

Background: Abnormal accumulation of glucosylceramide (GlcCer), a fundamental membranous glycolipid, causes numerous diseases.
Results: Activation of AMP-activated protein kinase (AMPK) decreases intracellular GlcCer levels and synthase activity.
Conclusion: AMPK negatively regulates GlcCer synthesis by decreasing UDP-glucose, a precursor of GlcCer.
Significance: AMPK activation may be a new target for treating diseases caused by the accumulation of GlcCer.
Keywords: AMP-activated kinase (AMPK), Gaucher disease, glycolipid, lipid, metformin, sphingolipid, glucosylceramide, glycosphingolipid, sugar nucleotide, UDP-glucose pyrophosphatase
Abstract
The membrane glycolipid glucosylceramide (GlcCer) plays a critical role in cellular homeostasis. Its intracellular levels are thought to be tightly regulated. How cells regulate GlcCer levels remains to be clarified. AMP-activated protein kinase (AMPK), which is a crucial cellular energy sensor, regulates glucose and lipid metabolism to maintain energy homeostasis. Here, we investigated whether AMPK affects GlcCer metabolism. AMPK activators (5-aminoimidazole-4-carboxamide 1-β-d-ribofuranoside and metformin) decreased intracellular GlcCer levels and synthase activity in mouse fibroblasts. AMPK inhibitors or AMPK siRNA reversed these effects, suggesting that GlcCer synthesis is negatively regulated by an AMPK-dependent mechanism. Although AMPK did not affect the phosphorylation or expression of GlcCer synthase, the amount of UDP-glucose, an activated form of glucose required for GlcCer synthesis, decreased under AMPK-activating conditions. Importantly, the UDP-glucose pyrophosphatase Nudt14, which degrades UDP-glucose, generating UMP and glucose 1-phosphate, was phosphorylated and activated by AMPK. On the other hand, suppression of Nudt14 by siRNA had little effect on UDP-glucose levels, indicating that mammalian cells have an alternative UDP-glucose pyrophosphatase that mainly contributes to the reduction of UDP-glucose under AMPK-activating conditions. Because AMPK activators are capable of reducing GlcCer levels in cells from Gaucher disease patients, our findings suggest that reducing GlcCer through AMPK activation may lead to a new strategy for treating diseases caused by abnormal accumulation of GlcCer.
Introduction
Glycosphingolipids (GSLs)2 are amphipathic compounds consisting of oligosaccharides and ceramide moieties. They are ubiquitous in the outer leaflet of the plasma membrane and believed to be involved in a large number of cellular processes, including signal transduction, membrane trafficking, cytoskeletal organization, and pathogen entry (1, 2). Because most mammalian GSLs are generated from glucosylceramide (GlcCer), GlcCer synthesis is an important step for determining cellular GSL levels. GlcCer is synthesized by GlcCer synthase (UDP-glucose:ceramide glucosyltransferase (UGCG), EC 2.4.1.80) from ceramide and uridine diphosphate-glucose (UDP-Glc) at the cytosolic surface of the Golgi apparatus (3).
GlcCer is a fundamental GSL found in organisms ranging from mammals to fungi. In vivo studies demonstrate that UGCG plays critical roles in development, differentiation, and energy homeostasis (4–7). For instance, ugcg knock-out mice die in utero, because gastrulation is blocked by ectodermal apoptosis (4, 8). The forebrain neuron-specific deletion of UGCG in mice results in obesity, hypothermia, and lower sympathetic activity (9). This study demonstrated that UGCG expression in neurons of the adult central nervous system (CNS) regulates the leptin signaling pathway and controls energy homeostasis at the whole-animal level.
On the other hand, abnormal accumulation of GlcCer and GSLs is known to be closely related to several diseases or disorders, such as Gaucher disease, Parkinson disease, insulin resistance, breast cancer, and polycystic kidney disease (PKD) (10–16). In ob/ob mice, an animal model of type II diabetes, GlcCer levels are increased in several tissues, such as liver and muscle. Interestingly, insulin sensitivity, glucose homeostasis, and adipocyte function are improved by treating the mice with an UGCG inhibitor (17, 18). The expression of ugcg mRNA is significantly increased in tumors of the breast, small intestine, cervix, and rectum compared with that in normal human tissues (11). ugcg overexpression is associated with drug resistance in several cancer cells and the maintenance of pluripotency in breast cancer stem cells (19, 20). Suppression of UGCG expression sensitizes cancer cells to anticancer agents (21).
Previous studies indicated that GlcCer levels should be strictly regulated to appropriate levels to maintain biological activities. How cells regulate the activity of UGCG is yet to be clarified. Elucidating the mechanisms underlying the regulation of UGCG may lead to new and more effective treatment options for diseases caused by the abnormal accumulation of GlcCer/GSLs.
The hydrolysis of ATP drives all energy-requiring processes in living cells. To maintain ATP at a sufficient level, eukaryotic cells have an important nutrient and energy sensor, AMP-activated protein kinase (AMPK) (22). AMPK is a heterotrimeric serine/threonine kinase that enhances signaling in ATP-generating pathways, such as glycolysis or fatty acid oxidation, while inhibiting anabolic processes, such as biosynthesis of fatty acids, cholesterol, glycogen, and triacylglycerol, under energy-reducing conditions (i.e. increasing AMP/ATP or ADP/ATP ratios). Because AMPK is closely involved in the metabolism of glucose and fatty acids, which are components of GlcCer, we expected that AMPK could also control GlcCer metabolism in mammalian cells. To test this possibility, we assessed the intracellular UGCG activity and cellular GlcCer levels under AMPK-activating conditions.
In the present study, we found that AMPK affects the GlcCer biosynthesis pathway. Intracellular GlcCer levels and UGCG activity were reduced by AMPK-activating drugs, such as 5-aminoimidazole-4-carboxamide 1-β-d-ribofuranoside (AICAR) and the anti-diabetic drug metformin. On the other hand, an AMPK inhibitor and AMPK siRNA overrode the reduced GlcCer synthase activity or cellular GlcCer levels under AMPK-activating conditions, indicating that AMPK is a negative regulator of GlcCer synthesis. The expression or phosphorylation levels of UGCG were unchanged under AMPK-activating and AMPK-inhibiting conditions. Instead, we found that cellular sugar nucleotides, including UDP-Glc, a precursor of GlcCer synthesis, were decreased by AMPK-activating compounds. In addition, we found that UDP-Glc-degrading enzyme, UDP-Glc pyrophosphatase Nudt14, is phosphorylated and activated by AMPK, which is partly, but significantly, involved in the reduction of UDP-Glc.
The present study provides mechanistic insights into the regulation of GlcCer synthesis by AMPK, by which UDP-Glc levels were decreased through the activation of UDP-Glc pyrophosphatase. Importantly, AMPK activators were capable of reducing cellular GlcCer levels of cells derived from patients with Gaucher disease. Our findings suggest that reduction of GlcCer via AMPK activation may serve as the basis for new treatment options for diseases caused by the accumulation of GlcCer.
Experimental Procedures
Materials
Ammonium formate, AICAR, 6-[4-(2-piperidin-1-ylethoxy)phenyl]-3-pyridin-4-ylpyrazolo[1,5-a]pyrimidine(Compound C (CC)), and anti-α-tubulin antibody were purchased from Sigma-Aldrich. Precoated silica gel 60 TLC plates were purchased from Merck. Metformin HCl was purchased from LKT Laboratories, Inc. (St. Paul, MN). Tetrabutylammonium hydrogen sulfate was purchased from Tokyo Chemical Industry Co., Ltd. Formic acid, methanol, chloroform, and acetonitrile were purchased from Nacalai Tesque. Pro-Q Diamond phosphoprotein gel stain, SYPRO® Ruby protein gel stain, 7-nitro-2,1,3-benzoxadiazole (NBD) C6-ceramide, NBD C6-ceramide conjugated to bovine serum albumin (BSA), anti-β-actin mouse antibody, Alexa Fluor 568 anti-mouse IgG, and Alexa Fluor 488 anti-rabbit IgG were purchased from Life Technologies, Inc. NBD C6-GlcCer was purchased from Matreya, LLC (State College, PA). Antibodies against DYKDDDDK (FLAG) tag, AMPKα1/2, phospho-AMPKα1/2 (Thr-172), ACC, phospho-ACC (Ser-79), phospho-(Ser/Thr) AMPK substrate, and LKB1 were purchased from Cell Signaling Technology (Beverly, MA). Nudt14 antibody and donkey anti-goat IgG HRP were obtained from Santa Cruz Biotechnology, Inc. GM130 antibody was purchased from BD Biosciences.
Construction of Expression Vectors
PCR was carried out by Phusion polymerase (Thermo Scientific) using mouse kidney cDNA (GenoStaff, Japan) as a template and primers listed in Table 1 for amplification of Nudt14 and UGP2. Phosphorylation site-deleted mutants, Nudt14 T141A and UGP2 S448A, were generated using mutation primers listed in Table 1 (Nudt14 T141A-S, Nudt14 T141A-A, UGP2 S448A-S, and S448A-A). The amplified products were inserted into HindIII-digested p3xFLAG-CMV-10 (Sigma-Aldrich) to generate N-terminal 3xFLAG-tagged proteins by using an In-Fusion HD cloning kit (Clontech). The expression vectors of constitutively active and dominant negative forms of AMPK α1 were prepared as described previously (23). C-terminal FLAG-tagged UGCG expression vector was a gift from Dr. Shun Watanabe (RIKEN).
TABLE 1.
