

Background: A native Kir channel in a distant relative of vertebrates interacts weakly with PIP2.
Results: Mutagenesis restores the vertebrate channel sensitivity to PIP2 in sponge channels.
Conclusion: A basic residue in the tether helix of Kir is required for high affinity PIP2 regulation.
Significance: Evolution conferred a high affinity interaction of vertebrate Kir channels with PIP2, which is lacking in a distant relative.
Keywords: evolution, gating, inositol phospholipid, molecular modeling, phosphatase, IRK channel, TEVC, inwardly rectifying, patch clamp, sponge
Abstract
All vertebrate inwardly rectifying potassium (Kir) channels are activated by phosphatidylinositol 4,5-bisphosphate (PIP2) (Logothetis, D. E., Petrou, V. I., Zhang, M., Mahajan, R., Meng, X. Y., Adney, S. K., Cui, M., and Baki, L. (2015) Annu. Rev. Physiol. 77, 81–104; Fürst, O., Mondou, B., and D'Avanzo, N. (2014) Front. Physiol. 4, 404–404). Structural components of a PIP2-binding site are conserved in vertebrate Kir channels but not in distantly related animals such as sponges and sea anemones. To expand our understanding of the structure-function relationships of PIP2 regulation of Kir channels, we studied AqKir, which was cloned from the marine sponge Amphimedon queenslandica, an animal that represents the phylogenetically oldest metazoans. A requirement for PIP2 in the maintenance of AqKir activity was examined in intact oocytes by activation of a co-expressed voltage-sensing phosphatase, application of wortmannin (at micromolar concentrations), and activation of a co-expressed muscarinic acetylcholine receptor. All three mechanisms to reduce the availability of PIP2 resulted in inhibition of AqKir current. However, time-dependent rundown of AqKir currents in inside-out patches could not be re-activated by direct application to the inside membrane surface of water-soluble dioctanoyl PIP2, and the current was incompletely re-activated by the more hydrophobic arachidonyl stearyl PIP2. When we introduced mutations to AqKir to restore two positive charges within the vertebrate PIP2-binding site, both forms of PIP2 strongly re-activated the mutant sponge channels in inside-out patches. Molecular dynamics simulations validate the additional hydrogen bonding potential of the sponge channel mutants. Thus, nature's mutations conferred a high affinity activation of vertebrate Kir channels by PIP2, and this is a more recent evolutionary development than the structures that explain ion channel selectivity and inward rectification.
Introduction
Inwardly rectifying potassium (Kir)2 channels represent a superfamily of one pore domain and two transmembrane domain channels. Kir channels are present in organisms from bacteria to humans and are found throughout the animal kingdom. In animal cells, they play an important role in regulating the resting membrane potential, K+ homeostasis, and membrane excitability. An ancestral precursor to all Kir channels is predicted to have originated in prokaryotes followed by changes in channel structure and function at the prokaryotic-eukaryotic transition and, most likely, throughout the time course of animal evolution (3, 4).
Like many ion channels and transporters, the activity of Kir channels from vertebrates, including mammalian and nonmammalian species, is dependent on a membrane lipid, phosphatidylinositol 4,5-bisphosphate (PIP2) (1, 2, 5–7). PIP2 is found primarily in the inner leaflet of the plasma membrane (8), and its availability is dynamically controlled by neurotransmitter, hormone, and growth factor receptors that couple to phospholipase C (9, 10). Signaling that activates phospholipase C is expected to lower the availability of PIP2, which reduces or prevents the activation of vertebrate Kir channels. Understanding how channels are regulated by PIP2 is of fundamental physiological importance to cellular signaling and K+ homeostasis in eukaryotic cells.
The evolutionary origin of PIP2-mediated regulation of Kir channels is unknown. Prokaryotic Kir channels are inhibited by PIP2 (11, 12), whereas Kir channels from all eukaryotic cells studied to date are activated by PIP2 (2, 6, 10, 13). This is an interesting difference, but PIP2 is not present in the prokaryotic cell membrane (2, 6), and thus its inhibitory effect on bacterial Kir channels lacks physiological relevance. Among eukaryotes, the structural basis for Kir channel activation by PIP2 has been clarified by many studies (1, 2), culminating in the co-crystallization of PIP2 with chicken Kir2.2 (7). Among the critical components of the protein-lipid interaction are several basic residues within the channel that provide an electrostatically favorable interaction with the negative charges on the phosphate groups of PIP2. However, despite the tremendous insight gained by many years of physiology and structural biology experiments, the functional priority of the basic residues in the Kir channel and whether they are sufficient to explain high affinity interactions of the lipid and channel remain untested outside the confines of vertebrate species such as rat, mouse, human, and chicken. Because there is a 400–600 million year evolutionary gap between humans and basal invertebrates such as sponges (14), there is a good possibility that evolutionary pressures resulted in structural changes to regulatory sites on ion channels (15).
To explore the impact of evolutionary pressure on structural changes in ion channel proteins, we studied PIP2 regulation of AqKir, a K+ channel with strong inward rectification, which was previously cloned from the marine sponge Amphimedon queenslandica (16). Sponges are an important comparative group for understanding the relationships between extant unicellular protists and the first multicellular organisms from which all animals evolved (14, 17–20). By studying the physiological and structural impact of “nature's mutations,” we provide a window into the functional consequences of evolution that is represented in distantly related animal species. A Kir channel cloned from a marine sponge (AqKir) lacks two positive charges that are important for high affinity interaction with PIP2 in vertebrate Kir2 channels (7). We report that, following time-dependent rundown, AqKir channels are insensitive to re-activation by PIP2 added to the internal surface of an excised membrane patch, despite additional evidence from intact oocytes that suggests that PIP2 may be required for this channel's function. In addition, AqKir channels, which we engineered to possess the two positive charges present in the PIP2-binding site of vertebrate Kir2 channels, showed strong activation by diC8 PIP2, with a sensitivity that was even 3-fold greater than that for vertebrate Kir2.1 channels. Our results confirm that certain positively charged residues (corresponding to residues 80 and 189 in cKir2.2) are absolutely necessary to confer high sensitivity to PIP2 and represent evolutionary changes that are more recent than the evolution of structures that confer K+ selectivity and inward rectification, which are conserved from sponge to human (16). Finally, we propose that other structures present in distant relatives of vertebrate Kir channels must facilitate the activation properties conferred by PIP2 such that restoration of even a single positive charge (N180K in AqKir) recapitulates the high sensitivity to PIP2 present in vertebrate Kir channels.
Experimental Procedures
Functional Expression in Oocytes
Wild-type channel constructs included AqKir (16) (GenBankTM accession number FJ375323, previously named AmqKirA), chicken Kir2.2 (cKir2.2, GenBankTM accession number F1NHE9), mKir2.1 (GenBankTM accession number X73052), and mKir2.3 (GenBankTM accession number S71382). A plasmid containing CiVSP (GenBankTM accession number AB183035) was used, and mutation C363S was prepared to destroy the enzymatic activity (21). Mutants were constructed using QuikChange (Agilent Technologies, catalogue no. 200523) and confirmed by DNA sequencing. The preparation of cRNA, isolation of oocytes, and injections were performed using standard methods (22). In brief, plasmids containing sponge channel cDNAs were linearized and capped RNAs synthesized using Ambion (Austin, TX) mMessage Machine RNA polymerase kits. RNA was purified by use of the RNAid kit (Bio 101, Vista, CA) or LiCl precipitation, and concentrations were determined by Nanodrop (ThermoScientific) spectrophotometry. Oocytes were surgically harvested from anesthetized female Xenopus laevis (Xenopus I, Dexter, MI) frogs. Oocytes were released and defolliculated by gentle agitation in 0.5 mg/ml collagenase A (Sigma) dissolved in a Ca2+-free solution containing (in mm) 96 NaCl, 2 KCl, 1 MgCl2, 5 HEPES, pH 7.4. Oocytes were then washed and selected, and stage V/VI oocytes were microinjected with 36–50 nl of cRNA dissolved in diethyl pyrocarbonate-treated water (0.05–15 ng of cRNA/oocyte), depending on functional expression levels (23–25). Oocytes were maintained for 2–12 days at 14–17 °C in a solution containing (in mm) 96 NaCl, 1 KCl, 1 CaCl2, 2 MgCl2, 10 HEPES, 0–5 sucrose, and 2 sodium pyruvate, pH 7.4, with 50 units/ml penicillin G and 50 μg/ml streptomycin.
