

Background: Two enzymes from Mycobacterium tuberculosis involved in aromatic amino acid biosynthesis form a hetero-octameric complex.
Results: Complex formation boosts the catalytic activity of both enzymes and greatly extends the allosteric effector sensitivity.
Conclusion: Enzyme interactions allow complex allosteric machinery of one of the complex partners to be shared.
Significance: Sophisticated allosteric responses are delivered through protein-protein interactions, allowing enhanced metabolic control.
Keywords: allosteric regulation, enzyme catalysis, Mycobacterium tuberculosis, protein complex, protein-protein interaction, 3-deoxy-d-arabino-heptulosonate 7-phosphate synthase, TB, chorismate mutase, oligomer, shikimate
Abstract
Allostery, where remote ligand binding alters protein function, is essential for the control of metabolism. Here, we have identified a highly sophisticated allosteric response that allows complex control of the pathway for aromatic amino acid biosynthesis in the pathogen Mycobacterium tuberculosis. This response is mediated by an enzyme complex formed by two pathway enzymes: chorismate mutase (CM) and 3-deoxy-d-arabino-heptulosonate 7-phosphate synthase (DAH7PS). Whereas both enzymes are active in isolation, the catalytic activity of both enzymes is enhanced, and in particular that of the much smaller CM is greatly enhanced (by 120-fold), by formation of a hetero-octameric complex between CM and DAH7PS. Moreover, on complex formation M. tuberculosis CM, which has no allosteric response on its own, acquires allosteric behavior to facilitate its own regulatory needs by directly appropriating and partly reconfiguring the allosteric machinery that provides a synergistic allosteric response in DAH7PS. Kinetic and analytical ultracentrifugation experiments demonstrate that allosteric binding of phenylalanine specifically promotes hetero-octameric complex dissociation, with concomitant reduction of CM activity. Together, DAH7PS and CM from M. tuberculosis provide exquisite control of aromatic amino acid biosynthesis, not only controlling flux into the start of the pathway, but also directing the pathway intermediate chorismate into either Phe/Tyr or Trp biosynthesis.
Introduction
Interactions between proteins play a central role in biological processes. Oligomerization to form homomeric and heteromeric assemblies confers protein stability and extends protein functionality, including enhancing enzyme catalytic rates and bestowing allosteric properties to the assemblies (1–4). Multifunctional enzymes and complex allosteric machinery have previously been described for the enzymes of the shikimate pathway, which is ultimately responsible for the biosynthesis of the aromatic compounds in plants, microorganisms, and apicomplexans (Fig. 1) (5, 6). In these studies, we explore the role of hetero-oligomeric complex formation between two key enzymes in the shikimate pathway from Mycobacterium tuberculosis in providing a sophisticated allosteric response.
FIGURE 1.
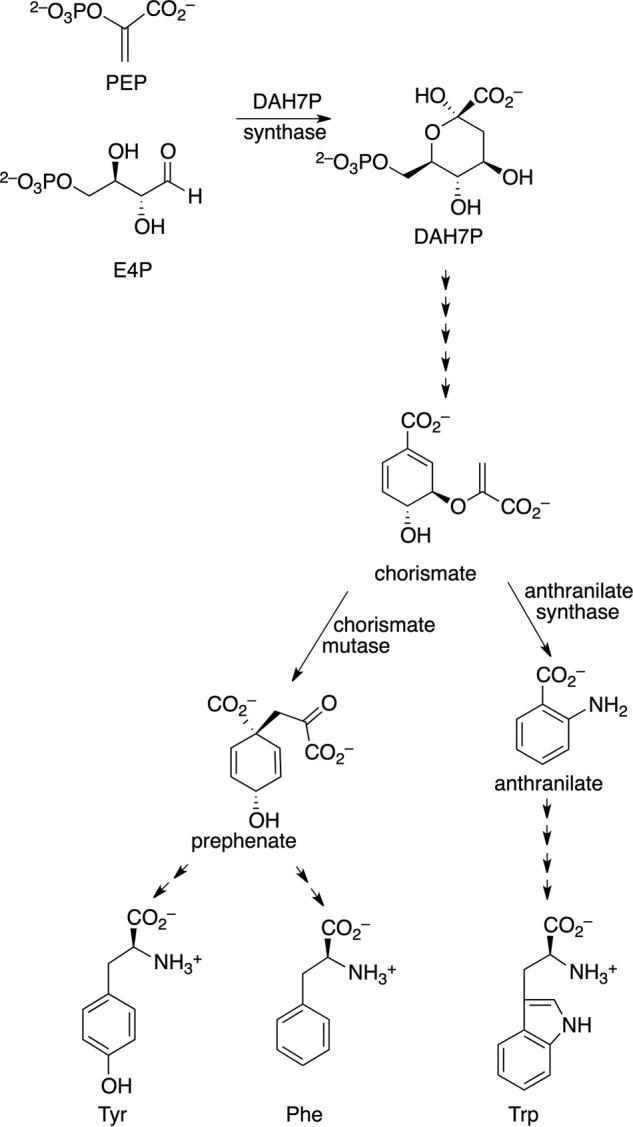
Aromatic amino acid biosynthesis.
The shikimate pathway, which commits metabolites from central metabolism to the biosynthesis of aromatic compounds, leads initially to the formation of chorismate (Fig. 1). Chorismate is the precursor for a range of aromatic metabolites, including the aromatic amino acids Trp, Phe, and Tyr. The first step of the shikimate pathway is catalyzed by 3-deoxy-d-arabino-heptulosonate 7-phosphate synthase (DAH7PS).3 Six further enzyme-catalyzed steps lead to chorismate. Chorismate mutase (CM) catalyzes the Claisen rearrangement of chorismate to prephenate, a step located at the branch point of aromatic metabolite biosynthesis that commits chorismate to Phe or Tyr biosynthesis. The enzymes for the production of the aromatic amino acids are absent in mammals yet are required for the viability of a number of organisms including M. tuberculosis, the pathogen responsible for tuberculosis, making these enzymes potential targets for novel anti-mycobacterial drugs (7).
Because of its critical location at the start of the pathway, DAH7PS activity is usually tightly controlled by feedback regulation by pathway end products (8). M. tuberculosis DAH7PS (MtuDAH7PS) is distinct from DAH7PS enzymes from other sources in that it possesses a highly sophisticated allosteric response, demonstrating inhibition by binary and ternary combinations of all three aromatic amino acids (9–11). The homotetrameric protein has three distinct inhibitor binding sites per protein chain, each responsible for binding preferentially one of the aromatic amino acids (Fig. 2). The individual amino acids Phe, Trp, and Tyr alone are not inhibitory, yet combinations that include Trp, especially the ternary combination, significantly inhibit DAH7PS activity. Because no significant conformational change of the protein is observed on inhibitor binding, except in the near vicinity of the Phe binding site, inhibition of MtuDAH7PS is consistent with a change in protein dynamics, causing altered substrate binding (12).
FIGURE 2.

MtuCM-MtuDAH7PS hetero-octameric complex and MtuDAH7PS allosteric binding sites. A, complex (Protein Data Bank code 2W19) of MtuDAH7PS in blue (Phe/Tyr binding sites in red, and Trp binding site in yellow) with MtuCM in magenta. B, MtuDAH7PS (Protein Data Bank code 3KGF) showing Phe/Tyr and Trp binding sites (ligands shown as orange spheres, and active site manganese shown as green spheres) provided by extra subdomains at the N terminus (red) or C terminus (yellow) of the catalytic (β/α)8 barrel (blue). C, Phe binding site (Protein Data Bank code 3KGF). D, Trp binding site (Protein Data Bank code 3KGF). E, Tyr binding site (Protein Data Bank code 2YPP). Whereas there are four Phe and Trp binding sites per tetramer, there are only two Tyr-binding sites per tetramer.
