

Abstract
Tuberculosis is a severe disease affecting millions worldwide. Unfortunately, treatment strategies are hampered both by the prohibitively long treatment regimen and the rise of drug-resistant strains. Significant effort has been expended in the search for new treatments, but few options have successfully emerged, and new treatment modalities are desperately needed. Recently, there has been growing interest in the synergistic antibacterial effects of copper ions (CuII/I) in combination with certain small molecular compounds, and we have previously reported development of a drug screening strategy to harness the intrinsic bactericidal properties of CuII/I. Here, we describe the copper-dependent antimycobacterial properties of disulfiram, an FDA-approved and well-tolerated sobriety aid. Disulfiram was inhibitory to mycobacteria only in the presence of CuII/I and exerted its bactericidal activity well below the active concentration of CuII/I or disulfiram alone. No other physiologically relevant bivalent transition metals (e.g., FeII, NiII, MnII, and CoII) exhibited this effect. We demonstrate that the movement of the disulfiram-copper complex across the cell envelope is porin independent and can inhibit intracellular protein functions. Additionally, the complex is able to synergistically induce intracellular copper stress responses significantly more than CuII/I alone. Our data suggest that by complexing with disulfiram, CuII/I is likely allowed unfettered access to vulnerable intracellular components, bypassing the normally sufficient copper homeostatic machinery. Overall, the synergistic antibacterial activity of CuII/I and disulfiram reveals the susceptibility of the copper homeostasis system of Mycobacterium tuberculosis to chemical attacks and establishes compounds that act in concert with copper as a new class of bacterial inhibitors.
INTRODUCTION
In 2013, 9 million people worldwide were estimated to have developed tuberculosis (TB), resulting in 1.5 million deaths globally (1). Major obstacles to global control efforts include (i) the complete lack of convenient and easy-to-comply-with short-term or single-drug treatment options, (ii) the ability of Mycobacterium tuberculosis to persist in the human host, asymptomatically and undetectably, during and after treatment with a 10% lifetime chance to reactivate eventually (2), (iii) adverse drug interactions in the cotreatment of HIV and M. tuberculosis infections (3, 4), (iv) a short supply of first-line drugs (5), and (v) the rise of multidrug-resistant (MDR), extensively drug-resistant (XDR), and even totally drug-resistant (TDR) M. tuberculosis strains (6). Only two new antitubercular drugs, bedaquiline (Sirturo, TMC207) and delamanid (Deltyba, OPC-67683), were approved in recent years for use against MDR- and XDR-TB in the United States and Europe, respectively, with phase III clinical trials still ongoing to elucidate potentially serious side effects that may limit the utility of these drugs (7–9). Additional antituberculosis drugs are still desperately needed. However, the approval of novel inhibitors for any treatment is a lengthy and costly process. Efficacy studies and the establishment of comprehensive safety profiles may take up to 6 years per drug, with time estimates for a new four-drug cocktail approaching 25 years (10). Repurposing of already approved drugs for new medical uses is therefore a faster and more economical approach as safety profiles in humans have already been established, and there are multiple promising candidates (11).
An example of potential drug repurposing is disulfiram (DSF), currently FDA approved for the treatment of chronic alcoholism. Studies to evaluate the potency of disulfiram against certain types of cancer (12–14), Menkes disease (15), and HIV-1 infections are ongoing or have just been completed (16, 17). Of additional interest to the medical community are its antinematode (18), antifungal (19), and, importantly, its antibacterial properties (11, 20–24).
Chemically, disulfiram belongs to the group of dithiocarbamates. While stable at neutral or slightly alkaline pH (7 to 7.9), disulfiram quickly breaks down to diethyldithiocarbamate (DETC) in acidic or copper-rich environments (25–27). Two molecules of DETC chelate a single copper ion (Fig. 1A). Though copper ions have been found to play a role in disulfiram's anticancer activity (26), whether or not they are also essential for the previously reported antibacterial properties of disulfiram against M. tuberculosis (21, 28) and other microbes (20, 23) has not been sufficiently addressed.
FIG 1.

Formation of transition metal complexes by disulfiram and DETC. (A) At an acidic pH or in the presence of copper, disulfiram reductively splits into 2 DETC molecules (27, 54), which are strong metal ion-complexing agents. The copper complex of disulfiram is identical to the DETC-copper complex CuII(DETC)2. (B) Complex formation of disulfiram with copper and the lack thereof in the presence of other metabolically relevant trace metal ions. (C) Metal complexes of DETC (from left to right: CuII, CoII, FeII, NiII, MnII, ZnII). Reports of the overall stability constants (log β2) of the copper-DETC complex range from 18.7 (39) to 25 (37), but copper is consistently found to form the most stable complex with DETC.
Recently, we described copper ions as an important modulator/activator of the antibacterial properties of small molecules (24, 29). Here, we investigate the role of copper in the antimycobacterial activity of disulfiram. We identified a significant copper dependency of the antimycobacterial properties of disulfiram. Further, we demonstrate its ability to inhibit intracellular proteins and induce a copper stress response. We present disulfiram both as a promising therapeutic compound and as an excellent example of a new class of compounds harnessing copper ions as an environmental insult to enhance antibacterial activity.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The Mycobacterium smegmatis strains SMR5 (wild type [wt]) (30) and ML16 (SMR5 derivative; ΔmspA ΔmspC ΔmspD) (31) and M. tuberculosis strains H37Rv (wt) and mc26230 (H37Rv derivative; ΔpanCD ΔRD1) (32) were regularly grown in Middlebrook 7H9 (BD Difco) or Hartmans-de Bont minimal medium (HdB) (33) or on Middlebrook 7H10 agar (BD Difco). Our modified, copper-free HdB consists of 30 μM EDTA, 500 μM MgCl2, 7 μM CaCl2, 0.8 μM NaMoO4, 1.68 μM CoCl2, 5.49 μM MnCl2, 6.95 μM ZnSO4, 20 μM FeSO4, 15 mM (NH4)2SO4, 6 mM K2HPO4, 6 mM NaH2PO4, 0.5% glucose, and 0.02% tyloxapol. Hygromycin (50 μg/ml), 10% oleic acid-albumin-dextrose-catalase (OADC) (Remel), 1% casamino acids, 24 μg/ml pantothenate, or combinations thereof were supplemented as needed. When indicated, strains carried the empty expression vector pMS2 (34) or its derivative pML775 expressing luxAB under the control of the pwmyc promoter (34). Copper-reduced media (HdB, 7H9, or 7H10) were prepared from scratch using a copper salt-free recipe and deionized water.
Resazurin assay.
Multiple dose-response curves were obtained as described previously (29). Briefly, bacteria were grown in copper-free HdB medium. M. smegmatis strains grew overnight and M. tuberculosis strains for up to 14 days not to exceed an optical density at 600 nm (OD600) of 2.0. Cultures were diluted to an OD600 of 0.04 in 2-fold concentrated copper-free HdB medium right before the assay was performed. Test compounds were diluted at 2-fold concentrations (starting at 20 μM disulfiram or the equivalent of 40 μM DETC) in 2-fold increments in 96-well plates using deionized water with or without 20 μM copper sulfate as indicated. An equal volume of 2-fold concentrated HdB medium with cells was then added to each well. Unless stated otherwise, the final copper sulfate concentration per well was 10 μM, and compounds were diluted down from 10 μM. Resazurin dye was added to a final concentration of ∼90 μM resazurin and 0.01% Tween 80 after a 16- to 24-h incubation period at 37°C for M. smegmatis or after 7 to 10 days of incubation for M. tuberculosis. Over the course of several hours, metabolically active cells convert resazurin (blue) to resorufin (pink). The fluorescence intensity of resorufin (excitation, 530 nm; emission, 590 nm) was determined on a Cytation 3 imaging reader (BioTek) and analyzed using Gen5 software (BioTek) and Excel (Microsoft). All conditions were analyzed in triplicate, and all experiments were repeated at least twice. Plates contained a medium-only control to determine the background dye fluorescence in the absence of cells and a no-treatment condition to determine dye conversion rates under optimal growth (100% growth). All data points were normalized to plate-specific controls prior to the comparison of results from different plates.
