

Background: Neuronal axons use dense clusters of voltage-gated ion channels to conduct rapid electrical signals called action potentials.
Results: We mapped sites on a cytoskeletal protein, ankyrin-G, which binds and clusters sodium and potassium channels.
Conclusion: Channel binding is dually regulated by protein kinase CK2 phosphorylation and the ankyrin-G sequence.
Significance: The new mechanisms identified control axonal membrane excitability and are drug development candidates.
Keywords: ankyrin, axon, evolution, potassium channel, sodium channel, M-current, axon initial segment, node of Ranvier
Abstract
In many mammalian neurons, fidelity and robustness of action potential generation and conduction depends on the co-localization of voltage-gated sodium (Nav) and KCNQ2/3 potassium channel conductance at the distal axon initial segment (AIS) and nodes of Ranvier in a ratio of ∼40 to 1. Analogous “anchor” peptides within intracellular domains of vertebrate KCNQ2, KCNQ3, and Nav channel α-subunits bind Ankyrin-G (AnkG), thereby mediating concentration of those channels at AISs and nodes of Ranvier. Here, we show that the channel anchors bind at overlapping but distinct sites near the AnkG N terminus. In pulldown assays, the rank order of AnkG binding strength is Nav1.2 ≫ KCNQ3 > KCNQ2. Phosphorylation of KCNQ2 and KCNQ3 anchor domains by protein kinase CK2 (CK2) augments binding, as previously shown for Nav1.2. An AnkG fragment comprising ankyrin repeats 1 through 7 (R1–7) binds phosphorylated Nav or KCNQ anchors robustly. However, mutational analysis of R1–7 reveals differences in binding mechanisms. A smaller fragment, R1–6, exhibits much-diminished KCNQ3 binding but binds Nav1.2 well. Two lysine residues at the tip of repeat 2–3 β-hairpin (residues 105–106) are critical for Nav1.2 but not KCNQ3 channel binding. Another dibasic motif (residues Arg-47, Arg-50) in the repeat 1 front α-helix is crucial for KCNQ2/3 but not Nav1.2 binding. AnkG's alternatively spliced N terminus selectively gates access to those sites, blocking KCNQ but not Nav channel binding. These findings suggest that the 40:1 Nav:KCNQ channel conductance ratio at the distal AIS and nodes arises from the relative strength of binding to AnkG.
Introduction
Action potentials are the principal, defining, rapid long distance signal in neurons (1). Although vertebrate action potentials consist mainly in an abrupt increase and fall in inward Na+ current, diverse outward K+ currents powerfully influence action potential properties including threshold, shape, and adaptation. A recently appreciated example of such collaboration is the functional interaction of small numbers of voltage-gated KCNQ2/3 and more abundant Nav3 channels in action potential generation at the axon initial segment (AIS) and conduction at nodes of Ranvier (2–5). Nav channels (including Nav1.1, 1.2, and 1.6 α-subunits, depending on cell type) and KCNQ channels (either KCNQ2/KCNQ3 heteromers, or KCNQ2 homomers) are concentrated at AISs, and nodes of Ranvier are concentrated by interaction of their intracellular domains with a scaffolding protein AnkG (Fig. 1). AnkG not only plays a central role in establishing and maintaining the specialized electrical activity of AISs and nodes but is essential for neuronal axodendritic polarity (6–10). AnkG is a very large molecule with diverse splice isoforms (190–480 kDa; see the schematic structure, Fig. 1C) known to be capable of recognizing many ligands including Nav and KCNQ2/3 channels via its membrane binding (MB) domain (6, 11, 12). The MB domain consists of 24 ankyrin repeats encoded by 22 exons. A canonical ankyrin repeat contains 33 amino acid residues. Ankyrin repeat structures have been extensively studied (13, 14).
FIGURE 1.
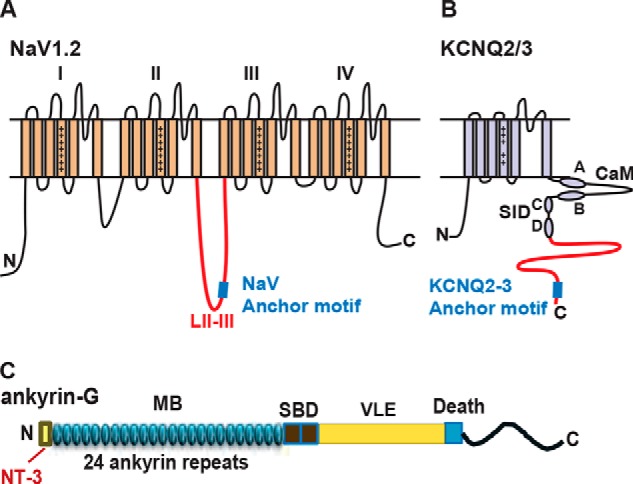
Schematics showing the domain structures of Nav1.2, KCNQ2/3, and Ankyrin-G. A, Nav1.2 possesses four homologous domains (I-IV); the anchor motif recognizing AnkG (blue square) is located near the midpoint of the loop between domains II and III. The region fused to GST for pulldown assays is indicated in red. B, KCNQ2/3 subunits possess six transmembrane segments and an unusually long intracellular C terminus. Two short α-helical segments representing sites for interaction with calmodulin (CaM, helices A and B) and two for subunit-subunit interactions (subunit interaction domain (SID), helices C and D) are labeled. The region used for GST fusions is shown in red; at the distal end is the AnkG binding motif (blue square). C, AnkG is a large multimodular protein containing a membrane binding (MB) domain, a spectrin binding domain (SBD), a C-terminal region including a very large exon domain (VLE), and a death domain.
Understanding the mechanisms determining the clustering of AIS and nodal channels is of both biological and medical interest. The protein complex of AnkG and its axonal channel ligands is implicated in psychiatric and neurological diseases. Variation in ANK3, the gene encoding AnkG, is strongly associated with bipolar disorder, and Nav blocking drugs are very commonly used as mood stabilizers (15). Mutations in genes for both Nav and KCNQ2 channels cause severe forms of epilepsy associated with developmental impairment and autism (16–19). Studies combining pharmacology, electrophysiology, and computational modeling suggest that relatively minor changes in the ratio of AIS KCNQ and Nav conductance can have a large impact on neuronal responsiveness (5, 20, 21).