The genome of M. tuberculosis encodes two CM proteins (13, 14). One is highly active and secreted into the periplasmic compartment, and its biological role is yet to be determined (14). The cytosolic homodimeric CM is a poor catalyst on its own; however, on complexation with the tetrameric MtuDAH7PS, to generate a hetero-octameric complex, the catalytic efficiency of this protein is dramatically enhanced (15). In contrast to the large tetrameric MtuDAH7PS (over 200 kDa), the MtuCM homodimer is small, and complex formation is achieved by the MtuCM homodimers capping the MtuDAH7PS tetramer interfaces (Fig. 2). More recently the C-terminal region of the MtuCM has been demonstrated to be important for the interaction with and activation by MtuDAH7PS (16).
The three binding sites on MtuDAH7PS for aromatic amino acids are formed by discrete extensions to the core catalytic barrel of the protein and comprise the allosteric machinery of the enzyme (Fig. 2) (9). Key Arg residues, Arg-171 and Arg-256, are responsible for forming interactions with Phe and Tyr, respectively. Substitution of these Arg residues by Ala impairs the allosteric response of MtuDAH7PS activity to Phe and Tyr without compromising catalytic activity (9). We now demonstrate that hetero-octameric complex formation between MtuDAH7PS and MtuCM not only provides an activity boost for MtuCM, but, very significantly, also allows this enzyme to share the allosteric machinery of its much larger complex partner. Our results illuminate an allosteric relay in which Phe binding is linked directly to complex formation and MtuCM activation. Together, the complex formation delivers another level of sophistication to the control of aromatic amino acid biosynthesis and significantly extends the function of the DAH7PS allosteric machinery.
Experimental Procedures
Expression and Purification of MtuCM and MtuDAH7PS
MtuDAH7PS and MtuDAH7PS variants were prepared and purified as described previously (9, 11, 17). An MtuCM synthetic gene (Geneart), optimized for Escherichia coli codon usage, was transferred into pDEST 15 using Gateway cloning technology (Invitrogen). This plasmid was then transformed into Escherichia coli BL21 (Star) cells. Transformants containing the recombinant plasmids were grown in Luria-Bertani medium at 37 °C in the presence of ampicillin (100 μg ml−1) until the A600 reached ∼0.6. Protein expression was induced by addition of isopropyl β-d-thiogalactopyranoside (Roche) (0.5 mm) and incubated overnight at 23 °C. Cells were lysed in buffer A (10 mm phosphate buffer, pH 7.5) by sonication. Clarified supernatant was applied to a GSTrap HP (GE Healthcare) and eluted with an increasing gradient of reduced-glutathione (Sigma). Fractions containing GST-tagged MtuCM were pooled and treated with TEV protease and reapplied to the GSTrap HP to remove the cleaved GST tag. Buffer A was supplemented with 1% glycerol to aid tag removal. The protein was further purified by size exclusion chromatography (Superdex 75 16/60) in size exclusion chromatography buffer (10 mm 1,3-bis[tris(hydroxymethyl)methylamino]propane (Sigma), 150 mm NaCl, 200 μm each of tris-(2-carboxyethyl) phosphine hydrochloride (Sigma), MnSO4 (Sigma), and PEP (Research Chemicals). MtuCM protein concentration was determined via Bradford assay using bovine serum albumin as a standard. MtuDAH7PS and MtuDAH7PS variant concentrations were determined using the A280 absorbance via a NanoDrop ND1000 spectrophotometer.
Kinetic Assays
The kinetic parameters were derived from measurements of MtuDAH7PS activity obtained by monitoring the loss of PEP at 232 nm (ϵ = 2.8 × 103 m−1·cm−1 at 303 K) in the presence of E4P using a Varian Cary 1 UV-visible spectrophotometer using 1-cm quartz cuvettes and standard assay conditions (9, 10). The assay solutions contained PEP (fixed at 300 μm or varied to determine apparent PEP Km) (Research Chemicals), E4P (fixed at 300 μm or varied to determine apparent E4P Km) (Sigma), and MnSO4 (100 μm) (Sigma) in assay buffer (50 mm 1,3-bis[tris(hydroxymethyl)methylamino]propane (Sigma), pH 7.5, and 1 mm tris-(2-carboxyethyl) phosphine hydrochloride). Kinetic parameters of MtuCM activity were determined by monitoring the loss of chorismate (added as chorismic acid; Sigma) at 274 nm (ϵ = 2.630 m−1·cm−1 at 303 K) in the same assay buffer as MtuDAH7PS, MnSO4 (100 μm), PEP (150 μm), and chorismate varied to determine chorismate Km. Chorismate, PEP, and E4P solutions were made up in assay buffer, and the MnSO4 solution was made up in ultrapure water. Initial reaction rates were determined by a least squares fit of the initial rate data. Apparent Km, Km, and kcat values were determined by fitting the data to the Michaelis-Menten equation by nonlinear regression using software GraFit (Erithacus software). For DAH7PS activity, kcat is taken from both sets of data with either PEP or E4P as the variable substrate. These measured values were within experimental error. For the complexes, CM activity was determined with a 1:10 ratio for CM to DAH7PS, and DAH7PS activity was determined with a 10:1 ratio of CM to DAH7PS. All assay components (except initiating substrate, chorismate, or E4P respectively) were incubated at 303 K for 5 min.
Inhibition Studies with Aromatic Amino Acids
Solutions of l-Phe (Sigma), l-Tyr (Sigma), and l-Trp (Sigma) in ultrapure water were added to standard assay reaction mixtures to give a concentration of 200 μm in inhibitor studies. The concentrations of Trp, Phe, and Tyr were varied between 20 μm and 4 mm to determine the effect of increasing aromatic amino acid concentration on MtuCM activity. Steady-state kinetic assays were conducted in the presence of none, one, two, or all three aromatic amino acids. The assay solutions contained PEP (150 μm), E4P (150 μm), and MnSO4 (100 μm) in assay buffer for measurements of DAH7PS activity and chorismate (25 or 150 μm as stated), PEP (150 μm), and MnSO4 (100 μm) in assay buffer for CM activity. Reactions were initiated by the addition of E4P or chorismic acid, respectively. Measurements for CM alone required 3 μm CM, those involving the complex CM activity were determined with a 1:10 ratio of CM to DAH7PS, and the CM concentration was held at 60 nm. The concentration of MtuDAH7PS was held at 40 nm in the presence or absence of MtuCM to measure DAH7PS activity. DAH7PS activity of the complex was determined with a 10:1 ratio of CM to DAH7PS. The half-maximal effective inhibitor concentration values (EC50) were determined by fitting triplicate data to the log[inhibitor] versus response using a nonlinear regression curve and globally fitting in GraphPad Prism6®.
Determination of Kinetic Parameters for CM Activity of the Complex with Added Phe and/or Tyr, or Trp
Solutions of l-Phe, l-Trp, or l-Tyr were added to the standard assay conditions used to determine the chorismate Km to give a final concentration of either 100 or 200 μm. All assay components were left to incubate (except initiating substrate) for 5 min. Because of high absorbance readings at 274 nm of Tyr, 1-mm-path length cuvettes were used for the measurements in the presence of 200 μm Tyr and 200 μm Tyr and Phe. The smaller cuvettes required an increase in MtuCM concentration to 120 nm to detect CM activity. The ratio of CM to DAH7PS was kept at 1:10.
