

Abstract
Annually, medical device infections are associated with >250,000 catheter-associated bloodstream infections (CLABSI), with up to 25% mortality. Staphylococcus aureus, a primary pathogen in these infections, is capable of biofilm production, allowing organism persistence in harsh environments, offering antimicrobial protection. With increases in S. aureus isolates with reduced susceptibility to current agents, ceftaroline (CPT) offers a therapeutic alternative. Therefore, we evaluated whether CPT would have a role against biofilm-producing methicillin-resistant S. aureus (MRSA), including those with decreased susceptibilities to alternative agents. In this study, we investigated CPT activity alone or combined with daptomycin (DAP) or rifampin (RIF) against 3 clinical biofilm-producing MRSA strains in an in vitro biofilm pharmacokinetic/pharmacodynamic (PK/PD) model. Simulated antimicrobial regimens were as follows: 600 mg of CPT every 8 h (q8h) (free maximum concentration of drug [fCmax], 17.04 mg/liter; elimination half-life [t1/2], 2.66 h), 12 mg/kg of body weight/day of DAP (fCmax, 14.7 mg/liter; t1/2, 8 h), and 450 mg of RIF q12h (fCmax, 3.5 mg/liter; t1/2, 3.4 h), CPT plus DAP, and CPT plus RIF. Samples were obtained and plated to determine colony counts. Differences in log10 CFU/cm2 were evaluated by analysis of variance with Tukey's post hoc test. The strains were CPT and vancomycin susceptible and DAP nonsusceptible (DNS). CPT displayed activity throughout the experiment. DAP demonstrated initial activity with regrowth at 24 h in all strains. RIF was comparable to the drug-free control, and little benefit was observed when combined with CPT. CPT plus DAP displayed potent activity, with an average log10 CFU/cm2 reduction of 3.33 ± 1.01 from baseline. CPT demonstrated activity against biofilm-producing DNS MRSA. CPT plus DAP displayed therapeutic enhancement over monotherapy, providing a potential option for difficult-to-treat medical device infections.
INTRODUCTION
The Infectious Diseases Society of America has recognized several pathogens for which novel therapies are needed, including Staphylococcus aureus (1). Not only are rates of S. aureus infections increasing, but the frequency of methicillin resistance is as well, ultimately promoting vancomycin as a first-line therapy to combat these infections (2). This increased usage is a driving force of resistance, as S. aureus isolates are now demonstrating decreased susceptibility to vancomycin (3–5).
In addition to increasing resistance rates and prevalence, S. aureus also has the propensity to produce bacterial biofilm, a serious concern in patients with prosthetic material, including intravenous catheters (6). Essentially, biofilm encapsulates the microorganism, creating an outer layer of glycocalyx, a layer of actively dividing cells, and a layer of stationary cells. Antimicrobials are often rendered ineffective, not only due to lack of penetration into the cellular component, but also because several agents, most notably β-lactams, are dependent upon active cellular division for activity. As such, stationary cells are inherently resistant to cell wall-active agents (7, 8). Therefore, combination therapy is often employed (9).
Combination therapy against MRSA-infected prostheses typically employs rifampin as the synergistic agent. However, potent in vitro synergy has been identified with the combination of β-lactam antibiotics plus either daptomycin or vancomycin (10, 11). Data evaluating either cefazolin or ceftaroline plus daptomycin, vancomycin, or rifampin against biofilm-producing MRSA have been described in time-kill methodologies, and these combinations may be effective, although pharmacokinetic/pharmacodynamic (PK/PD) modeling has not yet been performed (12). Therefore, our objective was to evaluate ceftaroline alone and in combination against biofilm-producing MRSA strains in an in vitro PK/PD model of bacterial biofilms.
MATERIALS AND METHODS
Bacterial strains and culture media.
Three methicillin-resistant and daptomycin-nonsusceptible S. aureus strains were evaluated in this study. These strains included a vancomycin-susceptible strain and an isogenic strain pair consisting of a heterogeneous vancomycin-intermediate S. aureus (hVISA) parent strain and a VISA mutant (isolates R6911 and R6913, respectively). Tryptic soy agar (TSA) (Difco, Detroit, MI) plates were used to grow isolates and perform bacterial quantification. Tryptic soy broth supplemented with 10% and 1% glucose (gSTSB) was used for the 24-h and 16-h portions of the conditioning phases, respectively (13). Mueller-Hinton broth supplemented with 50 mg/liter calcium and 12.5 mg/liter magnesium was used for all in vitro PK/PD experiments and susceptibility testing due to the calcium-dependent mechanism of daptomycin.
Antimicrobial agents.
Ceftaroline analytical powder was provided by Actavis Pharmaceuticals (New York, NY). Daptomycin (Cubist Pharmaceuticals, Lexington, MA), rifampin, and vancomycin (Sigma Chemical Co., St. Louis, MO) were purchased commercially.
Susceptibility testing.
Susceptibility testing of all antimicrobials was performed in duplicate by broth microdilution, according to CLSI guidelines (14). Biofilm MIC testing was carried out utilizing the modified Calgary Biofilm Device method, as previously described (15).
In vitro PK/PD model.
A CDC biofilm reactor model (BioSurface Technologies, Bozeman, MT) was set up with polyurethane coupons inserted into eight rods, as previously described (13). A 40-h conditioning phase was performed prior to antimicrobial exposure to stimulate biofilm production. The conditioning phase consisted of 24 h of incubation at 37°C of inoculated 1% gSTSB, followed by a 16-h continuous flow with 10% supplied via peristaltic pumps (Masterflex; Cole-Parmer Instrument Co., Chicago, IL) set at a rate of 13.3 ml/min to achieve a 30-min residence time. Post-continuous flow, inflow medium with Mueller-Hinton broth (MHB) was used for antibiotic simulations, and antibiotic bolus doses were injected into the reactor. The regimens evaluated included 600 mg of ceftaroline every 8 h (free drug peak concentration [fCmax], 17.04 mg/liter; elimination half-life [t1/2], 2.66 h; protein binding, 20%) (16), 12 mg/kg of body weight of daptomycin every 24 h (fCmax, 14.7 mg/liter; t1/2, 8 h; protein binding, 92%) (17), 450 mg of rifampin every 12 h (fCmax, 3.5 mg/liter; t1/2, 3.4 h; protein binding, 90%) (18–20), ceftaroline plus daptomycin, and ceftaroline plus rifampin. Peristaltic pumps were set up to simulate the half-lives of the antibiotics. For combination models, the daptomycin and rifampin doses were supplemented to account for the shorter half-life of ceftaroline (21). All model experiments were completed in duplicate to ensure reproducibility.
PD analysis.
One coupon from each model was aseptically removed at 0, 4, 8, 24, 32, 48, 56, and 72 h. Each coupon was washed twice in sterile normal saline to remove excess planktonic cells. Each coupon was then placed in a sterile tube containing 10 ml of normal saline. Biofilm-embedded cells were recovered via three alternating 60-s cycles of vortexing and sonication at 20 Hz (Bransonic 12; Branson Ultrasonics Corporation). Both biofilm-embedded cells and planktonic samples derived from the model were serially diluted in sterile normal saline and spiral plated onto TSA utilizing an automated spiral plater (Don Whitley Scientific Ltd., West Yorkshire, England). TSA plates were then incubated at 37°C for 24 h, and colony counts were determined using a laser colony counter (ProtoCOL; Symbiosis, Cambridge, England). Enhancement of activity by the addition of a drug was defined as a ≥2-log10 CFU/cm2 increase in kill compared to that with the most active single agent of the combination. Combinations that resulted in a ≥1-log10 CFU/cm2 increase in bacterial growth in comparison to the least-active single agent were considered to represent antagonism (22).
PK analysis.
Pharmacokinetic samples were obtained through the injection port of each model at 0.5, 2, 4, 8, 24, 32, 48, 56, and 72 h in duplicate for verification of target antimicrobial concentrations. All samples were stored at −80°C until analysis. Ceftaroline and rifampin concentrations were determined by a bioassay, as previously described (13, 23). Blank 0.25-in. disks were placed on preswabbed (Escherichia coli strain ATCC 25922 for ceftaroline PK and Kocuria rhizophila [formerly Micrococcus luteus] strain ATCC 9394 for rifampin PK) agar plates and spotted with 10 μl of the standards (1.88, 3.75, 7.5, 15, and 30 mg/liter for ceftaroline and 0.625, 1.25, 2.5, 5, and 7.5 mg/liter for rifampin) or experimental samples. Each standard and sample were tested in duplicate. Plates were incubated for 24 h at 37°C, at which time the zones of inhibition were measured using an automated plate reader (ProtoCOL; Synoptics Ltd., Frederick, MD). The t1/2, fCmax, and free minimum drug concentration (fCmin) were determined from the concentration-versus-time plots using PK Analyst software (version 1.10; MicroMath Scientific Software, Salt Lake City, UT). The time above the MIC for ceftaroline and rifampin Cmax was calculated using first-order elimination concepts. Daptomycin concentrations were determined using a validated high-performance liquid chromatography (HPLC) assay that conforms to the guidelines set forth by the College of American Pathologists. The area under the concentration-time curve (AUC) was determined by the trapezoidal method utilizing the PK Analyst Software (version 1.10; MicroMath Scientific Software).
Confocal laser scanning microscopy analysis.
The coupons were placed in a flow cell chamber, and fluorescent dyes were injected into the chamber for 20 min and then flushed with sterile water. Syto-9 (1.3 μM final concentration) was applied to identify viable cells in the biofilm, and propidium iodide (PI) (4.0 μM final concentration) was added to stain dead cells. The biofilm was imaged using a Zeiss 510 Meta confocal laser scanning microscopy (CLSM) with an Achroplan 40 by 0.8 numerical aperture (NA) water dipping objective. The Syto-9 and PI fluorophores were excited with an argon laser at 488 nm, and the emission bandpass filters used for Syto-9 and PI were 515 ± 15 nm and 630 ± 15 nm, respectively. Confocal images were processed using the Velocity software (Improvision, Lexington, MA).
Scanning electron microscopy of biofilms with antibiotic exposure from a static well plate assay.
A modified kill curve method was developed as a follow-up to the most potent PK/PD model therapy to more rapidly assess antibiotic synergy against bacterial biofilms. Sterile polyurethane coupons similar to those used in the CDC reactor were placed in a 24-well plate containing 2 ml of 1% gSTSB medium per well. A 0.5 McFarland standard was diluted 100-fold into each well and grown for 18 to 24 h in an orbital shaker at 150 rpm to allow for biofilm formation on the coupon. The coupons were then washed gently with 1× phosphate-buffered saline (PBS) to remove nonadherent cells and placed into a new plate containing 2 ml of Mueller-Hinton broth supplemented with 50 mg/liter calcium and 12.5 mg/liter magnesium. Experimental conditions were no antibiotic, ceftaroline plus daptomycin at 1× the organism MIC (standard initial kill curve assessment concentrations), and ceftaroline plus daptomycin at fCmax (17 and 14.7 μg/ml, respectively). The free concentrations were selected to mimic those targeted in the PK/PD model for comparison. All experiments were performed in triplicate over 24 h. Bacterial quantification was done on two coupons per time point using methods described earlier.
The third coupon in the experiment was used for scanning electron microscopy (SEM) imaging. After removal, the coupon was washed in 1× PBS to remove nonadherent cells and immersion fixed in a solution of 2.5% glutaraldehyde and 2% paraformaldehyde in 0.1 M sodium phosphate buffer. The coupons were then dehydrated in a graded ethanol series and carbon coated at 30 Å for 3 min by using a SeeVac Conduct Vac IV sputter coater. The coupons were imaged using a Hitachi S570 SEM at 2,000× magnification and evaluated for the presence and characteristics of biofilm and cells.
Emergence of resistance.
Development of resistance was evaluated at 72 h. Samples of 100 μl were plated on brain heart infusion agar (BHIA) or calcium-supplemented Mueller-Hinton agar (MHA) containing 3× the MIC of the respective antibiotic to assess for increases in MIC. The plates were examined for growth after 48 h of incubation at 37°C. Broth microdilution MICs, in accordance with CLSI guidelines, were determined for any isolate observed to grow on the drug-containing agar plates.
Statistical analysis.
Changes in log10 CFU/cm2 for biofilm-embedded and planktonic bacteria for the PK/PD model at 72 h were evaluated for each regimen by analysis of variance with Tukey's post hoc test. A P value of ≤0.05 was considered statistically significant. All statistical analyses were performed using SPSS software (release 21.0; SPSS, Inc., Chicago, IL).
RESULTS
Susceptibility testing.
The MICs for the DNS strains R6067, R6911, and R6913 were 2, 2, and 4 mg/liter to vancomycin and 0.5, 1, and 0.5 mg/liter to ceftaroline, respectively. All three isolates were DNS, with MICs of 4, 2, and 4 mg/liter, respectively, and rifampin resistant (MIC, >4 mg/liter). The biofilm MICs were 0 to 1 dilutions higher for ceftaroline, 1 to 2 dilutions higher for daptomycin, and 1 to 2 dilutions higher for vancomycin.
In vitro PK/PD model.
The quantitative changes in log10 CFU/cm2 for the evaluated regimens against the 3 strains are displayed in Fig. 1. Against all strains, the most potent reduction in bacterial density was observed with the combination of ceftaroline plus daptomycin, with an average reduction ± standard deviation of 3.33 ± 1.01 log10 CFU/cm2 within 72 h. This combination was ultimately bactericidal in 2 (R6911 and R6913) of the 3 strains evaluated. Inconsistent results were demonstrated with the combination of ceftaroline and rifampin. Against strain R6911, the addition of rifampin to ceftaroline provided no benefit and resulted in similar kill to that with ceftaroline monotherapy (P = 0.997). Against R6067, the combination appeared to be antagonistic, with minimal change in bacterial density compared to that in the starting inoculum. For monotherapy regimens, ceftaroline alone displayed the most consistent activity in reducing the initial inoculum against all 3 strains compared to other monotherapy runs (P ≤ 0.001). While bactericidal activity was only noted against R6913, reductions of 2.65 ± 0.50 and 1.79 ± 0.31 log10 CFU/cm2 were observed for strains R6911 and R6067, respectively. Despite daptomycin alone achieving initial kill within the first 8 h for all 3 strains, due to daptomycin nonsusceptibility, bacterial regrowth was noted at subsequent time points. The results with rifampin monotherapy and drug-free controls were comparable in all strains tested.
FIG 1.