What is the role of AnkG in setting the number and ratio of AIS and nodal Nav and KCNQ channels? Previously Pan et al. (3) identified a highly conserved ∼80-amino acid ankyrin binding domain at the distal end of the intracellular C terminus of KCNQ2 and KCNQ3. Within this larger domain, a 9-amino acid segment mimics the Nav channel ankyrin-G binding sequence (3, 22, 23). Hill et al. (24) found that although these Nav and KCNQ channel sequences for binding ankyrin-G are similar, the Nav sequence appeared 50–100 million years earlier during vertebrate evolution. The sequence similarity suggests that Nav and KCNQ channels might bind at similar or identical locations on the AnkG polypeptide. Here we characterize AnkG association with Nav and KCNQ2/3 channels; our results show that both Nav and KCNQ2/3 channels preferentially bind to an overlapped area on AnkG within the first seven ankyrin repeats. However, Nav channels bind much more tightly to AnkG than KCNQ2/3 channels. Although Nav and KCNQ2/3 channels share a binding region, different residues are critical for binding. Furthermore, the AnkG N terminus selectively interferes with KCNQ2/3 interaction but has no effect on Nav channel interaction. We argue that these results indicate that AnkG recruits membrane Nav and KCNQ channels and precisely maintains the Nav:KCNQ ratio via 1) differential binding of the channel ligands and 2) N terminus regulation, thus underlying normal neuronal excitability.
Experimental Procedures
Reagents
Antibodies were purchased: mouse anti-GFP (clone N86/8, antibody registry #AB_10671444, UC Davis/NIH NeuroMab Facility), rabbit anti-GFP (Invitrogen), rat monoclonal anti-HA (Roche Applied Science). Secondary antibodies highly purified to minimize cross-reactivity were from Jackson ImmunoResearch (West Grove, PA). A cDNA encoding the 270-kDa isoform of rat ankyrin-G was obtained from Vann Bennett (25). Cloned human KCNQ2 and KCNQ3 cDNAs were obtained from Thomas Jentsch (26, 27).
cDNA Constructs
AnkG-MB-GFP, KCNQ2-Neurofascin-HA, and CD4-NavII-III constructs have been described previously (3, 22). Constructs encoding various fragments of the AnkG-MB domain were generated by PCR with Pfu polymerase. Those PCR products were inserted into EcoRI and SalI sites of pEGFP-N1 (Invitrogen). GST-Nav1.2 DII-III loop was generated by PCR amplification using the rat Nav1.2 cDNA clone (28) as template. The PCR product, corresponding to residues 989–1203 of the domain II-III intracellular loop, was inserted into BamHI+SalI sites of pGEX4T-1. The distal portion of the C-terminal intracellular domains of human KCNQ2 and KCNQ3 were cloned into BamHI+NotI sites of pGEX6p-1 to make GST-Q2C (571–844) and GST-Q3C (578–872). To generate clones encoding alternatively used AnkG N termini NT1 and NT2, we exploited a unique PstI site within the first repeat (R1) of the rat AnkG cDNA. We synthesized cDNAs for NT1-R1 or NT2-R1, including a BamHI site at 5′ end, and cloned this fragment into pUC57 (Genewiz). The NT1-R1, NT2-R1 fragments were digested with BamHI and PstI and inserted into pEGFP-AnkG N-R7 digested with BamHI and PstI.
Deletion and alanine substitution mutations in AnkG were made using QuikChange (Stratagene) using rat AnkG-NT3 cDNA as template. The β hairpin tip alanine mutations were introduced at positions corresponding to the first and last codon of exons, i.e. hairpin 1 (H1): 39,40SD-AA; H2: 71,72NQ-AA; H3: 105,106KK-AA; H4: 138,139QN-AA; H5: 171,172ED-AA; H6: 204,205PA-AA; H7: 241,242ES-AA. The alanine substitution mutations within the AnkG-NT3 non-ankyrin N terminus were at MB N-R7 E15, EEE19–21, EEETE19–23; KKKK24–27, RKR29–31, and KKK36–38. Other alanine substitution mutations were at R47, R50 (Arg-47 and Arg-50) in repeat 1. To mimic AnkG splice isoforms lacking exon 7 (29), codons 234–241 encoding residues MVVNRATE were deleted. All constructs were verified by DNA sequencing before use. Additional cloning details are provided in supplemental Table S1.
Fusion Protein Expression and Purification
GST fusion proteins were expressed in Escherichia coli strain BL21 (DE3) and induced by 1 mm isopropyl β-d-1-thiogalactopyranoside. Overexpressed GST fusion proteins were affinity-purified by glutathione-Sepharose 4B according to the manufacturer's manual (GE Healthcare). The protein eluted was dialyzed against 1 liter of phosphate-buffered saline (PBS, diluted from 10× PBS, Thermo Scientific) using 10-kDa molecular weight cutoff dialysis cassettes (Pierce). Protein concentration was estimated by colloidal Coomassie Blue staining (Invitrogen) using a standard curve generated using bovine serum albumin (BSA). Purified proteins were aliquoted and snap-frozen in liquid nitrogen and stored (−80 °C).
In Vitro Protein Kinase (CK2) Phosphorylation
Phosphorylation reactions were performed by incubating purified GST fusion proteins (2 μg) with 1× CK2 buffer containing 200 μm ATP and 10 units of CK2 (New England BioLabs) for 1 h at 30 °C. For the control conditions, protein samples were treated identically except CK2 was omitted.
Cell Culture and Transfection
Human embryonic kidney (HEK) 293 cells were cultured in minimum Eagle's medium containing 10% fetal bovine serum (Invitrogen). X-tremeGENE 9 (Roche Applied Science) or Lipofectamine 2000 (Invitrogen) was used for transfection following the manufacturer's manual.