Activation of CM as a Function of MtuDAH7PS Concentration
The kinetic parameters were determined by measuring CM activity at various concentrations of MtuDAH7PS under the standard assay conditions described above, in the absence of ligand or presence of 200 μm Phe, Trp or Tyr. The MtuCM concentration was held at 10 nm (90 nm in the absence of MtuDAH7PS). For each MtuDAH7PS concentration, at least seven points were used to determine the Michaelis-Menten kinetic parameters and to find the catalytic efficiency (kcat/Km) of MtuCM activity. Each set of Michaelis-Menten kinetics was conducted in duplicate. The Kd,app was derived using the equations adapted from Taira and Benkovic (18) as outlined by Sasso et al. and assumes a 1:1 stoichiometry of MtuCM:MtuDAH7PS (15, 18). The data were fitted to equation E1, using υCM·DS and Kd,app as the fitting parameters, by the iterative curve-fitting software KaleidaGraph (Synergy Software). Sasso et al. (15) adapted work by Taira and Benkovic (18) to derive this equation, allowing the determination of the apparent MtuCM·MtuDAH7PS dissociation constant for a 1:1 stoichiometry.
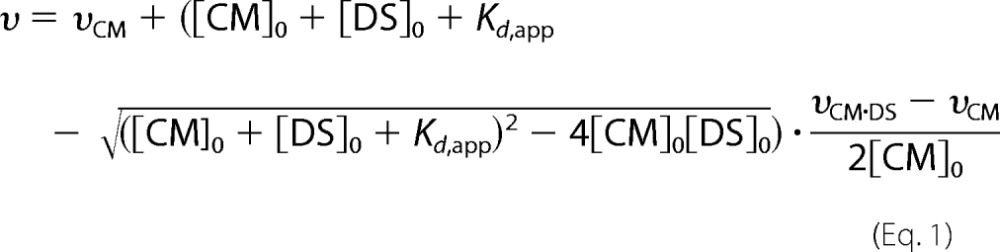 |
Parameters υ, υCM, and υCM·DS are the corresponding catalytic efficiencies, and [CM]0 and [DS]0 are the total concentrations of MtuCM and MtuDAH7PSWT, respectively. Kd,app is the apparent dissociation constant of the complex.
Analytical Ultracentrifugation
Analytical ultracentrifugation samples of MtuDAH7PS, MtuCM, and combinations of these proteins were dialyzed for 2–3 h against a 50 mm Tris, 150 mm NaCl, and 150 μm MnSO4 buffer at pH 7.5 prior to each experiment. The dialysis buffer was used as the buffer blank.
Sedimentation velocity experiments were performed in a Beckman Coulter model XL-I analytical ultracentrifuge equipped with UV-visible scanning optics. Reference buffer solution and sample solutions were loaded into 12-mm double-sector cells with quartz windows, and the cells were then mounted in an An-60 Ti 4-hole rotor. Samples at different protein concentrations 0.2 mg·ml−1, 0.4 mg·ml−1, and 0.6 mg·ml−1 (380 μl) and reference (400 μl) were centrifuged at 50,000 rpm at 20 °C, and absorbance data were collected in continuous mode at 278 or 279 nm without averaging. The data were fitted to a continuous size distribution model (19) using the program SEDFIT. The partial specific volume (v̄) of the proteins, buffer density (1.005 g·ml−1) and buffer viscosity (1.021 cp) were calculated using the program SEDNTERP (20). The partial specific volume of the complex was calculated using Equation 2,
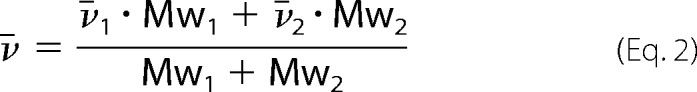 |
where, respectively, v̄1 and Mw1 are the partial specific volume and molecular weight of MtuDAH7PS, and v̄2 and Mw2 are the partial specific volume and molecular weight of MtuCM.
For the sedimentation equilibrium experiments, MtuDAH7PSWT concentrations of 0.2, 0.4, and 0.6 mg·ml−1 (3.9, 7.9, and 11.9 μm, respectively, based on the molecular weight of monomer) were used. Protein solutions (110 μl) and the reference buffer solutions (120 μl) were loaded into the cells and centrifuged at 6,000, 8,000, 10,000, and 12,000 rpm until sedimentation equilibrium was attained, as determined by matching the root mean square differences between the last scans. At sedimentation equilibrium, the final absorbance versus radial position profile was collected at 20 °C at 279 and 288 nm for MtuDAH7PSWT and 4:1 mixtures of MtuCM and MtuDAH7PSWT, respectively. Different wavelengths were chosen to optimally collect data at different concentrations. A global fit to the multispeed data were performed using a species analysis model in SEDPHAT (21).
Results
The Formation of a Hetero-octameric Complex Enhances the Activity of Both MtuCM and MtuDAH7PS
MtuCM alone demonstrates very low catalytic activity (Table 1). The inclusion of MtuDAH7PS in the assays, however, significantly enhances the catalytic activity of the enzyme, increasing the specific activity by over 120-fold in accordance with previous studies (15). The enhancement arises from both a decrease in the Michaelis constant and an increase in the turnover number. MtuDAH7PS activity was also enhanced in the presence of MtuCM, although the level of activation, as expressed by the increase in kcat/Km ratio on association with MtuCM, was far more modest.
TABLE 1.
Enhancement of MtuCM and MtuDAH7PS activities by complex formation
| Protein | CM activity |
DAH7PS activity |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| kcat | Km | kcat/Km | Activation factora | kcat | KmPEPb | KmE4Pb | kcat/Km | kcat/Km | Activation factora | |
| s−1 | μm | mm−1 s−1 | s−1 | μm | μm | mm−1 s−1 | mm−1 s−1 | |||
| MtuCM | 0.42 ± 0.02 | 500 ± 30 | 0.84 ± 0.09 | |||||||
| MtuDAH7PSWT | 4.7 ± 0.1 | 37 ± 4 | 28 ± 2 | 130 ± 20 | 170 ± 20 | |||||
| MtuCM-MtuDAH7PSWTc | 5.4 ± 0.1 | 52 ± 4 | 104 ± 9 | 120 ± 20 | 6.4 ± 0.1 | 27 ± 2 | 20 ± 2 | 240 ± 20 | 320 ± 40 | 1.8 ± 0.4/1.9 ± 0.5 |
| DAH7PSR171A | 7.0 ± 0.3 | 60 ± 6 | 35 ± 3 | 120 ± 20 | 200 ± 30 | |||||
| MtuCM-MtuDAH7PSR171Ac | 6.5 ± 0.2 | 64 ± 6 | 100 ± 1 | 120 ± 20 | 7.9 ± 0.1 | 15 ± 1 | 48 ± 4 | 530 ± 40 | 170 ± 10 | 4 ± 1/0.9 ± 0.2 |
| DAH7PSR256A | 5.5 ± 0.1 | 16 ± 2 | 33 ± 1 | 340 ± 50 | 170 ± 10 | |||||
| MtuCM-MtuDAH7PSR256Ac | 5.5 ± 0.1 | 53 ± 4 | 104 ± 9 | 120 ± 20 | 5.6 ± 0.1 | 10 ± 1 | 23 ± 1 | 560 ± 70 | 240 ± 20 | 1.6 ± 0.5/1.4 ± 0.2 |
a Activation factors are calculated from the ratios of kcat/Km with and without the complex partner present. For DAH7PS, this gives two values with respect to each substrate: PEP and E4P, respectively.
b For KmPEP and KmE4P, the values reported are apparent Km values when the concentration of the nonvariable substrate was held at 300 μm.
c For the complexes CM activity was determined with a 1:10 ratio for CM to DAH7PS and DAH7PS activity was determined with a 10:1 ratio of CM to DAH7PS.
