. Chiamvimonvat N., Pérez-García M. T., Ranjan R., Marban E., Tomaselli G. F. (1996) Depth asymmetries of the pore-lining segments of the Na+ channel revealed by cysteine mutagenesis. Neuron 16, 1037–1047 [DOI] [PubMed] [Google Scholar]
- 73. Karlin A., Akabas M. H. (1998) Substituted-cysteine accessibility method. Methods Enzymol. 293, 123–145 [DOI] [PubMed] [Google Scholar]
- 74. Rotin D., Grinstein S. (1989) Impaired cell volume regulation in Na+-H+ exchange-deficient mutants. Am. J. Physiol. 257, C1158-C1165 [DOI] [PubMed] [Google Scholar]
- 75. Orlowski J. (1993) Heterologous expression and functional properties of the amiloride high affinity (NHE-1) and low affinity (NHE-3) isoforms of the rat Na/H exchanger. J. Biol. Chem. 268, 16369–16377 [PubMed] [Google Scholar]
- 76. Franchi A., Perucca-Lostanlen D., Pouyssegur J. (1986) Functional expression of a human Na+/H+ antiporter gene transfected into antiporter-deficient mouse L cells. Proc. Natl. Acad. Sci. U.S.A. 83, 9388–9392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Thomas J. A., Buchsbaum R. N., Zimniak A., Racker E. (1979) Intracellular pH measurements in Ehrlich ascites tumor cells utilizing spectroscopic probes generated in situ. Biochemistry 18, 2210–2218 [DOI] [PubMed] [Google Scholar]
- 78. Aharonovitz O., Zaun H. C., Balla T., York J. D., Orlowski J., Grinstein S. (2000) Intracellular pH regulation by Na+/H+ exchange requires phosphatidylinositol 4,5-bisphosphate. J. Cell Biol. 150, 213–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Shrode L. D., Gan B. S., D'Souza S. J., Orlowski J., Grinstein S. (1998) Topological analysis of NHE1, the ubiquitous Na+/H+ exchanger using chymotryptic cleavage. Am. J. Physiol. 275, C431-C439 [DOI] [PubMed] [Google Scholar]
- 80. Tang X. B., Fujinaga J., Kopito R., Casey J. R. (1998) Topology of the region surrounding Glu681 of human AE1 protein, the erythrocyte anion exchanger. J. Biol. Chem. 273, 22545–22553 [DOI] [PubMed] [Google Scholar]
- 81. Xu M., Akabas M. H. (1993) Amino acids lining the channel of the γ-aminobutyric acid type A receptor identified by cysteine substitution. J. Biol. Chem. 268, 21505–21508 [PubMed] [Google Scholar]
- 82. Doyle D. A., Morais Cabral J., Pfuetzner R. A., Kuo A., Gulbis J. M., Cohen S. L., Chait B. T., MacKinnon R. (1998) The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science 280, 69–77 [DOI] [PubMed] [Google Scholar]
- 83. Schneider H., Scheiner-Bobis G. (1997) Involvement of the M7/M8 extracellular loop of the sodium pump α subunit in ion transport. Structural and functional homology to P-loops of ion channels. J. Biol. Chem. 272, 16158–16165 [DOI] [PubMed] [Google Scholar]
- 84. Tsushima R. G., Li R. A., Backx P. H. (1997) P-loop flexibility in Na+ channel pores revealed by single- and double-cysteine replacements. J. Gen. Physiol. 110, 59–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Tikhonov D. B., Zhorov B. S. (2005) Modeling P-loops domain of sodium channel: homology with potassium channels and interaction with ligands. Biophys. J. 88, 184–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Doktor H. S., Benjamin N., Todd S. D., Ritter J. M. (1991) Sodium dependence of sodium-proton exchange in platelets from patients with essential hypertension. J. Hum. Hypertens. 5, 161–165 [PubMed] [Google Scholar]
- 87. Wu M. L., Vaughan-Jones R. D. (1997) Interaction between Na+ and H+ ions on Na-H exchange in sheep cardiac Purkinje fibers. J. Mol. Cell Cardiol. 29, 1131–1140 [DOI] [PubMed] [Google Scholar]
- 88. Semplicini A., Spalvins A., Canessa M. (1989) Kinetics and stoichiometry of the human red cell Na+/H+ exchanger. J. Membr. Biol. 107, 219–228 [DOI] [PubMed] [Google Scholar]
- 89. Fuster D., Moe O. W., Hilgemann D. W. (2008) Steady-state function of the ubiquitous mammalian Na/H exchanger (NHE1) in relation to dimer coupling models with 2Na/2H stoichiometry. J. Gen. Physiol. 132, 465–480 [DOI] [PMC free article] [PubMed] [Google Scholar]