Surface Redistribution Assay and Immunostaining
The AnkG surface redistribution screening assay (see Fig. 4) has been previously described (3, 25, 30). In brief, HEK293 cells were transfected with plasmids AnkG-MB GFP together and HA-tagged NF-KCNQ2 C-terminal (residues 571–844) plasmids and replated onto coverslips at low density. Cells were fixed 2 days after transfection. After washing 3 times, the cells were blocked in 10% normal goat serum, 2% BSA, 1× PBS for 1 h, then incubated with rat anti-HA antibody at 4 °C overnight. Cells were washed 5 times with PBS then incubated with Cy3-conjugated anti-rat IgG antibody for 2 h at room temperature. Coverslips were mounted using Prolong Gold (Invitrogen) and photographed at high magnification (60×) under epifluorescence. Cells included in groups photographed and analyzed further fulfilled several criteria. They were well isolated and spindle-shaped or multipolar. Small cells lacking a clearly defined cytoplasmic compartment were excluded. Under red imtk;1munofluorescence, cells that gave anti-HA-positive staining with a clearly resolved edge distribution were photographed, and then the distribution of green fluorescence was analyzed. For each experimental condition, images of 20 or more cells from three separate transfection experiments were collected. Imaged cells were classified as either possessing (edge positive) or lacking (edge negative) an easily detected green fluorescence surface “edge” that co-localized with the red anti-HA staining. Intensity histogram line positions were chosen so they would cross positions where the red HA surface edge and underlying cytoplasmic compartment were readily detected.
FIGURE 4.
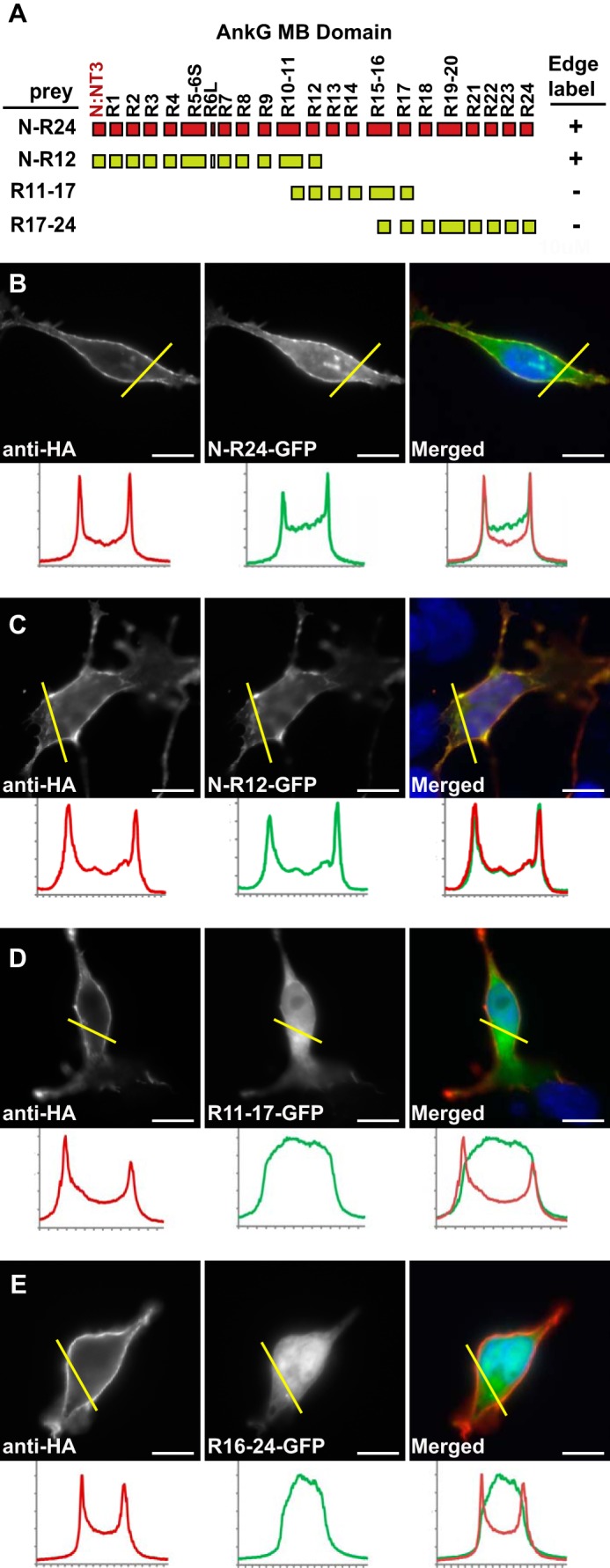
The N-terminal half of AnkG-MB (N-R12, ∼400 residues) contains the KCNQ2 determinant(s) responsible for surface redistribution. A, schematic showing MB domain exonic structure, encoded ankyrin repeats, and the overlapping truncations used to generate GFP fusion proteins. + indicates surface redistribution of GFP signal to the surface membrane was observed; − indicates GFP signal remained homogeneously cytoplasmic. B–E, surface redistribution assay results. The left panels show cells labeled using an extracellular HA tag on the neurofascin-KCNQ2 fusion protein bait, and middle panels show the distribution of AnkG-GFP fusion proteins, right panels show overlays (AnkG-GFP, green; anti-HA, red). Below each panel, intensity histograms show signals along the yellow line. B and C, AnkG-MB (green) and N-R12 truncation (green) are redistributed to the cell surface. D and E, MB R11–17 (green) and MB R16–24 (green) remain cytoplasmic. Scale bars, 10 μm.
Cellular Localization Correlation Analysis
To analyze KCNQ2/3 and Nav interaction with AnkG in a cellular environment, we used a localization correlation approach (31–33). NF-Q2C or CD4-NavII-III constructs were co-transfected in HEK293 cells with GFP-fused AnkG MB fragments. Two day after transfection, cells were fixed, washed 3 times, permeabilized with 1× PBS containing 0.2% Triton X-100, blocked in 10% normal goat serum, 2% BSA, 1× PBS, 0.02% Triton X-100 for 1 h, and then incubated with rat anti-HA antibody at 4 °C overnight. Cells were washed 5 times with PBS with 0.02% Triton X-100, incubated with Cy3-conjugated anti rat IgG antibody for 2 h at room temperature, and then mounted and imaged. Images for analysis were acquired from three independent transfections. The co-localization of red and green fluorescence was quantified using NIS Elements AR (Nikon Instruments). Regions of interest selected were based on overlapped/merged green and red channels.
CK2 Activity Inhibition
TBB and TBCA (4,5,6,7-tetrabromo-2-azabenzimidazole, and ((E)-3-(2,3,4,5-tetrabromophenyl)acrylic acid; Sigma) were used as CK2 inhibitors. TBB was dissolved in DMSO at 10 mm; TBCA was dissolved in DMSO at 5 mm. HEK293 cells were co-transfected with MB-GFP and NF-Q2C. One day after transfection, 50 μm TBB or 25 μm TBCA was added to culture medium for either 6 or 24 h. DMSO alone was added to controls.