Electrophysiology
Potassium currents were recorded from oocytes using standard electrophysiological methods (22–26). For two-electrode voltage clamp (TEVC), we used a Geneclamp 500B amplifier (Axon Instruments, Foster City, CA) or an OC-725C amplifier (Warner Instruments, Hamden, CT). Voltage-measuring and current-passing electrodes were backfilled with 3 m KCl and had resistances between 0.3 and 1.0 megohms. Currents were sampled at 5–10 kHz and filtered at 1–2 kHz. All recordings were obtained at room temperature (about 23 °C), and the bath was perfused continuously during recordings. The standard external solution contained (in mm) 96 KCl, 4 NaCl, 1 CaCl2, 2 MgCl2, 10 HEPES, pH 7.3–7.4.
For inside-out patch clamp recordings, we used an A-M Systems Model 2400 or an Axon Instruments 200B patch clamp amplifier. Macropatches were excised from oocytes following manual removal of the vitelline membrane (24–26). Electrodes were pulled from borosilicate glass using a Sutter P-97 puller and gave a tip diameter of 5–15 μm and a resistance of 0.5–2 megohms when filled with a solution containing (in mm) 96 KMeSO3, 1 MgCl2, and 10 HEPES, pH 7.4. The standard bath (internally facing) high K+ (“HK”) solution contained 96 KCl, 5 EGTA, and 10 HEPES, pH 7.4, unless otherwise mentioned. Currents were elicited with a voltage ramp protocol from −100 to +100 mV over 1 s. Signals were low pass-filtered at 10 kHz and digitized at 20-μs intervals.
Electrophysiological data were recorded on computers equipped with Digidata 1320A (Axon Instruments) A/D hardware. Axon's Clampex acquisition and Clampfit analysis software (versions 7–9) were used.
Data Analysis and Curve Fitting
Data were also transferred to Microsoft Excel and Microcal Origin (Northampton, MA) for additional analysis, curve-fitting, and the production of figures. Results were calculated from cells with negligible background currents and for TEVC, only from cells with membrane potentials not more depolarized than −30 mV in 5 mm external K+. Results are expressed as mean ± S.E. with n = number of oocytes tested with a minimum of three batches of oocytes per experiment. The EC50 value of PIP2 effects for each channel was obtained by fitting concentration-response data to a Hill Equation:
where I/Imax is the normalized current; [X]i is the PIP2 concentration applied from the intracellular side; n is the Hill coefficient, and EC50 is the concentration required to reach half of the maximum activation.
In cases with multiple comparisons to a single set of control values, a single factor ANOVA with a post-hoc Dunnett's test was used with p < 0.05 considered to be significant. Other statistical evaluations included paired or unpaired Student's two-tailed t-tests, as appropriate, with p < 0.05 considered to be significant.
Chemicals
AASt (“long chain”) PIP2 was purchased from Avanti Polar Lipids (Alabaster, AL). diC8 PIP2 was purchased from Avanti Polar Lipids or Echelon Biosciences (Salt Lake City, UT). The lipids were prepared as described previously (23, 25, 27). All other chemicals were purchased from Sigma or VWR.
Modeling of AqKir/PIP2 Channel Complexes
The AqKir channel model was constructed based on the chicken Kir2.2 x-ray crystal structure template (PDB code 3SPI (7)). The sequence alignment between Kir2.2 and AqKir channels was generated by ClustalW2. The sequence identity between Kir2.2 and AqKir is ∼40%. We used the program MODELLER 9 version 12 (28) to generate 10 initial homology models. The model assessment and selection were based on the internal objective function, 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine, and GA341 assessment scores in the MODELLER program to select models with the lowest free energy potentials. The full-length PIP2 molecules were added into the AqKir model by superimposition with the short-chain derivative of PIP2 in the Kir2.2 crystal structure (7). Models for AqKir channel mutants were constructed by a mutation function using Discovery Studio version 3.5 molecular modeling program (Accelrys Software Inc., San Diego).
Molecular Dynamics Simulations
The PIP2-AqKir channel complexes were immersed in an explicit palmitoyloleoylphosphatidylcholine bilayer generated from the VMD membrane package (29). After being solvated with SPC water molecules, neutralized by KCl (150 mm) as the counter ions and including K+ located in the selectivity filter as obtained from the crystal structures, each system involved ∼140,000 atoms in the MD simulations. GROMACS version 4.5.4 (30) was used to conduct the simulation with the GROMOS96 53a6 force field (31). The parameters for PIP2 and lipid were generated as described previously (32). Long range electrostatics were calculated using the particle mesh Ewald (PME) method (33) with a 12 Å cutoff. van der Waal interactions were modeled using Lennard-Jones 6–12 potentials with 14-Å cutoff. All simulations were conducted at a constant temperature of 300 K using the Berendsen thermostat. The system pressure was coupled at isotropic (X + Y, Z) directions referenced to 1 bar using the Berendsen method (34). All bonds were constrained with the LINCS algorithm (35). The time step was 2 fs, and the neighboring list was updated every 10 time steps.
Prior to production runs, energy minimization of 3000 steps of the steepest descent were carried out on each system followed by a 0.5-ns two-step equilibration process. In the first 0.2 ns, channels, K+ ions, and PIP2 were position-restrained using a constant force of 1000 kJ/mol/nm2, allowing lipid and water molecules to move freely. The restraint was weakened to 10 kJ/mol/nm2 in the following 0.3-ns equilibration. An electrical field of 0.02857 V/nm was applied in this step as well as the production run, along the z axis of the box to maintain the lower potentials in the intracellular side. A 40-ns production run was conducted on each system, and coordinates were saved every 10 ps for analysis.
Interaction Energy Calculations
The Discovery Studio 3.5 molecular modeling program (Accelrys Software Inc.) was used to conduct energy minimizations and calculate the interaction energy between the PIP2 and the AqKir channels. All structures were subjected to energy minimization using Smart Minimizer algorithm (2000 steps) and Generalized Born implicit solvent model using the CHARMm force field. Interaction energies between the PIP2 and the channel were calculated using a distance-dependent dielectric constant (ϵ = 5r) implicit solvent model. The decomposed interaction energy contribution of residues to the total interaction energy was analyzed to identify the critical interacting residues in the protein complexes. A residue was considered to be critical if it bound the ligand with an interaction energy lower than −1 kcal/mol.
Results
Sequence Comparisons
An amino acid sequence comparison between sponge and vertebrate Kir channels shows that AqKir is missing two canonical positively charged residues at positions 78 and 180, within the outer and tether helices, respectively (Fig. 1A), of the PI(4,5)P2-binding site in the crystal structure of chicken Kir2.2 (7) and is represented in a homology model of AqKir (Fig. 1B). Both positive charges are present in members of all but one of the seven vertebrate Kir channel subfamilies (Fig. 1A). Thus, AqKir is the only known functional Kir channel that lacks multiple residues previously shown to be important for regulation by PIP2 (27, 36). A BLAST search for additional distant relatives of the vertebrate Kir channels found three putative Kir channels in the genome of the sea anemone Nematostella vectensis (Nv). All three lack a positive charge within the tether helix region of the PIP2-binding site in cKir2.2, although the positive charge in the outer helix region, which is missing in the sponge channel, is preserved in the three sequences from sea anemone (Nv1, His; Nv2–3, Arg; Fig. 1A). As both sponges and sea anemones are modern day representatives of more primitive metazoans, these structural similarities provide evidence for the impact of evolution on the function of vertebrate Kir channels.
FIGURE 1.

Alignment of Kir channels within critical PIP2-binding regions. A, ClustalW2 alignment of the outer and tether helices of the sponge channel, AqKir, with representative members of all vertebrate Kir channel subfamilies. Three Kir channels predicted from the genome (52) of the sea anemone, N. vectensis are shown for comparison. Important residues within the PIP2 interacting region of vertebrate Kir channels are underlined. AqKir lacks a positively charged amino acid at residue 78, which is otherwise present in the outer helix of the vertebrate channels (Arg) and in the three N. vectensis sequences (His or Arg). At residue 180, AqKir lacks the positively charged amino acid present in the tether helix of all vertebrate channels (Lys or Arg) except Kir7.1, which has asparagine (Asn). This positively charged amino acid in the tether helix is also missing from the sea anemone (Nv1–3) channel sequences (Glu, Asp, and Leu). The numbers in the figure refer to the last residue shown in each line. B, schematic using homology model of AqKir showing location of Ile-78 (magenta) and Asn-180 (green) in the channel structure. In the tetramer (box), four molecules of AASt-PIP2 are docked (acyl tails shown in blue); in the enlarged image, a single subunit is shown to highlight the location of the PIP2 molecule, Ile-78 and Asn-180. The GenBankTM accession numbers for the sequence data are as follows: AqKir (FJ375323); rKir1.1 (NP_058719.1); rKir2.1 (NP_058992.1); cKir2.2 (F1NHE9.1); rKir2.3 (NP_446322.2); mKir3.1 (NP_032452.1); mKir3.4 (NP_034735.3); rKir4.1 (NP_113790.2); rKir5.1 (NP_445766.2); rKir6.1 (BAA96237.1); and rKir7.1 (NP_446060.1). The names Nv1, -2, and -3 are arbitrary and represent sequence data using GenBankTM accession numbers XP_001629058, XP_001630074, and XP_001636991, respectively.
Experiments on Intact Cells
Based on the sequence differences between AqKir and vertebrate Kir channels, we used TEVC on whole oocytes to probe the requirement for PIP2 in the maintenance of the AqKir current. We reduced the PIP2 concentrations in whole oocytes in three ways (37). First, activation of a co-expressed voltage-sensing phosphatase (CiVSP (38)) reversibly reduced the inward AqKir current in a time-dependent fashion (Fig. 2, A and B). CiVSP is known to dephosphorylate PI(4,5)P2 upon membrane depolarization (38, 39). The time to reach 50% inhibition by CiVSP (Fig. 2A) was 2.66 ± 1.03 s for AqKir (n = 4), which was 10-fold faster than that measured for Kir2.1 (25.3 ± 6.10 s; n = 6). Likewise, the t50 for Kir current recovery (Fig. 2B), using hyperpolarizing pulses to de-activate the enzyme, was about 3-fold slower for AqKir (57.2 ± 13.2 s; n = 4) than for Kir2.1 (22.0 ± 1.39 s; n = 2). In several experiments, we tested a catalytically inactive mutant of CiVSP (C363S (21) and observed no change in Kir current amplitudes upon membrane depolarization (data not shown). As a second test for PIP2 dependence, the amplitude of resting Kir currents was measured after preincubation with 20 μm wortmannin, a concentration that inhibits phosphatidylinositol 4-kinase activity and thus lowers endogenous levels of PI(4,5)P2 (40). Compared with time-matched, vehicle-treated (DMSO) controls using oocytes from the same frogs, wortmannin pretreatment resulted in a significant reduction in the resting AqKir and Kir2.3 currents but not Kir2.1 currents (Fig. 2C). Notably, Kir2.1 has a higher affinity for PIP2 interaction than does Kir2.3 (41). Third, when muscarinic (M1) acetylcholine receptors were co-expressed with AqKir and activated by acetylcholine, the inward AqKir K+ currents were inhibited (Fig. 2, D and E), whereas Kir2.1 currents were not inhibited (Fig. 2E). The muscarinic inhibition of AqKir mimics the established effect on Kir2.3 currents by M1 receptor activation of a G protein-coupled phospholipase C (41). Together, these three results from intact oocytes suggest that, despite the lack of positively charged residues in key locations of the vertebrate Kir2 channel PIP2-binding site (Fig. 1), AqKir demonstrates known characteristics of PIP2-dependent channel regulation. However, the effects of CiVSP and wortmannin suggest that the AqKir-PIP2 interaction is weaker than that for the Kir2.1-PIP2 interaction.
FIGURE 2.

AqKir channels possess common characteristics of Kir channels that require PIP2. A, time course of inhibition of Kir channel currents by activation of CiVSP in TEVC recordings. Inhibition was mediated by strong depolarizations to activate CiVSP in oocytes injected with a mixture of cRNA for a Kir channel and CiVSP. B, time course of recovery of Kir current by deactivation of CiVSP using hyperpolarizing pulses in TEVC recordings. Currents in A and B were normalized to the steady state maximal inward current at −80 mV for each cell recorded in a bath solution of 96 mm K+. Data points represent mean ± S.E. for n > 3 cells. C, in TEVC, the amplitude of resting Kir currents measured after preincubation (40–50 min) with 20 μm wortmannin; current amplitudes were normalized to the average current in time-matched vehicle (DMSO)-treated controls using oocytes from the same frogs. An upaired two-tailed t test showed significance at p < 0.05 (*). Bars represent mean ± S.E., n = 5–10. D, activation of a co-expressed muscarinic receptor (M1) by 1 and 10 μm acetylcholine (ACh) to AqKir currents in TEVC; the effects of 1 μm acetylcholine were saturating. E, comparison of the effect of 1 and 10 μm acetylcholines on AqKir and Kir2.1 currents; bars represent mean ± S.E., n = 6–8. Currents in C–E were measured at −100 mV in a bath solution of 50 mm K+. The dotted line in D is the zero current level, and a block by 2 mm Ba2+ was used to confirm that the inward current was due to Kir channels.
Loss of Activity in Excised Patches
To further test the prediction of PIP2 dependence of AqKir channels, we examined the impact of membrane patch excision on current activity of AqKir channels. In oocytes expressing AqKir currents, patch excision first resulted in a transient increase in current amplitude, which was quickly followed by decay of the current such that channel activity was lost within 2 min (Fig. 3A). In 90% of the patches, the “rundown” of AqKir current demonstrated two phases, as observed in Fig. 3A (black circles). Overall, the loss of AqKir current was faster than the well known, time-dependent decay of vertebrate Kir channels, cKir2.2, and particularly Kir2.1, measured under the same ionic conditions. We found that the time to decay to 50% of the initial on-cell AqKir current was 12-fold faster than for Kir2.1 and 4-fold faster than for cKir2.2 currents (Fig. 3B). As rundown of Kir channels is commonly explained by the loss of PIP2 from the excised patch due to phosphatase-mediated dephosphorylation of PIP2 (42), the faster rate of AqKir current decay suggests that a higher concentration of PIP2 is needed to sustain the sponge Kir channels than the vertebrate Kir channels.
FIGURE 3.
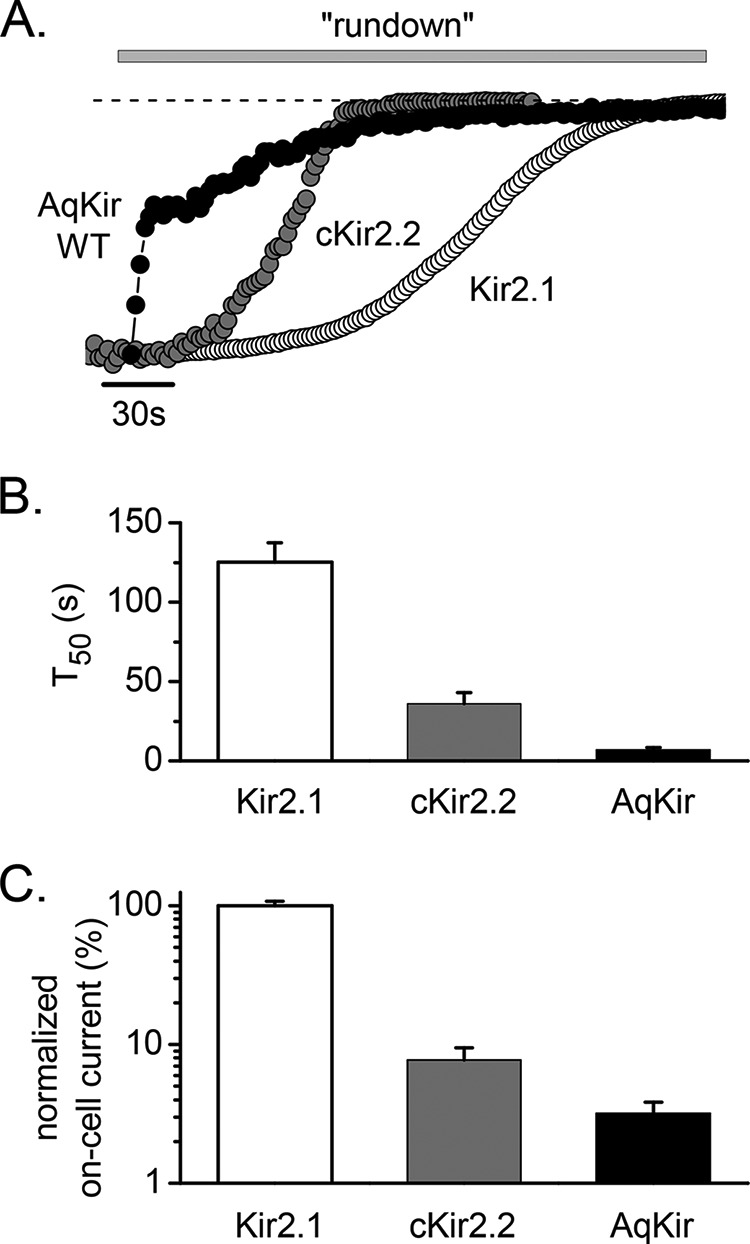
AqKir currents show rundown in excised inside-out patch recordings. A, inward K+ currents measured at −100 mV following patch excision demonstrate the time course of rundown in HK solution (gray bars indicate the application of HK + 2 mm MgCl2) for representative recordings from oocytes expressing AqKir WT (wild type, filled circles) and the vertebrate homologs, chicken Kir2.2 (gray circles), and mouse Kir2.1 (open circles). The dotted line indicates zero current. B, time to 50% of maximal rundown (t50) was measured for each construct (n = 6–15). C, magnitude of the on-cell current prior to patch excision is compared by normalizing to the average magnitude for Kir2.1 currents (n = 6–15). The cRNAs for Kir2.1 and cKir2.2 were injected into oocytes at the same concentration, but AqKir was injected at ∼4-fold higher concentration, as needed to obtain measurable currents.
On-cell Current Magnitude
On-cell patch recordings obtained before patch excision showed that the average magnitude of the AqKir channel currents was 24-fold lower than that found for Kir2.1 (Figs. 3C and 4C). Likewise, the whole-cell current amplitudes measured in TEVC showed a markedly lower current level for AqKir (−1.9 ± 0.39 μA) when compared with cKir2.2 (−4.6 ± 1.0 μΑ) or Kir2.1 (−60 ± 4.8 μA; recordings in HK solution). However, a double mutant I78R/N180K restored large AqKir current amplitudes in TEVC (−65 ± 10 μA; n = 7–17) to values comparable with the Kir2.1 channels. The reduced magnitude of the wild-type AqKir channel activity in the on-cell patch and in whole-cell recordings is consistent with the hypothesis that wild-type AqKir channel activity is limited by weaker interactions of PIP2 with the wild-type AqKir channel than with mutant AqKir channels.
FIGURE 4.

Long chain PIP2 fails to reactivate AqKir channels following rundown in excised patches. A, Kir2.1 rundown following inside-out patch excision (arrow i/o) into HK + 2 mm MgCl2 solution; the solid bar shows the application of 10 μm AASt-PIP2 without Mg2+. B, using the same protocol and lipid, the time course of AqKir WT (wild type) current rundown is shown followed by incomplete reactivation by 25 μm AASt PIP2, even with a long duration application to the patch. C, comparison of on-cell current amplitudes (in nA) at −100 mV following injection of the same concentrations of RNA for AqKir (n > 10) and Kir2.1 (n = 6). D, comparison of amplitudes of AASt-PIP2 (10 μm)-reactivated current (black bars) normalized to the on-cell current (gray bars) for Kir2.1 and AqKir WT prior to patch excision. Bars in C and D represent the mean ± S.E. for n = 5–10 patches.
Application of PIP2 to Inside-out Patches
To further test this hypothesis, we used direct application of two forms of PIP2 to the inside-facing surface of inside-out membrane patches. The lipids were prepared fresh from frozen stocks and applied in the HK bath solution using a gravity-driven perfusion system. Long chain (AASt) PIP2 contains the 20-carbon polyunsaturated fatty acid, arachidonic acid (AA) and the 18-carbon saturated stearic acid (St) in its two acyl tails. The hydrophobic nature of AASt-PIP2 limits its use to single dose applications to membranes or bilayers. In contrast, water-soluble diC8 PIP2 contains two 8-carbon unsaturated fatty acid tails and can be applied reversibly, multiple times; it is most useful in quantifying the concentration dependence of PIP2 and comparing the relative sensitivities for the lipids in different channel constructs.
The experiment in Fig. 4 tests the ability of direct application of AASt PIP2 to re-activate Kir currents following rundown due to patch excision (i/o arrow). At a concentration of 10 μm, AASt PIP2 completely restored the Kir2.1 current (Fig. 4A) but even 25 μm incompletely re-activated AqKir (Fig. 4B). In these recordings, the on-cell current magnitude was 24-fold higher for Kir2.1 when compared with AqKir (Fig. 4C), similar to previous results shown in Fig. 3C. To negate the impact of the initial (on-cell) current value on the strength of the re-activation by PIP2 in each construct, we compared the magnitude of the recovered current after AASt PIP2 (Fig. 4D, black bars) with the on-cell current amplitude before rundown (Fig. 4D, gray bars). AASt-PIP2 (10 μm) reactivated all of the Kir2.1 current but only about 25% of the AqKir WT current (Fig. 4D). This incomplete reactivation of the sponge Kir channel activity is consistent with results from our TEVC experiments suggesting that PIP2 interactions with AqKir are weaker than for Kir2 channels.
Direct application of diC8 PIP2 to the intracellular face of the membrane patches expressing AqKir also did not restore channel activity (Fig. 5). The lowest tested concentrations of diC8 PIP2 (0.1 and 1 μm) showed only very small increases in AqKir currents after rundown (Fig. 5A), and higher concentrations showed no activity. Clearly, diC8 PIP2 could not restore the sponge channel activity. On average, the current activity with diC8 PIP2 was so low that AqKir WT current amplitudes reached, at most, 10% of the on-cell patch current. Thus, a concentration-response relationship for channel activation could not be determined for the wild-type sponge Kir channel (see Fig. 6C, black circles), although Kir2.1 showed characteristic re-activation by diC8 PIP2 using the same recording conditions and same preparations of the lipid (Fig. 5, B and D). Current-voltage relationships for the two constructs showed that the strongly rectifying inward AqKir currents were not recovered by even 100 μm diC8 PIP2 (Fig. 5C), whereas Kir2.1 was maximally activated by 10 μm diC8 PIP2 (Fig. 5D).
FIGURE 5.

Water-soluble diC8 PIP2 fails to re-activate AqKir currents in inside-out patches. A, inward AqKir WT currents measured at −100 mV in on-cell patch mode and after patch excision at the time indicated (arrow i/o). HK + 2 mm MgCl2 (open bars) was interspersed with applications of increasing concentrations of diC8 PIP2 (0.1 −100 μm in HK without Mg2+, gray bars) to test for recovery of current. B, under the same conditions, the dose-dependent reactivation of Kir2.1 by diC8 PIP2 is shown. A and B, the dotted lines indicate the zero current level. C, current-voltage relationships elicited by voltage ramps from −100 to +100 mV for a representative AqKir WT on-cell recording (black trace), immediately following patch excision (red trace), and following application of diC8 PIP2 as follows: 1 μm (blue), 10 μm (pink), and 100 μm (teal). D, similar recordings for Kir2.1 show the currents in the on-cell patch (black trace) and after re-activation by diC8 PIP2 as follows: 0.3 μm (teal), 1 μm (pink), 3 μm (blue), and 10 μm (red) following washout/rundown.