Primers used in this study
| Primer name | Sequence (5′–3′) |
|---|---|
| 3xFLAG-UGP2-S | TGACGATGACAAGCTTTCGAGATTTGTACAAGATCTTAG |
| 3xFLAG-UGP2-A | TCGCGGCCGCAAGCTTTCAGTGATCCAGGATCCG |
| UGP2 S448A-S | AGGTTTGAAGCTATACCCGATATG |
| UGP2 S448A-A | CATATCGGGTATAGCTTCAAACC |
| 3xFLAG-Nudt14-S | TGACGATGACAAGCTTGAGCGCATCGACGGGGTGGCC |
| 3xFLAG-Nudt14-A | TCGCGGCCGCAAGCTTTCACTGGAGACTCAAATGGGGAACC |
| Nudt14 T141A-S | AGAGTTGCCGCATACATGTCTG |
| Nudt14 T141A-A | CAGACATGTATGCGGCAACTC |
Cell Cultures and Gene Transfection
NIH3T3 (3T3), human embryonic kidney 293 (HEK293), HeLa, Madin-Darby canine kidney (MDCK), mouse embryonic fibroblast (MEF), and Chinese hamster ovary (CHO) cells were cultured in Dulbecco's modified Eagle's medium (DMEM), containing 10% fetal bovine serum (FBS), 100 units/ml penicillin, and 100 μg/ml streptomycin at 37 °C in 5% CO2. The cell lines ugcg(lox/lox) MEF and ugcg(−/−) MEF, which were established from ugcg(lox/lox) MEF by expressing Cre recombinase, were kindly donated by Dr. Shun Watanabe (RIKEN) (24). Type 1, 2, and 3 fibroblasts derived from Gaucher disease patients (GM00372, GM00877, and GM00852, respectively) were purchased from the Coriell Institute. These fibroblast cells were cultured in Eagle's minimum essential medium with Earle's salts and non-essential amino acids, containing 15% FBS, 100 units/ml penicillin, and 100 μg/ml streptomycin at 37 °C in 5% CO2. Cells were transfected with expression vectors by using TurboFect transfection reagent (Thermo Scientific) according to the manufacturer's instructions.
Immunoblot Analysis
Cells were lysed in ice-cold lysis buffer (20 mm Tris-HCl buffer (pH 7.5), containing 150 mm NaCl, 1 mm EDTA, 1% Nonidet P-40, and phosphatase and protease inhibitor mixtures (Roche Diagnostics)) by sonication for 45 s. Cell debris was removed by centrifugation (18,000 × g for 10 min at 4 °C). The amount of protein in the supernatant was determined by bicinchoninic acid protein assay (Pierce), with BSA as a standard. For immunoprecipitation (IP) of FLAG-tagged protein, the protein extract prepared in lysis buffer was subjected to IP with anti-FLAG M2-agarose affinity gel (Sigma-Aldrich). Then the gel was washed with PBS three times, and the FLAG-tagged protein was eluted with lysis buffer containing 100 μg/ml of 3xFLAG peptide (Sigma-Aldrich). Equal amounts of protein were separated by SDS-PAGE using Mini-PROTEAN TGX Gel (Bio-Rad) and then blotted onto PVDF membranes by Trans Blot Turbo (Bio-Rad). The membranes were blocked in Blocking One-P solution (Nacalai Tesque) and incubated with Signal Enhancer HIKARI diluent (Nacalai Tesque) containing antibodies against AMPKα1/2 (1:2000), phospho-AMPKα1/2 (Thr-172) (1:2000), ACC (1:2000); phospho-ACC (Ser-79) (1:2000), LKB1 (1:3000); DYKDDDDK tag (1:6000), phospho-(Ser/Thr) AMPK substrate (1:1000), α-tubulin (1:20,000), Nudt14 (1:250), or β-actin (1:20,000). The blots were washed with TBS-T and then incubated with HRP-conjugated anti-mouse or anti-rabbit IgG antibody (1:10000) (Cell Signaling Technology) or anti-goat IgG antibody. The blots were washed again with TBS-T. The protein was detected by chemiluminescence using Luminate Forte Western HRP substrate (Millipore) and then analyzed with an LAS-3000 luminescence image analyzer (Fujifilm). To avoid dephosphorylation of the AMPK α subunit and ACC, cells treated with or without AMPK-activating compounds were immediately frozen with liquid nitrogen after incubation.
Immunostaining
To determine the subcellular localization of Nudt14 or Nudt14 T141A, 3T3 cells were transfected with plasmids encoding FLAG-Nudt14 or FLAG-Nudt14 T141A. After transfection, cells were treated with AICAR (1 mm) or metformin (8 mm), fixed in 3% paraformaldehyde for 15 min, and then washed with PBS. The fixed cells were blocked in Blocking One Histo (Nacalai Tesque) and incubated with antibodies against FLAG tag (1:500) and GM130 (1:500), a Golgi marker, for 12 h at 4 °C. After incubation, cells were washed with PBS and incubated with Alexa Fluor 568 anti-mouse IgG (1:500) and Alexa Fluor 488 anti-rabbit IgG (1:500) for 1 h. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole, dihydrochloride (DAPI) for 5 min. Cells were mounted in Immu-Mount reagent (Thermo Scientific) and observed under a confocal laser-scanning microscope (FV1000, Olympus).
Measurement of Intracellular UGCG and Glucosylceramidase Activity
The assays were performed as described previously with modification (25). Cells grown in 24-well plates containing DMEM plus 10% FBS were exposed to AICAR, metformin, CC, or AMPK α1/2 siRNA for an appropriate period. The culture medium was switched to 250 μl of DMEM plus 10% FBS containing 0.5 μm NBD C6-ceramide conjugated to BSA (Invitrogen). After 90 min of incubation at 37 °C, cells were rinsed with ice-cold Dulbecco's PBS (Nacalai Tesque), scraped from the wells using 50 μl of ice-cold water, and transferred to 1.5-ml tubes. Lipids were extracted by the addition of 190 μl of chloroform/methanol (1:2, v/v). After incubation at room temperature for 10 min, 62.5 μl of water and chloroform were added, and then the tubes were centrifuged at 12,000 × g for 5 min. The lower phase was collected and dried with a SpeedVac concentrator. The dried sample was dissolved in 20 μl of chloroform/methanol (1:2, v/v), and 3 μl of the sample was applied onto a TLC plate, which was developed with chloroform/methanol/water (65:25:4, v/v/v). The reaction products were visualized with an LAS-3000 image analyzer equipped with a blue light-emitting diode (460-nm EPI) and a Y515-Di filter and then quantified with ImageJ version 1.43u software (National Institutes of Health). The extent of synthesis of NBD-labeled GlcCer was calculated with the equation, synthesis (%) = (peak area for NBD C6-GlcCer) × 100/(peak area for NBD C6-ceramide + peak area for NBD C6-GlcCer + peak area for NBD C6-sphingomyelin). Under this experimental condition, NBD fatty acid, which is generated from ceramide by ceramidase (26), was not detected. To measure intracellular glucosylceramidase (GCase) activity, we used BSA-conjugated NBD C6-GlcCer, which was prepared as described previously (27), instead of NBD C6-ceramide. The extent of degradation of NBD-labeled GlcCer was calculated with the equation, GCase activity (%) = (peak area for NBD C6-ceramide) × 100/(peak area for NBD C6-ceramide + peak area for NBD C6-GlcCer).
Lipid Extraction and Quantification of Sphingolipids by LC-ESI MS/MS
Cells were washed once with cold PBS, collected in 100 μl of cold PBS, and sonicated with a Handy Sonic Disruptor (Tomy Seiko Co.) for 10 s. Part of the sample (5 μl) was used in the bicinchoninic acid protein assay to determine the amount of protein. Total lipids were extracted by adding 375 μl of chloroform/methanol (1:2, v/v) containing 62.5 pmol of each component of Ceramide/Sphingoid Internal Standard Mixture II (Avanti Polar Lipids, Inc.). The single-phase mixture was incubated at 48 °C overnight. After cooling, 125 μl of water and chloroform were added. The lower phase was collected and dried with a SpeedVac concentrator. The dried sample was suspended in chloroform/methanol (2:1, v/v) containing 0.1 n KOH, incubated for 2 h at 37 °C, and neutralized with an equal amount of acetic acid, and then water was added. The lower phase was withdrawn and dried and then resuspended in 200 μl of methanol, sonicated for 10 s, and centrifuged at 14,000 × g for 5 min. The supernatant was transferred to autoinjector vials. The amounts of GlcCer, LacCer, Cer, and sphingomyelin were analyzed by LC-ESI MS using a triple quadruple mass spectrometer 4000Q TRAP (AB SCIEX) coupled to an Agilent 1100 series HPLC system (Agilent). A binary solvent gradient with a flow rate of 0.2 ml/min was used to separate sphingolipids by normal phase chromatography using an InertSustain NH2 column (2.1 × 150 mm, 5-μm bead size, GL Science, Japan). The gradient was started at 20% buffer B (methanol/water/formic acid (89:9:1, v/v/v) with 20 mm ammonium formate) in buffer A (acetonitrile/methanol/formic acid (97:2:1, v/v/v) with 5 mm ammonium formate). The gradient reached 100% B in 4 min and was maintained at 100% B for 2 min. Finally, the gradient was returned to the starting conditions, and the column was equilibrated for 5 min before the next run. Sphingolipids containing C16:0, C18:0, C20:0, C22:0, C24:0, and C24:1 fatty acids were detected using a multiple-reaction-monitoring method, as described previously (28).
RNA Interference
Stealth RNAi duplexes (Life Technologies) were designed to mouse AMPK α1 (5′-CCAGGUCAUCAGUACACCAUCUGAU-3′) and α2 (5′- AAGUGAAGAUUGGAGAACACCAAUU) using the BLOCK-iT RNAi Designer. Predesigned Stealth RNAi siRNA (MSS287847, Life Technologies) was used to knock down Nudt14. Stealth RNAi siRNA Negative Control Med GC or Low GC were used as control siRNAs for knockdown of AMPK α subunits and Nudt14. The reverse transfection method was used to transfect Stealth RNAi into 3T3 cells. Stealth RNAi duplexes (30 pmol) were mixed with 5 μl of Lipofectamine RNAiMAX reagent (Life Technologies) in 500 μl of Opti-MEM I medium (Gibco) in the wells of 6-well plates and incubated for 20 min at room temperature. Cells in complete growth medium were seeded into 6-well plates at 150,000 cells/well and incubated for 48 h, and then the medium was replaced with flesh medium.
Quantification of Nucleotide Sugars
The quantification method was performed as described previously with some modifications (29). Cells were washed with cold PBS, collected in 300 μl of cold PBS, and spiked with GDP-Glc as an internal standard and then sonicated with a Handy Sonic Disruptor (Tomy Seiko Co.) for 10 s. Part of the sample (5 μl) was used in the bicinchoninic acid protein assay. Ice-cold ethanol (900 μl) was added to the sample. The extract was centrifuged at 16,000 × g for 10 min at 4 °C, and then the supernatant was lyophilized. The freeze-dried sample was applied to an Envi-Carb column (Supelco Inc.) and purified as described previously (29). After purification, nucleotide sugars were separated and detected by ion pair reverse-phase HPLC using an Inertsil ODS-3 column (particle size = 3 μm, 4.6 × 150 mm; GL Science, Tokyo, Japan). For HPLC analysis, buffer C (100 mm potassium phosphate buffer (pH 6.4), supplemented with 8 mm tetrabutylammonium hydrogen sulfate) and buffer D (70% buffer C with 30% acetonitrile) were used. The gradient was started at 0% buffer D and reached 77% buffer D in 22 min and 100% buffer D in 23 min and maintained at 100% buffer D for 14 min at a flow rate of 1.0 ml/min. Nucleotide sugars were detected with a Hitachi Elite LaChrom UV detector L-2400 (Hitachi, Tokyo, Japan) set to a wavelength of 254 mm.
Measurement of Nudt14 Activity
An aliquot of 50 nmol of UDP-Glc was incubated at 37 °C for 2 h with a suitable amount of cell lysates derived from mock-, Nudt14-, or Nudt14 T141A-transfected HEK293 cells in 50 μl of 50 mm Tris-HCl buffer (pH 8), containing 10 mm MgCl2. After incubation, reaction mixtures were neutralized by adding 50 μl of 1 m Tris-HCl, pH 6.5. The enzymatic reaction was stopped by adding 100 μl of MeOH, the sample was centrifuged (13,000 × g for 3 min), and the supernatant was transferred to autoinjector vials. The enzymatic product (UMP) and substrate (UDP-Glc) were separated and detected by ion pair reverse-phase HPLC with an Inertsil ODS-3 column using an isocratic mobile phase (70% buffer C with 30% buffer D) at a flow rate of 1.0 ml/min.
Quantitative Real-time PCR
Total RNA was extracted from cells using TRIzol reagent (Invitrogen) according to the manufacturer's instructions. Two micrograms of total RNA were used to obtain cDNA via MultiScribe Reverse Transcriptase (Applied Biosystems). Real-time PCR was performed using an ABI7900HT real-time PCR system (Applied Biosystems). The expression of the ugcg gene was analyzed using specific predesigned TaqMan probes (Applied Biosystems) and normalized against gapdh expression as a standard.
Statistical Analysis
All statistical analyses used unpaired two-tailed Student's t tests, and all data were expressed as means and S.D. values from at least three experiments. Statistical significance in the figures is indicated as follows: *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Results
Intracellular UGCG Activity and Sphingolipid Content in AMPK-activating Cells
To activate intracellular AMPK, we used two representative AMPK activators, metformin and AICAR. The AMPK α subunit is phosphorylated and activated by upstream kinase liver kinase B1 (LKB1) in response to metformin and AICAR (30). We consistently observed increased phosphorylation of the AMPK α subunit and acetyl-CoA carboxylase (ACC), a representative substrate of AMPK, after metformin and AICAR treatment (Fig. 1A). To better understand the effect of AMPK activation on intracellular GlcCer metabolism, we performed a cell-based GlcCer synthase assay. This was accomplished by using a cell-permeable fluorescent ceramide, which is incorporated into GlcCer and sphingomyelin (25). Although fluorescent GlcCer was detected in control MEF cells, no corresponding product was observed in ugcg(−/−) MEF cells (Fig. 1B). This result indicates that incorporated fluorescent ceramide was converted to GlcCer by UGCG and that other hexosylated ceramides, such as GalCer, were not synthesized in MEF cells. Therefore, to determine intracellular UGCG activity in this study, we primarily used 3T3 cells, an MEF-derived cell line (Fig. 1B).
FIGURE 1.

Cellular UGCG activity and sphingolipid levels under AMPK-activating conditions. A, immunoblot analysis showing protein expression and phosphorylation levels of AMPK α subunit and ACC in 3T3 cells. After treatment with or without metformin (8 mm) or AICAR (1 mm) for 8 h, proteins were extracted and subjected to immunoblotting with antibodies against P-S79 ACC, ACC, P-T172 AMPK α1/2, AMPK α1/2, and β-actin. B, TLC analysis showing intracellular UGCG and sphingomyelin synthase activities. Lipids were extracted from ugcg(lox/lox) MEF, ugcg(−/−) MEF, and 3T3 cells that were treated without (−) or with (+) NBD C6-ceramide conjugated to BSA for 1 h, and then they were applied to TLC developed with a solution of chloroform/methanol/water (65:25:4, v/v/v). The reaction products were visualized with an LAS-3000 image analyzer. C, effects of metformin and AICAR on intracellular UGCG and sphingomyelin synthase (SMS) activities. After treatment with several concentrations of metformin and AICAR for 8 h, NBD C6-ceramide conjugated to BSA was added to cells and incubated for 90 min and then analyzed by TLC as described above. D, quantification of cellular GlcCer, LacCer, ceramide, and sphingomyelin levels under AMPK-activating conditions by LC-ESI MS analysis. The bar graphs show the amount of cellular GlcCer (top left), LacCer (top right), ceramide (bottom left), and sphingomyelin (bottom right). Lipids were extracted from cells treated with metformin (8 mm) or AICAR (1 mm) for 16 h, and then they were applied to LC-ESI MS, as described under “Experimental Procedures.” Bars, means ± S.D. (error bars) of at least three experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
The cell-based UGCG assay revealed that intracellular UGCG activity decreased in response to metformin and AICAR in a concentration-dependent manner (Fig. 1C). On the other hand, sphingomyelin synthase activity was unchanged under AMPK-activating conditions (Fig. 1C). Next, we measured sphingolipid content of cells under AMPK-activating conditions by using LC-ESI MS/MS analysis (28). In agreement with the reduced enzymatic activity, the amount of GlcCer and LacCer, which is synthesized from GlcCer and UDP-galactose (UDP-Gal), was significantly decreased after metformin and AICAR treatment (Fig. 1D). By contrast, cellular sphingomyelin levels were hardly affected by either AMPK activator (Fig. 1D). On the other hand, ceramide levels were decreased by the AMPK activators, especially by AICAR treatment (Fig. 1D). AMPK phosphorylates and inhibits ACC, which is the late limiting enzyme for fatty acid synthesis, to promote fatty acid oxidation and suppress fatty acid synthesis, leading to a reduction of cellular fatty acid levels. Ceramide is composed of a sphingoid base and a fatty acid; therefore, the reduction of ceramide is probably due to the reduction of its components under AMPK-activating conditions. Taken together, these results suggest that the activity of UGCG is decreased under AMPK-activating conditions, leading to a reduction of intracellular GlcCer and LacCer levels.
Effects of an AMPK Inhibitor and RNAi on GlcCer Levels and UGCG Activity
In contrast to intracellular GlcCer synthase activity (Fig. 1C), in vitro UGCG activity that was measured using cell lysates was unaffected by adding several concentrations of metformin and AICAR (Fig. 2A), suggesting that the effect of these compounds on UGCG activity was AMPK-dependent but indirectly so. To verify whether the reduction of UGCG activity and GlcCer levels by AMPK-activating compounds is dependent on AMPK, we examined the effect of an AMPK inhibitor and AMPK siRNA on GlcCer synthesis. AMPK and ACC phosphorylation levels in AICAR-treated cells were restored to control levels by adding CC, an AMPK inhibitor (Fig. 2B). Importantly, CC failed to affect in vitro UGCG activity, indicating that CC itself did not affect UGCG activity (Fig. 2A). On the other hand, the decreased intracellular UGCG activity in AICAR-treated cells was restored to control levels by adding CC (Fig. 2B). Consistent with this result, the decreased GlcCer and LacCer levels in AMPK-activating cells were restored by CC (Fig. 2C).
FIGURE 2.

Effect of AMPK inhibitor on UGCG activity and GlcCer levels. A, effect of metformin, AICAR, and CC on in vitro UGCG activity. In vitro UGCG activity was measured by using fluorescent ceramide as described previously (55). Several concentrations of metformin, AICAR, and CC were added to the reaction mixture. B, immunoblot analysis showing the effect of AICAR and CC on AMPK α subunit and ACC phosphorylation levels. After incubation with AICAR (1 mm) or AICAR (1 mm) plus CC (10 μm) for 3 h, proteins were extracted from 3T3 cells and subjected to immunoblot with antibodies against P-S79 ACC, ACC, P-T172 AMPK α1/2, AMPK α1/2, and β-actin. C, effect of CC-associated AMPK inhibition on intracellular UGCG activity of AICAR-treated cells. After treatment with AICAR or AICAR plus several different concentrations of CC for 8 h, NBD C6-ceramide conjugated with BSA was added to cells and incubated for 90 min and then analyzed by TLC. D, effect of CC-associated AMPK inhibition on cellular GlcCer and LacCer levels in AICAR-treated cells. Lipids were extracted from cells treated with AICAR (1 mm) or AICAR (1 mm) plus CC (40 μm) for 8 h and then applied to LC-ESI MS as described under “Experimental Procedures.” Bars, means ± S.D. (error bars) of at least three experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Next, we performed an siRNA-mediated knockdown experiment to verify the results obtained with the AMPK inhibitor. AMPK α1 siRNA reduced the expression and phosphorylation levels of the AMPK α1 subunit, indicating that siRNA-mediated knockdown suppressed AMPK (Fig. 3A). Because siRNA of the AMPK α2 subunit failed to affect the protein expression levels of the AMPK α subunit (Fig. 3A), we concluded that the cells used in this experiment predominantly express the α1 subunit. In agreement with the results of the AMPK inhibitor experiment, knockdown of the AMPK α1 subunit restored intracellular UGCG activity in AICAR-treated cells (Fig. 3B). GlcCer and LacCer levels in AICAR-treated cells were also restored by knockdown of the AMPK α1 subunit (Fig. 3, C and D).
FIGURE 3.

Effect of AMPK α subunit knockdown on UGCG activity and GlcCer levels. A, immunoblot analysis showing the expression and phosphorylation levels of AMPK α subunit in siRNA-mediated AMPK α subunit-disrupted cells. Short interfering RNAs against AMPK α1 or α2 subunits were transfected into 3T3 cells and incubated for 72 h, and then AMPK α1-attenuated cells were treated with (−) or without (+) AICAR for 3 h. B, intracellular UGCG activity of AMPK α1 subunit-attenuated cells. After treatment with AMPK α1 siRNA, cells were incubated with (+) or without (−) AICAR (1 mm) for 8 h, and then NBD C6-ceramide conjugated with BSA was added to cells and incubated for 90 min and analyzed by TLC. C, quantification of cellular GlcCer and LacCer levels of AMPK-attenuated cells. Lipids were extracted from cells treated with (+) or without (−) AICAR (1 mm) for 8 h and then applied to LC-ESI MS, as described under “Experimental Procedures.” Bars, means ± S.D. (error bars) of at least three experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
In addition to the inhibitor and knockdown experiments, we used LKB1-deficient HeLa cells (31) to verify the dependence of the AMPK pathway on UGCG activity and cellular glycolipid levels. LKB1 is the upstream kinase necessary for the AICAR- or metformin-mediated activation of AMPK (32). As expected, no changes in phosphorylation levels of AMPK α subunit and ACC were detected in AICAR-treated HeLa cells, in which LKB1 expression was not detected (Fig. 4, A and B). By contrast, phosphorylation of AMPK α subunit and ACC were enhanced by AICAR treatment in 3T3 cells that express LKB1 (Fig. 4, A and B). Consistent with these results, AICAR treatment failed to decrease intracellular UGCG activity in HeLa cells but did decrease it in 3T3 cells (Fig. 4C). Moreover, AICAR treatment did not affect the intracellular GlcCer and LacCer levels in HeLa cells (Fig. 4D). These results demonstrate that intracellular UGCG activity is regulated by the LKB1-AMPK pathway.
FIGURE 4.

Contribution of LKB1, an upstream kinase of the AMPK pathway, on UGCG activity and GlcCer levels. A, immunoblot analysis showing the protein expression and phosphorylation levels of AMPK α subunit and ACC in 3T3 and HeLa cells. After treatment with AICAR (1 or 2 mm), proteins were extracted from the cells and subjected to immunoblot with antibodies against phospho-Ser-79 (P-S79) ACC, ACC, phospho-Thr-172 (P-T172) AMPK α1/2, AMPK α1/2, and β-actin. B, immunoblot analysis showing a deficiency of LKB1 in HeLa cells. C, intracellular UGCG activity of LKB1-deficient cells. HeLa and 3T3 cells were incubated with several different concentrations of AICAR for 6 h, and then NBD C6-ceramide conjugated with BSA was added to the cells and incubated for 90 min. Fluorescent sphingolipids were extracted from cells and then analyzed by TLC. D, quantification of cellular GlcCer and LacCer levels of LKB1-deficient cells. Lipids were extracted from HeLa cells treated with metformin (8 mm) or AICAR (1 mm) for 16 h and then applied to LC-ESI MS as described under “Experimental Procedures.” Bars, means ± S.D. (error bars) of at least three experiments.*, p < 0.05; ***, p < 0.001.
Intracellular Sugar Nucleotide Content in AMPK-activated Cells
Although no AMPK phosphorylation sites were found in the UGCG amino acid sequence, we examined changes in UGCG phosphorylation levels by using a ProQ Diamond phosphoprotein gel stain reagent to clarify the mechanism underlying AMPK-mediated reduction of UGCG activity. As expected, phosphorylated UGCG was not detected in AICAR-treated cells (Fig. 4A). UGCG protein expression levels in AICAR- or CC-treated cells was nearly identical to that of control cells (Fig. 4A). In addition, quantitative real-time PCR revealed that AICAR or CC treatment did not affect UGCG RNA expression levels (Fig. 4B). Taken together, these results suggest that UGCG is not the direct substrate for AMPK and that UGCG activity is not regulated by its protein or mRNA expression levels or modification of phosphorylation under AMPK-activating conditions.
Cellular GlcCer levels are controlled not only by UGCG but also by GCase that degrades GlcCer, resulting in the generation of glucose and ceramide. In mammals, GlcCer is mainly catabolized by lysosomal GCase, also called acid β-glucosidase 1 (GBA1) (33) and endoplasmic reticulum/Golgi-localized non-lysosomal GCase, GBA2 (34–36). We confirmed that intracellular GCase activity, which might reflect the activity of GBA1 and/or GBA2, was not affected by AICAR or CC treatment (Fig. 5C). A deficiency in GBA1 activity due to biallelic mutations in the Gba1 gene leads to the most common inherited sphingolipidosis, Gaucher disease (10). We found that intracellular UGCG activity and GlcCer levels were still reduced by AICAR or metformin treatment in Gaucher disease patient-derived fibroblasts (Fig. 5, D and E). These results suggested that reduction of GlcCer by AMPK was caused by changes in the activity of UGCG, not by GCase. In fact, no AMPK phosphorylation sites were found in the amino acid sequences of GBA1 and GBA2, indicating that GCase activity may not be regulated by AMPK.
FIGURE 5.

Exploration of the mechanisms by which AMPK affects UGCG activity. A, examination of phosphorylation of UGCG. CHO cells expressing C-terminal FLAG-tagged UGCG were incubated with AICAR or CC for 3 h. The recombinant UGCG was immunoprecipitated from cell lysates by FLAG antibody and then subjected to immunoblot analysis and stained with ProQ Diamond, which detects phosphorylated proteins. B, expression levels of ugcg mRNA in AICAR- or CC-treated cells. The mRNA was extracted from 3T3 cells after treatment with AICAR or CC for 3 h and then quantified as described under “Experimental Procedures.” C, effects of AICAR and CC on intracellular GCase activity. After treatment with AICAR or CC, NBD C6-GlcCer conjugated to BSA was added to cells and incubated, and then NBD C6-ceramide released from GlcCer was measured by TLC. D, effects of metformin and AICAR on intracellular UGCG activity of fibroblasts derived from Gaucher disease patients. Fibroblasts derived from type 1, 2, and 3 Gaucher disease patients were incubated with metformin or AICAR for 8 h and then were incubated with C6-NBD Cer conjugated with BSA for 90 min. Fluorescent sphingolipids were extracted from the cells and then analyzed by TLC. E, quantification of cellular GlcCer levels of fibroblasts derived from a Gaucher disease patient (type 2). Lipids were extracted from fibroblasts treated with metformin (8 mm) or AICAR (1 mm) for 16 h and were then subjected to LC-ESI MS, as described under “Experimental Procedures.” F, sugar nucleotide levels in metformin- or AICAR-treated cells. Sugar nucleotides were extracted from 3T3 cells treated with metformin (8 mm) or AICAR (1 mm) for 8 h and then subjected to ion pair reverse-phase HPLC, as described under “Experimental Procedures.” G, effect of CC on sugar nucleotide levels in AICAR-treated cells. Sugar nucleotides were extracted from 3T3 cells treated with AICAR (1 mm) or AICAR (1 mm) plus CC (20 μm) for 8 h. Bars, means ± S.D. (error bars) of at least three experiments. **, p < 0.01; ***, p < 0.001.
It should be noted that exogenous fluorescent ceramide was used for determining intracellular UGCG activity; thus, the level of endogenous ceramide could be excluded as a reason for the reduction in UGCG activity. In addition, in contrast to GlcCer, ceramide levels changed little following metformin treatment (Fig. 1D). Therefore, we speculated that the amount of cellular UDP-Glc, the precursor of GlcCer, might be changed under AMPK-activating conditions, thereby affecting cellular GlcCer levels. Ion pair reverse-phase HPLC was used to determine sugar nucleotide levels under AMPK-activating conditions. AICAR and metformin significantly decreased sugar nucleotide levels, including UDP-Glc and UDP-Gal (Fig. 5F). Interestingly, the reduction of sugar nucleotide levels in AICAR-treated cells could be blocked with CC (Fig. 5G), suggesting that activation of AMPK decreased UDP-Glc levels, leading to the suppression of intracellular UGCG activity and GlcCer synthesis.
Phosphorylation of Sugar Nucleotide-metabolizing Enzymes by AMPK
UDP-Glc is synthesized from glucose 1-phosphate and UTP by UDP-Glc pyrophosphorylase UGP2 (EC 2.7.7.9) (37) and is degraded by UDP-Glc pyrophosphatase Nudt14 (EC 3.6.1.45) to release glucose 1-phosphate and UMP (38, 39) (Fig. 6A). We hypothesized that intracellular UDP-Glc levels are probably regulated by UGP2 and/or Nudt14 and that AMPK may phosphorylate one or both enzymes to regulate their activities. Interestingly, we found that UGP2 and Nudt14 contain the consensus phosphorylation motif of AMPK (40, 41), which is well conserved in higher eukaryotes. However, in Nudt14, some of the amino acids in this motif are replaced with similar amino acids (Fig. 6, B and C). In the case of humans and rodents, Ser-448 in UGP2 and Thr-141 in Nudt14 were predicted to be AMPK phosphorylation sites (Fig. 6, B and C). To address the possibility that AMPK phosphorylates these UDP-Glc-metabolizing enzymes, we first examined whether we could detect phosphorylation of UGP2 or Nudt14 by using an antibody against the phospho-(Ser/Thr) AMPK substrate that recognizes proteins bearing the LXRXX(pS/pT) motif (where pS and pT represent phosphoserine and -threonine, respectively). FLAG-tagged UGP2 or Nudt14 was expressed in HEK293 cells, which were treated with or without metformin. Although the phospho-(Ser/Thr) AMPK substrate antibody recognized both UGP2 and Nudt14 immunoprecipitated by FLAG antibody from HEK293 cell lysates, UGP2 phosphorylation levels remained unchanged following metformin treatment (Fig. 6D). Because the replacement of Ser-448 by Ala (S448A) reduced UGP2 phosphorylation levels, Ser-448 was identified as a phosphorylation site. However, it seems that AMPK is not the kinase responsible for phosphorylating UGP2 (Fig. 6D). Metformin treatment markedly enhanced Nudt14 phosphorylation levels, suggesting that Nudt14 is a direct substrate of AMPK (Fig. 6E). To identify the phosphorylation site of Nudt14, Thr-141 of Nudt14 was replaced with Ala (T141A) via site-directed mutagenesis. Notably, replacing T141A completely abolished phosphorylated Nudt14, indicating that Thr-141 of Nudt14 is the phosphorylation site for AMPK (Fig. 6E).
FIGURE 6.

Phosphorylation of UDP-Glc pyrophosphatase Nudt14 by AMPK. A, enzymes involved in the synthesis and degradation of UDP-Glc. UDP-Glc was synthesized by UGP2 from UTP and glucose 1-phosphate (Glc-1P) and degraded by Nudt14 to generate UMP and glucose 1-phosphate. B, alignment of UGP2 amino acid sequences from various animals. Ser-448 (S448) was predicted as a potential phosphorylation site for AMPK. C, alignment of amino acid sequences of Nudt14 from various animals. Thr-141 (T141) was predicted as a potential phosphorylation site for AMPK of human or rodent Nudt14. D and E, immunoblot analysis showing the phosphorylation of UGP2 and Nudt14. HEK293 cells transfected with mock-, FLAG-UGP2-, FLAG-UGP2 S448A-, FLAG-Nudt14-, or FLAG-Nudt14 T141A-expressing vectors were incubated with (+) or without (−) metformin. Cell lysates and immunoprecipitates resulting from incubation with anti-FLAG antibody were subjected to immunoblotting with the indicated antibodies. Anti-phospho-AMPK substrate antibody recognizes proteins bearing the LXRXX(pS/pT) motif. F, effect of CC on phosphorylation of Nudt14 in metformin-treated HEK293 cells expressing FLAG-Nudt14. Cells were treated with 10 mm metformin and several different concentrations of CC. G, effect of co-expression with CA or DN forms of AMPK on phosphorylation of Nudt14. The smaller ∼37 kDa band on the immunoblot probed with AMPK α1/2 antibody represents AMPK CA expression. AMPK DN has a molecular mass similar to that of the WT, ∼65 kDa.
To determine whether AMPK is responsible for phosphorylating Nudt14, we examined how the AMPK inhibitor CC and co-expression with a constitutively active form (CA) or dominant negative form (DN) of AMPK α subunit affect Nudt14 phosphorylation levels. CC treatment attenuated metformin-potentiated phosphorylation of Nudt14 in a concentration-dependent manner (Fig. 6F). The α subunit is the catalytic subunit of AMPK; it comprises a kinase domain, an autoinhibitory sequence, and a C-terminal domain, which is required for forming a complex with the β and γ subunits (42). AMPK CA is made by substituting Thr-172 with Asp, which mimics the phosphorylation by upstream kinases, such as LKB1, and truncation of the autoinhibitory sequence and C-terminal domain; AMPK DN has the D157A mutation, which inactivates kinase activity (23). The co-expression of AMPK CA and DN were confirmed by immunoblot analysis using AMPK α1/2 antibody (Fig. 6G). Importantly, we found that phosphorylation of Thr-141 of Nudt14 was facilitated by AMPK CA but prevented completely by AMPK DN induction (Fig. 6G). These results strongly suggest that AMPK phosphorylates Thr-141 of Nudt14.
Effect of Phosphorylation on the Activity of Nudt14
To determine the impact of the phosphorylation of Nudt14 by AMPK, we measured the UDP-Glc pyrophosphatase activity of Nudt14 by analyzing released UMP and UDP-Glc by HPLC analysis (Fig. 7A). HEK293 cells were transfected with mock-, Nudt14-, or Nudt14 T141A mutant-expressing vectors. The cells were then lysed, and the lysates were mixed with UDP-Glc. The peak intensity of UMP released from UDP-Glc was markedly increased in cell lysates from cells overexpressing Nudt14 and Nudt14 T141A compared with those expressing the mock transfectant (Fig. 7A). Importantly, greater UDP-Glc pyrophosphatase activity was detected in the Nudt14 wild type (WT) than in the T141A mutant (Fig. 7B), suggesting that phosphorylation modification may affect Nudt14 activity. In fact, metformin treatment increased the UDP-Glc pyrophosphatase activity of Nudt14 WT; however, treating the T141A mutant with or without metformin produced no difference (Fig. 7B). These results indicate that phosphorylation by AMPK may potentiate Nudt14 activity, although the extent of activation is small.
FIGURE 7.

Effect of phosphorylation of Nudt14 by AMPK on UDP-Glc pyrophosphatase activity. A, chromatogram showing UMP generated from UDP-Glc via Nudt14. UDP-Glc reacted with lysates derived from HEK293 cells transfected with mock, FLAG-Nudt14, or FLAG-Nudt14 T141A expression vectors for 120 min at 37 °C. The enzymatic product (UMP) and substrate (UDP-Glc) were separated and detected by ion pair reverse-phase HPLC. B, effect of phosphorylation on UDP-Glc pyrophosphatase activity of Nudt14. HEK293 cells transfected with Nudt14-FLAG or Nudt14 T141A-FLAG expression vectors were treated with (+) or without (−) metformin (10 mm) for 14 h. Cell lysates derived from these cells were reacted with UDP-Glc for 120 min at 37 °C. C, and D, UDP-Glc and UDP-Gal levels of Nudt14- or Nudt14 T141A-overexpressing cells. After treatment with metformin (10 mm) for 2 or 8 h, sugar nucleotides were extracted from HEK293 cells that were transfected with mock, FLAG-Nudt14, or FLAG-Nudt14 T141A expression vectors. Extracts were then subjected to ion pair reverse-phase HPLC, as described under “Experimental Procedures.” Bars, means ± S.D. (error bars) of at least three experiments. F, localization of FLAG-Nudt14 and FLAG-Nudt14 T141A in the absence or presence of AMPK-activating compounds. Cells transfected with FLAG-Nudt14 or FLAG-Nudt14 T141A were incubated with metformin (8 mm) or AICAR (1 mm) and then immunostained with anti-FLAG antibody or anti-GM130 antibody (Golgi body), as described under “Experimental Procedures.” Nuclei were stained with DAPI. Scale bar, 10 μm. *, p < 0.05; **, p < 0.01; ***, p < 0.001; n.s., not significant.
Next, we examined the effect of Nudt14 WT or T141A mutant overexpression on intracellular sugar nucleotide levels. Although both Nudt14 WT- and T141A-overexpressing cells displayed increased in vitro UDP-Glc pyrophosphatase activity relative to that of mock transfectant cells (Fig. 7A), UDP-Glc and UDP-Gal levels of these cells were quite similar under metformin-free conditions (Fig. 7, C and D). Thus, we hypothesized that Nudt14 might be localized in a specific cellular compartment under normal conditions in order to isolate it from UDP-Glc, thus preventing excess degradation. However, regardless of whether it is phosphorylated by AMPK, Nudt14 was found in the cytosol, where UDP-Glc is synthesized (Fig. 7E), indicating that Nudt14 must be accessible to UDP-Glc. These results suggest that intracellular Nudt14 may be strictly regulated by an unidentified factor, which maintains appropriate UDP-Glc levels under normal AMPK-inactivated conditions. Thus, overexpressed Nudt14 was not fully functional in our setup. On the other hand, in Nudt14 WT-overexpressing cells, UDP-Glc and UDP-Gal decreased more rapidly as a result of metformin treatment than did mock- and T141A mutant-transfected cells (Fig. 7, C and D), indicating that AMPK phosphorylation may activate intracellular Nudt14 activity.
Sugar Nucleotide Content in Nudt14-depleted Cells
To verify the contribution of Nudt14 in reducing UDP-Glc levels in AMPK-activated cells, Nudt14 expression in 3T3 cells was attenuated by siRNA-mediated knockdown (Fig. 8A). Unexpectedly, Nudt14 siRNA knockdown did not decrease UDP-Glc pyrophosphatase activity, although Nudt14 expression levels were markedly down-regulated (Fig. 8B). Consistent with this result, cellular sugar nucleotide levels, including UDP-Glc levels, were hardly affected by the knockdown of Nudt14 (Fig. 8C). Furthermore, Nudt14 siRNA knockdown did not restore the reduced sugar nucleotide content in AICAR-treated cells in these Nudt14-depleted cells (Fig. 8C). These results suggest that some other UDP-Glc pyrophosphatases in addition to Nudt14 are present in mammalian cells, and some of them may be affected by AMPK (Fig. 8D).
FIGURE 8.
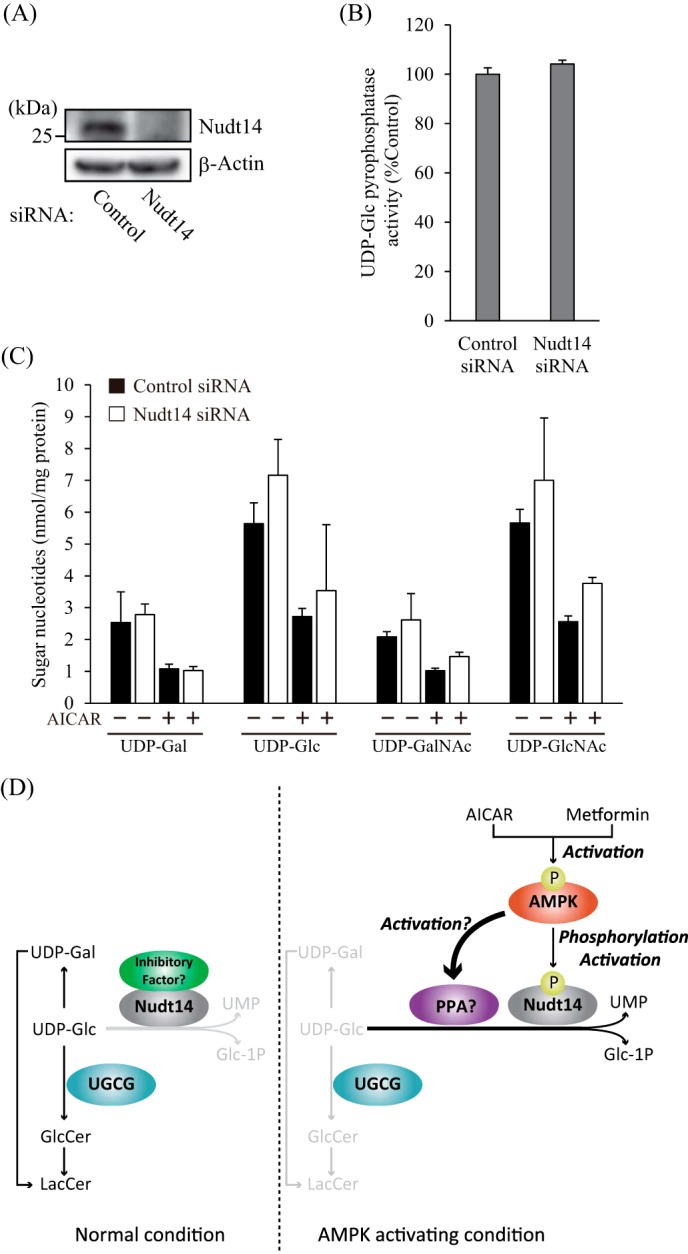
Contribution of Nudt14 to the reduction of cellular sugar nucleotide levels under AMPK-activating conditions. A, immunoblots showing the expression of Nudt14 in siRNA-mediated Nudt14-depleted cells. Nudt14 siRNA and control siRNA were transfected into 3T3 cells and incubated for 72 h. B, UDP-Glc pyrophosphatase activity of Nudt14-depleted cells. Lysates derived from 3T3 cells that were transfected with Nudt14 siRNA were reacted with UDP-Glc for 120 min. C, sugar nucleotide levels of Nudt14-depleted cells. After treatment with (+) or without (−) AICAR (1 mm) for 8 h, sugar nucleotides were extracted from 3T3 cells, in which Nudt14 was attenuated by siRNA and then subjected to ion pair reverse-phase HPLC, as described under “Experimental Procedures.” D, predicted mechanism of AMPK-mediated reduction of UDP-Glc and GlcCer levels. PPA, pyrophosphatase.
Discussion
Under energy-reducing conditions, AMPK stimulates ATP-generating pathways, such as the glycolytic pathway or the pathway of fatty acid oxidation, but inhibits anabolic processes, such as those involved in the biosynthesis of fatty acids, cholesterol, glycogen, and triacylglycerol, in order to maintain cellular energy homeostasis (42).
In the present study, we showed that GlcCer and LacCer synthesis is negatively regulated under AMPK-activating conditions. Under these conditions, the levels of sugar nucleotides are reduced, including those of UDP-Glc, the precursor of GlcCer (Fig. 8D). We demonstrated that the reduced GlcCer synthesis that occurs under AMPK-activating conditions could be restored by an inhibitor of AMPK or by knockdown of the AMPK α1 subunit. In addition, the reduction of GlcCer synthesis by AMPK-activating compounds did not occur in LKB1-defective HeLa cells, indicating that the phenomenon was induced by an LKB1-AMPK-dependent mechanism.
What is the physiological importance of AMPK-mediated reduction of GlcCer? GlcCer is composed of glucose, fatty acid, and sphingoid base generated from l-serine, which is made from an intermediate of glycolysis. Thus, one possibility is that glucose and fatty acids can be alternatively used for generating ATP by inhibiting the synthesis of GlcCer under AMPK-activating conditions. Another possibility may be linked to our previous study in the fruit fly, in which we showed that GlcCer levels are closely related to energy metabolism in the fat body, a tissue that is equivalent to adipose tissue and liver in mammals (43). Interestingly, overexpression of UGCG increases triacylglycerol and glycogen levels, whereas knockdown of UGCG reduces these energy-storing compounds in the fat body of the fruit fly. This suggests that AMPK-mediated reduction of GlcCer may serve as a “switch” that restores the consumption of triacylglycerol and glycogen, leading to ATP production. As described above, GlcCer comprises compounds that are closely related to energy homeostasis. Therefore, we surmise that changes in intracellular GlcCer levels may reflect an animal's cellular energy state and may affect glycogen and triacylglycerol metabolism to control energy homeostasis.
Because sugar nucleotides are used not only for the synthesis of GlcCer and GSLs but also for glycoconjugates such as glycogen, glycoproteins, and proteoglycans, we predict that glycoconjugate levels may also be affected by AMPK-activating compounds. In the present study, we focused on GlcCer metabolism and only measured (glyco)sphingolipid levels. Nakajima et al. (44) recently developed an LC-MS-based method to analyze sugar nucleotide metabolism by using glucose labeled with a stable isotope. This method would provide important information for the fate of sugar nucleotides and their derivatives under AMPK-activating conditions. It should be noted that GlcCer synthesis occurs at the cytosolic face of the Golgi apparatus via UGCG (45), and sugar nucleotides are also synthesized in the cytosol. Therefore, we hypothesize that GlcCer synthesis may be highly influenced by the reduction of sugar nucleotide levels relative to glycoproteins that are synthesized at the luminal side of the Golgi, where sugar nucleotides are transported from the cytosol.
In the present study, we found that AMPK phosphorylates the UDP-Glc pyrophosphatase Nudt14 at the Thr-141 site. Nudt14 belongs to the Nudix hydrolase superfamily, which mainly comprises pyrophosphatases that hydrolyze a wide range of substrates, such as nucleoside di- and triphosphates, dinucleoside and diphosphoinositol polyphosphates, nucleotide sugars, and RNA caps (46). Mammals have ∼24 Nudix genes. However, the phosphorylation site for AMPK found in Nudt14 is not conserved among Nudix proteins, suggesting that Nudt14 is the sole Nudix family enzyme that may be controlled by AMPK. According to the crystal structure of Nudt14, which has already been determined to be a homodimeric protein (Protein Data Bank code 3Q91), Thr-141 is located on β-strand 4 of the outer portion of Nudt14, where Nudt14 is faced with a dimeric partner and is far from the catalytic domain. Thus, it seems that the phosphorylation of Thr-141 may not be involved in the direct activation of Nudt14 but rather affects the formation of a dimeric state or complex with other proteins. Surprisingly, Nudt14 overexpression did not reduce cellular UDP-Glc levels under normal conditions, although in vitro UDP-Glc pyrophosphatase activity was remarkably increased. We found that Nudt14 consistently localized in the cytosol, regardless of whether it was phosphorylated by AMPK. This indicates that Nudt14 was accessible to UDP-Glc synthesized in the cytosol. Thus, we hypothesize that, in mammalian cells, Nudt14 may be negatively regulated by a factor that prevents excess degradation of UDP-Glc and UDP-Gal, which are utilized for the synthesis of functional glycoconjugates, such as glycolipids, glycogen, proteoglycans, and glycoproteins (Fig. 8D). Under AMPK-activating conditions, Nudt14 undergoes a phosphorylation-mediated conformational changethat may cause the inhibitory factor to leave or detach from Nudt14. This change would allow Nudt14 to exert proper activity toward UDP-Glc (Fig. 8D). In fact, greater UDP-Glc pyrophosphatase activity and faster reduction of UDP-Glc levels were detected in Nudt14-overexpressing cells incubated under AMPK-activating conditions. Because in vitro UDP-Glc pyrophosphatase activity was still observed in lysates derived from cells overexpressing the phosphorylation site null Nudt14 mutant (Nudt14 T141A), the inhibitory factor(s) may have detached from the enzyme during the processing of the cell lysates, although in this mutant, Nudt14 is not phosphorylated. Even if inhibitory factors that bind Nudt14 do exist, their avidity is probably weak, thus explaining why we were unable to isolate them during our IP experiment in the present study.
We considered that AMPK-mediated phosphorylation and activation of Nudt14 were the cause of the reduction of UDP-Glc levels. Unexpectedly, we found that UDP-Glc pyrophosphatase activity was not decreased and that the reduction of sugar nucleotide levels was not restored by siRNA-mediated knockdown of Nudt14. Thus, the AMPK-associated mechanisms responsible for reducing sugar nucleotides, especially UDP-Glc, remain to be worked out.
We surmise that mammalian cells possess some unidentified UDP-Glc pyrophosphatases (other than Nudt14) whose expression levels may be up-regulated to compensate for the absence of Nudt14 (Fig. 8D). We found that UGP2, the sole enzyme responsible for UDP-Glc synthesis in mammals according to the KEGG PATHWAY database, was not phosphorylated by AMPK. Thus, we speculate that AMPK may regulate UDP-Glc-degrading pathways, including the unidentified UDP-Glc pyrophosphatases (Fig. 8D), causing the reduction of UDP-Glc under AMPK-activating conditions. The results of our Nudt14 knockdown experiment also indicate that UDP-Glc is not the only endogenous substrate of Nudt14 in mammalian cells. Because Nudt14 is categorized as a Nudix family enzyme, certain organic pyrophosphates are candidate endogenous substrates of Nudt14. In order to test this hypothesis, quantification of total organic pyrophosphate levels in Nudt14-overexpressing or Nudt14-depleted cells would be required.
Because UDP-Gal is synthesized by UDP-Glc-4-epimerase from UDP-Glc, the reduction of UDP-Glc directly affects UDP-Gal levels. On the other hand, UDP-N-acetylglucosamine (GlcNAc) and UDP-N-acetylgalactosamine (GalNAc) are not derived from UDP-Glc. Therefore, reduction of UDP-GlcNAc and UDP-GalNAc under AMPK-activating conditions is carried out by mechanisms other than those responsible for reducing UDP-Glc and UDP-Gal. Eguchi et al. (47) showed that AMPK phosphorylates and decreases the activity of GFPT1 (glutamine-fructose-6-phosphate amidotransferase), which is the rate-limiting enzyme for amino sugar production. This AMPK-mediated phosphorylation reaction may result in the reduction of amino sugar nucleotides, such as UDP-GlcNAc or GalNAc.
PKD is one of the most common genetic disorders and is accompanied by renal cystic growth and massive kidney enlargement (48). GlcCer and ganglioside GM3 levels in kidney are increased in PKD patients and mouse models of PKD (16). Because the inhibition of UGCG activity effectively prevents cystogenesis in mouse models of PKD, the accumulation of GlcCer/GSLs is believed to be an important factor contributing to disease progression. Importantly, metformin inhibits kidney cystogenesis in a mouse model of PKD and in MDCK cells (49), which were used as an ex vivo model of renal cystogenesis (50). We confirmed that cellular UGCG activity of MDCK cells was decreased by metformin (data not shown), suggesting that reducing GlcCer and GSLs via AMPK activation may partly contribute to the beneficial effect of metformin on PKD.
In mammalian cells, GSLs are degraded by sequential glycosidases, and by acid ceramidase in lysosomes, to generate free sugars, fatty acids, and sphingoid bases (33). A deficiency of the GlcCer-degrading enzyme GBA1 leads to the abnormal accumulation of GlcCer in lysosomes and eventually is manifested as Gaucher disease (10). In Gaucher disease patients, GlcCer accumulates in lysosomes of macrophages, causing liver and spleen enlargement. Gaucher disease has three phenotypic variants (types 1–3) that can be distinguished based on neurological symptoms and severity. Enzyme replacement therapy (i.e. supplementation of GBA1 to lysosomes) is effective for the type 1 variant, which is characterized by non-neurological symptoms (51). However, because the replacement enzyme has difficulty crossing the blood brain barrier, enzyme replacement therapy is ineffective for treating the type 2 variant, which is characterized by CNS impairment. Importantly, we observed that metformin or AICAR reduced UGCG activity and cellular GlcCer levels in fibroblasts derived from Gaucher disease patients displaying type 1 or type 2 disease (Fig. 5, C and D). This finding indicates that GlcCer reduction by AMPK is caused by GBA1-independent mechanisms and that GlcCer reduction through AMPK activation may also serve as a new treatment option for Gaucher disease.
Recently, substrate (i.e. GlcCer) reduction therapy using UGCG inhibitors (e.g. eliglustat tartrate (Genz-112638)) has become a very effective treatment for Gaucher disease (52, 53). Because both AICAR and metformin do not directly inhibit UGCG activity (Fig. 2A), we hypothesize that GlcCer reduction may be synergistically induced by combining them with direct inhibitors of UGCG, such as eliglustat tartrate. Due to its safety and efficacy, metformin is now used as a first choice oral antidiabetic drug to suppress blood glucose levels, and it is prescribed to more than 100 million people worldwide (54). Although the in vivo effects of metformin on GlcCer metabolism were not examined in the present study, its safety profile would make it possible to use metformin as a therapy for Gaucher disease or other diseases caused by GlcCer accumulation.
Author Contributions
Y. I. and Y. H. designed the study and wrote the paper. Y. I. designed, performed, and analyzed all of the experiments in this study. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Drs. Kazuki Nakajima, Takuji Nabetani, Shota Sakai, and Takamitsu Sano (RIKEN); Dr. Kota Zama (Hokkaido University); and Dr. Junko Matsuda (Kawasaki Medical School) for technical advice. We are deeply grateful to Drs. Toshihide Kobayashi and Francoise Hullin-Mastuda (RIKEN) for donating MDCK cells and to Dr. Shun Watanabe for donating ugcg(lox/lox) MEF, ugcg(−/−) MEF, and UGCG-FLAG expression vectors. We are grateful to the Support Unit for Bio-material Analysis, RIKEN Brain Science Institute Research Resources Center, for help with the quantitative real-time PCR and nucleotide sequencing analyses.
This work was supported in part by grants from the RIKEN Brain Science Institute (to Y. H.) and RIKEN Special Postdoctoral Researchers Program (to Y. I.), a Grant-in-Aid for Young Scientists (B) (to Y. I.), the Research Activity Start-up (to Y. I.), and “Deciphering sugar chain-based signals regulating integrative neuronal functions” (to Y. H.) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan. The authors declare that they have no conflicts of interest with the contents of this article.
- GSL
- glycosphingolipid
- ACC
- acetyl-CoA carboxylase
- AICAR
- 5-aminoimidazole-4-carboxamide 1-β-d-ribofuranoside
- AMPK
- AMP-activated protein kinase
- CA
- constitutively active
- CC
- Compound C (6-[4-(2-piperidin-1-ylethoxy)phenyl]-3-pyridin-4-ylpyrazolo[1,5-a]pyrimidine)
- Cer
- ceramide
- DN
- dominant negative
- GalNAc
- N-acetylgalactosamine
- GBA1 and -2
- acid β-glucosidase 1 and 2, respectively
- GCase
- glucosylceramidase
- GlcCer
- glucosylceramide
- GlcNAc
- N-acetylglucosamine
- GM3
- NeuAc α 2–3Gal β 1–4Glc β 1–1′Cer
- HEK
- human embryonic kidney
- IP
- immunoprecipitation
- LacCer
- lactosylceramide
- LKB1
- liver kinase B1
- MDCK
- Madin-Darby canine kidney
- NBD
- 7-nitro-2,1,3-benzoxadiazole
- UDP-Glc
- uridine diphosphate-glucose
- UGCG
- UDP-glucose:ceramide glucosyltransferase
- PKD
- polycystic kidney disease
- 3T3
- NIH3T3
- ESI
- electrospray ionization
- MEF
- mouse embryo fibroblast.
References
- 1. Jennemann R., Gröne H. J. (2013) Cell-specific in vivo functions of glycosphingolipids: lessons from genetic deletions of enzymes involved in glycosphingolipid synthesis. Prog. Lipid Res. 52, 231–248 [DOI] [PubMed] [Google Scholar]
- 2. Hanada K. (2005) Sphingolipids in infectious diseases. Jpn. J. Infect. Dis. 58, 131–148 [PubMed] [Google Scholar]
- 3. Ishibashi Y., Kohyama-Koganeya A., Hirabayashi Y. (2013) New insights on glucosylated lipids: metabolism and functions. Biochim. Biophys. Acta 1831, 1475–1485 [DOI] [PubMed] [Google Scholar]
- 4. Yamashita T., Wada R., Sasaki T., Deng C., Bierfreund U., Sandhoff K., Proia R. L. (1999) A vital role for glycosphingolipid synthesis during development and differentiation. Proc. Natl. Acad. Sci. U.S.A. 96, 9142–9147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kohyama-Koganeya A., Sasamura T., Oshima E., Suzuki E., Nishihara S., Ueda R., Hirabayashi Y. (2004) Drosophila glucosylceramide synthase: a negative regulator of cell death mediated by proapoptotic factors. J. Biol. Chem. 279, 35995–36002 [DOI] [PubMed] [Google Scholar]
- 6. Marza E., Simonsen K. T., Faergeman N. J., Lesa G. M. (2009) Expression of ceramide glucosyltransferases, which are essential for glycosphingolipid synthesis, is only required in a small subset of C. elegans cells. J. Cell Sci. 122, 822–833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nomura K. H., Murata D., Hayashi Y., Dejima K., Mizuguchi S., Kage-Nakadai E., Gengyo-Ando K., Mitani S., Hirabayashi Y., Ito M., Nomura K. (2011) Ceramide glucosyltransferase of the nematode Caenorhabditis elegans is involved in oocyte formation and in early embryonic cell division. Glycobiology 21, 834–848 [DOI] [PubMed] [Google Scholar]
- 8. Yamashita T., Wada R., Proia R. L. (2002) Early developmental expression of the gene encoding glucosylceramide synthase, the enzyme controlling the first committed step of glycosphingolipid synthesis. Biochim. Biophys. Acta 1573, 236–240 [DOI] [PubMed] [Google Scholar]
- 9. Nordström V., Willershäuser M., Herzer S., Rozman J., von Bohlen Und Halbach O., Meldner S., Rothermel U., Kaden S., Roth F. C., Waldeck C., Gretz N., de Angelis M. H., Draguhn A., Klingenspor M., Gröne H. J., Jennemann R. (2013) Neuronal expression of glucosylceramide synthase in central nervous system regulates body weight and energy homeostasis. PLoS Biol. 11, e1001506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Grabowski G. A. (2008) Phenotype, diagnosis, and treatment of Gaucher's disease. Lancet 372, 1263–1271 [DOI] [PubMed] [Google Scholar]
- 11. Liu Y. Y., Patwardhan G. A., Xie P., Gu X., Giuliano A. E., Cabot M. C. (2011) Glucosylceramide synthase, a factor in modulating drug resistance, is overexpressed in metastatic breast carcinoma. Int. J. Oncol. 39, 425–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fuller M. (2010) Sphingolipids: the nexus between Gaucher disease and insulin resistance. Lipids Health Dis. 9, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sidransky E., Lopez G. (2012) The link between the GBA gene and parkinsonism. Lancet Neurol. 11, 986–998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tsuang D., Leverenz J. B., Lopez O. L., Hamilton R. L., Bennett D. A., Schneider J. A., Buchman A. S., Larson E. B., Crane P. K., Kaye J. A., Kramer P., Woltjer R., Kukull W., Nelson P. T., Jicha G. A., Neltner J. H., Galasko D., Masliah E., Trojanowski J. Q., Schellenberg G. D., Yearout D., Huston H., Fritts-Penniman A., Mata I. F., Wan J. Y., Edwards K. L., Montine T. J., Zabetian C. P. (2012) GBA mutations increase risk for Lewy body disease with and without Alzheimer disease pathology. Neurology 79, 1944–1950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mazzulli J. R., Xu Y. H., Sun Y., Knight A. L., McLean P. J., Caldwell G. A., Sidransky E., Grabowski G. A., Krainc D. (2011) Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 146, 37–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Natoli T. A., Smith L. A., Rogers K. A., Wang B., Komarnitsky S., Budman Y., Belenky A., Bukanov N. O., Dackowski W. R., Husson H., Russo R. J., Shayman J. A., Ledbetter S. R., Leonard J. P., Ibraghimov-Beskrovnaya O. (2010) Inhibition of glucosylceramide accumulation results in effective blockade of polycystic kidney disease in mouse models. Nat. Med. 16, 788–792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Aerts J. M., Ottenhoff R., Powlson A. S., Grefhorst A., van Eijk M., Dubbelhuis P. F., Aten J., Kuipers F., Serlie M. J., Wennekes T., Sethi J. K., O'Rahilly S., Overkleeft H. S. (2007) Pharmacological inhibition of glucosylceramide synthase enhances insulin sensitivity. Diabetes 56, 1341–1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhao H., Przybylska M., Wu I. H., Zhang J., Siegel C., Komarnitsky S., Yew N. S., Cheng S. H. (2007) Inhibiting glycosphingolipid synthesis improves glycemic control and insulin sensitivity in animal models of type 2 diabetes. Diabetes 56, 1210–1218 [DOI] [PubMed] [Google Scholar]
- 19. Liu Y. Y., Hill R. A., Li Y. T. (2013) Ceramide glycosylation catalyzed by glucosylceramide synthase and cancer drug resistance. Adv. Cancer Res. 117, 59–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gupta V., Bhinge K. N., Hosain S. B., Xiong K., Gu X., Shi R., Ho M. Y., Khoo K. H., Li S. C., Li Y. T., Ambudkar S. V., Jazwinski S. M., Liu Y. Y. (2012) Ceramide glycosylation by glucosylceramide synthase selectively maintains the properties of breast cancer stem cells. J. Biol. Chem. 287, 37195–37205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Morad S. A., Cabot M. C. (2013) Ceramide-orchestrated signalling in cancer cells. Nat. Rev. Cancer 13, 51–65 [DOI] [PubMed] [Google Scholar]
- 22. Hardie D. G., Ross F. A., Hawley S. A. (2012) AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 13, 251–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Woods A., Azzout-Marniche D., Foretz M., Stein S. C., Lemarchand P., Ferré P., Foufelle F., Carling D. (2000) Characterization of the role of AMP-activated protein kinase in the regulation of glucose-activated gene expression using constitutively active and dominant negative forms of the kinase. Mol. Cell. Biol. 20, 6704–6711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Watanabe S., Endo S., Oshima E., Hoshi T., Higashi H., Yamada K., Tohyama K., Yamashita T., Hirabayashi Y. (2010) Glycosphingolipid synthesis in cerebellar Purkinje neurons: roles in myelin formation and axonal homeostasis. Glia 58, 1197–1207 [DOI] [PubMed] [Google Scholar]
- 25. Gupta V., Patwardhan G. A., Zhang Q. J., Cabot M. C., Jazwinski S. M., Liu Y. Y. (2010) Direct quantitative determination of ceramide glycosylation in vivo: a new approach to evaluate cellular enzyme activity of glucosylceramide synthase. J. Lipid Res. 51, 866–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ito M., Okino N., Tani M. (2014) New insight into the structure, reaction mechanism, and biological functions of neutral ceramidase. Biochim. Biophys. Acta 1841, 682–691 [DOI] [PubMed] [Google Scholar]
- 27. Martin O. C., Pagano R. E. (1994) Internalization and sorting of a fluorescent analogue of glucosylceramide to the Golgi apparatus of human skin fibroblasts: utilization of endocytic and nonendocytic transport mechanisms. J. Cell Biol. 125, 769–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shaner R. L., Allegood J. C., Park H., Wang E., Kelly S., Haynes C. A., Sullards M. C., Merrill A. H., Jr. (2009) Quantitative analysis of sphingolipids for lipidomics using triple quadrupole and quadrupole linear ion trap mass spectrometers. J. Lipid Res. 50, 1692–1707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nakajima K., Kitazume S., Angata T., Fujinawa R., Ohtsubo K., Miyoshi E., Taniguchi N. (2010) Simultaneous determination of nucleotide sugars with ion-pair reversed-phase HPLC. Glycobiology 20, 865–871 [DOI] [PubMed] [Google Scholar]
- 30. Mihaylova M. M., Shaw R. J. (2011) The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 13, 1016–1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nguyen H. B., Babcock J. T., Wells C. D., Quilliam L. A. (2013) LKB1 tumor suppressor regulates AMP kinase/mTOR-independent cell growth and proliferation via the phosphorylation of Yap. Oncogene 32, 4100–4109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shackelford D. B., Shaw R. J. (2009) The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat. Rev. Cancer 9, 563–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schulze H., Kolter T., Sandhoff K. (2009) Principles of lysosomal membrane degradation: cellular topology and biochemistry of lysosomal lipid degradation. Biochim. Biophys. Acta 1793, 674–683 [DOI] [PubMed] [Google Scholar]
- 34. Yildiz Y., Matern H., Thompson B., Allegood J. C., Warren R. L., Ramirez D. M., Hammer R. E., Hamra F. K., Matern S., Russell D. W. (2006) Mutation of β-glucosidase 2 causes glycolipid storage disease and impaired male fertility. J. Clin. Invest. 116, 2985–2994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Boot R. G., Verhoek M., Donker-Koopman W., Strijland A., van Marle J., Overkleeft H. S., Wennekes T., Aerts J. M. (2007) Identification of the non-lysosomal glucosylceramidase as β-glucosidase 2. J. Biol. Chem. 282, 1305–1312 [DOI] [PubMed] [Google Scholar]
- 36. Körschen H. G., Yildiz Y., Raju D. N., Schonauer S., Bönigk W., Jansen V., Kremmer E., Kaupp U. B., Wachten D. (2013) The non-lysosomal β-glucosidase GBA2 is a non-integral membrane-associated protein at the endoplasmic reticulum (ER) and Golgi. J. Biol. Chem. 288, 3381–3393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Duggleby R. G., Chao Y. C., Huang J. G., Peng H. L., Chang H. Y. (1996) Sequence differences between human muscle and liver cDNAs for UDPglucose pyrophosphorylase and kinetic properties of the recombinant enzymes expressed in Escherichia coli. Eur. J. Biochem. 235, 173–179 [DOI] [PubMed] [Google Scholar]
- 38. Yagi T., Baroja-Fernández E., Yamamoto R., Muñoz F. J., Akazawa T., Hong K. S., Pozueta-Romero J. (2003) Cloning, expression and characterization of a mammalian Nudix hydrolase-like enzyme that cleaves the pyrophosphate bond of UDP-glucose. Biochem. J. 370, 409–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Heyen C. A., Tagliabracci V. S., Zhai L., Roach P. J. (2009) Characterization of mouse UDP-glucose pyrophosphatase, a Nudix hydrolase encoded by the Nudt14 gene. Biochem. Biophys. Res. Commun. 390, 1414–1418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Banko M. R., Allen J. J., Schaffer B. E., Wilker E. W., Tsou P., White J. L., Villén J., Wang B., Kim S. R., Sakamoto K., Gygi S. P., Cantley L. C., Yaffe M. B., Shokat K. M., Brunet A. (2011) Chemical genetic screen for AMPKα2 substrates uncovers a network of proteins involved in mitosis. Mol. Cell 44, 878–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gwinn D. M., Shackelford D. B., Egan D. F., Mihaylova M. M., Mery A., Vasquez D. S., Turk B. E., Shaw R. J. (2008) AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 30, 214–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hardie D. G. (2007) AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat. Rev. Mol. Cell Biol. 8, 774–785 [DOI] [PubMed] [Google Scholar]
- 43. Kohyama-Koganeya A., Nabetani T., Miura M., Hirabayashi Y. (2011) Glucosylceramide synthase in the fat body controls energy metabolism in Drosophila. J. Lipid Res. 52, 1392–1399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nakajima K., Ito E., Ohtsubo K., Shirato K., Takamiya R., Kitazume S., Angata T., Taniguchi N. (2013) Mass isotopomer analysis of metabolically labeled nucleotide sugars and N- and O-glycans for tracing nucleotide sugar metabolisms. Mol. Cell. Proteomics 12, 2468–2480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Marks D. L., Wu K., Paul P., Kamisaka Y., Watanabe R., Pagano R. E. (1999) Oligomerization and topology of the Golgi membrane protein glucosylceramide synthase. J. Biol. Chem. 274, 451–456 [DOI] [PubMed] [Google Scholar]
- 46. McLennan A. G. (2006) The Nudix hydrolase superfamily. Cell. Mol. Life Sci. 63, 123–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Eguchi S., Oshiro N., Miyamoto T., Yoshino K., Okamoto S., Ono T., Kikkawa U., Yonezawa K. (2009) AMP-activated protein kinase phosphorylates glutamine: fructose-6-phosphate amidotransferase 1 at Ser243 to modulate its enzymatic activity. Genes Cells 14, 179–189 [DOI] [PubMed] [Google Scholar]
- 48. Wilson P. D. (2004) Polycystic kidney disease. N. Engl. J. Med. 350, 151–164 [DOI] [PubMed] [Google Scholar]
- 49. Takiar V., Nishio S., Seo-Mayer P., King J. D., Jr., Li H., Zhang L., Karihaloo A., Hallows K. R., Somlo S., Caplan M. J. (2011) Activating AMP-activated protein kinase (AMPK) slows renal cystogenesis. Proc. Natl. Acad. Sci. U.S.A. 108, 2462–2467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mangoo-Karim R., Uchic M., Lechene C., Grantham J. J. (1989) Renal epithelial cyst formation and enlargement in vitro: dependence on cAMP. Proc. Natl. Acad. Sci. U.S.A. 86, 6007–6011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Desnick R. J., Schuchman E. H. (2012) Enzyme replacement therapy for lysosomal diseases: lessons from 20 years of experience and remaining challenges. Annu. Rev. Genomics Hum. Genet. 13, 307–335 [DOI] [PubMed] [Google Scholar]
- 52. Lukina E., Watman N., Arreguin E. A., Banikazemi M., Dragosky M., Iastrebner M., Rosenbaum H., Phillips M., Pastores G. M., Rosenthal D. I., Kaper M., Singh T., Puga A. C., Bonate P. L., Peterschmitt M. J. (2010) A phase 2 study of eliglustat tartrate (Genz-112638), an oral substrate reduction therapy for Gaucher disease type 1. Blood 116, 893–899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cox T. M., Drelichman G., Cravo R., Balwani M., Burrow T. A., Martins A. M., Lukina E., Rosenbloom B., Ross L., Angell J., Puga A. C. (2015) Eliglustat compared with imiglucerase in patients with Gaucher's disease type 1 stabilised on enzyme replacement therapy: a phase 3, randomised, open-label, non-inferiority trial. Lancet 385, 2355–2362 [DOI] [PubMed] [Google Scholar]
- 54. Rena G., Pearson E. R., Sakamoto K. (2013) Molecular mechanism of action of metformin: old or new insights? Diabetologia 56, 1898–1906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ichikawa S., Sakiyama H., Suzuki G., Hidari K. I., Hirabayashi Y. (1996) Expression cloning of a cDNA for human ceramide glucosyltransferase that catalyzes the first glycosylation step of glycosphingolipid synthesis. Proc. Natl. Acad. Sci. U.S.A. 93, 4638–4643 [DOI] [PMC free article] [PubMed] [Google Scholar]