Image Acquisition and Analysis
Epifluorescence images were collected using a Nikon 80i microscope, a 60×, 1.4 numerical aperture Plan Apo oil immersion objective, and a Exi Aqua CCD camera (QImaging) driven by NIS Elements (Nikon) software.
Sequential Pulldown and Western Blot
For each condition, equal amounts (2 μg) of purified GST fusion proteins either phosphorylated or non-phosphorylated by CK2 were incubated with glutathione beads for 2 h at 4 °C. After extensive washing, GST fusion protein-coated beads were further incubated with cell lysate from HEK cells expressing various AnkG-MB domain truncations fused with GFP, and then, after another extensive wash, the pulldown protein complex was eluted using Laemmli sample buffer (2×) with DTT for 5 min at 95 °C. The samples were resolved in SDS-PAGE and transferred to nitrocellulose membranes (Bio-Rad) then subject to Western blot and enhance chemiluminescent detection using the ECL kit (GE Healthcare).
Results
Phosphorylation by Protein Kinase CK2 (CK2) Enhances KCNQ2 and KCNQ3 Association with AnkG
Although expressed ubiquitously, CK2 is enriched at AISs and nodes of Ranvier (34). Four conserved CK2 consensus sites lie within or immediately C-terminal to the Nav channel anchor motif; phosphorylation of these sites has been shown to markedly enhance binding of AnkG (34, 35). Although Nav and KCNQ2/3 sequences have low similarity outside of the short anchor motifs (Fig. 2A, red asterisks), the C-terminal intracellular domains of KCNQ2 and KCNQ3 nonetheless have, respectively, three and four consensus CK2 sites in or C-terminal to their anchor motifs (Fig. 2A, arrows). To determine if CK2-mediated phosphorylation regulates KCNQ2/3 binding of AnkG, we performed sequential in vitro CK2 phosphorylation and pulldown experiments using GST-fused channel baits and soluble, heterologously expressed AnkG prey. The Nav1.2 bait included the entire intracellular loop between homology domains II and III (shown in red in Fig. 1A; Ref. 34); the KCNQ2/3 baits extended from the tetramerization domain tail (36) to the C terminus (KCNQ2 residues 571–844, KCNQ3 residues 578–872, red in Fig. 1B). Purified baits (GST-Q2C, GST-Q3C, GST-Nav II-III, or GST) were incubated with purified CK2 and ATP; in controls, CK2 was omitted. After 30 min, CK2-treated and control fusion proteins were collected on glutathione beads, washed extensively, and then mixed with lysates of HEK cells expressing AnkG membrane binding domain green fluorescence protein fusion proteins (AnkG-MB-GFP). Western blots showed that, without CK2 phosphorylation, KCNQ2 and KCNQ3 baits pulled down AnkG-MB-GFP very weakly but detectably, whereas unphosphorylated Nav bait yielded a much greater signal (Fig. 2B). For all the channel fusion baits, but not GST, CK2 phosphorylation markedly enhanced binding of AnkG-MB-GFP.
FIGURE 2.
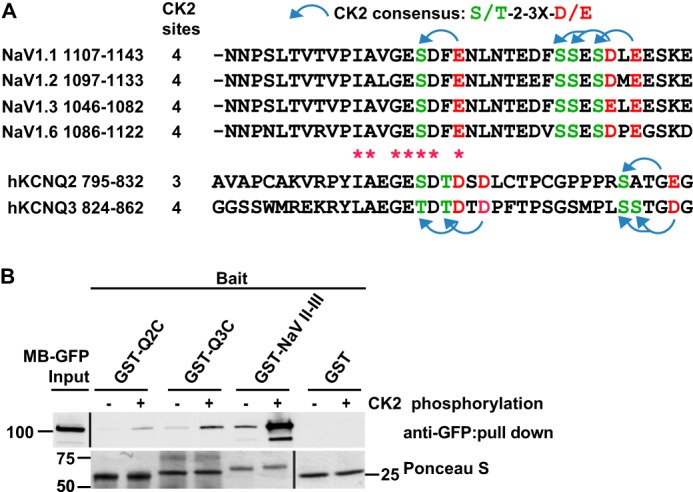
GST-Nav1.2 DII-III loop, GST-Q2C and GST-Q3C binding to AnkG-MB is markedly enhanced by phosphorylation of the channel polypeptides by CK2. A, alignment of AnkG binding regions of Nav and KCNQ2/3 channels. There are four predicted CK2 sites near the Nav anchor motif, four in KCNQ3, and three in KCNQ2. Blue arched arrow, CK2 consensus sites; red asterisks, identical or same group amino acid between Nav and KCNQ2/3 channels. B, pulldown results. The indicated baits were first phosphorylated by CK2 (+ lanes). The baits were then mixed with lysates of HEK cells expressing AnkG-MB-GFP protein. Upper row, AnkG-MB-GFP in lysate (left lane) and the indicated pulldowns, detected by Western blot. Signal strength, Nav1.2 ≫ KCNQ3 > KCNQ2. After CK2 phosphorylation, pulldown by all the three baits is increased. At this exposure, CK (−) pulled down bands for KCNQ2 and KCNQ3 are barely detected. Lower row, Ponceau S staining of transfer membrane before Western blot shows loading of GST fusion protein baits.
CK2 Inhibitor Treatment Decreases Co-localization of MB-GFP and NF-Q2C
The selective CK2 inhibitors TBB and TBCA have been used previously to study CK2 effects on Nav channel localization and KCNQ2 channel function (34, 37, 38). To further analyze the role of CK2 in regulation of AnkG and KCNQ channel interactions, we treated HEK cells expressing AnkG MB and either Nav or KCNQ anchor-bearing fusion constructs. Six hours of treatment with TBCA, but not TBB, induced a significant reduction of co-localization of NF-Q2C and AnkG compared with treated vehicle controls (Fig. 3, A–C and G). After 24 h of exposure to either TBB or TBCA, co-localization was significantly reduced (Fig. 3, D–G). These results indicate that CK2 activity inhibition alters interaction between the KCNQ2 and AnkG MB in intact HEK293 cells.
FIGURE 3.

