

Abstract
Patients suffering from cystic fibrosis (CF) are commonly affected by chronic Pseudomonas aeruginosa biofilm infections. This is the main cause for the high disease severity. In this study, we demonstrate that P. aeruginosa is able to efficiently colonize murine solid tumors after intravenous injection and to form biofilms in this tissue. Biofilm formation was evident by electron microscopy. Such structures could not be observed with transposon mutants, which were defective in biofilm formation. Comparative transcriptional profiling of P. aeruginosa indicated physiological similarity of the bacteria in the murine tumor model and the CF lung. The efficacy of currently available antibiotics for treatment of P. aeruginosa-infected CF lungs, such as ciprofloxacin, colistin, and tobramycin, could be tested in the tumor model. We found that clinically recommended doses of these antibiotics were unable to eliminate wild-type P. aeruginosa PA14 while being effective against biofilm-defective mutants. However, colistin-tobramycin combination therapy significantly reduced the number of P. aeruginosa PA14 cells in tumors at lower concentrations. Hence, we present a versatile experimental system that is providing a platform to test approved and newly developed antibiofilm compounds.
INTRODUCTION
Most bacterial infections are effectively treated by potent antimicrobial agents and can be quickly overcome. Nevertheless, bacterial infections continue to be a serious threat—mostly when onset of antimicrobial therapy is delayed, resistance of the microbes emerges, or chronic persistence develops (1–3). Although chronic infections are substantially mitigated by intensified antimicrobial therapy, antibiotics usually fail to resolve the infection, and very limited treatment options remain (4, 5). In this phase, bacteria often adopt a biofilm mode of growth: i.e., the bacterial community produces a protective extracellular matrix against antimicrobial therapy and the defense mechanisms of the host (6, 7). As a consequence, intensified treatment becomes insufficient, and surgery is often the ultimate means of infection control.
Despite the medical need, no effective antibiofilm treatment regimens are presently available in the clinic (8, 9). This is largely due to the fact that conventional treatment strategies fail to resolve biofilm infections. Therefore, to prevent biofilms or resolve them, understanding the mode of action of commonly used antibiotics against biofilm-grown bacteria in vivo is an essential requirement to develop more effective antimicrobial treatment regimens (10–12). This includes the evaluation of the synergistic activity of antibiotic combinations and the optimization of dosing regimens in vivo to maximize their effectiveness. Furthermore, novel compounds with an antibiofilm activity need to be developed. However, most of the novel therapeutic strategies for inhibition of biofilm growth have been developed in vitro (13). Obviously, identification of an in vitro antibiofilm strategy needs to be followed by the demonstration of in vivo efficacy in a suitable animal infection model (14).
Of major clinical relevance are chronic infections of the lung of cystic fibrosis (CF) patients by Pseudomonas aeruginosa. Most CF patients acquire P. aeruginosa during early childhood and develop chronic biofilm infections with repeated courses of exacerbation (15). Despite intensified antimicrobial therapy, progressive infection and inflammation lead to deterioration of pulmonary function. Thus, chronic P. aeruginosa infection remains the major reason for lung failure in these patients (16). To improve present antibiofilm therapies and to test their in vivo efficacy, a small-animal model should be employed. However, although the applied models to date (17) result in reduced bacterial clearance as one hallmark of chronic infections, most of them do not exert bacterial growth within biofilms or exhibit a low efficiency of bacterial colonization. This limits their use to study P. aeruginosa biofilm infections in vivo. We have recently described a model that might accomplish the requirements. It employs transplantable solid tumors in mice that are preferentially colonized by P. aeruginosa as well as by other bacteria after systemic administration (18). The microenvironment of the colonized tumor leads to biofilm formation by the bacteria (18–20). Apparently, this microenvironment of the murine tumor resembles very closely that found in the CF lung or in burn wounds (21).
In the present work, we carefully calibrated our murine tumor model and infected mouse tumors with the biofilm-forming PA14 wild-type (WT) strain as well as with two biofilm-defective mutants. Those mutants not only formed poor biofilms in vitro, but—albeit they were able to colonize the tumor at comparable CFU—they did not form biofilms in vivo within the tumor tissue. This is an important prerequisite for the robust analysis of antibiofilm and antibacterial effects of antimicrobial compounds in vivo.
By applying the model, we could demonstrate that ciprofloxacin, tobramycin, and colistin significantly reduced CFU counts of the biofilm-defective P. aeruginosa mutants within the tumor tissue, whereas an antibiofilm activity against biofilm-forming wild-type bacteria was observed only at antibacterial concentrations that exceed the normal doses used in the clinics. Importantly, a combination of tobramycin and colistin showed activity already at much lower concentrations, as previously demonstrated (22), indicating synergistic antibiofilm activity. Our results demonstrate that with the mouse tumor model, a versatile model is available that allows the in vivo characterization of antimicrobial compounds already in use as well as the evaluation of novel antibacterial substances.
MATERIALS AND METHODS
Bacterial strains and cell lines.