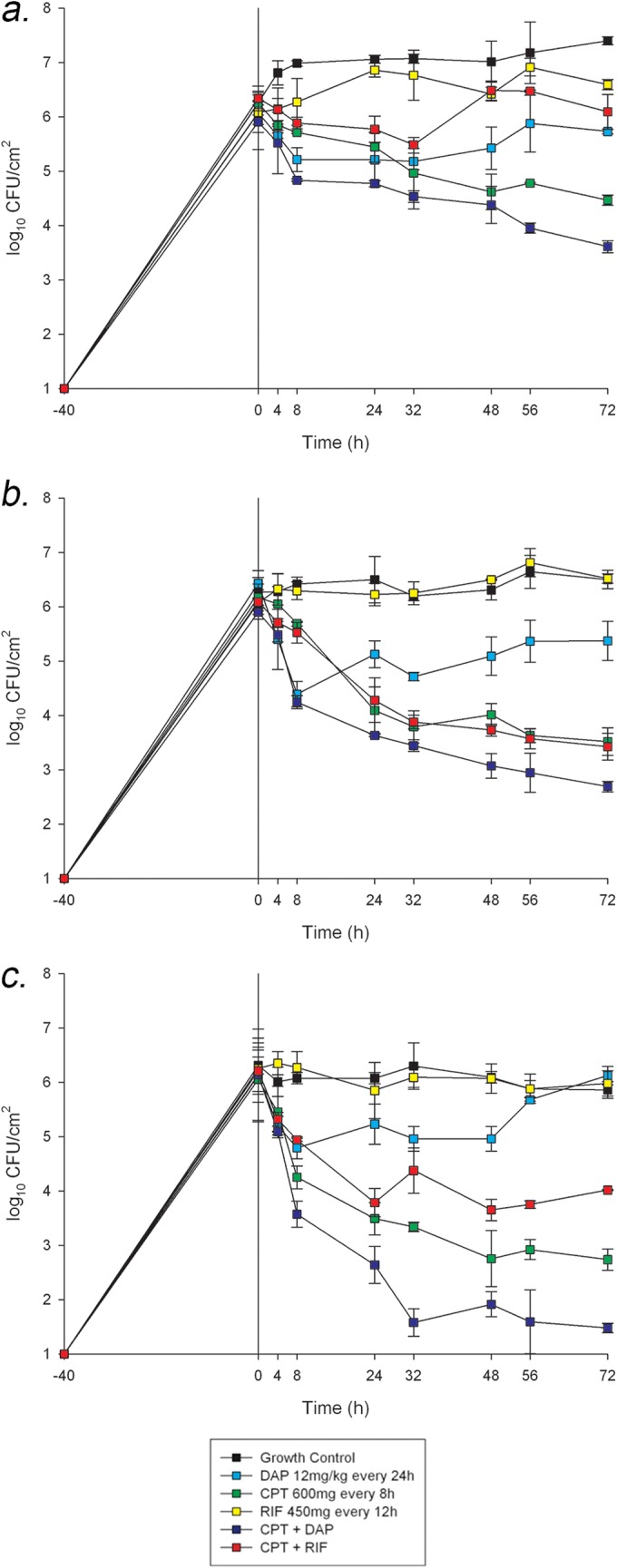
In vitro PK/PD biofilm model results. (A) Strain R6067 (vancomycin-susceptible S. aureus [VSSA]). (B) Strain R6911 (hVISA). (C) Strain R6913 (VISA).
Pharmacokinetics.
The observed PK parameters for ceftaroline, daptomycin, and rifampin were nearly equivalent to the target values. For ceftaroline, the observed fCmax was 17.04 ± 0.25 mg/liter (goal, 17.04 mg/liter), and the observed t1/2 was 2.39 ± 0.24 h (goal, 2.66 h). The ceftaroline free time above the MIC (fT>MIC) was maintained for 100% of the 8-h dosing interval for all 3 strains. The observed fCmax and average free trapezoidal area under the curve from 0 to 24 h (AUC0–24) values for daptomycin were 13.56 ± 0.43 mg/liter (goal, 14.7 mg/liter) and 142.98 ± 11.44 mg · h/ml, respectively. The observed t1/2 for daptomycin was 7.43 ± 0.59 h (goal, 8 h). Rifampin PK was as follows: the observed fCmax was 3.34 ± 0.20 mg/liter (goal, 3.5 mg/liter), and the observed t1/2 was 3.43 ± 0.16 h (goal, 2.66 h).
CLSM.
To further assess MRSA biofilm susceptibility to ceftaroline and the combination of ceftaroline plus daptomycin, biofilms exposed to these compounds were examined by confocal microscopy after LIVE/DEAD staining. The untreated control (Fig. 2) coupons displayed extensive biofilm coverage by live S. aureus. Concurrent with the MIC data, ceftaroline treatment alone resulted in reduced S. aureus surface colonization and the appearance of dead (red) cells. The combination of ceftaroline plus daptomycin exposure resulted in no large biofilm-like clusters on cells on the coupon, and no viable (green) S. aureus cells were observed.
FIG 2.