Formation of a hetero-octameric complex between CM and DAH7PS was observed by analytical ultracentrifugation (Fig. 3). Sedimentation velocity experiments for MtuDAH7PSWT and 4:1 mixtures of MtuCM and MtuDAH7PSWT were performed at three protein concentrations. For MtuDAH7PSWT, data fitting to a continuous size distribution model indicates the presence of a major species with a standardized sedimentation coefficient of 9.0 S (Fig. 3). The calculated mass of this species is 164 kDa, smaller than the expected mass of a homotetrameric species (predicted molecular mass, 204 kDa). When MtuCM was added to the MtuDAH7PS, a species with a larger sedimentation coefficient (10.2S) was observed at all three concentrations corresponding to a larger species of ∼220 kDa, consistent with the predicted molecular mass of the hetero-octameric species (245 kDa). A smaller species (3S) was also observed, corresponding to a molecular mass of ∼30 kDa. This observation of this species is consistent with the presence of homodimeric MtuCM (predicted mass of 20.7 kDa), which is present in excess but has a significantly reduced molar absorptivity in comparison to MtuDAH7PSWT.
FIGURE 3.
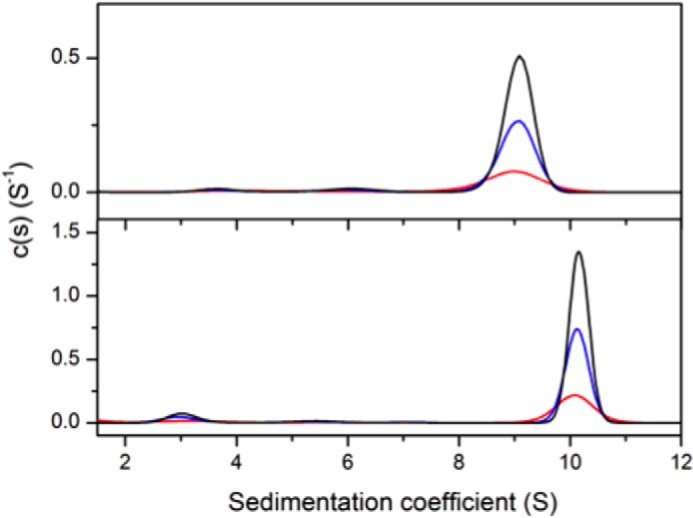
Sedimentation coefficient distribution of species. Shown are MtuDAH7PS (top panel) and a 4:1 mixture (molar ratio) of MtuCM-MtuDAH7PSWT (bottom panel). MtuDAH7PSWT was present at concentrations of 0.2 mg·ml−1 (red), 0.4 mg·ml−1 (blue), or 0.6 mg·ml−1 (black). Note that the molar extinction coefficient of MtuCM is 1,490 cm−1·mol−1, whereas that of MtuDAH7PSWT is 41,160 cm−1·mol−1 at a wavelength of 280 nm.
To confirm the oligomeric states of MtuDAH7PSWT and MtuCM-MtuDAH7PSWT, sedimentation equilibrium experiments were performed (Fig. 4, A–C). Global species analysis of the MtuDAH7PSWT data yields a major species with calculated molecular mass of 202 kDa, consistent with the expected tetrameric species (predicted mass of 204 kDa). In comparison, the experiments performed on the MtuCM-MtuDAH7PSWT combination establish the presence of two species of ∼21 and 236 kDa, consistent with the presence of excess homodimeric MtuCM and the hetero-octameric MtuDAH7PS-MtuCM complex (expected mass of 245 kDa).
FIGURE 4.

Sedimentation equilibrium experiments for MtuDAH7PSWT and the MtuDAH7PS-MtuCM complex. The experiments were performed at 6,000 (black), 8,000 (red), 10,000 (green), and 12,000 rpm (blue). A–C represent the data for the MtuDAH7PSWT at concentrations of 0.2, 0.4, and 0.6 mg·ml−1, respectively. D–F represent the data for the complex with the MtuDAH7PSWT concentrations of 0.2, 0.4, and 0.6 mg·ml−1, respectively, with a 4-fold molar excess of MtuCM at each concentration. The experimental data are represented using solid circles, and the fits using the species analysis model are shown as solid curves.
Hetero-octameric Complex Formation Bestows Allosteric Functionality to MtuCM
MtuDAH7PS alone is allosterically inhibited by binary or, especially, ternary combinations of aromatic amino acids, whereas MtuCM alone shows no response (Fig. 5) (9, 15). The substantial enhancement of CM activity for MtuCM in the presence of MtuDAH7PS is also accompanied by the acquisition of sensitivity to aromatic amino acids (Fig. 5A) (15). In the presence of MtuDAH7PS, aromatic amino acids were shown to inhibit the CM activity of the MtuCM-MtuDAH7PS complex. Phe was the most potent inhibitor of CM activity, whereas Trp was not inhibitory. Inhibition by Phe or Tyr was also augmented by the presence of Trp; this is congruous with the cross-talk known to occur between the Trp and Phe binding sites for MtuDAH7PS (12).
FIGURE 5.

Inhibition of MtuCM and MtuDAH7PS activity by aromatic amino acids. A, effect of aromatic amino acids on MtuCM activity alone (3 μm MtuCM, 150 μm chorismate) (purple) and in complex with MtuDAH7PS (60 nm MtuCM) at chorismate concentrations of 150 μm (pink) or 25 μm (gray). B, effect of aromatic amino acids on MtuDAH7PSWT activity (40 nm MtuDAH7PS) either in complex with MtuCM in a 1:10, a 1:50 ratio to MtuDAH7PS (red), or alone (blue). The effect of changing the concentration of each ligand present to 100 μm (orange) (from 200 μm) is also shown. C, effect of aromatic amino acids on MtuCM activity (60 nm MtuCM) in the presence of MtuDAH7PSWT (gray), MtuDAH7PSR171A (red), or MtuDAH7PSR256A (green). D, effect of increasing aromatic amino acid concentration on MtuCM (60 nm) activity in complex with MtuDAH7PS. Chorismate concentrations were held at either 150 μm (solid lines) or 25 μm (dashed lines). For complexes with MtuDAH7PS, CM activity was determined with a 1:10 ratio for CM to DAH7PS, and DAH7PS activity was determined with a 10:1 ratio of CM to DAH7PS. The ligands are denoted by single-letter amino acid codes (W, Trp; F, Phe; and Y, Tyr) with each individual letter also representing a concentration of 200 μm. Error bars represent the estimated standard deviation of triplicate measurements.
The response of the MtuCM activity of the MtuCM-MtuDAH7PS complex to increasing concentrations of Phe, Trp and Tyr was also investigated (Fig. 5D). Phe was clearly the most potent inhibitor with EC50 values of 160 ± 40 and 30 ± 10 μm at high (150 μm) and low (25 μm) chorismate concentrations, respectively. Although it is noted that the CM activity did not reduce completely at the higher chorismate concentration, Trp and Tyr were found to be significantly less effective, particularly at the higher chorismate concentration, with EC50 values of 560 ± 40 μm and 2.54 ± 0.07 mm, respectively. When the chorismate concentration was reduced, the EC50 values for Trp and Tyr reduced to 310 ± 40 and 450 ± 20 μm for Trp, respectively.