The Pseudomonas aeruginosa PA14 wild-type strain and transposon mutants PA14 pqsA::Tn (defective in 4-quinolone signaling) (23, 24) and PA14 pelA::Tn (defective in the production of the Pel exopolysaccharide) (25) were obtained from the Harvard transposon mutant library (26). The phenotype of such mutants was confirmed as shown in Fig. S1 and S2 in the supplemental material. The equal sensitivities of these strains toward the antibiotics employed have also been confirmed (see Table S1 in the supplemental material). CT26 colon carcinoma cells (ATCC CRL-2638) were grown as monolayers in Iscove's modified Dulbecco's medium (IMDM) (catalog no. 42200030; Gibco Life Technologies, Germany) supplemented with 10% (vol/vol) heat-inactivated fetal calf serum (FCS) (Integro) and 250 mmol liter−1 β-mercaptoethanol (Serva).
Animals.
Female BALB/c mice were purchased from Janvier (BALB/cByJRj) at 6 to 7 weeks of age and were allowed to acclimatize to the institutional animal facility for 1 week prior to experiments. All animal experiments were carried out in accordance with the institutional guidelines and are in agreement with the German Animal Welfare Act (Tierschutzgesetz, 1998) and international laws and policies governing the use of animals for scientific purposes (EC Council Directive 86/609, OJ L 358,1, 12 December 1987; and NIH Guide for the Care and Use of Laboratory Animals, U.S. National Research Council, 1996). The protocol was approved by the ethical board of the local authorities (Niedersächsisches Landesamt für Verbraucherschutz und Lebensmittelsicherheit [LAVES], Oldenburg, Germany). The permission number is 33.9.42502-04-050/09.
Infection of tumor-bearing mice.
Seven- to 8-week-old female BALB/c mice were subcutaneously (s.c.) inoculated with 5 × 105 CT26 cells into the abdomen or the flank (Fig. 1a). When the tumor reached an appropriate size, the mice were infected intravenously (i.v.) with 5 × 106 CFU of P. aeruginosa in phosphate-buffered saline (PBS).
FIG 1.

Infection of transplantable solid tumors in mice. (a) A total of 5 × 105 CT26 cells were injected subcutaneously into the abdomen/flank region. (b) After approximately 10 days, tumors reached a size of between 100 and 150 mm3. (c) Shortly after i.v. P. aeruginosa infection, tumors showed macroscopically visible necrotic areas, like the tumor presented here. (d) Induction of cytokines (exemplified by TNF-α) shortly after bacterial application. TNF-α is most likely the most important cytokine for this model system. (e) Overview of hematoxylin-and-eosin (HE)-stained tumor section colonized by P. aeruginosa. The black box indicates the area that is enlarged in panel f. N indicates the large necrotic area of the infected tumor, and V indicates the remaining viable part of the tumor after bacterial colonization. (A higher-resolution image was added to Fig. S4 in the supplemental material.) (f) Enlargement of the area from the P. aeruginosa-colonized tumor shown in panel e. Tumor-infiltrating neutrophils (black arrows) are identified by their nuclear shape. (g) Efficiency of tumor colonization using different infection doses. Bars represent individual mice. (h) Efficiency of tumor colonization depends on the size of the tumor. Bars represent individual mice.
Antibiotics and dosage.
Stock solutions of ciprofloxacin (Hexal, Holzkirchen, Germany), colistin (Grunenthal, Aachen, Germany), and tobramycin (Infectopharm Arzneimittel, Heppenheim, Germany) were prepared fresh in 0.9% saline at a final concentration of 2 mg/ml. Antibiotics were administered i.v. at the indicated time intervals and doses.
Determination of CFU within mouse tissue.
To determine CFU counts, mice were sacrificed and organs (tumor, liver, and gut) were homogenized in 0.1% (vol/vol) Triton X-100–PBS using a gentleMACS dissociator. The homogenates were serially diluted and plated on Luria-Bertani (LB) agar plates with ampicillin (0.1 mg/ml).
Histology.
Standard histology procedures were followed. Briefly, organs were fixed in 4% buffered formaldehyde for 24 to 48 h and embedded in paraffin, and 3-μm sections were obtained. The sections were stained with hematoxylin and eosin (HE). A combined periodic acid-Schiff-alcian blue reaction (PAS) was conducted according to a standard protocol. The stained sections were analyzed with an Olympus BX51 microscope, and images were captured with an Olympus U-CMAD3 camera using the software ZEN 2009.
FISH.
Fluorescence in situ hybridization (FISH) was carried out as described previously (27). In brief, tumor samples were fixed for 24 h at 4°C. Samples were embedded in cold polymerizing resin (methacrylate) and sectioned as described elsewhere (28). The hybridization buffer (20 μl) consisted of 0.9 M NaCl, 20 mM Tris-HCl, 0.01% sodium dodecyl sulfate (SDS), 20% formamide, and 4′,6-diamidino-2-phenylindole (DAPI). Probes were synthesized commercially and 5′ end labeled with a fluorochrome—either Cy3 (indocarbocyanine) or fluorescein isothiocyanate (FITC) (all from Biomers, Ulm, Germany) and used at a concentration of 20 pmol. Samples were incubated in a dark, humid chamber for 2 h at 50°C. The slides were then rinsed with sterile double-distilled water, dried, and mounted using Vectashield mounting medium (Vector Laboratories, Burlingame, CA) without DAPI. For visualization and identification of P. aeruginosa, the specific probe PSM was used (29). For each FISH experiment, positive and negative bacterial control strains were hybridized alongside the tumor samples to ensure specificity. These samples were further analyzed using an epifluorescence microscope (Axioplan 2; Carl Zeiss, Jena, Germany) equipped with narrow band filter sets (AHF Analysentechnik, Tübingen, Germany). Image acquisition was performed with an AxioCam MRm (Zeiss) making use of the AxioVision 4.4 software.