In some instances, small samples of 5 μl were removed from the wells and spotted on appropriate agar plates to determine the presence or absence of viable cells and to verify the results obtained from the resazurin assay.
Luciferase assay.
M. smegmatis cells carrying the control vector pMS2 or the luciferase vector pML775 were grown overnight in HdB medium and adjusted to an OD600 of 0.02. Cells for lysis were grown identically, but harvested, washed in phosphate-buffered saline (PBS), and resuspended at an OD600 of 0.1 and sonicated on ice with 1-s pulses. Both whole cells and lysates were treated with DETC, DETC-Cu, or Cu for 45 min at 37°C. A 100-μl sample was transferred to a 96-well plate in triplicate, and with use of the injector on the Cytation 3, 30 μl of 0.025% decanal was added. Luciferase activity was determined using the luminescence mode on the plate reader. Luminescence was recorded in 1-s intervals over 5 s, with the final recorded value as the integral of the curve. All samples were normalized to untreated control values. The data sets shown are representatives of at least two independent experiments with comparable trends.
Cellular copper content analysis.
We used inductively coupled plasma mass spectroscopy (ICP-MS), as previously described (24), and adapted our previously developed Phen Green FL (PGFL) assay (24) to determine treatment-dependent alterations of the cellular total copper content and the labile cellular copper pool, respectively. Mycobacteria were grown in copper-free HdB medium, washed in 10 mM HEPES buffer (pH 7.75), and then treated for 2 h (unless stated otherwise) at 37°C with Cu, DETC, or DETC-Cu at the indicated concentrations. Thereafter, cells were washed once in HEPES buffer containing 500 μM EDTA and then twice in plain HEPES buffer, and the pellet was resuspended and adjusted to an OD600 of 1. An aliquot of each sample was transferred into a 96-well plate, and a solution containing 2.5 μM PGFL was added. Fluorescence (excitation, 490 nm; emission, 520 nm) was determined on a Cytation 3 imaging reader, recorded by Gen5 software, and analyzed using Microsoft Excel and GraphPad Prism.
Induction and detection of MmcO expression in M. tuberculosis.
Cells were grown in copper-free HdB medium, harvested by centrifugation, adjusted to an OD600 of 1, and then treated for 2 days by addition of CuSO4 solution, disulfiram, or a mixture of both at the indicated concentrations. Upon completion of the incubation period, cells were washed with PBS containing 0.02% tyloxapol, centrifuged, resuspended in PBS containing 1% SDS, mechanically lysed in a fast prep benchtop homogenizer, and subsequently boiled for 10 min to extract membrane proteins. Insoluble debris was removed by centrifugation at 10,000 × g for 5 min. The protein content of the supernatant was determined using a Pierce bicinchoninic acid (BCA) protein assay kit (Thermo), following the instructions of the manufacturer. From each sample, 50 μg of protein extract was separated by SDS-PAGE and analyzed by Western blotting using custom-made polyclonal antibodies raised in rabbits against purified M. tuberculosis multicopper oxidase (MmcO) (35).
RESULTS
Copper complex formation by disulfiram and its major metabolite, DETC.
In contrast to many other dithiocarbamates and the prevailing opinion, disulfiram itself is a very poor metal chelator (36). The metal-complexing ability of disulfiram lies in the reduction of its disulfide bond, producing two molecules of diethyldithiocarbamate (DETC) (Fig. 1A), which is a very strong metal binder and subsequently coordinates copper or other divalent metal ions (37). In comparison to other bivalent transition metals, the reduction of disulfiram to DETC in the presence of copper ions is reasonably fast, producing the characteristic CuII(DETC)2 complex with its typical yellow/brownish color well within 1 h when disulfiram and copper ions are mixed together (Fig. 1B) or immediately if DETC is used instead (Fig. 1C). The Irving-Williams series describes the general stability of ligand complexes with first-row transition metal ions, predicting that copper generally forms the most stable complexes (38). Although DETC is also capable of coordinating a variety of other bivalent transition metals, including FeII, NiII, MnII, and CoII, as indicated by the distinctive and instantaneous change in color upon mixing (Fig. 1C) [the ZnII(DETC)2 complex is a colorless exception], copper complexes are in fact the most stable (39). Furthermore, the physiological transition metal ions neither interfered with the copper-mediated reduction of disulfiram nor did their presence prevent the formation of the characteristic yellow CuII(DETC)2 complex with an absorption peak at ∼435 nm, indicating that metal ions present in the growth medium used would not interfere with the formation and stability of the disulfiram-copper complex.
Antimycobacterial properties of disulfiram are copper dependent and copper specific.
We discovered the copper-dependent antimycobacterial properties of disulfiram in a small-scale pilot drug screen for compounds with copper-dependent antimycobacterial properties that was conducted on M. smegmatis, a well-established surrogate organism for M. tuberculosis. By constructing a dose-response curve, we confirmed that disulfiram was completely inactive on M. smegmatis under copper-depleted growth conditions but was an excellent growth inhibitor in the presence of copper with a 90% inhibitory concentration (IC90) of 0.6 μM (Fig. 2A). Disulfiram had an IC90 of 0.3 μM against M. tuberculosis mc26230 (ΔRD1 ΔpanCD; avirulent, biosafety level 2 [BSL2] classified derivative of M. tuberculosis H37Rv) (Fig. 2B) and M. tuberculosis H37Rv (virulent wild type) (Fig. 2C) and reached its maximum potency in the presence of only 0.3 μM copper (Fig. 2D). The equimolar stoichiometry between copper and disulfiram is consistent with the formation of a CuII(DETC)2 complex.
FIG 2.

Copper-dependent activity of disulfiram on mycobacteria. Disulfiram (DSF) exhibited strong, copper-dependent inhibitory effects on all mycobacterial strains tested, including M. smegmatis SMR5 (A), M. tuberculosis mc26230 (B), and M. tuberculosis H37Rv (C), at submicromolar concentrations. (D) A copper titration using 0.3 μM DSF exhibited complete inhibitory activity at a stoichiometrically equal concentration of 0.3 μM copper, indicating a synergistic interaction between both disulfiram and copper ions.
As mentioned above, the reduction of disulfiram to DETC in the presence of other transition metals, including ZnII, FeII, CoII, MnII, and NiII, is unfavorable as evidenced by the lack of a color change upon addition of these metals to disulfiram (Fig. 1B). Hence, we used DETC, the natural in vivo metabolite of disulfiram, to generate complexes with FeII, MnII, and NiII and purchased the colorless ZnII(DETC)2 complex from a commercial source (Sigma-Aldrich). None of these alternative metal complexes elicited a noteworthy growth inhibitory activity against M. tuberculosis (Fig. 3A).
FIG 3.

Copper-dependent antimycobacterial effects of disulfiram. (A) Against M. tuberculosis mc26230, DETC displayed significant antimycobacterial effects only in the presence of CuII, but not if other tested physiologically relevant transition metals (CoII, FeII, NiII, MnII, or ZnII) were provided instead. (B) A commercially obtained zinc-DETC complex had no inhibitory activity against M. tuberculosis mc26230, but the addition of copper to the ZnII(DETC)2 complex resulted in an inhibition profile identical to that for disulfiram in the presence of copper. (C) A CuII(DETC)2 complex has a spectrophotometric peak at ∼435 nm; complex formation was observed even when commercially purchased ZnII(DETC)2 was used as the DETC source, indicating that copper can outcompete other complexed metals. Control spectra, consisting of DMSO, copper, and DETC alone, as well as the ZnII(DETC)2 complex in the absence of copper, are displayed; none displayed absorbance over the given spectrum. (D) While M. tuberculosis mc26230 is susceptible to as little as 0.3 μM disulfiram in 0.3 μM Cu (Fig. 2D), addition of 10 μM bathocuproine (BCS), a membrane-impermeable copper chelator, completely ablates activity.