CK2 inhibition reduces the colocalization of overexpressed AnkG and KCNQ2 fusion proteins in HEK cells. A–F, representative images and fluorescence correlation profiles for HEK293 cells co-expressing NF-Q2C with MB-GFP, treated with DMSO (A and D, vehicle control), 50 μm TBB (B and E), or 25 μm TBCA (C and F); green versus red fluorescence correlation profiles are shown in rightmost column. In this profile, the x axis plots relative green fluorescence intensity (GFP, 0–100%); the y axis gives red fluorescence (Cy3 anti-HA, 0–100%). Region of interest selected are indicated with yellow lines (Merged panels). G, Pearson correlation coefficient factors for the conditions shown. The number of cells analyzed per condition, S.E., and t test results (***, p < 0.001; **, p < 0.01; *, p < 0.05) are indicated. Scale bars: 10 μm.
An AnkG-MB Fragment (N-R12, ∼400 residues) Contains the KCNQ2 Binding Site
Previously, Pan et al. (3) used AnkG-MB-GFP as a reporter in a cell-based surface redistribution assay to identify the homologous KCNQ2/KCNQ3 anchor motifs (3, 30). We generated expression constructs including three overlapping fragments of the AnkG-MB domain (Fig. 4A) and compared their ability to be concentrated at the cell membrane by interaction with the KCNQ2C polypeptide (residues 571–844; Fig. 1B). In 21 of 21 cells studied, AnkG-MB-GFP, containing the non-ankyrin repeat N terminus and all 24 ankyrin repeats, was efficiently redistributed to the cell membrane (Fig. 4B), as was an N-R12 truncation construct (32/33 cells studied; Fig. 4C). MB (R11–17) and MB (R16–24) constructs remained homogeneously distributed in the cytoplasm and never showed an edge appearance (0/27 and 0/22 cells, respectively; Fig. 4, D and E). We were unable to achieve efficient cytoplasmic expression of smaller AnkG N-terminal constructs (data not shown). Alternatively, we performed pulldown experiments using CK2-phosphorylated GST-Q3C and GST-Nav II-III fusion proteins baits and overlapping AnkG-MB fragments as prey. Although the Nav and KCNQ3 baits showed detectable CK2 phosphorylation-dependent binding of N-R17 and R7-R24 fragments, binding of the N-R17 fragment was much stronger (Fig. 5, A and B).
FIGURE 5.

AnkG-MB R1–7 is critical and sufficient for binding both Nav1.2 and KCNQ2/3 channels. A–D, GST pulldowns of AnkG-MB truncations. A and B, analysis of large fragments shows that interaction of both channel with AnkG is primarily mediated by the N terminus through R17 region and is CK2 phosphorylation-dependent. First through third lanes, anti-GFP Western blots of AnkG “prey” input. Fourth through ninth lanes, pulldown results. Upper row, CK2 phosphorylation-dependent binding to the GST-Nav1.2 DII-III loop (A) and GST-Q3C (B); lower row, Ponceau S staining shows equal loading of GST baits. C and D, further specification of the binding region using deletional analysis. For each AnkG-MB prey: top row, prey input; second row, prey pulled down by the CK2 phosphorylated GST-Nav1.2 DII-III loop; third row, Ponceau S stain showing equal loading of baits (note: for N-R12 and N-R7 lanes, prey pulled down by Nav1.2 is detectable); fourth row, prey pulled down by the CK2 phosphorylated GST-Q3C; fifth row, Ponceau S stain for GST-Q3C. E, summary of mapping results from A–D (R11–24 and N-R6, not shown). Pulldown band intensity was rated as very strongly positive (+++), strongly positive (++), positive (+), weak but still detectable (+/−), and barely detectable (−/+).
An N-R6 AnkG Fragment Is Sufficient for Robust Binding to the Nav Anchor, but a Larger N-R7 Fragment Is Required for Robust Binding to KCNQ3
To further map channel binding sites within the AnkG-MB, we performed additional pulldown experiments using smaller MB fragments as prey (Fig. 5). In a series of C-terminal truncations at exon-exon junctions, AnkG-MB N-R7 was the minimal fragment sufficient to bind the CK2-phosphorylated KCNQ3 anchor robustly (Fig. 5, C and D). A smaller fragment, N-R6, bound the Nav anchor well but bound the KCNQ3 anchor much more poorly (Fig. 5C).
Exon 7 in vertebrate ankyrins is atypical, only eight codons in length, and analysis of brain and heart tissues indicate it is alternatively spliced (29) (data not shown). As a result, the R6–7 β-hairpin has alternative forms possessing and lacking an eight-amino acid insert (termed here, R6L and R6S). Deletion of exon 7 had no effect on binding to either channel (Fig. 5D). Repeats 5 and 6 are coded on the same exon (exon 6 in vertebrates; Fig. 5E) and are the only repeats that break the canonical 33 residue per repeat pattern (39) and structure (14, 40); see “Discussion”). Deletion of exon 6/R5–6 resulted in an N-R4 fragment that showed markedly reduced binding to either anchor (Fig. 5D). Likewise, a deletion eliminating the non-ankyrin-repeat N terminus and R1 abolished binding to either channel (Fig. 5D).
The Third AnkG β-Hairpin Tip Is Required for Robust Binding to Nav but Not KCNQ2/3
The above findings indicate that the main sites for recognizing both channels are within the N-R7 region. Although N-R7 is a small portion of the whole AnkG polypeptide, it still offers a large potential binding surface. To further elucidate differences in Nav and KCNQ2/3 anchor binding, we used a combination of scanning and targeted point mutagenesis. The β-hairpin tips of ankyrin repeats are relatively variable in sequence and exposed to ligands and, therefore, often contribute to sites of specific binding. In AnkG, these tips are of additional interest as they correspond to exon-exon junction sites and thus may confer distinctive ligand binding through alternative splicing (13, 14, 29, 41). We, therefore, tested a series of mutant N-R7 constructs in which predicted β-hairpin tips were individually substituted by di-alanine mutations (Fig. 6, A-C). Pulldown experiments with CK2-phosphorylated GST-Nav II-III and GST-Q3C showed that mutation of β-hairpin 3 (H3) between repeats R2 and R3 exhibited a dramatic loss of binding to Nav II-III. The disrupted Nav II-III binding was observable by both Western blot and Ponceau staining (Fig. 6C, black arrow). Binding to phosphorylated KCNQ3-C was unchanged by the mutation of H3. Mutation at other β-hairpin tips had no effect on binding to either channel type. The H3 tip consists of two lysines (indicated in red, Fig. 6, A, E, and F), whereas anchor sequences of Nav and KCNQ2/3 channels possess multiple acidic and phosphorylated side chains. This suggests that electrostatic interactions involving this tip may be essential for binding of Nav channels to AnkG.