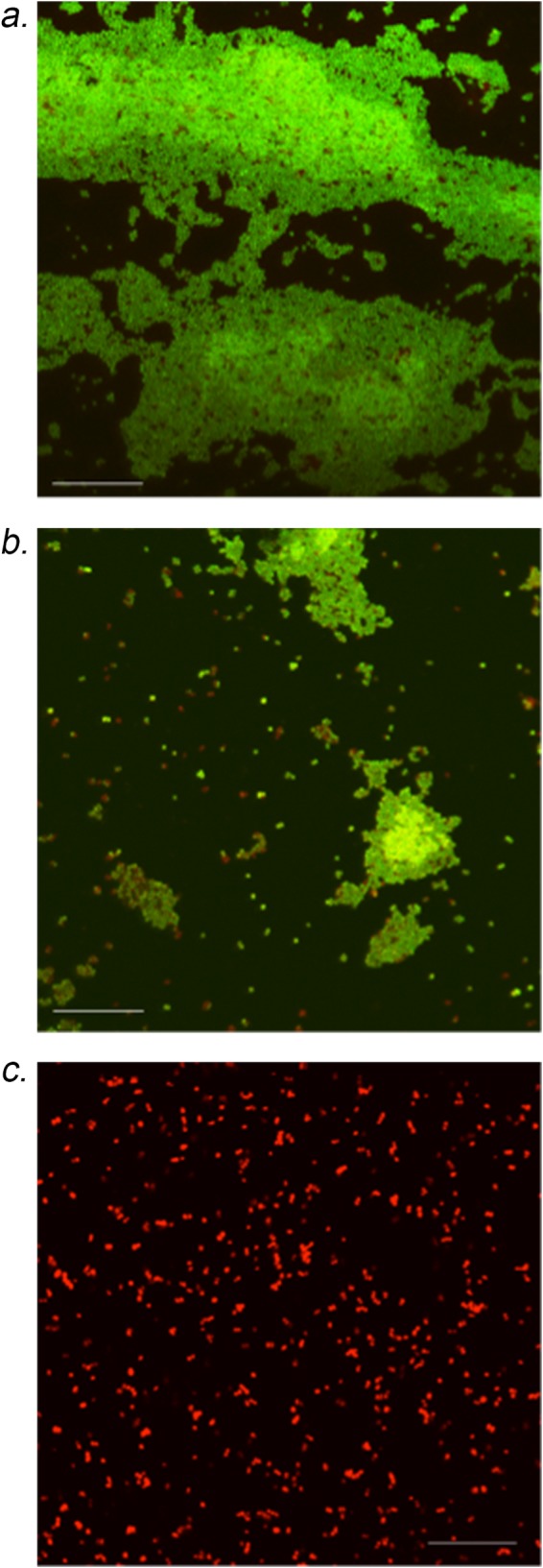
Confocal laser scanning microscopy analysis of coupons after 72 h in the in vitro PK/PD biofilm model for strain R6913. (A) Drug-free control, showing vast biomass with live cells. (B) Ceftaroline monotherapy, showing decreased biomass with live cells. (C) Ceftaroline plus daptomycin, showing minimal biomass with dead cells.
Antibiotic activity and SEM in coupons from the well plate assay.
The results of the PK/PD model were used to validate a more rapid assessment of antibiofilm activity in a 24-h well plate assay with polyurethane coupons. Figure 3 displays the result of bacterial quantification and development of biofilm by SEM analysis. Bacterial growth in this assay was similar to the PK/PD model at 24 h (6.85 ± 0.50 CFU/cm2). SEM imaging of the control displayed strong biofilm development on the coupon (Fig. 3B). When testing each at 1× the MIC, ceftaroline plus daptomycin was very synergistic, showing a 3.30 ± 0.35-log reduction in bacterial burden and biofilm clearance by SEM. An even greater synergistic effect was noted with the combination at fCmax for ceftaroline plus daptomycin. This combination had a 5.40 ± 0.64-log reduction in bacterial burden and scarce biofilm/organism remaining on the coupon by SEM visualization.
FIG 3.

Activity of ceftaroline (CPT) plus daptomycin (DAP) in combination with static concentrations on biofilms growth on polyurethane coupons in a well plate assay with isolate R6913. (A) Viable remaining bacteria recovered in biofilm after 24 h growth on coupon. (B) SEM images of biofilm/bacteria eradication on the coupon. Shown are growth (no antibiotic) (left), CPT and DAP at the MIC (0.5 and 4 μg/ml, respectively) (middle), and CPT plus DAP at fCmax (17 μg/ml and 15 μg/ml, respectively) (right).
Changes in susceptibility.
No increase in MIC to either ceftaroline or daptomycin was detected for any strain at any of the time points. Resistance was detected for rifampin monotherapy runs in all strains, with MICs of >64 mg/liter within 72 h of experimentation.
DISCUSSION
Bacteria, including S. aureus, are capable of adhering to foreign medical material and producing a biofilm matrix as protection from antimicrobial therapy. From 2009 to 2010, nearly 30,000 central line-associated bloodstream infections were reported, with Staphylococcus representing the most frequently isolated pathogen (2). Alarmingly, this number has increased by roughly 10,000 cases from 2 years before. It has been documented that antibiotics alone are ineffective at eradicating biofilm-producing organisms, and recurrence is frequently observed, even in patients who respond initially (24). Because of this, biofilm-associated infections have a large impact on patient outcomes, including cost of treatment.
The poor outcomes observed from biofilm-associated infections are multifactorial. Not only does decreased penetration into the biofilm matrix protect microorganisms, but the stationary growth phase of embedded organisms render many of the antimicrobial agents in our armamentarium useless (25). Cell wall-active agents, including the β-lactams, are unable to inhibit peptidoglycan synthesis when the cells are not actively replicating. However, not all cells embedded in biofilm are in stationary phase, which might explain the activity observed with ceftaroline. Additionally, biofilm formation protects against antimicrobial activity, with up to 1,000-fold decreases in drug susceptibility observed (20). This is evident when evaluating biofilm MICs that are often several dilutions higher than the MICs obtained by broth microdilution. The 1- to 2-fold increase in biofilm MIC compared to the standard broth microdilution MIC observed in our study was comparable to that in the literature (12, 26).
S. aureus, specifically MRSA, has been recognized by the Infectious Diseases Society of America as a pathogen for which new therapies are needed (1). Recently, several new Gram-positive agents have been approved with MRSA coverage, including ceftaroline (27). Previous studies conducted in our laboratory have demonstrated potent synergy against MRSA, including strains with decreased susceptibility to glyco- and lipopeptides, with combination therapy, particularly with ceftaroline plus daptomycin (10–12). However, these data were limited to one-compartment and simulated endocardial vegetation models, and to date, no studies have been conducted to explore these combinations against biofilm-embedded isolates using a biofilm PK/PD model.
In the present study, our results not only confirm significant activity for the combination of ceftaroline plus daptomycin but also provide evidence of the antibacterial activity within the biofilm. This combination was highly efficacious against biofilm-producing MRSA, with an average reduction in starting inoculum of 3.33 ± 1.01 log10 CFU/cm2 within 72 h. We demonstrated that ceftaroline monotherapy was active against biofilm-producing MRSA as well, with an overall reduction of 2.48 ± 0.6 log10 CFU/cm2 by 72 h. This activity is in part likely due to elevated vancomycin and daptomycin MICs observed in the biofilm-producing organisms, with ceftaroline previously demonstrating enhanced activity against isolates with reduced susceptibility to glycopeptides and lipopeptides (23, 28). Despite daptomycin nonsusceptibility, the addition of ceftaroline provides restoration of daptomycin activity, or potentially, the additional inhibition of the biofilm matrix provided by daptomycin may allow for the activity of ceftaroline against these organisms (29, 30). Imaging studies further confirmed the activity of the select agents evaluated. A decreased bacterial burden was observed with the combination of ceftaroline plus daptomycin versus either monotherapy or drug-free controls. Our results are consistent with previous imaging studies, which demonstrated the potency of daptomycin combinations against biofilm-producing MRSA (13). Unfortunately, we did not observe similar results with the other antimicrobial combination tested. In this experiment, the addition of rifampin to ceftaroline displayed minimal benefit and even antagonism, depending on the strain. While mechanistically we are uncertain of how this occurred, current literature supports a possible antagonistic effect between these two agents (31).
While other antimicrobials, including rifampin, gentamicin, and clarithromycin, have been used in combination with daptomycin against biofilm-producing MRSA, the isolates evaluated in these studies were susceptible to all agents tested (13, 32). Our study evaluated strains with decreased susceptibility, which is important when trying to extrapolate these findings to other strains. Clinically, biofilm MICs are not routinely assessed, which might have major impacts on patient outcomes. While a strain may appear to be susceptible, the concentration of antimicrobial needed to overcome the infection within the biofilm may not be adequate. Therefore, evaluating strains with already-elevated MICs would provide a worst-case scenario.
There are several limitations to our study that should be noted. We used only one type of material (polyurethane) commonly found in medical devices. While our results might simulate catheter-related biofilm-embedded organisms, different results may be obtained with other types of materials used in prostheses, such as titanium, Teflon, or steel. Additionally, our experiment duration was limited to 72 h. Ceftaroline alone may have reached bactericidal activity in the other strains, rather than just in R6913, with a longer experimental period.
In conclusion, we found that combination therapy with ceftaroline plus daptomycin improved killing of biofilm-producing staphylococci compared to that with monotherapy, with bactericidal activity obtained in all strains. Additionally, ceftaroline retained susceptibility despite biofilm formation and displayed activity against all strains, including reaching bactericidal activity in one strain at 72 h. While the results with rifampin combinations were inconsistent in vitro, clinical data suggest that the addition of this agent is useful against biofilm-associated medical device infections. Further investigations are warranted to determine the clinical impact of ceftaroline alone or combined with daptomycin against biofilm-producing MRSA.
ACKNOWLEDGMENTS
This work was funded by an investigator-initiated grant from Forest Pharmaceuticals (TEF-IT-34). B.R.B. was supported by grant R21AI111638 from the NIH NIAID.
M.J.R. has received partial support from R21 AI109266-01 from NIAID and has received grant support from, consulted for, or provided lectures for Cepheid, Cerexa, Cubist, Durata, Forest, Novartis, Theravance, and Trius. W.E.R. has received grant support from, consulted for, or provided lectures for Cubist, Theravance, The Medicines Company, and Visante, Inc. K.E.B., J.R.S., C.E.I., and B.R.B. declare no conflicts of interest.
REFERENCES
- 1.Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, Scheld M, Spellberg B, Bartlett J. 2009. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin Infect Dis 48:1–12. doi: 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- 2.Sievert DM, Ricks P, Edwards JR, Schneider A, Patel J, Srinivasan A, Kallen A, Limbago B, Fridkin S, National Healthcare Safety Network (NHSN) Team and Participating NHSN Facilities. 2013. Antimicrobial-resistant pathogens associated with healthcare-associated infections: summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2009–2010. Infect Control Hosp Epidemiol 34:1–14. doi: 10.1086/668770. [DOI] [PubMed] [Google Scholar]
- 3.Dhand A, Sakoulas G. 2012. Reduced vancomycin susceptibility among clinical Staphylococcus aureus isolates (‘the MIC Creep'): implications for therapy. F1000 Med Rep 4:4. doi: 10.3410/M4-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Hal SJ, Barbagiannakos T, Jones M, Wehrhahn MC, Mercer J, Chen D, Paterson DL, Gosbell IB. 2011. Methicillin-resistant Staphylococcus aureus vancomycin susceptibility testing: methodology correlations, temporal trends and clonal patterns. J Antimicrob Chemother 66:2284–2287. doi: 10.1093/jac/dkr280. [DOI] [PubMed] [Google Scholar]
- 5.Howden BP, Davies JK, Johnson PD, Stinear TP, Grayson ML. 2010. Reduced vancomycin susceptibility in Staphylococcus aureus, including vancomycin-intermediate and heterogeneous vancomycin-intermediate strains: resistance mechanisms, laboratory detection, and clinical implications. Clin Microbiol Rev 23:99–139. doi: 10.1128/CMR.00042-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Römling U, Balsalobre C. 2012. Biofilm infections, their resilience to therapy and innovative treatment strategies. J Intern Med 272:541–561. doi: 10.1111/joim.12004. [DOI] [PubMed] [Google Scholar]
- 7.Donlan RM, Costerton JW. 2002. Biofilms: survival mechanisms of clinically relevant microorganisms. Clin Microbiol Rev 15:167–193. doi: 10.1128/CMR.15.2.167-193.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hall-Stoodley L, Costerton JW, Stoodley P. 2004. Bacterial biofilms: from the natural environment to infectious diseases. Nat Rev Microbiol 2:95–108. doi: 10.1038/nrmicro821. [DOI] [PubMed] [Google Scholar]
- 9.Osmon DR, Berbari EF, Berendt AR, Lew D, Zimmerli W, Steckelberg JM, Rao N, Hanssen A, Wilson WR, Infectious Diseases Society of America. 2013. Diagnosis and management of prosthetic joint infection: clinical practice guidelines by the Infectious Diseases Society of America. Clin Infect Dis 56:e1–e25. doi: 10.1093/cid/cis803. [DOI] [PubMed] [Google Scholar]
- 10.Barber KE, Werth BJ, Rybak MJ. 2015. The combination of ceftaroline plus daptomycin allows for therapeutic de-escalation and daptomycin sparing against MRSA. J Antimicrob Chemother 70:505–509. doi: 10.1093/jac/dku378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Werth BJ, Sakoulas G, Rose WE, Pogliano J, Tewhey R, Rybak MJ. 2013. Ceftaroline increases membrane binding and enhances the activity of daptomycin against daptomycin-nonsusceptible vancomycin-intermediate Staphylococcus aureus in a pharmacokinetic/pharmacodynamic model. Antimicrob Agents Chemother 57:66–73. doi: 10.1128/AAC.01586-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barber KE, Werth BJ, McRoberts JP, Rybak MJ. 2014. A novel approach utilizing biofilm time-kill curves to assess the bactericidal activity of ceftaroline combinations against biofilm-producing methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother 58:2989–2992. doi: 10.1128/AAC.02764-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Parra-Ruiz J, Vidaillac C, Rose WE, Rybak MJ. 2010. Activities of high-dose daptomycin, vancomycin, and moxifloxacin alone or in combination with clarithromycin or rifampin in a novel in vitro model of Staphylococcus aureus biofilm. Antimicrob Agents Chemother 54:4329–4334. doi: 10.1128/AAC.00455-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clinical and Laboratory Standards Institute. 2012. Performance standards for antimicrobial susceptibility testing; 22nd informational supplement. CLSI document M100-S22. Clinical and Laboratory Standards Institute, Wayne, PA: http://antimicrobianos.com.ar/ATB/wp-content/uploads/2012/11/M100S22E.pdf. [Google Scholar]
- 15.Ceri H, Olson ME, Stremick C, Read RR, Morck D, Buret A. 1999. The Calgary Biofilm Device: new technology for rapid determination of antibiotic susceptibilities of bacterial biofilms. J Clin Microbiol 37:1771–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Saravolatz LD, Stein GE, Johnson LB. 2011. Ceftaroline: a novel cephalosporin with activity against methicillin-resistant Staphylococcus aureus. Clin Infect Dis 52:1156–1163. doi: 10.1093/cid/cir147. [DOI] [PubMed] [Google Scholar]