TEM.
For transmission electron microscopy (TEM), tumors were fixed in 5% formaldehyde and 2% glutaraldehyde in cacodylate buffer (0.1 M cacodylate, 0.01 M CaCl2, 0.01 M MgCl2, 0.09 M sucrose [pH 6.9]) overnight and then cut into cubes 3 to 5 mm in length. Samples were osmificated in 1% aqueous osmium tetroxide for 1 h and washed with cacodylate buffer. Samples were then dehydrated with graded series of acetone (10, 30, 50, 70, 90, and 100%) for 30 min each step. Dehydration in the 70% acetone step was done with 2% uranyl acetate overnight. Samples were then infiltrated with an epoxy resin (1 part acetone/1 part resin, 1 part acetone/2 parts resin, pure resin alternating over 2 days) according to Spurr's formula. Ultrathin sections were cut with a diamond knife, picked up with Formvar-coated grids, counterstained with uranyl acetate and lead citrate, and examined in a TEM910 transmission electron microscope (Carl Zeiss, Oberkochen, Germany) at an acceleration voltage of 80 kV. Images were taken at calibrated magnifications using a line replica. Images were recorded digitally with a ProScan slow-scan charge-coupled device (CCD) camera (1,024 by 1,024) (ProScan, Scheuring, Germany) with ITEM software (Olympus Soft Imaging Solutions, Münster, Germany) (30).
Statistics.
For all CFU counts and cytokine measurements, the mean values and standard deviations were calculated. Changes were analyzed using Student's t test in the statistical program GraphPad Prism version 5.0 (GraphPad Software, Inc., San Diego, CA).
RESULTS
A small-animal model for P. aeruginosa biofilm infections.
In previous publications, we could show that P. aeruginosa is able to efficiently colonize solid murine tumors and to form biofilm-like structures in such tissue (20, 21). Here, we aimed at calibrating the model system to evaluate the effectiveness of antimicrobial compounds and antimicrobial combinations against biofilm-grown bacteria in vivo. We therefore determined several parameters of bacterial tumor colonization, like the influence of the size of the inoculum or the tumor to establish robust infections.
Transplantable CT26 tumor cells were injected into either the abdomen or flank of syngeneic BALB/c mice (Fig. 1a). After 10 days, these mice were then infected intravenously (i.v.) with 5 × 106 P. aeruginosa PA14 cells (Fig. 1b). Roughly 2 to 3 h after application of the bacteria, the tumor changes its macroscopic appearance and turns black due to a severe hemorrhage (Fig. 1c). This reaction is most likely due to the induction of tumor necrosis factor alpha (TNF-α), which was found at high levels in the blood of P. aeruginosa-infected mice shortly after bacterial application (Fig. 1d). As a consequence, a large necrotic area forms in the center of the tumor, as shown by histology (Fig. 1e and f). Furthermore, histology revealed a strong inflammatory response and attraction of neutrophils in the colonized tumors (Fig. 1e and f) (20).
Approximately 10 days post-CT26 cell inoculation, tumors normally attain a size of between 100 and 150 mm3. This appears to be the optimal time point for infection. As depicted in Fig. 1h, tumors with a size smaller than 100 mm3 carried the risk of not being colonized by bacteria, while in larger tumors, we observed robust bacterial colonization at comparable CFU counts.
Optimal bacterial infection with an appropriate size of the inoculum is also extremely important for robust tumor colonization (Fig. 1g). An inoculum size too low will result in no tumor colonization or colonization of only part of the tumors (Fig. 1g). Probably, too little TNF-α is induced under these circumstances. The presence of too many bacteria, however, results in septicemia and death of the mice (data not shown). Interestingly, the extent of colonization was dependent on the size of the inoculum: i.e., lower colonization was observed when lower bacterial doses were used as the inoculum (Fig. 1g). This is different from other bacteria like Salmonella enterica serovar Typhimurium, where above a certain threshold, the colonization is mostly independent of the number of bacteria injected (31).
Standard laboratory strains of P. aeruginosa PAO1 and PA14 preferentially accumulate and multiply in the neoplastic tissue, while colonization of healthy organs, like liver and gut, is much lower (see Fig. S3a in the supplemental material). Fluorescent in situ hybridization (FISH) on tumor sections demonstrates that colonization of P. aeruginosa PA14 occurs mostly throughout region of the tumor tissue where still viable cells reside (see Fig. S3b). Biofilm-like structures develop shortly after tumor colonization. Scanning electron microscopy and transmission electron microscopy clearly show the embedment of the PA14 bacteria in a self-produced extracellular matrix (see Fig.S3c and d).
Biofilm-defective mutants colonize the tumor but do not form biofilms in the tumor tissue.
Several bacterial factors have been shown to be required for efficient formation of P. aeruginosa biofilms in vitro (24). However, whether those factors are also crucial for biofilm formation in vivo has remained elusive. Hence, we selected two mutants, both of which form poor biofilms in vitro. One (a pqsA transposon mutant) was defective in the production of the 4-quinolone signal molecules (see Fig. S2 in the supplemental material), and the second (a pelA transposon mutant) was defective in the production of the Pel exopolysaccharide (see Fig. S1 in the supplemental material). First, we elucidated whether the biofilm-defective mutants were able to colonize the tumor, liver, and gut at similar rates to the P. aeruginosa PA14 WT after i.v. application. The CFU counts (Fig. 2a) as well as the localization of the mutants within the tumors were comparable to those of WT bacteria (Fig. 2b and c). Most importantly, the in vitro biofilm-defective mutants were not able to produce an extracellular matrix in vivo, as judged by electron microscopy (Fig. 2b and c) (30). They largely remained as single cells in the tumor and were not surrounded by the biofilm-like structures seen for the WT bacteria (see Fig.S3 in the supplemental material).
FIG 2.

Tumor colonization of biofilm-defective P. aeruginosa mutants. (a) Wild-type P. aeruginosa PA14 and biofilm-defective mutants (PA14 pqsA::Tn and pelA::Tn) colonizing different tissues of mice. This experiment includes ≥5 mice per group and was performed at least two times. (b and c) Visualization of P. aeruginosa PA14 pqsA::Tn (b) and P. aeruginosa PA14 pelA::Tn (c) by FISH and TEM. Sections of tumor tissue samples were hybridized with a species-specific probe for P. aeruginosa (green) for FISH analysis. In TEM images, arrowheads indicate individual bacterial cells. No aggregate formation of bacteria and no visible matrix material were observed.
Efficacy of ciprofloxacin, colistin, and tobramycin against biofilm-grown P. aeruginosa.
Our test system to screen and characterize antibiofilm antibiotics consisted of the P. aeruginosa PA14 WT strain as well as the pqsA transposon mutant and was now extended by the pelA mutant, both of which are biofilm defective. The robustness of this system was challenged using anti-Pseudomonas antibiotics that are routinely used in clinics (ciprofloxacin, colistin, and tobramycin). Treatment of P. aeruginosa-infected tumor-bearing mice was initiated 48 h after infection. At this time point, PA14 and the two biofilm-deficient mutant isolates are expected to have reached CFU counts of approximately 108/g in the tumors and 100- to 1,000-fold lower counts in liver and gut. Antibiotics were administered i.v. in 3 individual doses at time intervals of 12 h before the mice were sacrificed, and CFU counts were determined for tumors as well as for liver and gut. Neither of the antibiotics successfully reduced bacterial counts of the PA14 wild type in the tumor tissue, despite treatment with a dose of 5 mg/kg ciprofloxacin, 5 mg/kg colistin, or 4 mg/kg tobramycin, respectively (Fig. 3a to c). On the other hand, ciprofloxacin, colistin, and tobramycin reduced the CFU counts very effectively in liver and gut, where lower bacterial numbers are present and the bacteria presumably do not form biofilms (Fig. 3a to c). In contrast, all three antibiotics proved to be effective against the two biofilm-defective mutants (Fig. 3a to c). We observed not only a reduction of CFU counts in liver and gut comparable to the wild type but also a significant reduction of bacterial counts in the tumor tissue (Fig. 3a to c). Interestingly, when the treatment doses of the antibiotics were increased to 10 mg/kg, the CFU counts of all strains, including the WT, were reduced at comparable rates even in the tumor (Fig. 4) (data not shown).
FIG 3.

Effect of antibiotics against biofilm-sufficient and biofilm-defective P. aeruginosa in vivo. Tumor-bearing mice were infected with PA14 WT, PA14 pqsA::Tn, and pelA::Tn. Forty-eight hours postinfection (p.i.), mice were treated i.v. with ciprofloxacin (CIP) at a dose of 5 mg/kg (a), colistin (CST) at a dose of 5 mg/kg (b), and tobramycin (TOB) at a dose of 4 mg/kg (c). Control mice received 0.9% saline solution i.v. as a placebo (PLA). One exemplary experiment out of at least three is presented, with n ≥ 4. **, P < 0.005; ***, P < 0.0005.
FIG 4.
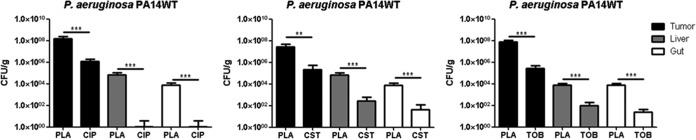
High antimicrobial doses kill biofilm P. aeruginosa in vivo. Tumor-bearing mice were infected with the PA14 WT strain, and at 48 h p.i., mice were treated with ciprofloxacin (CIP), colistin (CST), and tobramycin (TOB) i.v. at a dose of 10 mg/kg. Control mice were injected with 0.9% saline solution as a placebo (PLA). One exemplary experiment out of at least three is presented, with n ≥ 5. **, P < 0.005; ***, P < 0.0005.
Colistin-tobramycin combination therapy.
Colistin-tobramycin combinations have been shown previously to be superior to monotherapy against biofilm-grown P. aeruginosa in an in vitro system as well as in a rat lung infection model and in CF patients (22). We therefore tested the ability of colistin-tobramycin combinations to kill P. aeruginosa biofilms in our test system. Tumor-bearing mice were infected with the PA14 WT strain and the pqsA transposon mutant. Two days postinfection, the antibiotic treatment was initiated: colistin alone, tobramycin alone, or colistin and tobramycin in combination. Colistin and tobramycin alone did not show a significant effect on the P. aeruginosa PA14 WT strain (Fig. 5b and c). Therapy was clearly more effective for the pqsA transposon mutant defective in biofilm formation, despite a reduction of the administration dose to 2.5 mg/kg colistin and tobramycin, respectively. Again, the bacterial counts of liver and gut from antibiotic-treated groups were reduced significantly by 100- to 1,000-fold. Most interestingly, we found a significant reduction of WT bacterial counts from tumors when treated with a combination of colistin and tobramycin despite low individual antimicrobial doses (Fig. 5a).
FIG 5.

Effective combination therapy against biofilm-producing P. aeruginosa and the biofilm-defective pqsA::Tn mutant. Tumor-bearing mice were infected with PA14 WT or pqsA::Tn, and at 48 h p.i., mice were treated with 2.5 mg/kg of colistin and tobramycin alone or in combination. (a) Colistin-tobramycin in combination (CST+TOB). (b) Colistin only (CST). (c) Tobramycin only (TOB). Control mice were injected with 0.9% saline solution as a placebo (PLA). One exemplary experiment out of at least three is presented, with n ≥ 5. *, P < 0.05; **, P < 0.005; ***, P < 0.0005.
DISCUSSION
Despite intensive investigations, we are far from understanding the molecular mechanisms that underlie tolerance of biofilm bacteria to a large variety of adverse environmental conditions, including antimicrobial therapy and attacks by the immune system. Although intensified antimicrobial treatment is applied, chronic P. aeruginosa infections are rarely eradicated. In an attempt to more efficiently treat P. aeruginosa infections in CF, many European CF care centers start antibiotic treatment early after the first detection of the pathogen (32). Nevertheless, this did not eliminate chronic P. aeruginosa infections in CF patients. Therefore, alternative therapeutic strategies are needed. The use of combination therapy involving antibiotics with different modes of action against P. aeruginosa is one option in this context. Another option is to foster screens for novel compounds with an effective antibiofilm activity. However, the identification of an antibiofilm activity by in vitro systems needs to be followed by a series of steps that include the demonstration of efficacy in a suitable animal infection model (14).
Here, we demonstrate that our tumor model offers a robust and versatile experimental system to monitor the impact of antimicrobials against biofilm-producing P. aeruginosa. Matchable tumor growth and equivalent colonization of the tumor tissue with biofilm-forming bacteria can be achieved if standardized protocols are followed. We believe that the invasion of the tumors by P. aeruginosa is passive. Most likely, TNF-α produced early during the cytokine storm elicited by the bacterial application is opening the pathological blood vessels of the tumor. TNF-α is known for such activity and might preferentially act on the already leaky blood vessels of the tumor, as shown for Salmonella before (33). With the inflowing blood, the bacteria reach the tumor tissue and proliferate. Although the microorganisms reside among large numbers of immigrating neutrophilic granulocytes, these phagocytes did not harm the bacteria under the hypoxic conditions of the tumor. On the other hand, they might force the bacteria to form biofilms as a protective response to reactive oxygen species (ROS) or antimicrobial peptides produced by these immune cells.
Most importantly, bacteria that in vitro are non-biofilm producing colonize the tumor tissue at similar CFU counts as WT bacteria. We could show that two disparate bacterial factors important for biofilm formation in vitro are also of relevance in vivo. The PA14 pqsA transposon mutant is not able to synthesize 4-quinolone signal molecules involved in quorum sensing. The variant pelA is unable to synthesize sugars required for formation of the extracellular matrix (25). None of the variant bacteria exhibiting a defect in forming biofilms in vitro was able to form such structures in the tumor. This fact is based on electron microscopic analysis of tumor-colonizing P. aeruginosa. Thus, it is possible to specifically track biofilm-associated characteristics in vivo. This allows the evaluation of the efficacy of classical antimicrobial agents and combinations thereof, as well as novel active compounds against P. aeruginosa biofilm infections and to dissect in vivo antibacterial from antibiofilm activities.
Here, we tested three commonly used antibiotics, ciprofloxacin, colistin, and tobramycin, in the concentrations that were equivalent to doses used for human patients. Both biofilm-defective variants were sensitive against the activity of all three antibiotics, while the CFU counts of biofilm-forming wild-type bacteria in the tumor tissue did not decline. However, wild-type bacteria were sensitive to such antibiotics when not residing in biofilms, as could be concluded from the data obtained for liver and gut. Here, the CFU counts were strongly reduced as expected. Although the difference in CFU counts of wild-type bacteria in the tumor compared with those of the variants was sometimes only 10-fold, the results were reproducible. In addition, only three applications of the antibiotics were performed. A more extended application would most likely lead to a stronger reduction of the biofilm-defective mutants, while the wild-type bacteria are expected not to be affected. Thus, the most probable explanation for the differential resistance of the bacteria is indeed the ability to form biofilms. Since the two variants that are unable to form biofilms and the wild type are equally sensitive to the three antimicrobials when tested under planktonic conditions, we can conclude that the tumor model has proven to be a valid test model to dissect antibiotic sensitivity of biofilm P. aeruginosa.