Complex formation of MtuCM with MtuDAH7PS also results in some changes to the response of the DAH7PS activity to aromatic amino acids. Binary combinations Trp and Phe or Trp and Tyr or the presence of all three aromatic amino acids result in similar decreases in activity as is observed for MtuDAH7PS alone. Surprisingly, however, Trp alone and, to a lesser extent, Tyr or Phe alone, now activate the enzyme when MtuCM is present.
Phe Binding Results in an Increase in Chorismate Km and Weakens the Interaction between MtuCM and MtuDAH7PS
The inhibition of CM activity by Phe in the presence of MtuDAH7PSWT arises because of a significant increase in Km value for chorismate (Table 2). This increase is augmented by the inclusion of Tyr, and the Km value for chorismate when both Phe and Tyr are present approaches that of MtuCM in the absence of MtuDAH7PSWT. The addition of Trp to the kinetic assay resulted in no observable change in the CM catalytic efficiency.
TABLE 2.
Kinetic parameters for MtuCM activity in the presence of MtuDAH7PSWT alone or in combination with Phe, Tyr, or Trp
| Inhibitor (concentration) | kcat | Km | kcat/Km | Activation factor |
|---|---|---|---|---|
| s−1 | μm | mm−1 s−1 | ||
| None | 5.4 ± 0.1 | 52 ± 4 | 104 ± 9 | 1 |
| Phe (200 μm) | 6.3 ± 0.2 | 220 ± 20 | 29 ± 4 | 0.29 ± 0.06 |
| Phe (100 μm) and Tyr (100 μm) | 6.0 ± 0.3 | 340 ± 30 | 18 ± 2 | 0.17 ± 0.03 |
| Phe (200 μm) and Tyr (200 μm)a | 8.0 ± 0.3 | 480 ± 40 | 18 ± 2 | 0.16 ± 0.03 |
| Tyr (200 μm)a | 5.0 ± 0.1 | 84 ± 5 | 60 ± 5 | 0.57 ± 0.09 |
| Trp (200 μm) | 6.9 ± 0.2 | 66 ± 8 | 104 ± 10 | 1 ± 0.2 |
a Most assays used an MtuCM concentration of 30 nm and MtuDAH7PSWT concentration of 300 nm. For the indicated inhibitors, however, smaller 1-mm-path length cuvettes were used because of the high absorbance of ligands, which required higher enzyme concentrations of 120 nm MtuCM and 1200 nm MtuDAH7PSWT to detect activity. Kinetic analyses were conducted in 50 mm 1,3-bis[tris(hydroxymethyl)methylamino]propane buffer with 1 mm Tris-(2-carboxyethyl) phosphine hydrochloride, 100 μm MnSO4, and 150 μm PEP.
Phe was also shown to alter the affinity of MtuCM for MtuDAH7PS. In the absence of the aromatic amino acids, a Kd,app value of 300 ± 80 nm was calculated from activity measurements (Fig. 6A). The addition of Trp (200 μm) to the assay did not alter this affinity with a Kd,app of 300 ± 100 nm, and the addition of Tyr (200 μm) caused only a modest decrease in this affinity to 700 ± 200 nm. However, when Phe (200 μm) was present, the affinity decreased by almost a factor of 10 to 2.0 ± 0.6 μm under the same assay conditions. In addition, the maximal level of CM activity, reported as kcat/Km, was reduced by ∼70% in the presence of Phe, indicating that even when the complex is fully formed it has a significantly reduced CM activity.
FIGURE 6.

A, response of MtuCM catalytic efficiency (kcat/Km) to increasing concentrations of MtuDAH7PSWT in the absence of ligand (black dots) or in the presence of Phe (200 μm) (pink dots), Trp (200 μm) (purple dots), and Tyr (200 μm) (green dots). MtuCM concentration was held constant at a value of 10 nm. Error bars represent the spread of duplicate independent Michaelis-Menten kinetic measurements. B, complex formation between MtuCM and MtuDAH7PSWT, MtuDAH7PSR171A, and MtuDAH7PSR256A. Shown are the results of native PAGE (Amersham Biosciences ECL, using 50 mm Tris, 175 mm alanine running buffer, pH 9.2) of MtuDAH7PS, MtuDAH7PSR171A, and MtuDAH7PSR256A alone (lanes 1, 4, and 7, respectively), with 4-fold molar excess of MtuCM (lanes 2, 5, and 8, respectively), and with 4-fold molar excess of MtuCM and Phe (1 mm) (lanes 3, 6, and 9, respectively). Lanes marked M show the native molecular mass markers of stated masses.
Native PAGE provided additional evidence that Phe alters the interaction between MtuCM and MtuDAH7PS of the MtuCM-MtuDAH7PS complex in solution (Fig. 6B). When the two proteins are combined prior to conducting native PAGE, a band shift is observed relative to MtuDAH7PSWT alone, but in the presence of Phe (1 mm), the shifted band becomes very weak.
Analytical ultracentrifugation experiments also support a reduced affinity of the complex partners in presence of Phe. Sedimentation velocity experiments were performed on MtuDAH7PS, a 4:1 mixture of MtuCM-MtuDAH7PS, and 4:1 mixture of MtuCM-MtuDAH7PS in the presence of 100 μm Phe. The results demonstrate a shift in the sedimentation coefficient of the hetero-octameric complex to a smaller value (9.4 S) in the presence of Phe in comparison with the value of the hetero-octameric complex (10.1 S). This value is intermediate between that for the MtuDAH7PS tetramer and the value observed for the hetero-octamer (Fig. 7).
FIGURE 7.
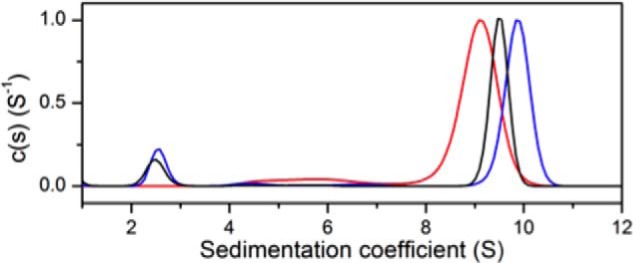
Normalized sedimentation coefficient distribution of species. Red, wild-type MtuDAH7PS; blue, MtuCM-MtuDAH7PS (molar ratio 4:1); black, MtuCM-MtuDAH7PS (molar ratio 4:1) in presence of 100 μm Phe. The concentration of MtuDAH7PSWT was 0.4 mg·ml−1 in all three experiments.
Variant MtuDAH7PS Proteins with Impaired Tyr and Phe Binding Activate MtuCM but Impair the Allosteric Functionality of the Complex
Variants of MtuDAH7PS, with mutations in the Phe (MtuDAH7PSR171A) or Tyr (MtuDAH7PSR256A) binding sites that disrupt Phe and Tyr binding, were examined for their ability to activate MtuCM (9). MtuCM was similarly activated by MtuDAH7PSWT or either of the variants (Table 1), indicating that these substitutions, at sites greater than 40 Å from the interface between the two enzymes, by themselves do not interfere with complex formation or CM activity activation. For all the enzymes, although the MtuCM activity was enhanced significantly, DAH7PS catalytic activity was only slightly enhanced by the presence of MtuCM (Table 1).
Whereas the activity of MtuCM was unaffected by complexation with either MtuDAH7PSR171A or MtuDAH7PSR256A, dramatic changes in CM activity in response to the aromatic amino acids were observed for this variant relative to wild-type enzyme (Fig. 5C). Inhibition by Phe was abolished for MtuCM activated by MtuDAH7PSR171A, revealing the importance of the Phe site of MtuDAH7PS for the allosteric response (Fig. 5C). Moreover, this complex was found to be unresponsive to any aromatic amino acid combination. In contrast, the CM activity of MtuCM-MtuDAH7PSR256A, which bears a mutation at the Tyr binding site of the DAH7PS protein, remained responsive to Phe but was unresponsive to Tyr. Furthermore, this complex was no longer able to display the synergistic response in combination of Phe with Tyr or Trp that was observed for the wild-type complex.