FIGURE 6.

Effects of AqKir mutations on re-activation by diC8 PIP2. A, application of diC8 PIP2 (0.01–100 μm) to inside-out patches from oocytes expressing AqKir WT (top) or the mutants N180K (middle) or I78R/N180K (bottom). Lipids were applied (horizontal bars) after rundown in a Mg2+-free HK solution. Currents were measured at −80 mV; the dotted line shows the zero current level. B, comparison of whole-cell current amplitudes (in μA) for AqKir WT and three mutants measured in TEVC at −80 mV in a bath solution of 96 mm K+. The RNA for AqKir WT was injected at 2–4 times the concentration for the mutants. Bars represent mean ± S.E. for n = 7–16 cells for each construct. C, fitted concentration-response data for diC8 PIP2 reactivation of Kir channels as follows: AqKir WT (solid black circles); N180K (open blue circles); I78R/N180K (open red circles); and Kir2.1 (solid gray squares). Currents were normalized to the maximum on-cell current amplitude in each patch. D, fitted EC50 values for diC8 PIP2 reactivation of AqKir WT, two AqKir mutants, and Kir2.1. C and D, values are mean ± S.E. for n = 3–7 patches for each construct.
Recapitulating Mutations in Nature in AqKir
Channel proteins that associate with PIP2 are known to engage in electrostatic interactions with the negatively charged headgroup of PIP2 and positively charged residues in the protein (2, 7). To test the critical nature of the two basic residues in vertebrate Kir channel-PIP2 interactions (Fig. 1), we used site-directed mutagenesis to add the positive charges to AqKir, individually and together. Although wild-type whole cell and on-cell AqKir currents were small and failed to be restored by diC8 applied to excised patches (Fig. 5A, 6A), even a single mutation, N180K, restored the capability of re-activation by diC8 PIP2 (Fig. 6, A and C). The effects of diC8 on N180K were dose-dependent, reversible, and saturated by 30 μm (Fig. 6, A and C); this mimicked the effect of diC8 on vertebrate channels such as Kir2.1 (Fig. 6C). We also tested a double mutant, I78R/N180K, which restored the two missing positive charges to AqKir and resulted in large Kir currents in whole-cell recordings (Fig. 6B, red bar). The double mutant was exquisitely sensitive to diC8 PIP2; the on-cell current amplitude was completely recovered with less than 10 μm of the lipid (Fig. 6, A and C). Fitted concentration-response relationships (Fig. 6D) showed that the single mutant AqKir N180K has an apparent affinity similar to the vertebrate Kir2.1 channel, whereas the double mutant I78R/N180K has a higher sensitivity to diC8 than Kir2.1, i.e. EC50 of 0.04 μm for the double mutant versus 2 μm for Kir2.1. Prior to the restoration of I78R/N180K currents by diC8 PIP2 as shown in Fig. 6, we also observed a slow time course of rundown in the double mutant (t50 of 250 ± 38.5 ms, n = 22; compared with 17.5 ± 3.86 ms for AqKir WT from Fig. 3B); AASt-PIP2 quickly reactivated the I78R/N180K current (data not shown).
An additive effect on diC8 sensitivity of I78R in the double mutant (Fig. 6C, red circles) points to an important role of this arginine residue although, in macropatches, we could not reliably measure currents from the single mutant, I78R, following diC8 application. However, the mutant I78R produced measurable currents for TEVC recordings (Figs. 6B and 7) and was capable of re-activation by AASt PIP2 in inside-out patch recordings (Fig. 8, C and D).
FIGURE 7.

Effects of AqKir mutations on PIP2 reduction by CiVSP. Time courses of inhibition (A) and recovery (B) of AqKir WT (black) and the AqKir mutants N180K (blue) and I78R/N180K (red) by activation and deactivation of CiVSP, respectively, in TEVC recordings. Inhibition was mediated by strong depolarizations to activate CiVSP, and recovery was mediated by hyperpolarizing pulses. Oocytes were injected with a 1:2 mixture of cRNA for the Kir channel and CiVSP. Currents in A and B were normalized to the steady state maximal inward current at −80 mV for each cell recorded in a bath solution of 96 mm K+. C, comparison of the time to 50% inhibition (C) or recovery (D) for CiVSP-mediated effects in AqKir WT and mutant constructs. Bars represent mean ± S.E. for n = 4–10 cells for each construct.
FIGURE 8.

Mutant AqKir channels reactivated by PIP2 have a lower sensitivity to inhibition by neomycin than Kir2.1 channels. Inward K+ currents recorded in excised patches in HK solution at −100 mV were briefly exposed to AASt PIP2 (+ 2 mm Mg-ATP) to stably reactivate the currents and then exposed to different concentrations of neomycin. A shows the PIP2-reactivated Kir2.1 current, and B, shows the PIP2-reactivated current expressed by the double mutant AqKir I78R/N180K. C, concentration dependence of neomycin inhibition of PIP2-reactivated currents for the following: Kir2.1 (as in A, open circles), AqKir WT (gray squares), the single mutants AqKir I78R (open triangles), and AqKir N180K (open squares), and the double mutant AqKir I78R/N180K (as in B, filled triangles). Data were normalized to the maximum inward current activated by PIP2 for each construct and were fitted to a standard Hill equation. D, comparison of the fitted Kd values for neomycin inhibition shown in C (plotted on a log scale due to the large differences in values). C and D, the data represent mean ± S.E. for n = 4–7 patches for each construct.
We also examined the AqKir mutant channels for changes in CiVSP-mediated inhibition of whole oocyte current. The previously documented fast inhibition of whole-cell wild-type AqKir currents by depolarization-mediated CiVSP activation (Fig. 2A) was slowed by single or double mutations at residues Ile-78 and Asn-180 (Fig. 7, A and C). The time course of recovery with hyperpolarization to de-activate CiVSP showed the inverse effect; recovery was slow in WT AqKir but faster with mutation of Ile-78 or Asn-180 or both (Fig. 7, B and D). Indeed, quantitative comparisons of the time to 50% inhibition (t50) and the t50 for recovery following inhibition point to an important role for each positive charge alone and an additive effect of the two mutations (Fig. 7, C and D). Overall, the data suggest that the loss of PIP2 due to phosphatase activity has a stronger impact on WT than on mutant AqKir channels, which further demonstrates that these critical positive charges support the high sensitivity of PIP2 interaction with the channel. Furthermore, our results suggest that PIP2 concentrations in the membrane of intact cells (frog oocytes) falls to very low levels upon CiVSP activation, less than that required to sustain the activity of the double mutant AqKir channels for which 1 μm diC8 PIP2 maximally activated them.
Neomycin Inhibition
Neomycin, an amino-glycosidic antibiotic, is a positively charged molecule that binds with high affinity to PIP2; its application to the inside surface of the membrane is expected to inhibit PIP2-dependent currents by binding to the anionic headgroup of the lipid (43, 44). Thus, we used neomycin to further examine the relative sensitivities to PIP2 of AqKir mutant channels and the vertebrate Kir2.1 channel. Kir channel currents were allowed to run down in inside-out patch recordings, and long chain AASt-PIP2 was applied to restore the Kir currents to sustained levels. We then measured the inhibition of Kir2.1 (Fig. 8A) and AqKir mutant channels (Fig. 8B) by increasing concentrations of neomycin in a stepwise fashion. The fractional current inhibition for five constructs was plotted as a function of neomycin concentration, and each data set fitted to a standard Hill equation (Fig. 8C). The fitted Kd values for neomycin inhibition quantify the strength of PIP2 binding (Fig. 8D). The strong neomycin inhibition measured for AqKirWT was made weaker with each of the single mutants, and the Kd value for neomycin inhibition of the lipid-reactivated double mutant channels was even higher than that measured for Kir2.1. These data are consistent with the strengthened diC8 PIP2 interactions measured for N180K and the double mutation in AqKir (Fig. 7).