The key advantages of this model are that first, the model mimics the natural steps of biofilm formation during the establishment of biofilm-associated P. aeruginosa infections. Second, the niche within the tumor largely reflects environmental characteristics of the chronically infected CF respiratory tract. The in vivo transcription profile of wild-type P. aeruginosa isolated from tumors was previously shown to match that of isolates from CF lungs and lesions of patients suffering from burn wounds (21). This provides evidence that the microenvironments of colonized solid tumors and CF lungs or burn wounds are very similar. Third, the transferability of in vitro findings on biofilm developmental processes to the in vivo situation has been demonstrated. The model therefore allows us to reconsider currently used therapies of chronic P. aeruginosa biofilm infections—e.g., in CF patients.
Biofilm infection by P. aeruginosa in lungs of CF patients is the leading cause of their morbidity and leads finally to functional failure of this organ (34, 35). Current therapies are facing serious problems due to antibiotic tolerance and resistance against therapy. Thus, this life-shortening infection needs the development of efficient treatment. Antibiotics combination therapy might be more effective under certain circumstances than monotherapies. For instance, a penicillin-aminoglycoside combination is able to eradicate P. aeruginosa from biofilms more efficiently than monotherapy (36, 37). In accordance, tobramycin and colistin showed promising activity against P. aeruginosa biofilms. This combination of antibiotics was able to eliminate biofilm-residing bacteria of different stages (13). Similarly, the complementary activity of colistin and tobramycin was reported to be superior in the treatment of chronically P. aeruginosa-infected CF patients (22). In our tumor model, the synergistic effect of the combination of colistin with tobramycin could be confirmed, although at antimicrobial concentrations that are far below of those required for an effective monotherapy, the combination of colistin and tobramycin significantly reduced the numbers of biofilm-forming WT bacteria in the tumor.
In conclusion, our model will be ideal to investigate steps of natural biofilm formation under in vivo conditions and confirm the importance of bacterial factors that have been shown to be critical for biofilm formation processes in in vitro systems. The model is also applicable for unraveling the complex interactions that occur during P. aeruginosa coinfections with other bacteria—typically with Staphylococcus aureus or anaerobes (38). Furthermore, it is possible to evaluate the efficacy of classical antimicrobial agents against P. aeruginosa biofilm infections and optimize the combinations and application regimens thereof. Moreover, in the last decade, great efforts have been put forth to find and characterize novel antimicrobial/biofilm compounds (18). Therefore, it becomes clear that murine solid tumors represent a versatile, easy to handle, and robust model system that should help to improve treatment of infections that represent a real threat in the clinics.
Supplementary Material
ACKNOWLEDGMENTS
Deutsche Krebshilfe, DFG, and BMBF supported this work. The Indo-German Science Centre for Infectious Diseases (IG-SCID) supported V.P. The HZI Grad School supported V.P. and N.K.
We thank Martina Krey, Regina Lesch, Susanne zur Lage, Ina Schleicher, Dino Kocijancic, and Anna-Maria Wiejak for assistance.
S.H., S.W., and V.P. wrote the manuscript. V.P., U.K., N.K., P.B., B.G., M.C.P., A.M., and M.R. did the experimental work. V.P., N.K., P.B., S.H., and S.W. designed experiments and analyzed experimental data.
The authors declare they have no competing financial interests.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.00194-15.
REFERENCES
- 1.Kumar A, Roberts D, Wood KE, Light B, Parrillo JE, Sharma S, Suppes R, Feinstein D, Zanotti S, Taiberg L, Gurka D, Kumar A, Cheang M. 2006. Duration of hypotension before initiation of effective antimicrobial therapy is the critical determinant of survival in human septic shock. Crit Care Med 34:1589–1596. doi: 10.1097/01.CCM.0000217961.75225.E9. [DOI] [PubMed] [Google Scholar]
- 2.Lodise TP, McKinnon PS, Swiderski L, Rybak MJ. 2003. Outcomes analysis of delayed antibiotic treatment for hospital-acquired Staphylococcus aureus bacteremia. Clin Infect Dis 36:1418–1423. doi: 10.1086/375057. [DOI] [PubMed] [Google Scholar]