Native PAGE analysis was consistent with the findings of the kinetic analysis of the interaction between MtuCM and the MtuDAH7PS variants. Gels demonstrated that both MtuDAH7PS variants underwent a band shift when combined with MtuCM (Fig. 6B), indicative of complex formation. However, in the presence of Phe, the MtuCM-MtuDAH7PSR256A complex band becomes weaker, whereas the MtuCM-MtuDAH7PSR171A complex band is still observed, indicating that the very weak binding of Phe to the MtuDAH7PSR171A species alone pertains also to the MtuCM-MtuDAH7PSR171A complex.
Discussion
Our understanding of how the thousands of enzyme-catalyzed reactions within the cellular environment are interconnected and regulated to meet the overall needs of the cell is continuously developing. Cells were once thought to consist of a collection of soluble proteins, substrates, cofactors, and products within the cellular matrix that relied primarily on diffusion for the right components to come together and react. However, as our understanding of cellular organization advances, the number of examples of strategic use of compartmentalization, membrane-bound systems, and formation of multimolecular complexes by cells to organize and control the enzymes of particular pathways is growing (22, 23).
The studies outlined in this report establish that two enzymes involved in the important biosynthesis of aromatic amino acids in M. tuberculosis are functionally connected through the formation of a hetero-octameric complex. This complex activates both enzymes, especially MtuCM, and allows the multifaceted allosteric machinery that is housed on the MtuDAH7PS to be shared with its smaller complex partner. One enzyme of this partnership, DAH7PS, sits at the very start of the aromatic amino acid biosynthetic pathway; the other, CM, sits on the dedicated branch for Phe and Tyr biosynthesis. The DAH7PS-CM complex sits within a growing number of examples where protein-protein interactions have been observed to impact on catalytic regulatory properties of key metabolic enzymes (24). Similarly to the system detailed here, the cysteine regulatory complex formed by an interaction between the enzymes of cysteine biosynthesis improves catalytic efficiency and alters the inhibitory response to cysteine (25). Dynamic assemblies of enzymes for efficient and regulated metabolism have been observed in purine metabolism (26).
Interactions between CM and DAH7PS have been previously observed, and a significant boost of the MtuCM activity by MtuDAH7PS has been previously reported by Sasso et al. (15). Although these observations of the complex have been made from in vitro experiments, the in vivo importance of the interaction and activity boost has been recently exposed in an elegant directed evolution study where the activity of MtuCM and the activity boost afforded by MtuDAH7PS are reported by an E. coli selection system (16). Similarly, interactions between DAH7PS and CM are reported for pairings from a number of organisms including Corynebacterium glutamicum, Amycolaptoposis methanolica, and Brevibacterium flavum (27–30). For these latter two organisms, enhanced CM catalytic activity was reported. Modeling of the C. glutamicum CM and DAH7PS predicted a similar interaction to that observed for M. tuberculosis (31). However, in contrast, the C. glutamicum CM activity was not enhanced by interaction with the DAH7PS, yet the DAH7PS was significantly activated and showed sensitivity to prephenate not shown by the other DAH7PS. This endowed regulatory sensitivity to prephenate draws parallels to the DAH7PS from Bacillus subtilis, which is regulated by chorismate and prephenate via a covalently linked CM domain (32). The DAH7PS enzymes display a remarkably wide variety of allosteric mechanisms delivered by variable allosteric machinery appended to the core catalytic barrel (8). Of these, MtuDAH7PS appears to show the most sophisticated and elaborate allostery (9).
CM displays a range of architectures and regulatory patterns in different organisms. Two unrelated types of CM are known: the relatively rare and unregulated AroH, which has a trimeric pseudo-α/β barrel structure, and AroQ, which is a homodimeric α-helical protein. Both the MtuCM and its periplasmic counterpart are members of the AroQ group. Some AroQ proteins are found as bifunctional proteins linked to the subsequent enzymes in the pathway, prephenate dehydratase (with and without a discrete regulatory domain), or prephenate dehydrogenase, as well as to DAH7PS. Allosteric control of the enzymes linked to the two prephenate-utilizing enzymes is known, although the precise molecular details are not clear. On the other hand, for the yeast CM, the details of allostery are well studied. For this enzyme, a complex allosteric response involving activation by Trp and inhibition by Tyr is mediated by changes in the relative positioning of the two domains of the homodimeric structure. The allosteric sites are found at the interface, illustrating the importance of protein-protein interactions in determining the activity of a remote active site (33–35).
Tetrameric MtuDAH7PS displays a complex synergistic system of allostery, which utilizes discrete yet functionally connected binding sites for inhibitors Trp, Phe, or Tyr (9). Ligand binding is not associated with significant changes in conformation of the protein, and the control of DAH7PS activity appears to be primarily entropically driven, mediated through changes in the dynamic properties of the protein (12). Variant MtuDAH7PSR171A, which has impaired Phe binding at the Phe-selective site, activates MtuCM; however, unlike wild-type protein, the CM activity of the complex is insensitive to Phe, unequivocally illustrating that the Phe binding site of the DAH7PS protein is directly involved in the acquired allostery. This Phe-selective binding site is located at the N-terminal region of the MtuDAH7PS protein, over 50 Å from the closest point of interaction with MtuCM.
The association between MtuDAH7PS and MtuCM occurs across the tetramer interface of MtuDAH7PS (Fig. 1) (15). Whereas MtuDAH7PS does not directly contribute residues to the MtuCM catalytic site, several MtuCM residues, most notably from the loop between helix 1 and 2, are repositioned on complex formation. In addition, residues of the C-terminal tail have been observed to be important for activating complex formation (15), and, moreover, activation by MtuDAH7PS of MtuCM variants appears to be correlated with Phe and Tyr inhibition (16). These rearrangements enhance the maximal rate and significantly lower the Michaelis constant of MtuCM. Our observations demonstrate that the presence of Phe disrupts the activating interactions between the two enzymes, impairing the activity boost and weakening the complex.
How is the impact of allosteric Phe binding over 50 Å away on CM activity manifested? The allosteric effects on DAH7PS are thought to be primarily entropically driven, and remote allosteric binding sites directly communicate their occupancy; Phe binding improves Trp binding at the tetramer interface, and likewise Trp binding enhances Phe binding (12). A change in conformational dynamics may also explain the altered interactions between MtuCM and MtuDAH7PS mediated by Phe (and enhanced by Trp and Tyr). We note that the only clear structural change on Phe binding to MtuDAH7PS is a two-residue change in the register of the two-stranded antiparallel beta-sheet made up from a pair of N-terminal chains (10, 12). This small conformational change at the Phe binding site, which helps create the Tyr binding sites, does not noticeably alter the relative arrangement of the chains or perturb the relative planarity of the MtuDAH7PS tetramer, at least as observed in the absence of MtuCM, but may alter the conformational dynamics of the tetrameric species and interfere with the activating interactions between the CM and DAH7PS. In this regard, it is important to note that when Phe is present, even when MtuCM tends to full saturation by MtuDAH7PS (Fig. 6A), the activity is significantly less than without Phe present. This observation implies that the changes in the complex on Phe binding result in less efficient activation of CM catalytic efficiency, as well as destabilizing the protein-protein interface promoting complex dissociation.
Binding of MtuCM to MtuDAH7PS significantly activates DAH7PS, and this activity is further enhanced in the presence of Phe or Tyr, and most remarkably by Trp, where the DAH7PS activity is more than doubled. The CM activity of the complex, on the other hand, is unaffected by Trp. Mobile loops bearing residues 371–381 and 413–443 are not defined in the MtuCM-MtuDAH7PS complex, in contrast to MtuDAH7PS alone, and, moreover, the beginning and end points of these loops point to different conformations, which may be responsible for the 2-fold enhancement of DAH7PS activity and catalytic efficiency delivered by the interaction with MtuCM. The response to Trp is intriguing, because alone, MtuDAH7PS activity is slightly inhibited by Trp. Thus, in the presence of excess Trp, and low levels of Phe or Tyr, DAH7PS activity is enhanced, and chorismate is directed to prephenate and to synthesis of Tyr and Phe, adding a further level of sophistication to the regulatory system. The activation by Phe is especially intriguing, given the observation that Phe promotes dissociation of the complex the MtuDAH7PS forms with MtuCM. It appears that the altered interaction that reduces the ability of MtuDAH7PS to activate MtuCM also has an impact on the DAH7PS activation. Dynamic changes have previously been predicted to play an important role in the MtuDAH7PS catalytic activity (12).
In contrast, Phe and Tyr together exert a synergistic down-regulation of CM activity greater than for Phe or Tyr alone. This then provides the critical regulation of Phe and Tyr synthesis in M. tuberculosis that is largely missing in DAH7PS alone and illustrates how the allosteric sites are repurposed for delivering control of either entry into the shikimate pathway or to direct metabolic traffic at the branch point on exiting the pathway.
Essentially, MtuDAH7PS has currently five distinct allosteric binding sites: Phe, Tyr, Trp, E4P (demonstrating homotropic cooperativity) (10), and CM. Moreover, the functional response to occupation of each of these sites is determined by the occupancy of other sites creating an interconnected communication network that allows a highly sophisticated response to the levels of allosteric effectors. The results of this study, the activation of MtuCM by binding of MtuDAH7PS and the inhibitions and activations wrought on MtuCM, MtuDAH7PS, and MtuCM-MtuDAH7PS activities by Phe, Tyr, and Trp, singly or in binary and ternary combinations, are summarized qualitatively in the linkage diagram in Fig. 8. Fig. 8C also shows the full complexity of the linkages that exist among MtuDAH7PS, MtuCM, Phe, Tyr, and Trp, omitting the homotropic allosteric effector E4P (10). Fig. 8 (A and B) shows the relative effects of MtuCM in the MtuCM-MtuDAH7PS complex on the activity of MtuDAH7PS (mildly activating in the presence of Phe and Trp) and conversely of MtuDAH7PS on the activity of MtuCM (strongly activating). In addition, the activation data suggest that the Phe-bound complex represents yet another species with altered DAH7PS and CM kinetic properties. We are unable as yet to find an enzyme of comparable size that offers so rich an allosteric regulation.
FIGURE 8.

The complex allostery of the MtuCM-MtuDAH7PS complex. A, summary of interactions which alter DAH7PS or CM activity of the MtuCM-MtuDAH7P complex or the enzymes without their complex partner present. Inhibitory effects are shown in red, activating effects in blue, and the thickness of the lines indicates qualitatively the magnitude of the effect. B, the expanded allosteric response of the MtuCM-MtuDAH7PS complex. C, linkage diagram showing the complexity of allosteric regulation in MtuDAH7PS, MtuCM, and MtuCM-MtuDAH7PS. In the interests of clarity, the additional linkages associated with the homotropic allosteric effector E4P are not shown.
In conclusion, CM is located at a critical biosynthetic branch point where chorismate is committed to the biosynthesis of Tyr or Phe, and its regulation allows the production of these essential metabolites to be tuned to metabolic demand. Complex formation of MtuCM with DAH7PS not only activates MtuCM but also allows the enzyme to acquire allosteric sensitivity to the aromatic amino acids. This response means that the sophisticated allosteric machinery, which evolved for the complex control of entry into the pathway, has also been efficiently harnessed to direct metabolic traffic at this branch point.
The importance of the physical organization of metabolic enzymes has been evident for some time. Protein-protein interactions are vital to this organization and feature in a wide range of pathways and a variety of organisms (23). It is clear that these interactions can be highly sophisticated and are themselves altered by the metabolic environment as is illustrated here by the interaction between a CM and DAH7PS. The details of the role of protein-protein interactions in regulating metabolic responses are only now starting to be more fully understood.
Author Contributions
N. J. B., M. N. W., R. D. H., and A. R. N. carried out experiments. N. J. B., A. R. N., G. B. J., and E. J. P. analyzed the results. E. J. P., E. N. B., and G. B. J. conceived the study and devised the experiments. N. J. B., G. B. J., and E. J. P. wrote the paper. E. N. B., N. J. B., G. B. J., and E. J. P. revised the paper. All authors approved the final version of the manuscript.
Acknowledgments
We gratefully acknowledge the use of the analytical ultracentrifugation facility of the School of Biological Sciences, University of Canterbury, operated by Dr. Renwick Dobson.
This work was supported in part by the Maurice Wilkins Centre for Biomolecular Discovery, by New Zealand Marsden Fund Grant UOC1105, and by a scholarship from the Maurice Wilkins Centre and the University of Canterbury (to N. J. B.). The authors declare that they have no conflicts of interest with the contents of this article.
- used: DAH7PS
- 3-deoxy-d-arabino-heptulosonate 7-phosphate synthase
- CM
- chorismate mutase
- E4P
- d-erythrose 4-phosphate
- Mtu
- M. tuberculosis
- PEP
- phosphoenolpyruvate.