We also found that in the presence of copper, disulfiram is bactericidal on M. tuberculosis at 0.3 μM, while disulfiram itself or the Zn complex has no such activity (Fig. 3B). Interestingly, addition of copper ions to the Zn complex killed M. tuberculosis as efficiently as the disulfiram-copper mix (Fig. 3B), suggesting that the copper complex forms even in the presence of zinc, which is the most abundant transition metal in the human body (40). Our spectrophotometric analysis of the ZnII(DETC)2-copper mix confirmed the replacement of ZnII by copper ions and the subsequent formation of a CuII(DETC)2 complex (Fig. 3C). These data are consistent with the much greater affinity of DETC for copper than for Zn (39). Additionally, bathocuproine (BCS), a very strong membrane-impermeable copper chelator, completely inhibited disulfiram's antibacterial activity in the presence of copper (Fig. 3D), indicating that complexation is necessary for activity. Alternatively, this phenotype could result from disulfiram inhibiting bacterial copper homeostasis systems, as BCS chelation of copper would prevent toxicity being realized through generalized copper overload mechanisms, although this seems unlikely. Multiple groups have extensively analyzed copper homeostasis mutants of M. tuberculosis, but such extreme susceptibility to only 0.3 μM Cu as we see with disulfiram (Fig. 2D) had never been reported (35, 41–45). Thus, it appears that the physical interaction of disulfiram or DETC with copper ions is a critical event for the antimycobacterial effects to take place.
The disulfiram-copper complex is bactericidal on replicating and nonreplicating M. tuberculosis cells.
While treatment of active M. tuberculosis infection has been complicated by the development of drug-resistant strains, the intrinsic drug resistance phenotype of persistent M. tuberculosis infections poses a huge challenge to TB drug development. During latency, M. tuberculosis persists in a nutrient-deprived but copper-rich environment and consequently rarely undergoes cell division, which contributes to the increased tolerance of latent M. tuberculosis to current anti-M. tuberculosis drugs (41, 46). For those reasons, drugs with the ability to sterilize M. tuberculosis in copper-rich in vivo environments and kill latent M. tuberculosis would be highly desirable (47). Therefore, we investigated the bactericidal properties of disulfiram in the presence of copper on replicating and nongrowing M. tuberculosis using a previously published and characterized starvation model of M. tuberculosis, which mimics dormant cells (28, 29). To induce starvation, M. tuberculosis mc26230 was cultured for 6 weeks in PBS buffer containing 0.02% tyloxapol. These cells were then treated with disulfiram in the presence and absence of copper ions for up to 8 days. Every 2 days, a 5-μl sample aliquot was transferred onto Middlebrook 7H10 solid medium to evaluate bacterial survival. Growth on plates was permitted for 16 days at 37°C. Rifampin was included as a control. In these experiments, we found that disulfiram at concentrations of 2.5 μM was able to kill nongrowing M. tuberculosis in the presence of copper (10 μM) (Fig. 4), while replicating cells were, as expected, susceptible to 0.3 μM disulfiram in the presence of copper. No killing was observed on either cell type in the absence of copper (Fig. 4).
FIG 4.

Disulfiram is active on nonreplicating M. tuberculosis when copper ions are present. Disulfiram at 0.3 μM and copper at 10 μM kill actively replicating M. tuberculosis mc26230 after 3 days of incubation, with 12 μM rifampin (RIF) shown as the bactericidal control. Nonreplicating bacilli were generated by incubating bacteria in PBS for 8 weeks; they were then exposed to the same conditions as the replicating bacilli. While 0.3 μM disulfiram was unable to inhibit growth after 3 days, 2.5 μM disulfiram, when acting in concert with 10 μM copper, was able to completely kill M. tuberculosis.
Taken together, our data suggest that both replicating and nonreplicating M. tuberculosis cells are highly susceptible to disulfiram and its major metabolite, DETC, at concentrations that are achievable in vivo (48).
Disulfiram-copper complex inhibits luciferase activity in the cytoplasm and penetrates the mycobacterial cell envelope in a porin-independent manner.
As the specific intracellular targets of the disulfiram-copper complex in mycobacteria are still unknown, we developed a luciferase reporter assay to demonstrate the potential of the copper complex to act upon intracellular targets. Although the precise mechanism of inhibition is still unknown, Vibrio harveyi luciferase contains a critical cysteine in its alpha chain, vulnerable to thiol-directed attacks (49). Given that both disulfiram and copper have affinity for thiol groups (50–52), we reasoned that the complex may target the critical cysteine, thereby inactivating the enzyme. We expressed V. harveyi luciferase from pML775 in M. smegmatis. We found that luciferase activity in lysates of M. smegmatis is highly susceptible to the disulfiram-copper complex but stable when challenged with an equivalent amount of copper ions or disulfiram alone (Fig. 5A). We then conducted the same experiment on whole cells expressing luciferase from pML775. Incubation with copper or disulfiram alone did not affect luciferase activity. However, combining disulfiram and copper resulted in a >85% decrease in luciferase activity (Fig. 5A). Inhibition of the cytoplasmic luciferase activity in whole cells is therefore indicative of the ability of the copper complex to effectively penetrate the mycobacterial cell envelope.
FIG 5.
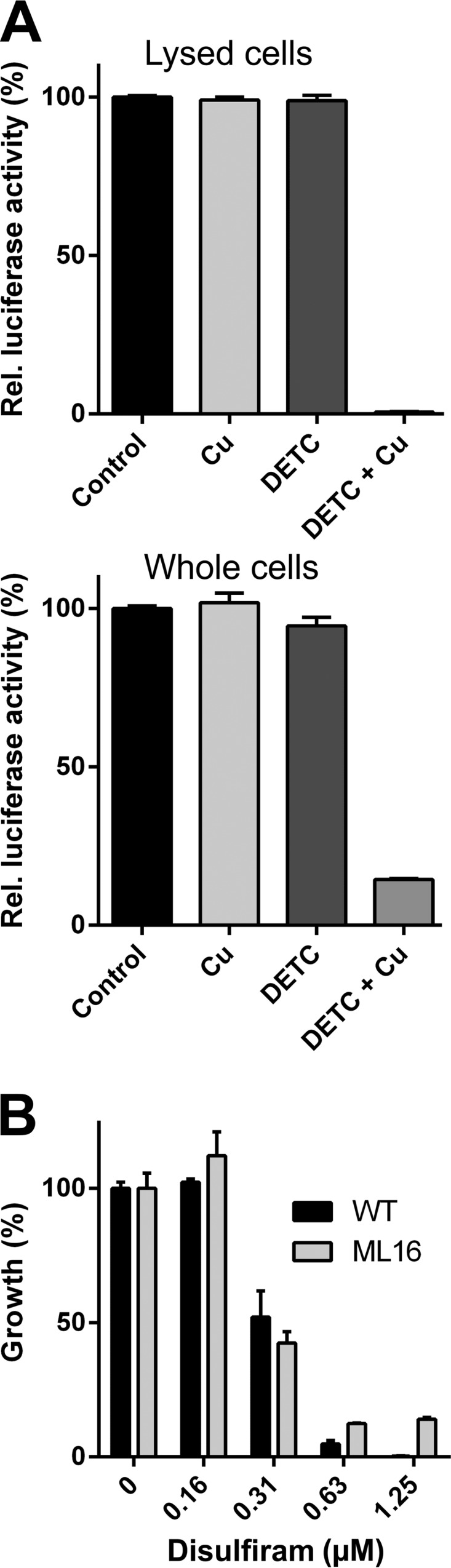
CuII(DETC)2 is able to pass through lipid membranes and inhibits intracellular targets. (A) Luciferase activity is sensitive to DETC-copper complex in lysed and whole cells of M. smegmatis SMR5. Luciferase was expressed from the mycobacterial vector pML775. Cu and DETC were provided at 0.3 μM and 0.6 μM, respectively. (B) The CuII(DETC)2 complex enters cells in a porin-independent manner, with no appreciable difference in inhibition between M. smegmatis SMR5 (wild type) and ML16 (ΔmspA ΔmspC ΔmspD).
There are two primary pathways by which most antibiotics cross the mycobacterial outer membrane: hydrophobic antibiotics directly diffuse through the lipid layers, while hydrophilic molecules utilize porins to enter the cell (53). The literature indicates that disulfiram and CuII(DETC)2 are highly lipophilic (54), which suggests that they cross the highly hydrophobic mycobacterial outer membrane in a porin-independent manner. To experimentally determine whether the uptake of the disulfiram-copper complex is a porin-independent process, we treated the triple-porin mutant of M. smegmatis, ML16, and the wild type with disulfiram in the presence or absence of copper. The use of ML16 for this purpose was previously validated by several studies (55–57). We found that the susceptibility of the triple-porin mutant to disulfiram in the presence of copper was similar to that of the wild type (Fig. 5B) despite the 15-fold differential in the numbers of outer membrane porin channels (31) and the much greater copper tolerance of ML16 (56). This result strongly suggests that CuII(DETC)2 uptake occurs in a porin-independent fashion. A similar conclusion can be reached from the comparison of the inhibitory concentrations between M. smegmatis and M. tuberculosis (Fig. 2). Both strains have comparable susceptibilities to CuII(DETC)2 despite the up to 40-fold lower membrane permeability of M. tuberculosis for small and hydrophilic solutes (58).
Disulfiram potentiates intracellular copper stress without increasing the total cellular copper content.
As copper ions are crucial for disulfiram's antibacterial properties, we analyzed the potential of the disulfiram-copper complex to induce a copper-dependent stress response. Several known copper homeostasis and resistance genes in M. tuberculosis are controlled by RicR (rv0190), a transcriptional repressor that enables gene expression in response to a rising intracellular copper content (59). Part of the RicR regulon is mmcO, which encodes a recently characterized periplasmic multicopper oxidase (35, 42). It was previously demonstrated that MmcO expression is dependent on the copper content of the medium. If the disulfiram-copper complex CuII(DETC)2 is capable of interfering with intracellular copper homeostasis, expression of mmcO should be induced. Direct Western blot analysis of M. tuberculosis mc26230-derived protein extracts revealed a significant increase in the MmcO protein after treatment with the disulfiram-copper complex within only 24 h of administration of the compound (Fig. 6A), while treatment with the individual components (copper or disulfiram) at appropriate concentrations failed to do so.
FIG 6.

The disulfiram-copper complex induces a copper stress response in M. tuberculosis. (A) Mycobacterial multicopper oxidase (MmcO), an indicator of copper stress, was heavily upregulated when M. tuberculosis mc26230 was challenged with both copper and disulfiram (DSF) but not when challenged with disulfiram or 1 μM Cu alone. RNA polymerase II (RNApol) was used to demonstrate that equal amounts of protein extract were analyzed. (B) M. smegmatis SMR5 cells incubated with copper and disulfiram showed no increase in total copper content compared to that with copper alone, as measured by ICP-MS. No increase in cell-associated copper levels was seen in EDTA-copper-treated cells. (C) Phen Green FL fluorescence was quenched significantly more by the DETC-copper complex than by copper alone, indicating that while total cellular copper content may be identical, intracellular copper is likely in a different, more labile, and accessible form in the presence of DETC. RFU, relative fluorescence units.
A previously reported property of copper complexes is the shuttling of copper ions into the cell by bypassing the normal homeostatic machinery and overloading the cell through increased concentrations of intracellular copper (60). To examine whether or not disulfiram acted in this manner, we subjected treated cultures to inductively coupled plasma mass spectrometry (ICP-MS) analysis, as well as the Phen Green FL assay, which we previously used with Staphylococcus aureus (24). Phen Green FL is taken up by cells and fluoresces, unless quenched by copper ions. Therefore, decreased fluorescence reflects an increase in accessible cellular copper. ICP-MS analysis showed no increase in the intracellular copper content of the disulfiram-copper-treated samples compared to copper-only treatment (Fig. 6B). However, when Phen Green FL was used to evaluate the properties of the intracellular copper pool, fluorescence quenching was observed in the DETC-Cu-treated sample as opposed to the copper-only group (Fig. 6C), indicating an increase in the labile pool of copper ions that are accessible to the dye. Together these data suggest that while the disulfiram-copper complex does not raise the total copper content of M. tuberculosis, the presence of disulfiram allows copper ions to stray from the proper intracellular copper handling and sequestration pathways, thereby inducing a RicR-dependent copper stress response in a concentration-dependent manner. In contrast to their sequestrated counterparts, these erratic copper ions may interfere with crucial cellular enzyme activities that, in the absence of disulfiram/DETC, are well protected by the cellular copper homeostasis and resistance machinery.
DISCUSSION
Disulfiram has been clinically prescribed for alcohol abuse for well over 60 years (25). Intensive research and numerous clinical studies established disulfiram as a safe drug lacking significant side effects. More recently, an extensive spectrum of potentially exploitable bioactivities beyond its initial clinical application has established disulfiram as an attractive candidate for multiple drug repositioning efforts (11, 12, 61–63). One of disulfiram's most promising traits is the ability to selectively target certain types of cancer, which is partially attributable to its ability to form a copper complex that inhibits proteasome functions (64, 65).
The antibacterial properties of disulfiram are less well studied, possibly because it was found to inhibit only a few select pathogens and only at in vitro concentrations much higher than those that can be achieved in humans. Nevertheless, in vivo activity of disulfiram against M. tuberculosis was previously established in both mouse and guinea pig models of infection, requiring doses of 80 and 20 mg/kg of body weight, respectively (21, 66). Faiman et al. reported for humans that with a daily dose of 250 mg of disulfiram, the blood concentration of its metabolite, DETC, is 0.77 μg/ml (∼5 μM), which is equivalent to ∼2.5 μM disulfiram (48). However, according to other reports, concentrations of 50 μM, 4.4 μM, or 27 μM, would be necessary to inhibit the growth of Pseudomonas aeruginosa (23), S. aureus (20), or M. tuberculosis (28) in vitro, respectively. Our reassessment of the antibacterial properties of disulfiram and its major metabolite, DETC, revealed that these compounds may be far more potent against at least M. tuberculosis than previously thought. We found that the antibacterial properties of disulfiram/DETC are strictly copper dependent with an active concentration of ∼0.3 μM toward growing and ∼2.5 μM toward nongrowing M. tuberculosis cells. Such a low MIC strongly supports its previously proposed consideration for repurposing as a novel antitubercular drug (11).
The natural antibacterial properties of copper ions are complex and, to date, not completely understood. The surrounding environment, both intra- and extracellular, may heavily modulate these effects. Anaerobic environments, for instance, induce far greater copper toxicity than aerobic environments. Some early evidence implied that redox cycling between CuII and CuI led to oxidative stress and DNA damage. However, newer studies have suggested that toxicity is more nuanced than simple oxidative damage. Macomber and Imlay were the first to change the paradigm, delivering compelling evidence that, at least in Escherichia coli, the primary targets of copper toxicity are solvent-exposed iron-sulfur clusters of branched-chain amino acid synthesis dehydrogenases, not DNA (67). The actual target may also shift, though, depending upon the physiology of the organism in question; copper toxicity has been reported against an array of aerobic and anaerobic metabolic targets containing or constructing iron-sulfur clusters (67–70). Given the different metabolic targets in these different systems, it seems possible that copper, with its broad chemical effects and ligand affinities, may actually target a wide variety of metabolic pathways. The reported toxic effects could simply represent the first disruption of homeostasis in each organism.