FIGURE 6.

Different residues within R1–7 are critical for Nav and KCNQ2/3 binding. A, schematic representing a single ankyrin repeat and the AnkG N-R7 fragment. Each 33-residue ankyrin repeat consists of a right-handed solenoid: half β-hairpin, two short antiparallel α-helices, a “loop” directed back toward the hairpin, and a second half β-hairpin. This pattern is followed in AnkG R1–7, except for R5-R6, which preserves the solenoidal periodicity but replaces the hairpin with a longer R6 front helix and longer loop (40). Locations of hairpin tips residues H1, H2, H4, H5, and H7 are labeled green; H3 tip KK implicated in Nav binding is labeled red. Arg-47 and Arg-50 residues implicated in KCNQ2/3 binding are labeled purple. B, amino acid sequence of MB (N-R7) fragment with ankyrin repeats aligned. R5-R6 secondary structure alignment is based on Wang et al. (40). C, AnkG-MB (N-R7) hairpin tip mutant pulldowns by CK2-phosphorylated GST-Nav DII-III loop. Upper and middle rows show prey input and prey pulldown, measured by anti-GFP Western blot. The H3 KK to AA mutation shows strongly reduced binding, whereas the other hairpin tip mutants bind similarly to the control. Lower row, Ponceau S staining of the filter. Upper bands (*) are the GST fusion baits; lower bands (arrow) are the wild-type and mutated AnkG prey. D, experiment performed in parallel with C, using CK2-phosphorylated GST- Q3C. The MB (N-R7) H3 KK to AA mutation shows very slight reduction of binding to KCNQ3, whereas the other hairpin tip mutants show no obvious effects. E, deletion of non-ankyrin repeat N terminus from AnkG-MB (N-R24) leads to increased KCNQ channel binding activity. Deletion of R2-R3 repeats of full-length MB leads to marked reduction of Nav channel binding activity. Nav and KCNQ3 baits were CK2-phosphorylated. F, summary of the results in E; scale is as in Fig. 5E.
When introduced into full-length (N-R24) AnkG-MB, the H3 di-alanine mutation had no effect on binding to GST-Nav II-III (data not shown). We hypothesized that because earlier experiments showed evidence of weak interaction with R7–24, change restricted to this hairpin tip might be compensated by other contributions from other sites or even by (physiologically irrelevant) simultaneous interactions with multiple bait molecules. We, therefore, introduced a larger disruption including H3, namely removal of R2-R3 with fusion of R1 and R4 at a potential native exon-exon junction site (Fig. 6F). The R2-R3 deletion decreased binding to GST-Nav II-III markedly but had little effect on binding to GST-KCNQ3C (Fig. 6, E, first versus third lanes, and F).
A Dibasic Sequence in Repeat 1 Contributes to KCNQ2/3 but Not Nav Channel Binding
MB N-R12 binds to both channel types, whereas MB R2–12 binds neither (Fig. 5D). To identify sites within the N-R1 region critical for binding Nav or KCNQ channels, we made additional alanine mutations, focusing on charged residues that might also interact electrostatically with the anchor domains. Previously, Abdi et al. (42) obtained evidence for interaction between an acidic sequence in the C terminus of ankyrin-B (amino acids 1597–1599, EED) and a basic sequence, RAAR, located in the front α-helix of ankyrin repeat 1 of Ankyrin B. This RAAR sequence is conserved in vertebrate and protostome ankyrins (data not shown). We noted that the ankyrin-B C-terminal ligand sequence EED resembles the key residues of the anchor motifs (ESD for KCNQ2 and Nav and ETD for KCNQ3), especially if the Ser and Thr residues are phosphorylated by CK2 (34, 43). Therefore, we generated R47A/R50A-mutated AnkG N-R7 (Figs. 6A and 7A) and performed pulldown assays. R47A/R50A mutation caused the complete loss of KCNQ2 and KCNQ3 binding activity (Fig. 7, B and C, lanes a and b) but had no detectable effect on Nav channel binding (Fig. 7B).
FIGURE 7.

Arg-47 and Arg-50, two arginines on the repeat 1 front α-helix, are critical for KCNQ2/3 but not Nav1.2 channel binding. A, the AnkG N terminus and first repeat, with residues mutated highlighted; positive charged residues (green shading), cluster including multiple acidic residues (red shading), single acidic residue (pink box). Shown are GST pulldowns of AnkG-MB (N-R7) and (R1–7) with and without R47A and R50A mutations (B and C) and of AnkG-MB (N-R7) with and without the indicated mutations in the N terminus (D and F). B, anti-GFP Western blots are shown; upper, prey input; middle, prey pulled down by CK2-phosphorylated GST-Nav1.2 DII-III loop; lower, prey pulled down by CK2-phosphorylated GST-Q3C. The R47A/R50A mutation abolishes KCNQ3 binding, but the Nav channel binding is unaffected. C, GST-Q2C pulldown AnkG-MB (N-R7), AnkG-MB (N-R7) Arg-47–Arg-50 mutation (R1–7), AnkG-MB (R1–7) Arg-47–Arg-50 mutations with or without CK2 activity. Upper, prey input; middle, prey pulled down by GST-Q2C; lower, prey pulled down by CK2-phosphorylated GST-Q2C. CK2 phosphorylation and N-terminal deletion additively increase binding. Lanes c′ and d′ are briefer exposures. D and F, mutations within the NT3 N-terminal have no affects. D, Western blots showing pull downs by GST-Nav 1.2 DII-III and GST-Q3C. E and F, pulldowns of additional mutant constructs showing both Western blots and Ponceau stains. In GST-Nav 1.2 DII-III, but not GST-Q3C pulldowns, Ponceau staining reveals both the bait (*) and the AnkG prey. For both baits, the mutations have no effect on pulldown results.
The initial N-terminal portion of all ankyrins consists of a non-ankyrin sequence encoded by one or two exons. The clone used here and in many previous studies by us and others (3, 25, 44) has a 39-residue N terminus bearing 3 clusters of basic amino acids and 1 cluster of acidic amino acids (Fig. 7A). We made alanine substitution mutations for several combinations of positive charged residues (KKKK24–27, RKR29-31, KKK36–38) and acidic residues (E15, EEE19–21, EEETE19–23). None of these mutant constructs changed the binding to either KCNQ3 or Nav baits (Fig. 7, D and F).