- 17.Benvenuto M, Benziger DP, Yankelev S, Vigliani G. 2006. Pharmacokinetics and tolerability of daptomycin at doses up to 12 milligrams per kilogram of body weight once daily in healthy volunteers. Antimicrob Agents Chemother 50:3245–3249. doi: 10.1128/AAC.00247-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Merrell Dow Pharmaceuticals, Inc. 2000. Rifadin(R), rifampin capsules and injection package insert. Merrell Dow Pharmaceuticals, Inc., Kansas City, MO. [Google Scholar]
- 19.Bennett WM, Aronoff GR, Morrison G, Golper TA, Pulliam J, Wolfson M, Singer I. 1983. Drug prescribing in renal failure: dosing guidelines for adults. Am J Kidney Dis 3:155–193. doi: 10.1016/S0272-6386(83)80060-2. [DOI] [PubMed] [Google Scholar]
- 20.Peloquin CA, Namdar R, Singleton MD, Nix DE. 1999. Pharmacokinetics of rifampin under fasting conditions, with food, and with antacids. Chest 115:12–18. doi: 10.1378/chest.115.1.12. [DOI] [PubMed] [Google Scholar]
- 21.Blaser J. 1985. In-vitro model for simultaneous simulation of the serum kinetics of two drugs with different half-lives. J Antimicrob Chemother 15(Suppl A):125–130. [DOI] [PubMed] [Google Scholar]
- 22.Pillai SK, Moellering RC Jr, Eliopoulos GM. 2005. Antimicrobial combinations, p 365–440. In Lorian V. (ed), Antibiotics in laboratory medicine, 5th ed Williams & Wilkins Co., Baltimore, MD. [Google Scholar]
- 23.Werth BJ, Steed ME, Kaatz GW, Rybak MJ. 2013. Evaluation of ceftaroline activity against heteroresistant vancomycin-intermediate Staphylococcus aureus and vancomycin-intermediate methicillin-resistant S. aureus strains in an in vitro pharmacokinetic/pharmacodynamic model: exploring the “seesaw effect.” Antimicrob Agents Chemother 57:2664–2668. doi: 10.1128/AAC.02308-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Falagas ME, Kapaskelis AM, Kouranos VD, Kakisi OK, Athanassa Z, Karageorgopoulos DE. 2009. Outcome of antimicrobial therapy in documented biofilm-associated infections: a review of the available clinical evidence. Drugs 69:1351–1361. doi: 10.2165/00003495-200969100-00005. [DOI] [PubMed] [Google Scholar]
- 25.Stewart PS, Costerton JW. 2001. Antibiotic resistance of bacteria in biofilms. Lancet 358:135–138. doi: 10.1016/S0140-6736(01)05321-1. [DOI] [PubMed] [Google Scholar]
- 26.Frank KL, Reichert EJ, Piper KE, Patel R. 2007. In vitro effects of antimicrobial agents on planktonic and biofilm forms of Staphylococcus lugdunensis clinical isolates. Antimicrob Agents Chemother 51:888–895. doi: 10.1128/AAC.01052-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sader HS, Fritsche TR, Kaniga K, Ge Y, Jones RN. 2005. Antimicrobial activity and spectrum of PPI-0903M (T-91825), a novel cephalosporin, tested against a worldwide collection of clinical strains. Antimicrob Agents Chemother 49:3501–3512. doi: 10.1128/AAC.49.8.3501-3512.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barber KE, Ireland CE, Bukavyn N, Rybak MJ. 2014. Observation of “seesaw effect” with vancomycin, teicoplanin, daptomycin and ceftaroline in 150 unique MRSA strains. Infect Dis Ther 3:35–43. doi: 10.1007/s40121-014-0023-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roveta S, Marchese A, Schito GC. 2008. Activity of daptomycin on biofilms produced on a plastic support by Staphylococcus spp. Int J Antimicrob Agents 31:321–328. doi: 10.1016/j.ijantimicag.2007.11.012. [DOI] [PubMed] [Google Scholar]
- 30.Dhand A, Bayer AS, Pogliano J, Yang SJ, Bolaris M, Nizet V, Wang G, Sakoulas G. 2011. Use of antistaphylococcal beta-lactams to increase daptomycin activity in eradicating persistent bacteremia due to methicillin-resistant Staphylococcus aureus: role of enhanced daptomycin binding. Clin Infect Dis 53:158–163. doi: 10.1093/cid/cir340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sundareshan V, Modi J, Bergman S, Koirala J. 2012. Possible antagonism with use of ceftaroline and rifampin to treat methicillin-resistant Staphylococcus aureus infection. Int J Infect Dis 16S:e421. doi: 10.1016/j.ijid.2012.05.583. [DOI] [Google Scholar]
- 32.LaPlante KL, Woodmansee S. 2009. Activities of daptomycin and vancomycin alone and in combination with rifampin and gentamicin against biofilm-forming methicillin-resistant Staphylococcus aureus isolates in an experimental model of endocarditis. Antimicrob Agents Chemother 53:3880–3886. doi: 10.1128/AAC.00134-09. [DOI] [PMC free article] [PubMed] [Google Scholar]