References
- 1. Nussinov R., Jang H. (2014) Dynamic multiprotein assemblies shape the spatial structure of cell signaling. Prog. Biophys. Mol. Biol. 116, 158–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Motlagh H. N., Wrabl J. O., Li J., Hilser V. J. (2014) The ensemble nature of allostery. Nature 508, 331–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gabizon R., Friedler A. (2014) Allosteric modulation of protein oligomerization: an emerging approach to drug design. Front. Chem. 2, 1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Perica T., Marsh J. A., Sousa F. L., Natan E., Colwell L. J., Ahnert S. E., Teichmann S. A. (2012) The emergence of protein complexes: quaternary structure, dynamics and allostery. Biochem. Soc. Trans. 40, 475–491 [DOI] [PubMed] [Google Scholar]
- 5. McConkey G. A., Pinney J. W., Westhead D. R., Plueckhahn K., Fitzpatrick T. B., Macheroux P., Kappes B. (2004) Annotating the Plasmodium genome and the enigma of the shikimate pathway. Trends Parasitol. 20, 60–65 [DOI] [PubMed] [Google Scholar]
- 6. Herrmann K. M., Weaver L. M. (1999) The shikimate pathway. Annu. Rev. Plant Physiol. Plant Mol. Biol. 50, 473–503 [DOI] [PubMed] [Google Scholar]
- 7. Parish T., Stoker N. G. (2002) The common aromatic amino acid biosynthesis pathway is essential in Mycobacterium tuberculosis. Microbiology 148, 3069–3077 [DOI] [PubMed] [Google Scholar]
- 8. Light S. H., Anderson W. F. (2013) The diversity of allosteric controls at the gateway to aromatic amino acid biosynthesis. Protein Sci. 22, 395–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Blackmore N. J., Reichau S., Jiao W., Hutton R. D., Baker E. N., Jameson G. B., Parker E. J. (2013) Three sites and you are out: ternary synergistic allostery controls aromatic amino acid biosynthesis in Mycobacterium tuberculosis. J. Mol. Biol. 425, 1582–1592 [DOI] [PubMed] [Google Scholar]
- 10. Webby C. J., Jiao W., Hutton R. D., Blackmore N. J., Baker H. M., Baker E. N., Jameson G. B., Parker E. J. (2010) Synergistic allostery, a sophisticated regulatory network for the control of aromatic amino acid biosynthesis in Mycobacterium tuberculosis. J. Biol. Chem. 285, 30567–30576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Webby C. J., Baker H. M., Lott J. S., Baker E. N., Parker E. J. (2005) The structure of 3-deoxy-d-arabino-heptulosonate 7-phosphate synthase from Mycobacterium tuberculosis reveals a common catalytic scaffold and ancestry for type I and type II enzymes. J. Mol. Biol. 354, 927–939 [DOI] [PubMed] [Google Scholar]
- 12. Jiao W., Hutton R. D., Cross P. J., Jameson G. B., Parker E. J. (2012) Dynamic cross-talk among remote binding sites: the molecular basis for unusual synergistic allostery. J. Mol. Biol. 415, 716–726 [DOI] [PubMed] [Google Scholar]
- 13. Kim S.-K., Reddy S. K., Nelson B. C., Robinson H., Reddy P. T., Ladner J. E. (2008) A comparative biochemical and structural analysis of the intracellular chorismate mutase (Rv0948c) from Mycobacterium tuberculosis H37R and the secreted chorismate mutase (y2828) from Yersinia pestis. FEBS J. 275, 4824–4835 [DOI] [PubMed] [Google Scholar]
- 14. Kim S. K., Reddy S. K., Nelson B. C., Vasquez G. B., Davis A., Howard A. J., Patterson S., Gilliland G. L., Ladner J. E., Reddy P. T. (2006) Biochemical and structural characterization of the secreted chorismate mutase (Rv1885c) from Mycobacterium tuberculosis H37Rv: an *AroQ enzyme not regulated by the aromatic amino acids. J. Bacteriol. 188, 8638–8648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sasso S., Okvist M., Roderer K., Gamper M., Codoni G., Krengel U., Kast P. (2009) Structure and function of a complex between chorismate mutase and DAHP synthase: efficiency boost for the junior partner. EMBO J. 28, 2128–2142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Roderer K., Neuenschwander M., Codoni G., Sasso S., Gamper M., Kast P. (2014) Functional mapping of protein-protein interactions in an enzyme complex by directed evolution. PLoS One 9, e116234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Webby C. J., Lott J. S., Baker H. M., Baker E. N., Parker E. J. (2005) Crystallization and preliminary x-ray crystallographic analysis of 3-deoxy-d-arabino-heptulosonate-7-phosphate synthase from Mycobacterium tuberculosis. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 61, 403–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Taira K., Benkovic S. J. (1988) Evaluation of the importance of hydrophobic interactions in drug-binding to dihydrofolate reductase. J. Med. Chem. 31, 129–137 [DOI] [PubMed] [Google Scholar]
- 19. Schuck P. (2000) Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys. J. 78, 1606–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lebowitz J., Lewis M. S., Schuck P. (2002) Modern analytical ultracentrifugation in protein science: a tutorial review. Protein Sci. 11, 2067–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vistica J., Dam J., Balbo A., Yikilmaz E., Mariuzza R. A., Rouault T. A., Schuck P. (2004) Sedimentation equilibrium analysis of protein interactions with global implicit mass conservation constraints and systematic noise decomposition. Anal. Biochem. 326, 234–256 [DOI] [PubMed] [Google Scholar]
- 22. Laursen T., Møller B. L., Bassard J. E. (2015) Plasticity of specialized metabolism as mediated by dynamic metabolons. Trends Plant Sci. 20, 20–32 [DOI] [PubMed] [Google Scholar]
- 23. Perkins J. R., Diboun I., Dessailly B. H., Lees J. G., Orengo C. (2010) Transient protein-protein interactions: structural, functional, and network properties. Structure 18, 1233–1243 [DOI] [PubMed] [Google Scholar]
- 24. Ouchi T., Tomita T., Horie A., Yoshida A., Takahashi K., Nishida H., Lassak K., Taka H., Mineki R., Fujimura T., Kosono S., Nishiyama C., Masui R., Kuramitsu S., Albers S. V., Kuzuyama T., Nishiyama M. (2013) Lysine and arginine biosyntheses mediated by a common carrier protein in Sulfolobus. Nat. Chem. Biol. 9, 277–283 [DOI] [PubMed] [Google Scholar]
- 25. Jez J. M., Dey S. (2013) The cysteine regulatory complex from plants and microbes: what was old is new again. Curr. Opin. Struct. Biol. 23, 302–310 [DOI] [PubMed] [Google Scholar]
- 26. An S., Kumar R., Sheets E. D., Benkovic S. J. (2008) Reversible compartmentalization of de novo purine biosynthetic complexes in living cells. Science 320, 103–106 [DOI] [PubMed] [Google Scholar]
- 27. Kloosterman H., Hessels G. I., Vrijbloed J. W., Euverink G. J., Dijkhuizen L. (2003) (De)regulation of key enzyme steps in the shikimate pathway and phenylalanine-specific pathway of the actinomycete Amycolatopsis methanolica. Microbiology 149, 3321–3330 [DOI] [PubMed] [Google Scholar]
- 28. Euverink G. J., Hessels G. I., Franke C., Dijkhuizen L. (1995) Chorismate mutase and 3-deoxy-d-arabino-heptulosonate 7-phosphate synthase of the methylotrophic actinomycete Amycolatopsis methanolica. Appl. Environ. Microbiol. 61, 3796–3803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sugimoto S., Shiio I. (1980) Purification and properties of dissociable chorismate mutase from Brevibacterium flavum. J. Biochem. 88, 167–176 [PubMed] [Google Scholar]
- 30. Sugimoto S., Shiio I. (1980) Purification and properties of bifunctional 3-deoxy-d-arabino-heptulosonate 7-phosphate synthetase-chorismate mutasse component A from Brevibacterium flavum. J. Biochem. 87, 881–890 [DOI] [PubMed] [Google Scholar]
- 31. Li P.-P., Li D.-F., Liu D., Liu Y.-M., Liu C., Liu S.-J. (2013) Interaction between DAHP synthase and chorismate mutase endows new regulation on DAHP synthase activity in Corynebacterium glutamicum. Appl. Microbiol. Biotechnol. 97, 10373–10380 [DOI] [PubMed] [Google Scholar]
- 32. Wu J., Woodard R. W. (2006) New insights into the evolutionary links relating to the 3-deoxy-d-arabino-heptulosonate 7-phosphate synthase subfamilies. J. Biol. Chem. 281, 4042–4048 [DOI] [PubMed] [Google Scholar]
- 33. Helmstaedt K., Krappmann S., Braus G. H. (2001) Allosteric regulation of catalytic activity: Escherichia coli aspartate transcarbamoylase versus yeast chorismate mutase. Microbiol. Mol. Biol. Rev. 65, 404–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Westfall C. S., Xu A., Jez J. M. (2014) Structural evolution of differential amino acid effector regulation in plant chorismate mutases. J. Biol. Chem. 289, 28619–28628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lin S. L., Xu D., Li A., Nussinov R. (1998) Electrostatics, allostery, and activity of the yeast chorismate mutase. Proteins Struct. Funct. Genet. 31, 445–452 [DOI] [PubMed] [Google Scholar]