To further assess the role of the repeat 1 Arg-47 and Arg-50 residues in a cellular context, we introduced R47A and R50A mutations into GFP-tagged AnkG MB and MB Ndel and assayed co-localization with HA-tagged NF-Q2C in intact HEK293 cells (Fig. 8). Rows A-G show GFP and HA fluorescence of representative cells that gave correlation values near the means plotted in panel H. As expected, GFP and NF-Q2C localization was poorly correlated, whereas AnkG MB, MB Ndel, or R1–7 localization was highly correlated with NF-Q2C. Interestingly, whereas AnkG MB was redistributed to the cell surface when co-expressed with NF-Q2C (Fig. 8, A and B), the N terminus-lacking fragments most effective in pulldown assays caused NF-Q2C to be redistributed intracellularly (Fig. 8, C and F). R47A/R50A-mutated MB Ndel (Fig. 8E) or R1–7 (Fig. 8G) diminished the intracellular staining for NF-Q2C and significantly reduced localization correlation (p < 0.001 and p < 0.01, respectively; Fig. 8H). R47A/R50A mutation gave no significant effect on co-localization of AnkG MB and NF-Q2C, suggesting that the presence of the N-terminal may weaken the impact of this mutation.
FIGURE 8.

R47A/R50A mutation repeat reduces AnkG colocalization with NF-Q2C. A–G, images and fluorescence correlation profiles for representative HEK293 cells co-expressing NF-Q2C with GFP fusion constructs. For each condition, images show (left to right) DAPI labeling of the nucleus, GFP fluorescence, immunofluorescence staining for the HA-NF-Q2C fusion protein, merged image, and the intensity correlation plot. A, GFP alone gives a diffuse pattern, unlike the punctate intracellular and surface edge pattern of NF-Q2C. This results in an intensity correlation profile (right) showing poor correlation. B, MB-GFP shows a punctate and surface pattern similar to NF-Q2C and high correlation. MB Ndel-GFP (C), MB R47A/R50A-GFP (D), MB Ndel R47A/R50A-GFP (E), MB R1–7-GFP (F), or MB R1–7 R47A/R50A-GFP (G). H, quantification of co-localization of NF-Q2C with GFP fused AnkG MB fragment mutants for cells in A–G. Shown are Pearson correlation coefficient factors for conditions shown. The number of cells analyzed per condition, S.E., and t test results (***, p < 0.001; **, p < 0.01) are indicated. Scale bars: 10 μm.
The Non-ankyrin Repeat N Terminus of AnkG Inhibits KCNQ2/3 but Not Nav Channel Binding
As targeted mutation of charged residues in the N terminus failed to mimic the effect of deleting N-R1, we generated a construct deleting all 39 residues of the N terminus. Surprisingly, this construct (R1–7) exhibited remarkably enhanced binding to both GST-Q3C (Fig. 7A) and GST-Q2C (Fig. 7B) compared with N-R7. Binding enhancement by removal of the N terminus was robust even without phosphorylation of the Q2C bait by CK2 (Fig. 7C). By contrast, N-R7 and R1–7 constructs bound to the Nav channel bait equally well. The selective importance of the R1 dibasic site (Arg-47 and Arg-50) for KCNQ2/3 binding was further underlined by introduction of alanine substitutions of those residues into R1–7 (Fig. 7, B and C, lanes c and d). The double mutation very strongly reduced KCNQ2/3 binding to R1–7 but had no effect on Nav binding.
To examine the contribution of the N terminus to binding in larger context, we introduced the deletion into full-length AnkG-MB and performed pulldown experiments. Consistent with results using the R1–7 fragment, the deletion increased binding to the KCNQ3 channel but not to the Nav bait (Fig. 6, E and F).
ANK3 Alternative N Termini Differentially Regulate KCNQ2/3 and Nav Channel Association with AnkG
ANK3 has at least three alternative promoters and corresponding first exons encoding non-ankyrin N termini variants (namely exon1a, exon1e, and exon1b, encoding NT1–3, respectively; Fig. 9, A and B) (6, 42, 45, 46). These promoters and alternative N termini exhibit tissue- and cell type-specific expression. Exons 1b and 1e are expressed in central neurons, whereas exon 1a is expressed in other tissues (6, 42). Our experiments all used the widely distributed NT3-bearing clone of rat AnkG (6). We, therefore, examined whether NT1 and NT2, like NT3, were able to inhibit KCNQ channel binding to AnkG. NT1, NT2, and NT3 are 33, 21, and 39 amino acids in length, respectively (Fig. 9A). We performed pulldown assays in parallel using three identical constructs except for the N termini (Fig. 9C). Whereas both NT2 and NT3 strongly inhibited AnkG association with the KCNQ3 bait, NT1 slightly inhibited binding. The three N termini had no effect on Nav channel binding. KCNQ2 and KCNQ3 are relatively new genes, arising first by gene duplications in an ancestor of the jawed vertebrates (24). Interestingly, the AnkG N-terminal sequences are also jawed vertebrate novelties absent from invertebrates (46); within gnathostomes, they are extremely well conserved, as illustrated by alignment of the NT3 residues of Danio rerio and Homo sapiens (Fig. 9A). We attempted to measure interaction between GST fused with the AnkG NT3 N terminus and the soluble R1–7-GFP fragment by similar pulldown assays, but no binding was detectable (data not shown).
FIGURE 9.