Unlike free copper ions, which by themselves are not toxic at concentrations needed to activate disulfiram (0.3 μM), very little is known about the underlying mechanism by which copper boosts the antibacterial properties of disulfiram or vice versa. Previously published evidence suggests that disulfiram and its metabolites may inhibit the betaine aldehyde dehydrogenases (23) of Pseudomonas aeruginosa via interaction with a catalytic cysteine. Disulfiram has also been reported to inhibit beta carbonic anhydrase from M. tuberculosis (23), presumably by ZnII cofactor coordination. These findings suggest that disulfiram can act specifically against selected targets. However, the preference of DETC for copper over zinc (37) and the requirement of copper complexation for activity on whole cells suggests that zinc replacement from the two beta carbonic anhydrases of M. tuberculosis (rv1284, rv3588c) is not the prime mechanism of the antibacterial properties of disulfiram/DETC. Instead, the complex may enable copper or DETC to directly interact with accessible cysteine residues or iron-sulfur clusters of other yet undefined intracellular proteins, which are typically inaccessible to copper ions entering the cells by canonical pathways. This idea is supported by our luciferase inhibition assay (Fig. 5A) and by the observed derepression of the RicR-controlled mmcO gene (rv0846c) (Fig. 6A).
The enhanced expression of copper resistance genes may further indicate that copper bound to DETC cannot be properly sequestered by intracellular copper resistance proteins like the metallothionein MymT (43). According to current models, it is believed that such intracellular copper resistance proteins bind free copper ions immediately upon their passing through the cytoplasmic membrane and before they interact with vulnerable proteins. This classical pathway of copper uptake and sequestration may be disturbed by the action of disulfiram/DETC. Most simply, disulfiram/DETC may act as a copper ionophore, shuttling copper ions into the cytoplasm, thereby bypassing regulated copper-handling pathways. The ultimate toxicity would then be realized by overloading the cell with copper ions and the subsequent iron-sulfur cluster degradation, Fenton-like chemistry, or metal cofactor displacement/replacement. Such an ionophore effect was reported for a copper-dependent therapeutic antifungal (60). However, our ICP-MS data directly contradict the possibility of cellular copper overload for disulfiram/DETC, showing similar concentrations of copper in complex-treated and copper-only samples (Fig. 6B). When combined with the Phen Green FL data, which demonstrated that cotreatment with disulfiram and copper resulted in greater quenching of the Phen Green dye than copper alone (Fig. 6C), it seems likely that the copper ions inside the cell are more labile in the presence of disulfiram/DETC, allowing them to bypass their usual sequestration pathways. Consequently, DETC molecules may facilitate the release of their copper payloads in certain microenvironments otherwise not accessible to copper ions as we already suggested for bis-thiosemicarbazone-copper complexes (24).
Disulfiram is known to be well tolerated in humans and has previously been shown to exert antitubercular activity in macrophages and the guinea pig model (21, 66). The conversion of disulfiram into a copper complex in blood (which contains typically between 15 and 25 μM copper) has been evaluated previously (36, 54). Because the formation of a disulfiram-copper complex in vivo is likely (21, 36, 54), and since we found no significant activity toward M. tuberculosis in the absence of copper, the previously reported in vivo activity of disulfiram in macrophage and animal models is likely associated with its ability to form a copper complex. It is therefore intriguing to imagine that the various infection-associated copper reservoirs may activate/potentiate the antibacterial properties of disulfiram/DETC through formation of disulfiram-derived copper complexes. If this scenario holds true, disulfiram may be of benefit in depleting M. tuberculosis from the copper-rich environments found in lung granulomas (41), which are vital sanctuaries for M. tuberculosis replication, dissemination, and persistence (71). Such a scenario would be supported by disulfiram's copper-dependent bactericidal action on nonreplicating M. tuberculosis cells (Fig. 4).
Overall, our data support a “Trojan Horse” model for disulfiram, by which complexation of copper with disulfiram/DETC appears to cloak the copper ions from the cell (Fig. 7). This allows the complex to penetrate the intrinsic cellular defenses, including the cell membrane, functioning as a permeability barrier, the cellular copper homeostasis machinery (reviewed in references 72 and 73), and the generally promiscuous drug resistance machinery, such as multidrug efflux pumps (74), and act upon its targets with impunity.
FIG 7.
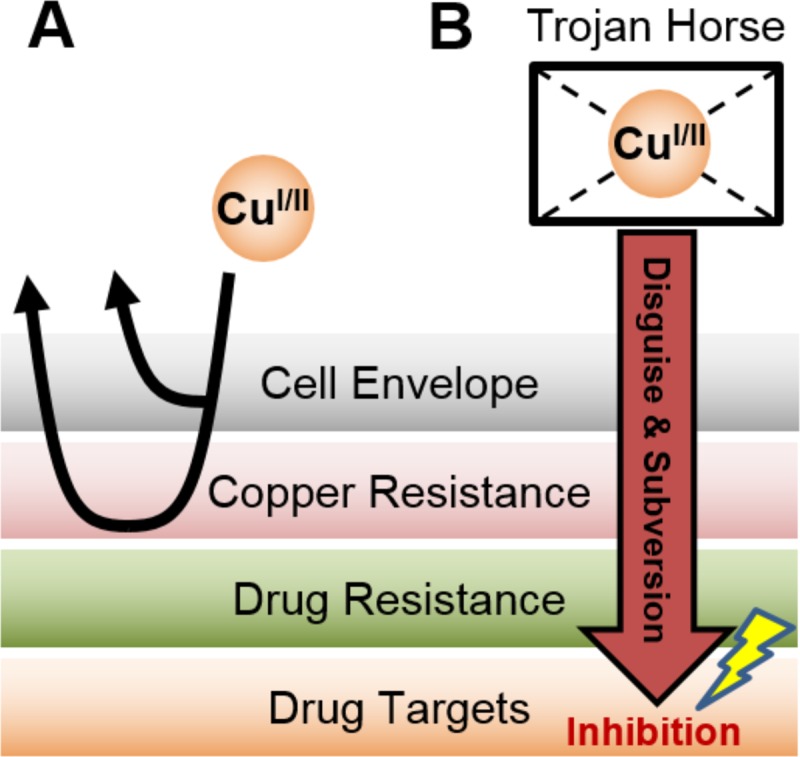
Disulfiram displays a “Trojan Horse” model of activity. (A) Copper ions normally are unable to penetrate the cell envelope, and those that do are rendered impotent by innate copper resistance mechanisms. (B) However, when complexed with a copper-boosting compound, such as disulfiram, copper ions enter the cell in a disguised manner. Unlike free copper ions, the copper complex is membrane permeable due to its greater lipophilicity, allowing copper to bypass the cell envelope, as well as subverting the canonical copper handling and resistance machinery. Disguised copper ions may be able to reach and interact with novel targets that are usually off limits for copper ions entering the cell in an orderly fashion.
Our description of disulfiram's copper-dependent antibacterial properties may also affect future drug screening and discovery efforts. The most frequently used medium for M. tuberculosis drug screening, Middlebrook 7H9, requires the addition of an albumin-containing supplement or bacteria will not grow. Albumin has the ability to sequester copper, thereby neutralizing the potential antibacterial properties of the copper content that is present in this medium (∼6.8 μM). However, albumin is exclusively produced by hepatocytes and secreted into the blood (75) and therefore is not present in the macrophage phagosomes or lung tissue. The use of albumin-containing medium has therefore precluded the discovery and investigation of the majority of copper-dependent antibacterial inhibitors by previous high-throughput operations. Conducting drug screening efforts in a manner that enables compound access to copper is thus an intriguing concept that may reveal novel interactions between small molecules and copper-dependent innate immune functions that would otherwise be unknown. In extension, this unconventional approach could lead to the discovery of new compound classes and activities that could significantly extend the compound diversity of the current M. tuberculosis drug pipeline, which has been a severe challenge over the past few decades.
ACKNOWLEDGMENTS
We are grateful to William R. Jacobs for providing the M. tuberculosis strain mc26230. We thank James B. Cochran and Saran Kupul for excellent technical support and Yaofang Zhang, David Hachey (Mass Spectrometry Research Center, Vanderbilt University), and Eric Skaar (Department of Pathology, Microbiology, and Immunology, Vanderbilt University) for providing ICP-MS services.
This research was supported by NIH grants AI083632, R01-AI104499, and R01-AI104952 awarded to M.N., O.K., and F.W., respectively. Further support was provided by the University of Alabama at Birmingham (UAB) Center for AIDS Research (CFAR), an NIH-funded program (P30 AI027767) that was made possible by the following NIH institutes: NIAID, NIMH, NIDA, NICHD, NHLBI, and NIA.
REFERENCES
- 1.World Health Organization. 2014. Global tuberculosis report 2014. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 2.Pawlowski A, Jansson M, Skold M, Rottenberg ME, Kallenius G. 2012. Tuberculosis and HIV co-infection. PLoS Pathog 8:e1002464. doi: 10.1371/journal.ppat.1002464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shankar EM, Vignesh R, Ellegard R, Barathan M, Chong YK, Bador MK, Rukumani DV, Sabet NS, Kamarulzaman A, Velu V, Larsson M. 2014. HIV-Mycobacterium tuberculosis co-infection: a ‘danger-couple model’ of disease pathogenesis. Pathog Dis 70:110–118. doi: 10.1111/2049-632X.12108. [DOI] [PubMed] [Google Scholar]
- 4.Semvua HH, Kibiki GS, Kisanga ER, Boeree MJ, Burger DM, Aarnoutse R. 2015. Pharmacological interactions between rifampicin and antiretroviral drugs: challenges and research priorities for resource-limited settings. Ther Drug Monit 37:22–32. doi: 10.1097/FTD.0000000000000108. [DOI] [PubMed] [Google Scholar]
- 5.Centers for Disease Control and Prevention. 2013. Impact of a shortage of first-line antituberculosis medication on tuberculosis control—United States, 2012-2013. MMWR Morb Mortal Wkly Rep 62:398–400. [PMC free article] [PubMed] [Google Scholar]
- 6.World Health Organization. 2014. Antimicrobial resistance: global report on surveillance 2014. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 7.Gler MT, Skripconoka V, Sanchez-Garavito E, Xiao H, Cabrera-Rivero JL, Vargas-Vasquez DE, Gao M, Awad M, Park SK, Shim TS, Suh GY, Danilovits M, Ogata H, Kurve A, Chang J, Suzuki K, Tupasi T, Koh WJ, Seaworth B, Geiter LJ, Wells CD. 2012. Delamanid for multidrug-resistant pulmonary tuberculosis. N Engl J Med 366:2151–2160. doi: 10.1056/NEJMoa1112433. [DOI] [PubMed] [Google Scholar]
- 8.Olaru ID, von Groote-Bidlingmaier F, Heyckendorf J, Yew WW, Lange C, Chang KC. 2015. Novel drugs against tuberculosis: a clinician's perspective. Eur Respir J 45:1119–1131. doi: 10.1183/09031936.00162314. [DOI] [PubMed] [Google Scholar]
- 9.Sotgiu G, Migliori GB. 2015. Facing multi-drug resistant tuberculosis. Pulm Pharmacol Ther 32:144–148. doi: 10.1016/j.pupt.2014.04.006. [DOI] [PubMed] [Google Scholar]
- 10.Ginsberg AM, Spigelman M. 2007. Challenges in tuberculosis drug research and development. Nat Med 13:290–294. doi: 10.1038/nm0307-290. [DOI] [PubMed] [Google Scholar]
- 11.Alsaad N, Wilffert B, van Altena R, de Lange WCM, van der Werf TS, Kosterink JGW, Alffenaar J-WC. 2014. Potential antimicrobial agents for the treatment of multidrug-resistant tuberculosis. Eur Respir J 43:884–897. doi: 10.1183/09031936.00113713. [DOI] [PubMed] [Google Scholar]
- 12.Triscott J, Pambid MR, Dunn SE. 2015. Bullseye: targeting cancer stem cells to improve the treatment of gliomas by repurposing disulfiram. Stem Cells 33:1042–1046. doi: 10.1002/stem.1956. [DOI] [PubMed] [Google Scholar]
- 13.Schweizer MT, Lin J, Blackford A, Bardia A, King S, Armstrong AJ, Rudek MA, Yegnasubramanian S, Carducci MA. 2013. Pharmacodynamic study of disulfiram in men with non-metastatic recurrent prostate cancer. Prostate Cancer Prostatic Dis 16:357–361. doi: 10.1038/pcan.2013.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Robinson TJ, Pai M, Liu JC, Vizeacoumar F, Sun T, Egan SE, Datti A, Huang J, Zacksenhaus E. 2013. High-throughput screen identifies disulfiram as a potential therapeutic for triple-negative breast cancer cells: interaction with IQ motif-containing factors. Cell Cycle 12:3013–3024. doi: 10.4161/cc.26063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ogawa E, Kodama H. 2012. Effects of disulfiram treatment in patients with Menkes disease and occipital horn syndrome. J Trace Elem Med Biol 26:102–104. doi: 10.1016/j.jtemb.2012.04.017. [DOI] [PubMed] [Google Scholar]
- 16.Doyon G, Zerbato J, Mellors JW, Sluis-Cremer N. 2013. Disulfiram reactivates latent HIV-1 expression through depletion of the phosphatase and tensin homolog. AIDS 27:F7–F11. doi: 10.1097/QAD.0b013e3283570620. [DOI] [PubMed] [Google Scholar]
- 17.Spivak AM, Andrade A, Eisele E, Hoh R, Bacchetti P, Bumpus NN, Emad F, Buckheit R III, McCance-Katz EF, Lai J, Kennedy M, Chander G, Siliciano RF, Siliciano JD, Deeks SG. 2014. A pilot study assessing the safety and latency-reversing activity of disulfiram in HIV-1-infected adults on antiretroviral therapy. Clin Infect Dis 58:883–890. doi: 10.1093/cid/cit813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hill DE, Fetterer RH. 1997. The effect of disulfiram on egg shell formation in adult Trichuris muris. J Parasitol 83:938–942. doi: 10.2307/3284293. [DOI] [PubMed] [Google Scholar]
- 19.Khan S, Singhal S, Mathur T, Upadhyay DJ, Rattan A. 2007. Antifungal potential of disulfiram. Nihon Ishinkin Gakkai Zasshi 48:109–113. doi: 10.3314/jjmm.48.109. [DOI] [PubMed] [Google Scholar]
- 20.Phillips M, Malloy G, Nedunchezian D, Lukrec A, Howard RG. 1991. Disulfiram inhibits the in vitro growth of methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother 35:785–787. doi: 10.1128/AAC.35.4.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Horita Y, Takii T, Yagi T, Ogawa K, Fujiwara N, Inagaki E, Kremer L, Sato Y, Kuroishi R, Lee Y, Makino T, Mizukami H, Hasegawa T, Yamamoto R, Onozaki K. 2012. Antitubercular activity of disulfiram, an antialcoholism drug, against multidrug- and extensively drug-resistant Mycobacterium tuberculosis isolates. Antimicrob Agents Chemother 56:4140–4145. doi: 10.1128/AAC.06445-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Velasco-García R, Zaldivar-Machorro VJ, Mujica-Jimenez C, Gonzalez-Segura L, Munoz-Clares RA. 2006. Disulfiram irreversibly aggregates betaine aldehyde dehydrogenase—a potential target for antimicrobial agents against Pseudomonas aeruginosa. Biochem Biophys Res Commun 341:408–415. doi: 10.1016/j.bbrc.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 23.Zaldívar-Machorro VJ, Lopez-Ortiz M, Demare P, Regla I, Munoz-Clares RA. 2011. The disulfiram metabolites S-methyl-N,N-diethyldithiocarbamoyl sulfoxide and S-methyl-N,N-diethylthiocarbamoyl sulfone irreversibly inactivate betaine aldehyde dehydrogenase from Pseudomonas aeruginosa, both in vitro and in situ, and arrest bacterial growth. Biochimie 93:286–295. doi: 10.1016/j.biochi.2010.09.022. [DOI] [PubMed] [Google Scholar]