Docking and Energy Analysis
To understand how the mutations affect the AqKir channel and PIP2 interactions, we developed homology models of AqKir, both wild-type and single and double mutants, in complex with PIP2. Fig. 9 depicts a snapshot of AASt PIP2 bound to AqKir WT (Fig. 9A) and AqKir I78R/N180K (Fig. 9B). In the double mutant, the positive charges on Lys-180 and Arg-78 orient more closely to the PIP2 molecule.
FIGURE 9.
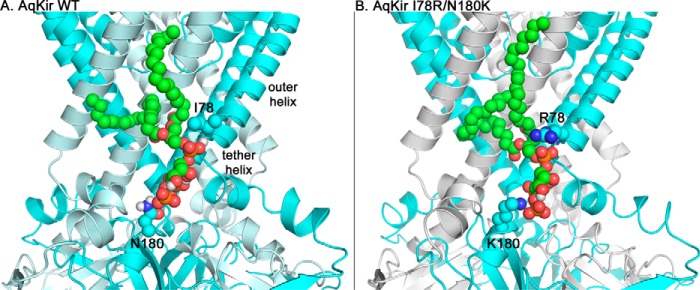
Homology models showing one AASt-PIP2 molecule bound to tetrameric Kir channels. Schematic representation is shown of AqKir WT (wild type) (A) or AqKir I78R/N180K channel (tetramers, two subunits are cyan and two subunits are pale cyan) (B) binding to AASt-PIP2 (spheres, green carbon backbone) illustrating the position of residues 78 and 180 adjacent to the phospholipid headgroup. The basic (blue) side chains of the mutant channel form additional, favorable intermolecular interactions to stabilize the channel-ligand interaction. A, the amine group of Ile-78 is part of the secondary structure of the outer helix and is not shown in this representation. The images are zoomed in for clarity and thus lack the regions that include the most extracellular portions of the channel and most of the cytoplasmic portions.
We conducted MD simulations on each model system in a membrane environment with explicit solvent and calculated the energy of the PIP2-channel interactions. Our results show that PIP2 docked in AqKir WT, at a site that maps to the Kir2.2 PIP2-binding site, has a total interaction energy of −90.6 kcal/mol. This interaction became more favorable with the I78R/N180K mutant, which had a total interaction energy of −127.4 kcal/mol. Each single mutant also improved the overall favorability of interaction with PIP2; the total interaction energies for I78R and N180K were −113.4 and −121.0 kcal/mol, respectively.
To assess quantitatively the importance of specific chemical bonds in the proposed interaction of PIP2 within the sponge Kir channel, we used energy decomposition analysis (Table 1). Addition of one basic residue to AqKir increases the total interaction energy ∼2-fold for I78R (from −6.77938 to −11.5338 kcal/mol) and ∼50-fold for N180K (from −0.51548 to −25.1471 kcal/mol). In each case, the electrostatic energy component nearly matched the total interaction energy at that residue. Thus, the more favorable interaction with PIP2 could be largely explained by improved electrostatic interactions. Comparatively, the mutant N180K, alone or in combination with I78R, had a stronger impact on energetic favorability than the positive charge added at residue 78. This is consistent with the higher priority for N180K in recapitulating the functional reactivation of AqKir by diC8 PIP2 (Fig. 6).
TABLE 1.
Interaction energies for AqKir and PIP2
Total, van der Waals, and electrostatic energies (in kcal/mol) were calculated by decomposition analysis at each residue in one of four constructs for the AqKir model with AASt-PIP2 in the binding pocket. See “Experimental Procedures” for details on the MD simulations.
| Construct and residue | Total | van der Waals | Electrostatic |
|---|---|---|---|
| WT | |||
| Residue Ile-78 | −6.77938 | −5.48137 | −1.29801 |
| Residue Asn-180 | −0.51548 | −0.46793 | −0.04755 |
| Mutant I78R | |||
| Residue Arg-78 | −11.5338 | −2.60115 | −8.93265 |
| Residue Asn-180 | −0.33572 | −0.53888 | 0.203162 |
| Mutant N180K | |||
| Residue Ile-78 | −2.45379 | −1.78845 | −0.66534 |
| Residue Lys-180 | −25.1472 | 0.879979 | −26.0272 |
| Double mutant I78R/N180K | |||
| Residue Arg-78 | −7.89737 | −2.88633 | −5.01104 |
| Residue Lys-180 | −26.2704 | −3.94693 | −22.3235 |
Fig. 10 provides a visual representation of the favorable changes in electrostatic interactions between PIP2 and the WT (Fig. 10A) and mutant (Fig. 10, B–D) AqKir channels. The sandwiching of the PIP2 headgroup between the basic side chains at positions 78 and 180 of the double mutant channel (Fig. 10D) provides additional potential for hydrogen bonding interactions between channel and ligand that are consistent with the enhanced total and electrostatic interaction energy of this mutant. The WT channel may form only one weak (3.2–4.0 Å) hydrogen bond with PIP2, whereas the mutants form a mixture of multiple weak and moderate (2.5–3.2 Å) hydrogen bonds with PIP2. Overall, each AqKir mutant has stronger hydrogen bonding potential than the WT AqKir channel, which is consistent with an increase in the apparent affinity for PIP2 in the mutant AqKir channels.
FIGURE 10.

Hydrogen bonding pattern for AqKir channels and the PIP2 headgroup. Stick representation of the hydrogen bonding pattern for AqKir residues 78 and 180 in WT (A), I78R (B), N180K (C), and I78R/N180K (D) channels to the PIP2 headgroup, based on MD simulations with homology models (see “Experimental Procedures” details). Distances between potential hydrogen bonding partners, demarcated by dotted lines, are given in green text. Atoms are colored by standard convention. The WT channel may form only one weak (3.2–4.0 Å) hydrogen bond with PIP2, whereas the mutants form a mixture of multiple weak and moderate (2.5–3.2 Å) hydrogen bonds with PIP2.
Discussion
To better understand the impact of evolutionary changes in ion channel structure and function, we studied the PIP2 regulation of a sponge Kir channel using mutagenesis, electrophysiology, and MD simulations using a homology model. PIP2 is a known regulator of many ion channel and transport molecules (1, 2). The solution of two crystal structures of vertebrate Kir channels complexed with PIP2 highlights the interaction of positively charged residues between the transmembrane domain and the C terminus with the negatively charged headgroup phosphates (7, 45). The sponge Kir channel, AqKir, a distant relative of vertebrate Kir channels, lacks two basic residues in the PIP2-binding site. This structural difference predicts low sensitivity to PIP2 regulation in the sponge Kir channel.
In whole-cell (TEVC) recordings, blocking PIP2 synthesis or stimulating PIP2 hydrolysis or dephosphorylation reduced AqKir currents, effects typically associated with a requirement for regulation by PIP2. Direct application of PIP2 to inside-out membrane patches failed to restore AqKirWT channel activity after rundown, consistent with an even lower apparent affinity for PIP2 following excision, as could occur due to loss of a post-translational modification that is not maintained under excised patch conditions (46). Consistent with this interpretation of our results, in TEVC and excised patches, AqKirWT currents were typically smaller than the AqKir mutant channels that displayed much higher affinity interactions with PIP2.
The partial reactivation of wild-type AqKir by long chain AASt PIP2, but not diC8 PIP2, suggests that hydrophobic interactions with the acyl chain of the lipid could be important in the mechanism of PIP2 activation of AqKir current, but alone are insufficient. It is also possible that other phospholipids maintain the WT current due to species-dependent differences in lipid specificity (47–49), which has not yet been examined in this channel.
Multiple positively charged residues located in the N and C termini of vertebrate Kir channels are important in coordinating with the negatively charged headgroup phosphates of PIP2 (1, 2, 7, 45). Do the various basic residues play equivalent roles? We have clarified the molecular significance by studying a channel that natively lacks these positively charged residues. Vertebrate levels of PIP2 sensitivity could be restored by mutational insertion of just one of the basic residues in AqKir, N180K (represented in 4-fold symmetry by the tetrameric channel). These results signify a particularly important role for this residue. MD simulations provided an explanation (see Fig. 10) in that N180K had the greatest impact on strengthening electrostatic interactions. Interestingly, a search for homologs of the sponge Kir channel in other distantly related metazoan species discovered putative Kir channels in sea anemones, which also lack the critical positive charge in the tether helix region. We suggest that mutation at this residue (Lys-K189 in cKir2.2) over the course of vertebrate evolution was perhaps the most critical final change that brought about the PIP2 dependence of vertebrate Kir channels.
Of course, measurements of channel activity in the excised patches following PIP2 application cannot determine directly whether the mutations affect PIP2 binding or channel gating. To this end, the MD simulations provide an explanation that highlights the more energetically favorable molecular interactions following recapitulation of one or both of the positive charges (Fig. 10 and Table 1).
Whether the sponge Kir channel is physiologically regulated by changes in PIP2 concentrations in its native environment is unknown, but the possibility surely exists. We ran a KEGG analysis (50) of the sponge genome and confirmed that all of the required components for inositol phosphate metabolism are present, and molecular cloning has previously reported that Gq, phospholipase C, and protein kinase C exist in sponges (51). Thus, the capability for PIP2 regulation of Kir channels or other proteins via phospholipase C signaling pathways is evolutionarily old, with origins at the earliest branch point of extant animal phyla.
Conclusion
Nature's mutations over the course of animal evolution have resulted in a high affinity PIP2 regulatory site within vertebrate Kir channels. However, critical features that confer high sensitivity to PIP2 in the vertebrate channels are missing in Kir channels from basal metazoans such as the channel from A. queenslandica studied here. The greatly enhanced PIP2 sensitivity of AqKir mutants containing two positive charges confirms the absolute necessity of these basic residues for PIP2 regulation, which is consistent with our homology model and MD simulations of AqKir complexed with PIP2. Furthermore, our results support a functional priority for a single positive charge (residue 180 in AqKir) in recapitulating the high sensitivity to PIP2.
Changes in the binding site over time, in response to forces that drive evolution, could include changes in the bioavailability of PIP2 (15) or molecules involved in cell-cell signaling. Conservation of Kir channel selectivity and inward rectification mechanisms (16) have been retained over millions of years of evolution, but acquisition of a new function, PIP2 regulation by high affinity interaction, is more recent, occurring after the prokaryotic-eukaryotic split.
Acknowledgments
We thank Heikki Vannanen, James Engesser, and Ariana Prinzbach for preparation of the frog oocytes; Rod MacKinnon for the cKir2.2 plasmid, and Yasushi Okamura for the CiVSP plasmid. We thank Zhe Zhang for help with molecular biology and Arco Paul for help with preliminary experiments.
This work was supported, in whole or in part, by National Institutes of Health Grants R15-GM096142 (to L. M. B.), HL059949 and HL090882 (to D. E. L.), and S10RR027411 from NCCR (to M. C.). This work was also supported by Grant 53051117 from the Jiangsu Specially Appointed Professor Program (to Q. Y. T.), undergraduate summer fellowships from the University of Richmond School of Arts and Sciences (to K. H. and E. Y.), and an undergraduate research grant from the University of Richmond School of Arts and Sciences (to E. Y.). The authors declare that they have no conflicts of interest with the contents of this article.
- Kir channel
- inwardly rectifying potassium channel
- Aq
- Amphimedon queenslandica
- PIP2
- phosphatidylinositol 4,5-bisphosphate
- TEVC
- two-electrode voltage clamp
- MD
- molecular dynamic
- CiVSP
- Ciona intestinalis voltage-sensing phosphatase
- HK
- high K+
- diC8
- dioctanoyl
- PI(4,5)P2
- phosphatidylinositol 4,5-bisphosphate.
References
- 1. Logothetis D. E., Petrou V. I., Zhang M., Mahajan R., Meng X. Y., Adney S. K., Cui M., Baki L. (2015) Phosphoinositide control of membrane protein function: a frontier led by studies on ion channels. Annu. Rev. Physiol. 77, 81–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fürst O., Mondou B., D'Avanzo N. (2014) Phosphoinositide regulation of inward rectifier potassium (Kir) channels. Front. Physiol. 4, 404–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jegla T., Salkoff L. (1994) Molecular evolution of K+ channels in primitive eukaryotes. Soc. Gen. Physiol. Ser. 49, 213–222 [PubMed] [Google Scholar]
- 4. Jegla T., Zmasek C., Batalov S., Nayak S. (2009) Evolution of the human ion channel set. Comb. Chem. High Throughput Screen. 12, 2–2-23 [DOI] [PubMed] [Google Scholar]
- 5. Suh B. C., Kim D. I., Falkenburger B. H., Hille B. (2012) Membrane-localized β-subunits alter the PIP2 regulation of high-voltage activated Ca2+ channels. Proc. Natl. Acad. Sci. U.S.A. 109, 3161–3166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Logothetis D. E., Jin T., Lupyan D., Rosenhouse-Dantsker A. (2007) Phosphoinositide-mediated gating of inwardly rectifying K+ channels. Pflugers Arch. 455, 83–95 [DOI] [PubMed] [Google Scholar]
- 7. Hansen S. B., Tao X., MacKinnon R. (2011) Structural basis of PIP2 activation of the classical inward rectifier K+ channel Kir2.2. Nature 477, 495–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Di Paolo G., De Camilli P. (2006) Phosphoinositides in cell regulation and membrane dynamics. Nature 443, 651–657 [DOI] [PubMed] [Google Scholar]
- 9. Logothetis D. E., Nilius B. (2007) Dynamic changes in phosphoinositide levels control ion channel activity. Pflugers Arch. 455, 1–3 [DOI] [PubMed] [Google Scholar]
- 10. Falkenburger B. H., Jensen J. B., Dickson E. J., Suh B. C., Hille B. (2010) Phosphoinositides: lipid regulators of membrane proteins. J. Physiol. 588, 3179–3185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Enkvetchakul D., Jeliazkova I., Nichols C. G. (2005) Direct modulation of Kir channel gating by membrane phosphatidylinositol 4,5-bisphosphate. J. Biol. Chem. 280, 35785–35788 [DOI] [PubMed] [Google Scholar]
- 12. Wang S., Lee S. J., Heyman S., Enkvetchakul D., Nichols C. G. (2012) Structural rearrangements underlying ligand-gating in Kir channels. Nat. Commun. 3, 617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. D'Avanzo N., Cheng W. W., Doyle D. A., Nichols C. G. (2010) Direct and specific activation of human inward rectifier K+ channels by membrane phosphatidylinositol 4,5-bisphosphate. J. Biol. Chem. 285, 37129–37132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Srivastava M., Simakov O., Chapman J., Fahey B., Gauthier M. E., Mitros T., Richards G. S., Conaco C., Dacre M., Hellsten U., Larroux C., Putnam N. H., Stanke M., Adamska M., Darling A., et al. (2010) The Amphimedon queenslandica genome and the evolution of animal complexity. Nature 466, 720–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. D'Avanzo N., Cheng W. W., Wang S., Enkvetchakul D., Nichols C. G. (2010) Lipids driving protein structure? Evolutionary adaptations in Kir channels. Channels 4, 139–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tompkins-Macdonald G. J., Gallin W. J., Sakarya O., Degnan B., Leys S. P., Boland L. M. (2009) Expression of a poriferan potassium channel: insights into the evolution of ion channels in metazoans. J. Exp. Biol. 212, 761–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Müller W. E., Schröder H. C., Skorokhod A., Bünz C., Müller I. M., Grebenjuk V. A. (2001) Contribution of sponge genes to unravel the genome of the hypothetical ancestor of Metazoa (Urmetazoa). Gene 276, 161–173 [DOI] [PubMed] [Google Scholar]
- 18. Sakarya O., Armstrong K. A., Adamska M., Adamski M., Wang I. F., Tidor B., Degnan B. M., Oakley T. H., Kosik K. S. (2007) A post-synaptic scaffold at the origin of the animal kingdom. PLoS ONE 2, e506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Larroux C. (2011) The genome of the sponge Amphimedon queenslandica is helping us to reconstruct our Precambrian ancestor. Med. Sci. 27, 138–141 [DOI] [PubMed] [Google Scholar]
- 20. Knoll A. H. (2011) The multiple origins of complex multicellularity. Annu. Rev. Earth Planet. Sci. 39, 217–239 [Google Scholar]
- 21. Liu L., Kohout S. C., Xu Q., Müller S., Kimberlin C. R., Isacoff E. Y., Minor D. L., Jr. (2012) A glutamate switch controls voltage-sensitive phosphatase function. Nat. Struct. Mol. Biol. 19, 633–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Boland L. M., Drzewiecki M. M. (2008) Polyunsaturated fatty acid modulation of voltage-gated ion channels. Cell Biochem. Biophys. 52, 59–84 [DOI] [PubMed] [Google Scholar]
- 23. Tang Q. Y., Zhang Z., Xia J., Ren D., Logothetis D. E. (2010) Phosphatidylinositol 4,5-bisphosphate activates Slo3 currents and its hydrolysis underlies the epidermal growth factor-induced current inhibition. J. Biol. Chem. 285, 19259–19266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wells G. D., Tang Q. Y., Heler R., Tompkins-MacDonald G. J., Pritchard E. N., Leys S. P., Logothetis D. E., Boland L. M. (2012) A unique alkaline pH-regulated and fatty acid-activated tandem pore domain potassium channel (K2P) from a marine sponge. J. Exp. Biol. 215, 2435–2444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tang Q. Y., Zhang Z., Meng X. Y., Cui M., Logothetis D. E. (2014) Structural determinants of phosphatidylinositol 4,5-bisphosphate (PIP2) regulation of BK channel activity through the RCK1 Ca2+ coordination site. J. Biol. Chem. 289, 18860–18872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tang Q. Y., Zeng X. H., Lingle C. J. (2009) Closed channel block of BK potassium channels by bbTBA requires partial activation. J. Gen. Physiol. 134, 409–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lopes C. M., Zhang H., Rohacs T., Jin T., Yang J., Logothetis D. E. (2002) Alterations in conserved Kir channel-PIP2 interactions underlie channelopathies. Neuron 34, 933–944 [DOI] [PubMed] [Google Scholar]
- 28. Sali A., Potterton L., Yuan F., van Vlijmen H., Karplus M. (1995) Evaluation of comparative protein modeling by MODELLER. Proteins 23, 318–326 [DOI] [PubMed] [Google Scholar]
- 29. Humphrey W., Dalke A., Schulten K. (1996) VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38 [DOI] [PubMed] [Google Scholar]
- 30. Hess B., Kutzner C., van der Spoel D., Lindahl E. (2008) GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 4, 435–447 [DOI] [PubMed] [Google Scholar]
- 31. Oostenbrink C., Villa A., Mark A. E., van Gunsteren W. F. (2004) A biomolecular force field based on the free enthalpy of hydration and solvation: the GROMOS force-field parameter sets 53A5 and 53A6. J. Comput. Chem. 25, 1656–1676 [DOI] [PubMed] [Google Scholar]
- 32. Meng X. Y., Zhang H. X., Logothetis D. E., Cui M. (2012) The molecular mechanism by which PIP2 opens the intracellular G-loop gate of a Kir3.1 channel. Biophys. J. 102, 2049–2059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Darden T., York D., Pedersen L. (1993) Particle mesh Ewald- an N.Log(n) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 [Google Scholar]
- 34. Berendsen H. J., Postma J. P., Vangunsteren W. F., Dinola A., Haak J. R. (1984) Molecular-dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690 [Google Scholar]
- 35. Hess B., Bekker H., Berendsen H. J., Fraaije J. G. (1997) LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 18, 1463–1472 [Google Scholar]
- 36. Soom M., Schönherr R., Kubo Y., Kirsch C., Klinger R., Heinemann S. H. (2001) Multiple PIP2 binding sites in Kir-2.1 inwardly rectifying potassium channels. FEBS Lett. 490, 49–53 [DOI] [PubMed] [Google Scholar]
- 37. Rohács T., Lopes C., Mirshahi T., Jin T., Zhang H., Logothetis D. E. (2002) Assaying phosphatidylinositol bisphosphate regulation of potassium channels. Methods Enzymol. 345, 71–92 [DOI] [PubMed] [Google Scholar]
- 38. Murata Y., Okamura Y. (2007) Depolarization activates the phosphoinositide phosphatase Ci-VSP, as detected in Xenopus oocytes coexpressing sensors of PIP2. J. Physiol. 583, 875–889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Halaszovich C. R., Schreiber D. N., Oliver D. (2009) Ci-VSP is a depolarization-activated phosphatidylinositol-4,5-bisphosphate and phosphatidylinositol-3,4,5-trisphosphate 5′-phosphatase. J. Biol. Chem. 284, 2106–2113 [DOI] [PubMed] [Google Scholar]
- 40. Nakanishi S., Catt K. J., Balla T. (1995) A wortmannin-sensitive phosphatidylinositol 4-kinase that regulates hormone-sensitive pools of inositolphospholipids. Proc. Natl. Acad. Sci. U.S.A. 92, 5317–5321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Du X., Zhang H., Lopes C., Mirshahi T., Rohacs T., Logothetis D. E. (2004) Characteristic interactions with phosphatidylinositol 4,5-bisphosphate determine regulation of Kir channels by diverse modulators. J. Biol. Chem. 279, 37271–37281 [DOI] [PubMed] [Google Scholar]
- 42. Pian P., Bucchi A., Robinson R. B., Siegelbaum S. A. (2006) Regulation of Gating and rundown of HCN hyperpolarization-activated channels by exogenous and endogenous PIP2. J. Gen. Physiol. 128, 593–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schulze D., Krauter T., Fritzenschaft H., Soom M., Baukrowitz T. (2003) Phosphatidylinositol 4,5-bisphosphate (PIP2) modulation of ATP and pH sensitivity in Kir channels. A tale of an active and a silent PIP2 site in the N terminus. J. Biol. Chem. 278, 10500–10505 [DOI] [PubMed] [Google Scholar]
- 44. Suh B. C., Hille B. (2007) Electrostatic interaction of internal Mg2+ with membrane PIP2 seen with KCNQ K+ channels. J. Gen. Physiol. 130, 241–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Whorton M. R., MacKinnon R. (2011) Crystal structure of the mammalian GIRK2 K+ channel and gating regulation by G proteins, PIP2, and sodium. Cell 147, 199–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Logothetis D. E., Lupyan D., Rosenhouse-Dantsker A. (2007) Diverse Kir modulators act in close proximity to residues implicated in phosphoinositide binding. J. Physiol. 582, 953–965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rohács T., Chen J., Prestwich G. D., Logothetis D. E. (1999) Distinct specificities of inwardly rectifying K+ channels for phosphoinositides. J. Biol. Chem. 274, 36065–36072 [DOI] [PubMed] [Google Scholar]
- 48. Telezhkin V., Reilly J. M., Thomas A. M., Tinker A., Brown D. A. (2012) Structural requirements of membrane phospholipids for M-type potassium channel activation and binding. J. Biol. Chem. 287, 10001–10012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Shyng S. L., Cukras C. A., Harwood J., Nichols C. G. (2000) Structural determinants of PIP2 regulation of inward rectifier K(ATP) channels. J. Gen. Physiol. 116, 599–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kanehisa M., Goto S. (2000) KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Koyanagi M., Ono K., Suga H., Iwabe N., Miyata T. (1998) Phospholipase C cDNAs from sponge and hydra: antiquity of genes involved in the inositol phospholipid signaling pathway. FEBS Lett. 439, 66–70 [DOI] [PubMed] [Google Scholar]
- 52. Putnam N. H., Srivastava M., Hellsten U., Dirks B., Chapman J., Salamov A., Terry A., Shapiro H., Lindquist E., Kapitonov V. V., Jurka J., Genikhovich G., Grigoriev I. V., Lucas S. M., Steele R. E., et al. (2007) Sea anemone genome reveals ancestral eumetazoan gene repertoire and genomic organization. Science 317, 86–94 [DOI] [PubMed] [Google Scholar]