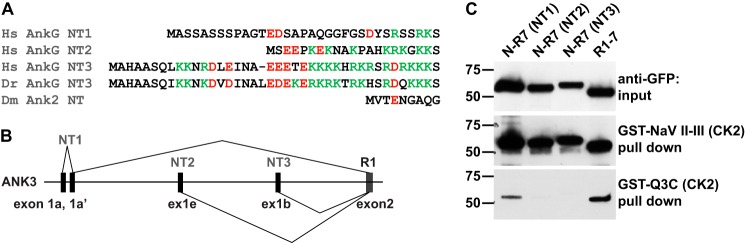
AnkG N termini NT1, NT2, and NT3 inhibit KCNQ2/3 but not Nav1.2 binding to AnkG. A, amino acid sequences of alternative N termini of human AnkG, N terminus of zebrafish Ank3a (ZDB-GENE-060621-1), and fly ankyrin-2 (GenBankTM number NP_001097533), aligned at their points of attachment to MB. The homologous NT3 termini are highly conserved in fish and man; the invertebrate N terminus shows no apparent homology. B, splicing diagram at the 5′ region of ANK3; this pattern is conserved in vertebrates, absent in invertebrates. C, Western blots of pull down experiments using AnkG R1–7 bearing different N termini (first through third lanes) or without an N terminus (fourth lane). Upper row, lysate inputs, detected using anti-GFP; middle row, prey pull downed by CK2-phosphorylated GST-Nav1.2 DII-III loop; lower row, prey pull downed by CK2-phosphorylated GST-Q3C. NT2 and NT3 strongly inhibit, and NT1 less strongly inhibits binding to GST-Q3C but not to GST-Nav1.2 DII-III.
These results imply that the non-ankyrin repeat N terminus can differentially regulate cargo binding activity of the adjoining ankyrin repeats. The N terminus selectively interferes with KCNQ3/2 channel but not Nav channel binding to AnkG. On the basis of our findings, we propose a “gated binding pocket” model for AnkG N-R7 interaction with Nav and KCNQ2/3 channel proteins (Fig. 10). Secondary structure prediction algorithms indicate that the N terminus is primarily α-helical (47, 48). The R1–7 repeats provide the binding surface recognized by both channels. The non-ankyrin repeat N terminus restricts access to the pocket, leaving it sufficiently open to be entered by the Nav channel's smaller anchor (Fig. 10C) but far less easily by the KCNQ2/3 anchor (Fig. 10D), thus leading to lower affinity. Removal of the N terminus, achieved experimentally by deletion but perhaps by conformational change and as yet unknown regulatory mechanisms in vivo, increases KCNQ2/3 anchor access without an effect on Nav channel access (Fig. 10E).
FIGURE 10.
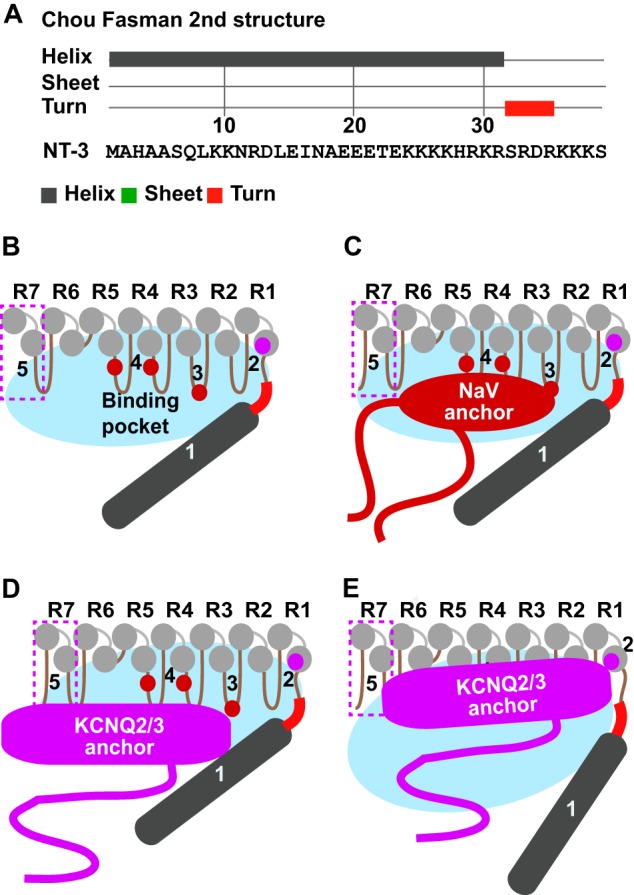
Gated binding pocket model for differential Nav and KCNQ2/3 channel binding to AnkG. A, Chou-Fasman secondary structure algorithm predicts the NT3 N terminus forms an α-helix (gray) followed by a three-four-residue turn (red) linked to downstream ankyrin repeats. B–E, model summarizing results of the current study, also considering results of Wang et al. (40). Five determinants of binding are shown: 1, the N terminus; 2, repeat 1 helix RAAR; 3, hairpin 3 KK; 4, two F residues on hairpins 4 and 5 (40); 5, repeat 7. The non-ankyrin N-terminal (dark gray) together with ankyrin repeats R1–7 (light gray) surround a binding pocket (blue oval). In the preferred “tight” conformation of the pocket (B–D), the smaller Nav anchor domain accesses its binding sites more easily than the larger KCNQ2/s3 anchor domain.
Discussion
Mammalian axons have evolved signaling systems that are reliable, fast-conducting, spatially compact, and energetically efficient (7, 24, 49, 50). This is achieved through genetic and developmental programs enabling protein deployment along the axon that is modular and elaborately polarized, radially and longitudinally. Recent studies have highlighted the central roles played by all three vertebrate ankyrins (ankyrin-G, -B, and -R) in axonal development, structure, and excitability (10, 44, 51, 52). Here we investigate AnkG biochemical interactions needed for the concentration of membrane voltage-gated sodium and KCNQ2/3 potassium channels at initial segments and nodes of Ranvier. We mapped the binding sites within AnkG for KCNQ2/3 and Nav1.2 and characterized molecular determinants for these interactions.
First, we found that CK2 phosphorylation markedly enhanced the ability of KCNQ2/3 C-terminal anchor domains to bind AnkG. Moreover, inhibition of CK2 activity diminished the colocalization of transfected AnkG and NF-Q2 fusion proteins co-expressed in HEK293 cells. CK2 has been shown to be enriched at the AIS, to regulate activity of KCNQ2 through phosphorylation of bound calmodulin, and to phosphorylate Nav channels and thereby enhance their binding to AnkG (34, 35, 37, 53, 54). Together, the new findings further support a “lock in place” mechanism where, after being synthesized in the soma or dendrites, Nav and KCNQ2/3 channels are transported to the axon hillock, admitted to the AIS, and phosphorylated at their anchor domains by local CK2. Once phosphorylated and captured within the diffusion barrier of the AIS/nodal membrane near abundant AnkG (55), exit from the AIS would be slowed even if the dimeric interaction between the ligand and AnkG was relatively weak. This appears to be the case for KCNQ2/3 channels; even under our in vitro conditions, binding is very substoichiometric (unlike Nav1.2; