

Abstract
Nonnucleoside reverse transcriptase inhibitors (NNRTIs) are important components of the highly active antiretroviral therapy (HAART) used to treat human immunodeficiency type 1 virus (HIV-1). However, because of the emergence of drug resistance and the adverse effects of current anti-HIV drugs, it is essential to develop novel NNRTIs with an excellent safety profile, improved activity against NNRTI-resistant viruses, and enhanced activity against clinical isolates of different subtypes. Here, we have identified 1-[(benzyloxy)methyl]-6-(3,5-dimethylbenzyl)-5-iodopyrimidine-2,4(1H,3H)-dione (WPR-6), a novel NNRTI with a 50% effective concentration (EC50) of 2 to 4 nM against laboratory-adapted HIV-1 strain SF33 and an EC50 of 7 to 14 nM against nucleoside reverse transcriptase inhibitor-resistant HIV-1 strain 7391 with a therapeutic index of >1 × 104. A panel of five representative clinical virus isolates of different subtypes circulating predominantly in China was highly sensitive to WPR-6, with EC50s ranging from 1 to 6 nM. In addition, WPR-6 showed excellent antiviral potency against the most prevalent NNRTI-resistant viruses containing the K103N and Y181C mutations. To determine whether WPR-6 selects for novel resistant mutants, in vitro resistance selection was conducted with laboratory-adapted HIV-1 strain SF33 on MT-4 cells. The results demonstrated that V106I and Y188L were the two dominant NNRTI-associated resistance mutations detected in the breakthrough viruses. Taken together, these in vitro data indicate that WPR-6 has greater efficacy than the reference HEPT analogue TNK651 and the marketed drug nevirapine against HIV-1. However, to develop it as a new NNRTI, further improvement of its pharmacological properties is warranted.
INTRODUCTION
Antiretroviral therapy (ART) has been successful in controlling HIV-1 replication, especially in suppressing HIV loads, restoring immune function, and improving longevity and the quality of life (1). Although ART controls actively replicating HIV, the virus persists in a stable latent reservoir in infected cells, and this remains the major barrier to HIV eradication (2). Recently, it has been shown that early initiation of ART can reduce the size of the latent reservoir, thus contributing to the possible curing of infected individuals by future eradication strategies (3, 4). In addition, emerging data have suggested that earlier ART initiation may prevent mother-to-child (5) and sexual (6) transmission, and this treatment-as-prevention approach is cost-effective (7). The highly active antiretroviral therapy (HAART) regimen, which is typically composed of three or more drugs with complementary mechanisms of action, has resulted in a dramatic increase in the life expectancy of patients infected with HIV-1. Consensus guidelines of HAART recommend the use of two nucleoside reverse transcriptase inhibitors (NRTIs) in combination with a non-NRTI (NNRTI), a protease inhibitor (PI), or an integrase inhibitor (2). HIV-1 reverse transcriptase (RT) is essential to the HIV-1 replication cycle, and it has no homologue in eukaryotic organisms (8, 9). Therefore, RT is an attractive target for the development of antiretroviral drug therapies against HIV-1 infection and AIDS. Two functionally distinct classes of HIV-1 RT inhibitors, nucleoside and nonnucleoside, have been discovered and are being used clinically. As important components of HAART, NNRTIs have gained a definite place in clinical use as a result of their unique antiviral potency based on a wide range of chemically diverse structures, favorable safety profiles, and high efficacy (10, 11). To date, five NNRTIs have been approved for clinical use: nevirapine (NVP) (12), delavirdine (DLV) (13), efavirenz (EFV) (14), etravirine (ETR) (15), and rilpivirine (RPV) (16).
Although NNRTIs can be key components of effective combination regimens, the therapeutic efficacy of NNRTIs is weakened by the very rapid development of drug-resistant mutants, in vitro and in vivo, both in monotherapy and in multidrug trials. A single amino acid change can lead to significantly reduced susceptibility, often to all of the available inhibitors within the same class (17–19). For instance, the two resistance-associated substitutions most prevalent today, K103N and Y181C, have reduced the efficacy of earlier NNRTIs (NVP, DLV, and EFV) (20). Although the currently approved NNRTIs ETR and RPV have made improvements in the treatment of HIV-1 cases, adverse effects and drug resistance continue to emerge in patients receiving ART (21). These data underscore the need to develop new NNRTIs as antiretroviral agents with improved resistance profiles and tolerability.
Great genetic diversity among HIV-1 subtypes (clades A to H, J, and K) and intersubtype recombinant strains in different geographic regions has been identified (22). The frequency and pattern of mutations that confer resistance to antiretroviral drugs differ among HIV-1 subtypes. Some in vitro and in vivo observations suggest that the various subtypes may display different levels of susceptibility to certain antiretroviral drugs, which could influence therapeutic outcomes (23). Therefore, the development of new drugs with strong antiviral activity independent of subtype will be important for improving the current HAART.
Extensive efforts have been made to develop novel NNRTIs possessing the desired antiviral potency and safety profile, especially for viruses carrying the K103N or Y181C substitution (24). In our previous study, 1-[(benzyloxy)methyl]-6-(3,5-dimethylbenzyl)-5-iodopyrimidine-2,4(1H,3H)-dione (Fig. 1) was identified as a novel HIV-1 RT inhibitor (50% inhibitory concentration [IC50] of 3 nM) through synthetic efforts based on modification of the C-6 benzyl moiety of the HEPT analogues (25). An excellent correlation was found between 50% effective concentrations (EC50s) for inhibition of HIV-1 infection and IC50s for inhibition of HIV-1 RT activity, confirming that WPR-6 acts as an inhibitor of HIV-1 RT. Here, we report on the in vitro antiviral activity of WPR-6 against wild-type (WT) and broader NNRTI-resistant HIV-1 strains, including viruses carrying the K103N and/or Y181C substitutions. To further explore the breadth and target mechanisms of the antiviral activity of WPR-6, we examined HIV strains of different subtypes circulating in China and performed an induction assay to clarify the interaction of the compound with the HIV pol sequence.
FIG 1.
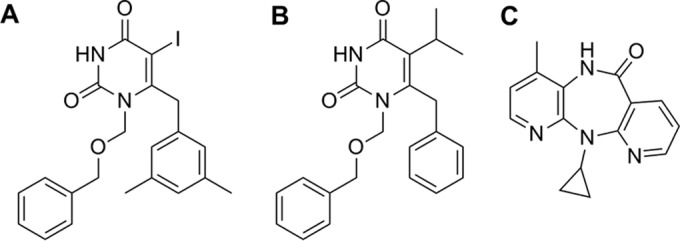
Chemical structures of WPR-6 (A), TNK651 (B), and NVP (C).
MATERIALS AND METHODS
Compounds.
WPR-6 is 1-[(benzyloxy)methyl]-6-(3,5-dimethylbenzyl)-5-iodopyrimidine-2,4(1H,3H)-dione; its molecular weight is 476.31, and its chemical formula is C21H21IN2O3. TNK651 is 6-benzyl-1-[(benzyloxy)methyl]-5-isopropylpyrimidine-2,4(1H,3H)-dione; its molecular weight is 364.44, and its chemical formula is C22H24N2O3. Both WPR-6 and TNK651 were synthesized at the Peking University Health Science Center as described previously (25). NVP was purchased from Shanghai DESANO Chemical Pharmaceutical Co., Ltd. For antiviral activity assays, inhibitors were dissolved in dimethyl sulfoxide (DMSO). The structures of the compounds are shown in Fig. 1.
Cells, viruses, and reagents.
TZM-bl and MT-4 cells and HIV-1 strains SF33 and 7391 were all obtained from the NIH AIDS Research and Reference Reagent Program (NIH, Bethesda, MD, USA). MT-4 and TZM-bl cells, respectively, were maintained in RPMI 1640 and Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin. PBMCs (peripheral blood mononuclear cells) from HIV-1-seronegative blood donors were isolated by standard density gradient centrifugation with Ficoll-Paque PLUS density gradient medium (GE Healthcare Biosciences Corp., Piscataway, NJ). The isolated PBMCs were cultured with phytohemagglutinin-stimulated PBMCs in RPMI 1640 medium supplemented with 10% fetal calf serum, 100 U/ml penicillin, 100 mg/ml streptomycin, 2.9 mg/ml l-glutamine, and 100 IU recombinant human interleukin-2 (rhIL-2; Roche Diagnostics). The Chinese clinical viruses were isolated from treatment-naive patients infected with the HIV-1 CRF07_BC and B′ strains circulating in China (26).
Site-directed mutagenesis assay.
Mutations encoding Y181C and K103A were introduced into the T-vector containing HIV RT gene regions by use of DNA polymerase (PrimerSTAR; TaKaRa) and proper site mutation primers, followed by digestion with BstEII and AgeI (NEB), purification by agarose gel electrophoresis, and ligation to BstEII- and AgeI-digested pNL4-3. DNA sequencing was performed in both directions across the entire RT coding region to verify the absence of spurious mutations and the presence of the desired mutations (27).
WT HIV-1 and HIV-1 with mutations were generated by transfection of plasmids into 293T/17 cells with Fugene 6 Transfection Reagent (Roche Applied Science) according to the manufacturer's instructions. Viral supernatants were harvested at 48 h posttransfection, filtered through a 0.45-μm filter to remove cellular debris, aliquoted, and stored at −80°C. The 50% tissue culture infective dose (TCID50) of a single thawed aliquot of each virus was determined in TZM-bl cells as described previously. The virus stock was quickly thawed; diluted with DMEM supplemented with 10% fetal calf serum, 100 U/ml penicillin, 100 mg/ml streptomycin, and 2.9 mg/ml l-glutamine; and added to a 96-well tissue culture plate in quadruplicate. TZM-bl cells were resuspended in complete medium with 30 mg/ml DEAE added and dispensed into all of the wells at 1 × 104 cells/well. After incubation for 48 h at 37°C and 5% CO2, luciferase activity was measured with luciferase assay reagent (Promega) and a Luminescence Counter (Perkin-Elmer) according to the manufacturers' instructions.
Assay of the inhibitory activity of compounds on HIV-1 SF33, 7391, and A17.
MT-4 cells and HIV-1 SF33, 7391, and A17 were obtained from the NIH AIDS Research and Reference Reagent Program (NIH, Bethesda, MD, USA). The inhibitory activities of the compounds on laboratory-adapted HIV-1 strain SF33, NRTI-resistant HIV-1 strain 7391, and NNRTI-resistant HIV-1 strain A17 were tested in TZM-bl cells, MT-4 cells, and PBMCs, respectively, as previously described (28). Briefly, cells (4 × 104/well) were infected by the addition of 200 TCID50s of HIV-1, followed by incubation for 2 h at 37°C before the addition of compounds at serial dilutions. After further incubation at 37°C for 7 days, p24 was measured with a commercial enzyme-linked immunosorbent assay kit (Vironostika HIV-1 Plus O Microelisa System; bioMérieux, Marcy l'Etoile, France). The EC50 was determined by nonlinear regression with GraphPad Prism 5.01.
Cytotoxicity assay.
The 2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide (XTT) assay was used as previously described to assess the cytotoxicity of the compounds tested for anti-HIV-1 activity. Briefly, graded concentrations of inhibitors were added to MT-4 cells, PBMCs, and TZM-bl cells at 5 × 104/well and incubated at 37°C for 3 days. Ten microliters of CCK-8 reagent was added to the cells. After incubation at 37°C for 4 h to allow color development of the XTT formazan product, the absorbance at 450 nm of each well was read in a Victor2 1420 Multilabel Counter (Wallace-PerkinElmer Life and Analytical Sciences Inc., Boston, MA). The percent cytotoxicity and CC50 (50% cytotoxic concentration) were calculated as previously described (28).
Assay to identify new drug-resistant mutants.
The in vitro selection of HIV-1 resistance to WPR-6 was performed as described previously (29, 30). Briefly, MT-4 cells were seeded at 1 × 104/ml of RPMI 1640 medium containing 10% fetal bovine serum into the wells of 12-well plates. The molecular clone of HIV-1 SF33 was used to infect the cells in the presence or absence of the inhibitor at an initial concentration of 2 TCID50s. The cells were incubated at 37°C with 5% CO2 until an extensive cytopathic effect was observed. Virus replication was monitored by observing the formation of syncytia by optical microscopy. At each virus breakthrough (massive syncytium formation), the inhibitor concentration was doubled. The culture supernatants were harvested and used for the next passage on fresh MT-4 cells. Cells and supernatants were harvested at regular time intervals and stored at −80°C. After an incubation period of 90 days, the final concentration of WPR-6 in the medium was 30 μM. For specific passage, viral RNA was extracted and purified from infected cells with the QIAamp Viral RNA kit (Qiagen).
Analysis and confirmation of mutations in the gene encoding RT.
RNA was extracted from the culture supernatant at different time point. The target sequence was amplified with One Step Reverse Transcription PCR reagents (Qiagen Inc., Hilden, Germany). Amplification steps were as follows: reverse transcription at 50°C for 30 min; predenaturation at 94°C for 5 min; 30 cycles of denaturation at 94°C for 30 s, annealing at 55°C for 30 s, and extension at 72°C for 2.5 min; and an additional extension at 72°C for 10 min. Nested PCR was performed with Taq PCR master mix (Qiagen Inc., Hilden, Germany). The cycling conditions were as follows: predenaturation at 94°C for 5 min; 30 cycles of denaturation at 94°C for 30 s, annealing at 63°C for 30 s, and extension at 72°C 2.5 min; and an additional extension at 72°C for 10 min. PCR products were visualized by 1% agarose gel electrophoresis and sequenced with the ABI 3730xl Automated DNA Analyzer (Applied Biosystems, Foster City, CA). Each step was carried out with negative controls (31). Sequences of the pol gene were compared with the consensus sequence by using HIV db software (Stanford University HIV Drug Resistance Database [http://hivdb.stanford.edu/]) to detect the mutations associated with viral resistance to PIs, NRTIs, and NNRTIs.
After analysis of the sequences from before and after in vitro selection of HIV-1 resistance to WPR-6, the substituted residues were identified. HIV-1 with the mutations was constructed by the site-directed mutagenesis method, and the WT HIV-1 strain was used as a control. The TCID50 of each virus was determined in TZM-bl cells as described previously.
RESULTS
WPR-6 has low toxicity for different cells used to test antiviral activity.
Cell growth inhibition (cytotoxicity) assessments of WPR-6 were done with human PBMCs, human T-cell leukemia virus type 1-immortalized leukemia cell line MT-4, and TZM-bl cells with TNK651 and NVP as reference compounds by the previously reported XTT assay (28). The toxicity of the compounds tested for these cells was demonstrated as CC50s and CC90s. WPR-6 did not display obvious toxicity to any of these cell types, with CC50s of 98.31, 137.80, and 76.41 μM on MT-4 cells, PBMCs, and TZM-bl cells, respectively. The reference compound TNK651 also displayed low cytotoxicity, with CC50s of 84.60, 166.57, and 65.81 μM on these same cell lines, respectively (Table 1). NVP did not display toxicity to any of the cell types tested, even at a high concentration of 1,000 μM. The CC50s of each compound were determined by using more than five different concentrations.
TABLE 1.
CC50s and CC90s of WPR-6, TNK651, and NVP for different cellsa
| Cell type | CC50, CC90 (μM)b |
||
|---|---|---|---|
| WPR-6 | TNK-651 | NVP | |
| MT-4 | 98.3 ± 2.7, 286.2 ± 4.0 | 84.6 ± 1.5, 166.6 ± 1.7 | >1,000, >1,000 |
| PBMCs | 137.8 ± 8.2, 297.6 ± 14.7 | 166.6 ± 1.7, 291.7 ± 6.0 | >1,000, >1,000 |
| TZM-bl | 76.4 ± 5.7, 360.4 ± 14.2 | 65.8 ± 0.7, 115.8 ± 2.2 | >1,000, >1,000 |
Each compound was tested in triplicate, and the data presented are mean values ± standard deviations from at least two independent experiments.
The XTT assay was used to determine CC50s and CC90s.
WPR-6 is effective at inhibiting laboratory-adapted strains of HIV-1 in different cell lines.
The activity of WPR-6 was evaluated in three fresh human cell lines, including PBMCs and TZM-bl and MT-4 cells, infected with commonly used laboratory-adapted HIV-1 strain SF33 and NRTI-resistant HIV-1 strain 7391. NVP and TNK651 were used as positive anti-HIV control compounds and were evaluated in parallel. The results are summarized in Table 2. WPR-6 was determined to be highly active against both HIV-1 strains SF33 and 7391 in MT-4 cells, PBMCs, and TZM-bl cells, exhibiting EC50s against HIV-1 SF33 ranging from 2 to 4 nM and EC50s against HIV-1 7391 ranging from 7 to 14 nM. The efficacy and cytotoxicity values obtained for WPR-6 yield a therapeutic index of >10,000. TNK651 inhibited the two virus isolates in these assays with EC50s ranging from 4 to 58 nM, which was about 2- to 10-fold and 4-fold weaker than WPR-6 against HIV-1 strains SF33 and 7391, respectively. In addition, NVP exhibited similar levels of activity against HIV-1 SF33 in PBMCs and TZM-bl cells and inhibited HIV-1 SF33 in MT-4 cells and HIV-1 7391 in PBMCs with EC50s of 68 and 143 nM, respectively. This result is consistent with the inhibitory activities of the drugs against HIV-1 RT (25), suggesting that WPR-6 demonstrated greater potency than the marketed nonnucleoside anti-HIV drug NVP in both RT-producing and laboratory-adapted HIV-1 strains.
TABLE 2.
Antiviral activity of WPR-6 against laboratory-adapted strains compared to that of other NNRTIs
| HIV-1 isolate and cell type | EC50 (μM)a (SI)b |
||
|---|---|---|---|
| WPR-6 | TNK651 | NVP | |
| SF33 | |||
| MT-4 | 0.004 ± 0.001 (24,578) | 0.016 ± 0.001 (3,845) | 0.068 ± 0.030 (>14,706) |
| PBMCs | 0.003 ± 0.001 (45,933) | <0.0024 (>41,643) | 0.690 ± 0.007 (>1,449) |
| TZM-bl | 0.002 ± 0.002 (38,205) | 0.022 ± 0.003 (2,991) | 0.634 ± 0.224 (>1,557) |
| 7391 | |||
| PBMCs | 0.007 ± 0.001 (19,686) | NDc (NCd) | 0.143 ± 0.005 (>6,993) |
| TZM-bl | 0.005 ± 0.001 (15,282) | 0.058 ± 0.047 (1,135) | ND (NC) |
EC50s against laboratory-adapted HIV-1 isolate SF33 and NRTI-resistant HIV-1 isolate 7391 were determined on MT-4 cells, TZM-bl cells, or PBMCs. Each compound was tested in triplicate, and the data presented are mean values ± standard deviations from at least two independent experiments.
SIs were calculated on the basis of the CC50s for PBMCs, MT-4 cells, and TZM-bl cells and the EC50 for inhibition of infection with HIV-1 SF33 and 7391, respectively. TNK651 and NVP were used as reference compounds.
ND, not determined.
NC, not calculated.
WPR-6 exhibits potential inhibitory activity against infection with primary HIV-1 isolates circulating in China.
B′ (Thailand B, a variant of subtype B), CRF_07BC, and CRF_01AE are three epidemic strains in China. Therefore, to verify the antiviral activity of WPR-6 against Chinese clinical isolates, B′, CRF_07BC, and CRF_01AE were chosen (32). WPR-6 was therefore tested in PBMCs and TZM-bl cells for antiviral activity against a panel of five representative clinical isolates of these subtypes and intersubtype recombinant strains obtained from drug-naive patients in Xinjiang, Beijing, and Anhui Provinces. The results are summarized in Table 3. All of the isolates of the representative subtypes tested were sensitive to WPR-6 at a nanomolar range (EC50s, 1 to 6 nM; EC90s, 7 to 302 nM), which was comparable to that of laboratory-adapted HIV-1 strain SF33 and NRTI-resistant HIV-1 strain 7391. The low toxicity of WPR-6 for TZM-bl cells and PBMCs yields high 50% selectivity indexes (SI50s; calculated by determining the CC50/EC50 ratio) of 10,000 to 80,000 and SI90s (calculated by determining the CC90/EC90 ratio) of about 1,000 to 60,000. It is noteworthy that the different cell-based assays displayed obviously different impacts of WPR-6 on the clinical isolates, as most of the isolates were tested on two different cell types, including PBMCs and TZM-bl cells. Specifically, WPR-6 exhibited greater potency against HIV-1 clinical isolates tested on TZM-bl cells than against HIV-1 clinical isolates tested on PBMCs, with EC90s ranging from 7 to 53 nM versus 103 to 302 nM, respectively. In addition to the cell type, it appears that the clinical isolates with either the CCR5 or the CXCR4 coreceptor for cell infection were susceptible to WPR-6, suggesting that the activity of WPR-6 was not dependent on virus tropism.
TABLE 3.
Activity of WPR-6 against clinical HIV-1 isolates circulating predominantly in China
| HIV-1 isolate | Virus subtype | Cell type | Tropisma | EC50 (μM)b | SI50c | EC90 (μM)b | SI90d |
|---|---|---|---|---|---|---|---|
| XJDC6291 | CRF07_BC | TZM-bl | CCR5 | 0.006 ± 0.001 | 12,735 | 0.025 ± 0.001 | 14,416 |
| XJDC6291 | CRF07_BC | PBMCs | CCR5 | 0.003 ± 0.001 | 45,933 | 0.103 ± 0.010 | 2,890 |
| 020100968 | B′ | TZM-bl | CCR5 | 0.004 ± 0.002 | 18,021 | 0.016 ± 0.003 | 22,329 |
| 020100968 | B′ | PBMCs | CCR5 | 0.002 ± 0.001 | 68,900 | 0.209 ± 0.014 | 1,424 |
| 020100311 | B′ | TZM-bl | CCR5/CXCR4 | 0.001 ± 0.001 | 76,410 | 0.007 ± 0.001 | 51,484 |
| 020100311 | B′ | PBMCs | CCR5/CXCR4 | 0.002 ± 0.001 | 68,900 | 0.119 ± 0.012 | 2,501 |
| CYM015 | CRF01_AE | TZM-bl | CCR5 | 0.001 ± 0.001 | 76,410 | 0.053 ± 0.009 | 6,800 |
| CYM015 | CRF01_AE | PBMCs | CCR5 | 0.006 ± 0.001 | 22,967 | 0.209 ± 0.014 | 1,424 |
| CYM033 | CRF01_AE | TZM-bl | CCR5 | 0.002 ± 0.001 | 39,386 | 0.014 ± 0.003 | 24,958 |
| CYM033 | CRF01_AE | PBMCs | CCR5 | 0.005 ± 0.001 | 27,560 | 0.302 ± 0.023 | 985 |
HIV-1 isolate uses CXCR4 (CXC chemokine receptor 4), CCR5 (chemokine receptor 5), or both for cell infection.
EC50s and EC90s were determined on the basis of the data from virus inhibition assays with fresh human PBMCs or TZM-bl cells. The experiment was performed in triplicate, and the data presented are mean values ± standard deviations from at least two independent experiments.
The SI is the ratio of the CC50s for PBMCs, MT-4 cells, and TZM-bl cells to the EC50s for inhibition of clinical HIV-1 isolates of different subtypes.
The SI is the ratio of the CC90s for PBMCs and TZM-bl cells to the EC90s for inhibition of clinical HIV-1 isolates of different subtypes.
WPR-6 can effectively inhibit NNRTI-resistant mutant HIV-1 strains in vitro.
As mentioned above, K103N and Y181C mutant viruses account for a majority of the NNRTI-resistant mutants detected in patients whose therapy with antiretroviral regimens containing NNRTIs failed. Therefore, WPR-6 was tested in the luminescence reduction assay against HIV-1 strains with site-directed mutations (SDMs) causing single or double amino acid substitutions (K103N, Y181C) and HIV-1 strain A17 bearing the K103N and Y181C mutations, which are known to confer NNRTI resistance. As shown in Table 4, WPR-6 displayed nanomolar potency in the inhibition of K103N and Y181C mutant viruses on MT-4 and TZM-bl cells, with low EC50s ranging from 21 to 151 nM. The reference compound TNK651 displayed diminished activity against most NNRTI resistance-associated mutant HIV-1 strains, which resulted in >100-fold reductions in susceptibility, compared to the laboratory-adapted viruses above. No antiviral activity against these NNRTI-resistant mutant viruses was observed in the presence of the marketed drug NVP, even at a high concentration of 10 μM. Notably, WPR-6 was more effective in suppressing the replication of the Y181C mutant virus, with EC50s of 21 and 22 nM on MT-4 and TZM-bl cells, respectively, than that of the K103N and K103N/Y181C double mutant viruses (EC50s ranging from 35 to 151 nM). Therefore, WPR-6 exhibited better overall potency than TNK651 and NVP against the most prevalent NNRTI-associated mutant viruses.
TABLE 4.
Antiviral activities of WPR-6 and TNK651 against NNRTI-resistant mutant viruses
| SDM(s) | Cell type | EC50 (μM)a |
||
|---|---|---|---|---|
| WPR-6 | TNK651 | NVP | ||
| K103N | MT-4 | 0.151 ± 0.004 | >10 | ND |
| K103N | TZM-bl | 0.035 ± 0.003 | NDb | >10 |
| Y181C | MT-4 | 0.021 ± 0.001 | 2.398 ± 0.246 | ND |
| Y181C | TZM-bl | 0.022 ± 0.005 | >10 | >10 |
| K103N, Y181C | MT-4 | 0.091 ± 0.013 | >10 | >10 |
| K103N, Y181C | TZM-bl | 0.122 ± 0.011 | >10 | >10 |
EC50s were determined by using MT-4 or TZM-bl cells in triplicate; the data presented are mean values ± standard deviations from at least two independent experiments.
ND, not determined.
Viruses with mutations encoding V106I and Y188L substitutions in RT were resistant to WPR-6.
When an anti-HIV agent is used as monotherapy, the emergence of variants resistant to the agent may be inevitable. In addition, when a particular anti-HIV drug is used in multidrug trials, it is critical to identify the types of resistant mutants that might evolve or whether such mutants are more resistant or more sensitive to other anti-HIV drugs (33). Therefore, in vitro selection of viruses resistant to WPR-6 was performed (Fig. 2). Laboratory-adapted HIV-1 strain SF33 was cultured in MT-4 cells in the presence of inhibitor at an initial concentration of 100 nM (CC50). Cells were passaged every 4 to 5 days, and virus replication was monitored microscopically by observing the formation of syncytia (34). At each virus breakthrough (massive syncytium formation), the concentration of the inhibitor was doubled. If an increase in syncytia, which is an indicator of reduced susceptibility to WPR-6, was observed by microscopy, proviral DNA was amplified and cloned and the RT coding region was sequenced. Additionally, the concentration of WPR-6 was increased to maintain the level of virus inhibition. At the end of the study (about 14 passages), the virus pool was fully resistant to WPR-6, when the concentration of WPR-6 reached 51,200 nM (more than 500-fold the initial concentration).
FIG 2.

Viral resistance to WPR-6. Virus became resistant to WPR-6 after serial passages in the presence of increasing concentrations of WPR-6.
Five mutations (K102Q, V106I, T139I, S162C, and Y188L) in the RT region were observed by alignment after a 14-passage period of WPR-6 selection. According to the HIV drug resistance database of the Stanford University data network, the V106I and Y188L substitutions were NNRTI resistance mutations and K102Q, T139I, and S162C, neither NRTI nor NNRTI resistance mutations, were other mutations.
In order to determine the levels of resistance to WPR-6 and other anti-HIV drugs conferred by WPR-6-selected mutations, we constructed single mutants with a V106I or Y188L mutation in RT by site-directed mutagenesis. The resulting viruses were tested for their replication capacity in the presence of WPR-6, NVP, and EFV (Table 5). The single mutation encoding V106I or Y188L confers significant (14-fold) or moderate (9-fold) resistance to WPR-6. Notably, the virus carrying the V106I substitution remained susceptible to both NVP (1.6-fold) and EFV (2-fold), while that bearing the Y188L substitution was highly resistant to NVP (>200-fold) and EFV (10-fold).
TABLE 5.
Sensitivities of molecular clones with RT mutations to WPR-6 and other anti-HIV agents
| WPR-6-selected mutation | EC50 (μM)c (fold differenced) |
||
|---|---|---|---|
| WPR-6 | NVP | EFV | |
| None (WT)a | 0.001 ± 0.0002 | 0.041 ± 0.001 | 0.0008 ± 0.00002 |
| Y188L | 0.009 ± 0.0004 (9) | 10 (>200) | 0.008 ± 0.0002 (10) |
| None (WT)b | 0.001 ± 0.0002 | 0.074 ± 0.015 | 0.001 ± 0.001 |
| V106I | 0.014 ± 0.003 (14) | 0.120 ± 0.009 (1.6) | 0.002 ± 0.001 (2) |
A WT HIV-1 strain was used as the control for the Y188L mutant molecular clone.
A WT HIV-1 strain was used as a control for the V106I mutant molecular clone.
EC50s were determined by virus inhibition assay using TZM-bl cells. The samples were tested in triplicate, and the data presented are mean values ± standard deviations from at least two independent experiments.
Fold change was determined by calculating the ratio of EC50s for mutant and WT viruses.
DISCUSSION
NNRTIs act by binding to a hydrophobic binding site that is distinct from the polymerase catalytic site, leading to conformational changes in the p66 subunit (35, 36). Novel HEPT derivatives targeting the NNRTI binding site have been rationally designed on the basis of the structure of the NNRTI binding pocket. Pharmacological research focused on modification of the C-6 benzyl substitutions of the HEPT analogues resulted in the synthesis of a variety of effective anti-HIV agents (25). One analogue, WPR-6, was found to be a unique and potent NNRTI of HIV-1 with favorable pharmacokinetic and toxicological profiles in preclinical investigations, and it was therefore selected for further development.
Here we tested the inhibitory activity of WPR-6 against infection with laboratory-adapted HIV-1 strains, including SF33 and 7391, as well as clinical isolates circulating in China. We found that WPR-6 was more potent than other NNRTIs that we tested, including NVP and EFV. When tested against purified HIV-1 RT, WPR-6 had an IC50 of 3 nM and was 1,000-fold more potent than NVP (25). Similarly, WPR-6 was 50- to 200-fold more potent than NVP and 2- to 10-fold more potent than TNK651, inhibiting the replication of laboratory-adapted HIV-1 strains SF33 and 7391 in different cell-based assays in vitro. The inhibition of HIV-1 replication did not depend on the host cells used (Table 2). WPR-6 exhibited low toxicity to cells that were used to test anti-HIV activity (Table 1), resulting in high SIs ranging from 10,000 to 50,000, suggesting, in turn, a potential for further development of WPR-6 as a new NNRTI.
HIV-1 is a genetically heterogeneous virus classified into three groups, M, N, and O. Group M has been divided into 10 subtypes, A to K (37). In China, B′, CRF_07BC, and CRF_01AE continue to be the most commonly transmitted HIV-1 subtypes across the country (38). Therefore, we focused on the antiviral activity of WPR-6 against Chinese epidemic HIV-1 subtype strains. The result showed that WPR-6 was active at nanomolar concentrations against infection with a broad spectrum of HIV-1 clinical isolates, including both R5 and X4 viruses, with low EC50s. WPR-6 demonstrated strong antiviral activity independent of subtype, with a median EC50 of 3.2 nM and a median EC90 of 105.7 nM. WPR-6 has effective antiviral activity on laboratory-adapted HIV strains, as well as clinical isolates from Chinese AIDS patients.
During the in vitro selection for viruses that were resistant to WPR-6, mutations encoding V106I and Y188L were selected, suggesting that V106I and Y188L may be the primary NNRTI resistance mutations selected by WPR-6. This was confirmed by determining the EC50s of WPR-6 against viruses carrying the V106I and/or Y188L substitutions. According to the Stanford University HIV Drug Resistance Database, V106I is a polymorphic accessory NNRTI-selected mutation. When it appears alone, V106I has little effect on NNRTI susceptibility or the virological response to an NNRTI-containing regimen. Y188L is a nonpolymorphic 2-bp mutation selected by NVP and EFV. It confers high-level resistance to NVP and EFV. The two selected variants are present in only 4 to 6% of NNRTI-resistant patient isolates. Therefore, mutant viruses containing the V106I and Y188L mutations may display resistance to other NNRTIs structurally distinct from WPR-6. Like K102Q, T139I and S162C were found in virus obtained after the 14th passage in the presence of WPR-6. However, according to the HIV drug resistance database of the Stanford University data network, K102Q, T139I, and S162C, neither NRTI nor NNRTI resistance mutations, are other mutations that may be accessory mutations or correlated mutations in our previous study (31, 39) and need to be researched further.
In summary, WPR-6 may be a novel NNRTI with greater potency than NVP and TNK651 against laboratory-derived strains of HIV-1 and the two most prevalent NNRTI-associated mutant viruses (with K103N and Y181C mutations). When tested against a broad panel of clinical isolates of HIV-1, WPR-6 displayed antiviral potency equivalent to that against WT HIV-1. Furthermore, V106I and Y188L were the dominant mutations in in vitro selection with escalating concentrations of WPR-6, and no K103N and Y181C mutants were observed. Taking these results together, the virologic profile of WPR-6 supports its further development as a new NNRTI for the treatment of HIV-1 infection and AIDS.
ACKNOWLEDGMENTS
We are grateful to the AIDS Research and Reference Reagent Program, NIAID, NIH, for providing TZM-bl cells, Ghost cell lines, and primary HIV strains.
This work was supported by grants from the National Natural Science Foundation of China (81172733, 81261120384) and the key project of the State Key Laboratory for Infectious Diseases Prevention and Control (2011SKLID102) to L.M. and the National Megaprojects of China for Infectious Diseases (2013ZX10001-006) to S.J. and L.M.
We have no competing interests to declare.
L.M. and X.W. conceived and designed the experiments. W.X., J.Z., J.S., Y.W., and X.W. performed the experiments. J.Z., L.M., Y.S., and S.J. analyzed the data and wrote the paper. All of us read and approved the final manuscript.
REFERENCES
- 1.Sax PE, Sypek A, Berkowitz BK, Morris BL, Losina E, Paltiel AD, Kelly KA, Seage GR III, Walensky RP, Weinstein MC, Eron J, Freedberg KA. 2014. HIV cure strategies: how good must they be to improve on current antiretroviral therapy? PLoS One 9:e113031. doi: 10.1371/journal.pone.0113031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tseng A, Seet J, Phillips EJ. 2015. The evolution of three decades of antiretroviral therapy: challenges, triumphs and the promise of the future. Br J Clin Pharmacol 79:182–194. doi: 10.1111/bcp.12403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Persaud D, Gay H, Ziemniak C, Chen YH, Piatak M Jr, Chun TW, Strain M, Richman D, Luzuriaga K. 2013. Absence of detectable HIV-1 viremia after treatment cessation in an infant. N Engl J Med 369:1828–1835. doi: 10.1056/NEJMoa1302976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Deng K, Siliciano RF. 2014. HIV: early treatment may not be early enough. Nature 512:35–36. [DOI] [PubMed] [Google Scholar]
- 5.Short CE, Douglas M, Smith JH, Taylor GP. 2014. Preterm delivery risk in women initiating antiretroviral therapy to prevent HIV mother-to-child transmission. HIV Med 15:233–238. doi: 10.1111/hiv.12083. [DOI] [PubMed] [Google Scholar]
- 6.Muessig KE, Smith MK, Powers KA, Lo YR, Burns DN, Grulich AE, Phillips AN, Cohen MS. 2012. Does ART prevent HIV transmission among MSM? AIDS 26:2267–2273. doi: 10.1097/QAD.0b013e328355713d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thornhill J, Fidler S, Frater J. 2015. Advancing the HIV cure agenda: the next 5 years. Curr Opin Infect Dis 28:1–9. doi: 10.1097/QCO.0000000000000123. [DOI] [PubMed] [Google Scholar]
- 8.Bacheler L, Jeffrey S, Hanna G, D'Aquila R, Wallace L, Logue K, Cordova B, Hertogs K, Larder B, Buckery R, Baker D, Gallagher K, Scarnati H, Tritch R, Rizzo C. 2001. Genotypic correlates of phenotypic resistance to efavirenz in virus isolates from patients failing nonnucleoside reverse transcriptase inhibitor therapy. J Virol 75:4999–5008. doi: 10.1128/JVI.75.11.4999-5008.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De Clercq E. 2007. The design of drugs for HIV and HCV. Nat Rev Drug Discov 6:1001–1018. doi: 10.1038/nrd2424. [DOI] [PubMed] [Google Scholar]
- 10.Tronchet JM, Seman M. 2003. Nonnucleoside inhibitors of HIV-1 reverse transcriptase: from the biology of reverse transcription to molecular design. Curr Top Med Chem 3:1496–1511. doi: 10.2174/1568026033451754. [DOI] [PubMed] [Google Scholar]
- 11.Tarby CM. 2004. Recent advances in the development of next generation non-nucleoside reverse transcriptase inhibitors. Curr Top Med Chem 4:1045–1057. doi: 10.2174/1568026043388295. [DOI] [PubMed] [Google Scholar]
- 12.Milinkovic A, Martínez E. 2004. Nevirapine in the treatment of HIV. Expert Rev Anti Infect Ther 2:367–373. doi: 10.1586/14787210.2.3.367. [DOI] [PubMed] [Google Scholar]
- 13.Scott LJ, Perry CM. 2000. Delavirdine: a review of its use in HIV infection. Drugs 60:1411–1444. doi: 10.2165/00003495-200060060-00013. [DOI] [PubMed] [Google Scholar]
- 14.Maggiolo F. 2009. Efavirenz: a decade of clinical experience in the treatment of HIV. J Antimicrob Chemother 64:910–928. doi: 10.1093/jac/dkp334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Johnson LB, Saravolatz LD. 2009. Etravirine, a next-generation nonnucleoside reverse-transcriptase inhibitor. Clin Infect Dis 48:1123–1128. doi: 10.1086/597469. [DOI] [PubMed] [Google Scholar]
- 16.Fernández-Montero JV, Vispo E, Anta L, de Mendoza C, Soriano V. 2012. Rilpivirine: a next-generation non-nucleoside analogue for the treatment of HIV infection. Expert Opin Pharmacother 13:1007–1014. doi: 10.1517/14656566.2012.667802. [DOI] [PubMed] [Google Scholar]
- 17.Calmy A, Pascual F, Ford N. 2004. HIV Drug Resistance. N Engl J Med 350:2720–2721. doi: 10.1056/NEJM200406243502621. [DOI] [PubMed] [Google Scholar]
- 18.Domaoal RA, Demeter LM. 2004. Structural and biochemical effects of human immunodeficiency virus mutants resistant to non-nucleoside reverse transcriptase inhibitors. Int J Biochem Cell Biol 36:1735–1751. doi: 10.1016/j.biocel.2004.02.026. [DOI] [PubMed] [Google Scholar]
- 19.Asahchop EL, Wainberg MA, Sloan RD, Tremblay CL. 2012. Antiviral drug resistance and the need for development of new HIV-1 reverse transcriptase inhibitors. Antimicrob Agents Chemother 56:5000–5008. doi: 10.1128/AAC.00591-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lindberg J, Sigurðsson S, Löwgren S, Andersson OH, Sahlberg C, Noréen R, Fridborg K, Zhang H, Unge T. 2002. Structural basis for the inhibitory efficacy of efavirenz (DMP-266), MSC194 and PNU142721 towards the HIV-1 RT K103N mutant. Eur J Biochem 269:1670–1677. doi: 10.1046/j.1432-1327.2002.02811.x. [DOI] [PubMed] [Google Scholar]
- 21.Famiglini V, La Regina G, Coluccia A, Pelliccia S, Brancale A, Maga G, Crespan E, Badia R, Riveira-Munoz E, Este JA, Ferretti R, Cirilli R, Zamperini C, Botta M, Schols D, Limongelli V, Agostino B, Novellino E, Silvestri R. 2014. Indolylarylsulfones carrying a heterocyclic tail as very potent and broad spectrum HIV-1 non-nucleoside reverse transcriptase inhibitors. J Med Chem 57:9945–9957. doi: 10.1021/jm5011622. [DOI] [PubMed] [Google Scholar]
- 22.Spira S. 2003. Impact of clade diversity on HIV-1 virulence, antiretroviral drug sensitivity and drug resistance. J Antimicrob Chemother 51:229–240. doi: 10.1093/jac/dkg079. [DOI] [PubMed] [Google Scholar]
- 23.Lai MT, Lu M, Felock PJ, Hrin RC, Wang YJ, Yan Y, Munshi S, McGaughey GB, Tynebor RM, Tucker TJ, Williams TM, Grobler JA, Hazuda DJ, McKenna PM, Miller MD. 2010. Distinct mutation pathways of non-subtype B HIV-1 during in vitro resistance selection with nonnucleoside reverse transcriptase inhibitors. Antimicrob Agents Chemother 54:4812–4824. doi: 10.1128/AAC.00829-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.de Béthune M-P. 2010. Non-nucleoside reverse transcriptase inhibitors (NNRTIs), their discovery, development, and use in the treatment of HIV-1 infection: a review of the last 20 years (1989–2009). Antiviral Res 85:75–90. doi: 10.1016/j.antiviral.2009.09.008. [DOI] [PubMed] [Google Scholar]
- 25.Wang X, Zhang J, Huang Y, Wang R, Zhang L, Qiao K, Li L, Liu C, Ouyang Y, Xu W, Zhang Z, Zhang L, Shao Y, Jiang S, Ma L, Liu J. 2012. Design, synthesis, and biological evaluation of 1-((2-benzyloxyl/alkoxyl)methyl)-5-halo-6-aryluracils as potent HIV-1 non-nucleoside reverse transcriptase inhibitors with an improved drug resistance profile. J Med Chem 55:2242–2250. doi: 10.1021/jm201506e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang Y, Wang X, Yu X, Yuan L, Guo Y, Xu W, Liu T, Liu J, Shao Y, Ma L. 2011. Inhibitory activity of 9-phenylcyclohepta[d]pyrimidinedione derivatives against different strains of HIV-1 as non-nucleoside reverse transcriptase inhibitors. Virol J 8:230. doi: 10.1186/1743-422X-8-230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van 't Wout AB, Schuitemaker H, Kootstra NA. 2008. Isolation and propagation of HIV-1 on peripheral blood mononuclear cells. Nat Protoc 3:363–370. doi: 10.1038/nprot.2008.3. [DOI] [PubMed] [Google Scholar]
- 28.Jiang S, Lu H, Liu S, Zhao Q, He Y, Debnath AK. 2004. N-substituted pyrrole derivatives as novel human immunodeficiency virus type 1 entry inhibitors that interfere with the gp41 six-helix bundle formation and block virus fusion. Antimicrob Agents Chemother 48:4349–4359. doi: 10.1128/AAC.48.11.4349-4359.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chong H, Qiu Z, Sun J, Qiao Y, Li X, He Y. 2014. Two M-T hook residues greatly improve the antiviral activity and resistance profile of the HIV-1 fusion inhibitor SC29EK. Retrovirology 11:40. doi: 10.1186/1742-4690-11-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chong H, Yao X, Qiu Z, Sun J, Qiao Y, Zhang M, Wang M, Cui S, He Y. 2014. The M-T hook structure increases the potency of HIV-1 fusion inhibitor sifuvirtide and overcomes drug resistance. J Antimicrob Chemother 69:2759–2769. doi: 10.1093/jac/dku183. [DOI] [PubMed] [Google Scholar]
- 31.Jiao Y, Li S, Li Z, Zhang Z, Zhao J, Li L, Wang L, Yin Q, Wang Y, Zeng Z, Shao Y, Ma L. 2014. HIV-1 transmitted drug resistance-associated mutations and mutation co-variation in HIV-1 treatment-naive MSM from 2011 to 2013 in Beijing, China. BMC Infect Dis. 14:689. doi: 10.1186/s12879-014-0689-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu X, Yuan L, Huang Y, Xu W, Fang Z, Liu S, Shao Y, Jiang S, Ma L. 2011. Susceptibility of HIV-1 subtypes B′, CRF07_BC and CRF01_AE that are predominantly circulating in China to HIV-1 entry inhibitors. PLoS One 6:e17605. doi: 10.1371/journal.pone.0017605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fujiwara T, Sato A, El-Farrash M, Miki S, Abe K, Isaka Y, Kodama M, Wu Y, Chen LB, Harada H, Sugimoto H, Hatanaka M, Hinuma Y. 1998. S-1153 inhibits replication of known drug-resistant strains of human immunodeficiency virus type 1. Antimicrob Agents Chemother 42:1340–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Z, Xu W, Koh YH, Shim JH, Girardet JL, Yeh LT, Hamatake RK, Hong Z. 2007. A novel nonnucleoside analogue that inhibits human immunodeficiency virus type 1 isolates resistant to current nonnucleoside reverse transcriptase inhibitors. Antimicrob Agents Chemother 51:429–437. doi: 10.1128/AAC.01032-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cihlar T, Ray AS. 2010. Nucleoside and nucleotide HIV reverse transcriptase inhibitors: 25 years after zidovudine. Antiviral Res 85:39–58. doi: 10.1016/j.antiviral.2009.09.014. [DOI] [PubMed] [Google Scholar]
- 36.Ivetac A, McCammon JA. 2009. Elucidating the inhibition mechanism of HIV-1 non-nucleoside reverse transcriptase inhibitors through multicopy molecular dynamics simulations. J Mol Biol 388:644–658. doi: 10.1016/j.jmb.2009.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Buonaguro L, Tornesello ML, Buonaguro FM. 2007. Human immunodeficiency virus type 1 subtype distribution in the worldwide epidemic: pathogenetic and therapeutic implications. J Virol 81:10209–10219. doi: 10.1128/JVI.00872-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Su L, Graf M, Zhang Y, von Briesen H, Xing H, Köstler J, Melzl H, Wolf H, Shao Y, Wagner R. 2000. Characterization of a virtually full-length human immunodeficiency virus type 1 genome of a prevalent intersubtype (C/B′) recombinant strain in China. J Virol 74:11367–11376. doi: 10.1128/JVI.74.23.11367-11376.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang Y, Li Z, Xing H, Jiao Y, Ouyang Y, Liao L, Jiang S, Armstrong R, Shao Y, Ma L. 2014. Identification of the critical sites of NNRTI-resistance in reverse transcriptase of HIV-1 CRF_BC strains. PLoS One 9:e93804. doi: 10.1371/journal.pone.0093804. [DOI] [PMC free article] [PubMed] [Google Scholar]