- 24.Haeili M, Moore C, Davis CJC, Cochran JB, Shah S, Shrestha TB, Zhang Y, Bossmann SH, Benjamin WH, Kutsch O, Wolschendorf F. 2014. Copper complexation screen reveals compounds with potent antibiotic properties against methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother 58:3727–3736. doi: 10.1128/AAC.02316-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eneanya DI, Bianchine JR, Duran DO, Andresen BD. 1981. The actions and metabolic fate of disulfiram. Annu Rev Pharmacol Toxicol 21:575–596. doi: 10.1146/annurev.pa.21.040181.003043. [DOI] [PubMed] [Google Scholar]
- 26.Cen D, Brayton D, Shahandeh B, Meyskens FL, Farmer PJ. 2004. Disulfiram facilitates intracellular Cu uptake and induces apoptosis in human melanoma cells. J Med Chem 47:6914–6920. doi: 10.1021/jm049568z. [DOI] [PubMed] [Google Scholar]
- 27.Lewis DJ, Deshmukh P, Tedstone AA, Tuna F, O'Brien P. 2014. On the interaction of copper(II) with disulfiram. Chem Commun (Camb) 50:13334–13337. doi: 10.1039/C4CC04767B. [DOI] [PubMed] [Google Scholar]
- 28.Byrne ST, Gu P, Zhou J, Denkin SM, Chong C, Sullivan D, Liu JO, Zhang Y. 2007. Pyrrolidine dithiocarbamate and diethyldithiocarbamate are active against growing and nongrowing persister Mycobacterium tuberculosis. Antimicrob Agents Chemother 51:4495–4497. doi: 10.1128/AAC.00753-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Speer A, Shrestha TB, Bossmann SH, Basaraba RJ, Harber GJ, Michalek SM, Niederweis M, Kutsch O, Wolschendorf F. 2013. Copper-boosting compounds: a novel concept for antimycobacterial drug discovery. Antimicrob Agents Chemother 57:1089–1091. doi: 10.1128/AAC.01781-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sander P, Meier A, Bottger EC. 1995. rpsL+: a dominant selectable marker for gene replacement in mycobacteria. Mol Microbiol 16:991–1000. doi: 10.1111/j.1365-2958.1995.tb02324.x. [DOI] [PubMed] [Google Scholar]
- 31.Stephan J, Bender J, Wolschendorf F, Hoffmann C, Roth E, Mailander C, Engelhardt H, Niederweis M. 2005. The growth rate of Mycobacterium smegmatis depends on sufficient porin-mediated influx of nutrients. Mol Microbiol 58:714–730. doi: 10.1111/j.1365-2958.2005.04878.x. [DOI] [PubMed] [Google Scholar]
- 32.Sambandamurthy VK, Derrick SC, Hsu T, Chen B, Larsen MH, Jalapathy KV, Chen M, Kim J, Porcelli SA, Chan J, Morris SL, Jacobs WR Jr. 2006. Mycobacterium tuberculosis DeltaRD1 DeltapanCD: a safe and limited replicating mutant strain that protects immunocompetent and immunocompromised mice against experimental tuberculosis. Vaccine 24:6309–6320. doi: 10.1016/j.vaccine.2006.05.097. [DOI] [PubMed] [Google Scholar]
- 33.Weber FJ, van Berkel WJ, Hartmans S, de Bont JA. 1992. Purification and properties of the NADH reductase component of alkene monooxygenase from Mycobacterium strain E3. J Bacteriol 174:3275–3281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kaps I, Ehrt S, Seeber S, Schnappinger D, Martin C, Riley LW, Niederweis M. 2001. Energy transfer between fluorescent proteins using a co-expression system in Mycobacterium smegmatis. Gene 278:115–124. doi: 10.1016/S0378-1119(01)00712-0. [DOI] [PubMed] [Google Scholar]
- 35.Rowland JL, Niederweis M. 2013. A multicopper oxidase is required for copper resistance in Mycobacterium tuberculosis. J Bacteriol 195:3724–3733. doi: 10.1128/JB.00546-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johansson B, Stankiewicz Z. 1985. Bis-(diethyldithiocarbamato) copper complex: a new metabolite of disulfiram? Biochem Pharmacol 34:2989–2991. doi: 10.1016/0006-2952(85)90026-7. [DOI] [PubMed] [Google Scholar]
- 37.Labuda J, Skatulokova M, Nemeth M, Gergely S. 1984. Formation and stability of diethyldithiocarbamate complexes. Chem Zvesti 38:597–605. [Google Scholar]
- 38.Irving H, Williams RJP. 1953. 637. The stability of transition-metal complexes. J Chem Soc (Resumed) 1953:3192–3210. doi: 10.1039/JR9530003192. [DOI] [Google Scholar]
- 39.Yamuna K, Ramana VV, Emmanuel KA, Saraswati K. 1992. Stability constants of metal complexes of dithiocarbamates. Asian J Chem 4:387–389. [Google Scholar]
- 40.Emsley J. 1998. The elements, 3rd ed Clarendon Press, Oxford, United Kingdom. [Google Scholar]
- 41.Wolschendorf F, Ackart D, Shrestha TB, Hascall-Dove L, Nolan S, Lamichhane G, Wang Y, Bossmann SH, Basaraba RJ, Niederweis M. 2011. Copper resistance is essential for virulence of Mycobacterium tuberculosis. Proc Natl Acad Sci U S A 108:1621–1626. doi: 10.1073/pnas.1009261108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shi X, Festa RA, Ioerger TR, Butler-Wu S, Sacchettini JC, Darwin KH, Samanovic MI. 2014. The copper-responsive RicR regulon contributes to Mycobacterium tuberculosis virulence. mBio 5(1):e00876-13. doi: 10.1128/mBio.00876-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gold B, Deng H, Bryk R, Vargas D, Eliezer D, Roberts J, Jiang X, Nathan C. 2008. Identification of a copper-binding metallothionein in pathogenic mycobacteria. Nat Chem Biol 4:609–616. doi: 10.1038/nchembio.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu T, Ramesh A, Ma Z, Ward SK, Zhang L, George GN, Talaat AM, Sacchettini JC, Giedroc DP. 2007. CsoR is a novel Mycobacterium tuberculosis copper-sensing transcriptional regulator. Nat Chem Biol 3:60–68. doi: 10.1038/nchembio844. [DOI] [PubMed] [Google Scholar]
- 45.Ward SK, Abomoelak B, Hoye EA, Steinberg H, Talaat AM. 2010. CtpV: a putative copper exporter required for full virulence of Mycobacterium tuberculosis. Mol Microbiol 77:1096–1110. doi: 10.1111/j.1365-2958.2010.07273.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Esmail H, Barry CE III, Wilkinson RJ. 2012. Understanding latent tuberculosis: the key to improved diagnostic and novel treatment strategies. Drug Discov Today 17:514–521. doi: 10.1016/j.drudis.2011.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lienhardt C, Raviglione M, Spigelman M, Hafner R, Jaramillo E, Hoelscher M, Zumla A, Gheuens J. 2012. New drugs for the treatment of tuberculosis: needs, challenges, promise, and prospects for the future. J Infect Dis 205(Suppl 2):S241–S249. doi: 10.1093/infdis/jis034. [DOI] [PubMed] [Google Scholar]
- 48.Faiman MD, Jensen JC, Lacoursiere RB. 1984. Elimination kinetics of disulfiram in alcoholics after single and repeated doses. Clin Pharmacol Ther 36:520–526. doi: 10.1038/clpt.1984.213. [DOI] [PubMed] [Google Scholar]
- 49.Baldwin TO, Christopher JA, Raushel FM, Sinclair JF, Ziegler MM, Fisher AJ, Rayment I. 1995. Structure of bacterial luciferase. Curr Opin Struct Biol 5:798–809. doi: 10.1016/0959-440X(95)80014-X. [DOI] [PubMed] [Google Scholar]
- 50.Lin J, Haffner MC, Zhang Y, Lee BH, Brennen WN, Britton J, Kachhap SK, Shim JS, Liu JO, Nelson WG, Yegnasubramanian S, Carducci MA. 2011. Disulfiram is a DNA demethylating agent and inhibits prostate cancer cell growth. Prostate 71:333–343. doi: 10.1002/pros.21247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vallari RC, Pietruszko R. 1982. Human aldehyde dehydrogenase: mechanism of inhibition of disulfiram. Science 216:637–639. doi: 10.1126/science.7071604. [DOI] [PubMed] [Google Scholar]
- 52.Cecconi I, Moroni M, Vilardo PG, Dal Monte M, Borella P, Rastelli G, Costantino L, Garland D, Carper D, Petrash JM, Del Corso A, Mura U. 1998. Oxidative modification of aldose reductase induced by copper ion. Factors and conditions affecting the process. Biochemistry 37:14167–14174. [DOI] [PubMed] [Google Scholar]
- 53.Niederweis M. 2003. Mycobacterial porins—new channel proteins in unique outer membranes. Mol Microbiol 49:1167–1177. doi: 10.1046/j.1365-2958.2003.03662.x. [DOI] [PubMed] [Google Scholar]
- 54.Johansson B. 1992. A review of the pharmacokinetics and pharmacodynamics of disulfiram and its metabolites. Acta Psychiatr Scand Suppl 369:15–26. [DOI] [PubMed] [Google Scholar]
- 55.Danilchanka O, Pavlenok M, Niederweis M. 2008. Role of porins for uptake of antibiotics by Mycobacterium smegmatis. Antimicrob Agents Chemother 52:3127–3134. doi: 10.1128/AAC.00239-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Speer A, Rowland JL, Haeili M, Niederweis M, Wolschendorf F. 2013. Porins increase copper susceptibility of Mycobacterium tuberculosis. J Bacteriol 195:5133–5140. doi: 10.1128/JB.00763-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Speer A, Rowland JL, Niederweis M. 2013. Mycobacterium tuberculosis is resistant to streptolydigin. Tuberculosis (Edinb) 93:401–404. doi: 10.1016/j.tube.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Song H, Niederweis M. 2012. Uptake of sulfate but not phosphate by Mycobacterium tuberculosis is slower than that for Mycobacterium smegmatis. J Bacteriol 194:956–964. doi: 10.1128/JB.06132-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Festa RA, Jones MB, Butler-Wu S, Sinsimer D, Gerads R, Bishai WR, Peterson SN, Darwin KH. 2011. A novel copper-responsive regulon in Mycobacterium tuberculosis. Mol Microbiol 79:133–148. doi: 10.1111/j.1365-2958.2010.07431.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Festa RA, Helsel ME, Franz KJ, Thiele DJ. 2014. Exploiting innate immune cell activation of a copper-dependent antimicrobial agent during infection. Chem Biol 21:977–987. doi: 10.1016/j.chembiol.2014.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Triscott J, Lee C, Hu K, Fotovati A, Berns R, Pambid M, Luk M, Kast RE, Kong E, Toyota E, Yip S, Toyota B, Dunn SE. 2012. Disulfiram, a drug widely used to control alcoholism, suppresses the self-renewal of glioblastoma and over-rides resistance to temozolomide. Oncotarget 3:1112–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Galkin A, Kulakova L, Lim K, Chen CZ, Zheng W, Turko IV, Herzberg O. 2014. Structural basis for inactivation of Giardia lamblia carbamate kinase by disulfiram. J Biol Chem 289:10502–10509. doi: 10.1074/jbc.M114.553123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cvek B. 2012. Nonprofit drugs as the salvation of the world's healthcare systems: the case of Antabuse (disulfiram). Drug Discov Today 17:409–412. doi: 10.1016/j.drudis.2011.12.010. [DOI] [PubMed] [Google Scholar]
- 64.Chen D, Cui QC, Yang H, Dou QP. 2006. Disulfiram, a clinically used anti-alcoholism drug and copper-binding agent, induces apoptotic cell death in breast cancer cultures and xenografts via inhibition of the proteasome activity. Cancer Res 66:10425–10433. doi: 10.1158/0008-5472.CAN-06-2126. [DOI] [PubMed] [Google Scholar]
- 65.Skrott Z, Cvek B. 2012. Diethyldithiocarbamate complex with copper: the mechanism of action in cancer cells. Mini Rev Med Chem 12:1184–1192. doi: 10.2174/138955712802762068. [DOI] [PubMed] [Google Scholar]
- 66.Jeney E, Zsolnai T. 1956. Experiments for the detection of new tuberculostatics. IV. Chemotherapeutic action of some hydrazine derivatives, and organic sulfur compounds on the experimental tuberculosis of guinea pigs. Zentralbl Bakteriol Orig 167:254–264. (In German.) [PubMed] [Google Scholar]
- 67.Macomber L, Imlay JA. 2009. The iron-sulfur clusters of dehydratases are primary intracellular targets of copper toxicity. Proc Natl Acad Sci U S A 106:8344–8349. doi: 10.1073/pnas.0812808106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chillappagari S, Seubert A, Trip H, Kuipers OP, Marahiel MA, Miethke M. 2010. Copper stress affects iron homeostasis by destabilizing iron-sulfur cluster formation in Bacillus subtilis. J Bacteriol 192:2512–2524. doi: 10.1128/JB.00058-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Azzouzi A, Steunou AS, Durand A, Khalfaoui-Hassani B, Bourbon ML, Astier C, Bollivar DW, Ouchane S. 2013. Coproporphyrin III excretion identifies the anaerobic coproporphyrinogen III oxidase HemN as a copper target in the Cu+-ATPase mutant copA− of Rubrivivax gelatinosus. Mol Microbiol 88:339–351. doi: 10.1111/mmi.12188. [DOI] [PubMed] [Google Scholar]
- 70.Johnson MD, Kehl-Fie TE, Rosch JW. 2015. Copper intoxication inhibits aerobic nucleotide synthesis in Streptococcus pneumoniae. Metallomics 7:786−794. doi: 10.1039/C5MT00011D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ramakrishnan L. 2012. Revisiting the role of the granuloma in tuberculosis. Nat Rev Immunol 12:352–366. doi: 10.1038/nri3211. [DOI] [PubMed] [Google Scholar]
- 72.Neyrolles O, Wolschendorf F, Mitra A, Niederweis M. 2015. Mycobacteria, metals, and the macrophage. Immunol Rev 264:249–263. doi: 10.1111/imr.12265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shi X, Darwin KH. 2015. Copper homeostasis in Mycobacterium tuberculosis. Metallomics 7:929–934. doi: 10.1039/c4mt00305e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Danilchanka O, Mailaender C, Niederweis M. 2008. Identification of a novel multidrug efflux pump of Mycobacterium tuberculosis. Antimicrob Agents Chemother 52:2503–2511. doi: 10.1128/AAC.00298-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hühmer AF, Biringer RG, Amato H, Fonteh AN, Harrington MG. 2006. Protein analysis in human cerebrospinal fluid: physiological aspects, current progress and future challenges. Dis Markers 22:3–26. doi: 10.1155/2006/158797. [DOI] [PMC free article] [PubMed] [Google Scholar]