- 3.Carmeli Y, Troillet N, Eliopoulos GM, Samore MH. 1999. Emergence of antibiotic-resistant Pseudomonas aeruginosa: comparison of risks associated with different antipseudomonal agents. Antimicrob Agents Chemother 43:1379–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stewart PS, Costerton JW. 2001. Antibiotic resistance of bacteria in biofilms. Lancet 358:135–138. doi: 10.1016/S0140-6736(01)05321-1. [DOI] [PubMed] [Google Scholar]
- 5.Costerton W, Veeh R, Shirtliff M, Pasmore M, Post C, Ehrlich G. 2003. The application of biofilm science to the study and control of chronic bacterial infections. J Clin Invest 112:1466–1477. doi: 10.1172/JCI20365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hall-Stoodley L, Stoodley P. 2009. Evolving concepts in biofilm infections. Cell Microbiol 11:1034–1043. doi: 10.1111/j.1462-5822.2009.01323.x. [DOI] [PubMed] [Google Scholar]
- 7.Fux CA, Costerton JW, Stewart PS, Stoodley P. 2005. Survival strategies of infectious biofilms. Trends Microbiol 13:34–40. doi: 10.1016/j.tim.2004.11.010. [DOI] [PubMed] [Google Scholar]
- 8.Wilkins M, Hall-Stoodley L, Allan RN, Faust SN. 2014. New approaches to the treatment of biofilm-related infections. J Infect 69(Suppl 1):S47–S52. doi: 10.1016/j.jinf.2014.07.014. [DOI] [PubMed] [Google Scholar]
- 9.Bjarnsholt T, Ciofu O, Molin S, Givskov M, Høiby N. 2013. Applying insights from biofilm biology to drug development—can a new approach be developed? Nat Rev Drug Discov 12:791–808. doi: 10.1038/nrd4000. [DOI] [PubMed] [Google Scholar]
- 10.Davies D. 2003. Understanding biofilm resistance to antibacterial agents. Nat Rev Drug Discov 2:114–122. doi: 10.1038/nrd1008. [DOI] [PubMed] [Google Scholar]
- 11.Mah TC, O'Toole GA. 2001. Mechanisms of biofilm resistance to antimicrobial agents. Trends Microbiol 9:34–39. doi: 10.1016/S0966-842X(00)01913-2. [DOI] [PubMed] [Google Scholar]
- 12.Brooun A, Liu S, Lewis K, Lewis KIM. 2000. A dose-response study of antibiotic resistance in Pseudomonas aeruginosa biofilms. Antimicrob Agents Chemother 44:640–646. doi: 10.1128/AAC.44.3.640-646.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pamp SJ, Gjermansen M, Johansen HK, Tolker-Nielsen T. 2008. Tolerance to the antimicrobial peptide colistin in Pseudomonas aeruginosa biofilms is linked to metabolically active cells, and depends on the pmr and mexAB-oprM genes. Mol Microbiol 68:223–240. doi: 10.1111/j.1365-2958.2008.06152.x. [DOI] [PubMed] [Google Scholar]
- 14.Bragonzi A. 2010. Murine models of acute and chronic lung infection with cystic fibrosis pathogens. Int J Med Microbiol 300:584–593. doi: 10.1016/j.ijmm.2010.08.012. [DOI] [PubMed] [Google Scholar]
- 15.Walker TS, Tomlin KL, Worthen GS, Poch KR, Lieber JG, Saavedra MT, Fessler MB, Malcolm KC, Vasil ML, Nick JA. 2005. Enhanced Pseudomonas aeruginosa biofilm development mediated by human neutrophils. Infect Immun 73:3693–3701. doi: 10.1128/IAI.73.6.3693-3701.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klepac-Ceraj V, Lemon KP, Martin TR, Allgaier M, Kembel SW, Knapp AA, Lory S, Brodie EL, Lynch SV, Bohannan BJM, Green JL, Maurer BA, Kolter R. 2010. Relationship between cystic fibrosis respiratory tract bacterial communities and age, genotype, antibiotics and Pseudomonas aeruginosa. Environ Microbiol 12:1293–1303. doi: 10.1111/j.1462-2920.2010.02173.x. [DOI] [PubMed] [Google Scholar]
- 17.van Heeckeren AM, Schluchter MD. 2002. Murine models of chronic Pseudomonas aeruginosa lung infection. Lab Anim 36:291–312. doi: 10.1258/002367702320162405. [DOI] [PubMed] [Google Scholar]
- 18.Pawar V, Crull K, Komor U, Kasnitz N, Frahm M, Kocijancic D, Westphal K, Leschner S, Wolf K, Loessner H, Rohde M, Häussler S, Weiss S. 2014. Murine solid tumours as a novel model to study bacterial biofilm formation in vivo. J Intern Med 276:130–139. doi: 10.1111/joim.12258. [DOI] [PubMed] [Google Scholar]
- 19.Crull K, Rohde M, Westphal K, Loessner H, Wolf K, Felipe-López A, Hensel M, Weiss S. 2011. Biofilm formation by Salmonella enterica serovar Typhimurium colonizing solid tumours. Cell Microbiol 13:1223–1233. doi: 10.1111/j.1462-5822.2011.01612.x. [DOI] [PubMed] [Google Scholar]
- 20.Komor U, Bielecki P, Loessner H, Rohde M, Wolf K, Westphal K, Weiss S, Häussler S. 2012. Biofilm formation by Pseudomonas aeruginosa in solid murine tumors—a novel model system. Microbes Infect 14:951–958. doi: 10.1016/j.micinf.2012.04.002. [DOI] [PubMed] [Google Scholar]
- 21.Bielecki P, Komor U, Bielecka A, Müsken M, Puchałka J, Pletz MW, Ballmann M, Martins dos Santos VA, Weiss S, Häussler S. 2013. Ex vivo transcriptional profiling reveals a common set of genes important for the adaptation of Pseudomonas aeruginosa to chronically infected host sites. Environ Microbiol 15:570–587. doi: 10.1111/1462-2920.12024. [DOI] [PubMed] [Google Scholar]
- 22.Herrmann G, Yang L, Wu H, Song Z, Wang H, Høiby N, Ulrich M, Molin S, Riethmüller J, Döring G. 2010. Colistin-tobramycin combinations are superior to monotherapy concerning the killing of biofilm Pseudomonas aeruginosa. J Infect Dis 202:1585–1592. doi: 10.1086/656788. [DOI] [PubMed] [Google Scholar]
- 23.Diggle SP, Winzer K, Chhabra SR, Worrall KE, Cámara M, Williams P. 2003. The Pseudomonas aeruginosa quinolone signal molecule overcomes the cell density-dependency of the quorum sensing hierarchy, regulates rhl-dependent genes at the onset of stationary phase and can be produced in the absence of LasR. Mol Microbiol 50:29–43. doi: 10.1046/j.1365-2958.2003.03672.x. [DOI] [PubMed] [Google Scholar]
- 24.Müsken M, Di Fiore S, Dötsch A, Fischer R, Häussler S. 2010. Genetic determinants of Pseudomonas aeruginosa biofilm establishment. Microbiology 156:431–441. doi: 10.1099/mic.0.033290-0. [DOI] [PubMed] [Google Scholar]
- 25.Friedman L, Kolter R. 2004. Genes involved in matrix formation in Pseudomonas aeruginosa PA14 biofilms. Mol Microbiol 51:675–690. [DOI] [PubMed] [Google Scholar]
- 26.Liberati NT, Urbach JM, Miyata S, Lee DG, Drenkard E, Wu G, Villanueva J, Wei T, Ausubel FM. 2006. An ordered, nonredundant library of Pseudomonas aeruginosa strain PA14 transposon insertion mutants Proc Natl Acad Sci U S A 103:2833–2838. doi: 10.1073/pnas.0511100103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mallmann C, Siemoneit S, Schmiedel D, Petrich A, Gescher DM, Halle E, Musci M, Hetzer R, Göbel UB, Moter A. 2010. Fluorescence in situ hybridization to improve the diagnosis of endocarditis: a pilot study. Clin Microbiol Infect 16:767–773. doi: 10.1111/j.1469-0691.2009.02936.x. [DOI] [PubMed] [Google Scholar]
- 28.Moter A, Leist G, Rudolph R, Schrank K, Choi B-K, Wagner M, Göbel UB. 1998. Fluorescence in situ hybridization shows spatial distribution of as yet uncultured treponemes in biopsies from digital dermatitis lesions. Microbiology 144:2459–2467. doi: 10.1099/00221287-144-9-2459. [DOI] [PubMed] [Google Scholar]
- 29.Braun-Howland EB, Vescio PA, Nierzwicki-Bauer SA. 1993. Use of a simplified cell blot technique and 16S rRNA-directed probes for identification of common environmental isolates. Appl Environ Microbiol 59:3219–3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Römling U, Rohde M, Olsén A, Normark S, Reinköster J. 2000. AgfD the checkpoint of multicellular and aggregative behaviour in Salmonella typhimurium regulates at least two independent pathways. Mol Microbiol 36:10–23. doi: 10.1046/j.1365-2958.2000.01822.x. [DOI] [PubMed] [Google Scholar]
- 31.Crull K, Bumann D, Weiss S. 2011. Influence of infection route and virulence factors on colonization of solid tumors by Salmonella enterica serovar Typhimurium. FEMS Immunol Med Microbiol 62:75–83. doi: 10.1111/j.1574-695X.2011.00790.x. [DOI] [PubMed] [Google Scholar]
- 32.Döring G, Hoiby N. 2004. Early intervention and prevention of lung disease in cystic fibrosis: a European consensus. J Cyst Fibros 3:67–91. [DOI] [PubMed] [Google Scholar]
- 33.Leschner S, Westphal K, Dietrich N, Viegas N, Jablonska J, Lyszkiewicz M, Lienenklaus S, Falk W, Gekara N, Loessner H, Weiss S. 2009. Tumor invasion of Salmonella enterica serovar Typhimurium is accompanied by strong hemorrhage promoted by TNF-α. PLoS One 4:e6692. doi: 10.1371/journal.pone.0006692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lyczak JB, Cannon CL, Pier GB. 2002. Lung infections associated with cystic fibrosis. Clin Microbiol Rev 15:194–222. doi: 10.1128/CMR.15.2.194-222.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rajan S, Saiman L. 2002. Pulmonary infections in patients with cystic fibrosis. Semin Respir Infect 17:47–56. doi: 10.1053/srin.2002.31690. [DOI] [PubMed] [Google Scholar]
- 36.Christensen LD, van Gennip M, Jakobsen TH, Alhede M, Hougen HP, Høiby N, Bjarnsholt T, Givskov M. 2012. Synergistic antibacterial efficacy of early combination treatment with tobramycin and quorum-sensing inhibitors against Pseudomonas aeruginosa in an intraperitoneal foreign-body infection mouse model. J Antimicrob Chemother 67:1198–1206. doi: 10.1093/jac/dks002. [DOI] [PubMed] [Google Scholar]
- 37.Høiby N. 2011. Recent advances in the treatment of Pseudomonas aeruginosa infections in cystic fibrosis. BMC Med 9:32. doi: 10.1186/1741-7015-9-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pustelny C, Komor U, Pawar V, Lorenz A, Bielecka A, Moter A, Gocht B, Eckweiler D, Müsken M, Grothe C, Lünsdorf H, Weiss S, Häussler S. 2015. Contribution of Veillonella parvula to Pseudomonas aeruginosa-mediated pathogenicity in a murine tumor model system. Infect Immun 83:417–429. doi: 10.1128/IAI.02234-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.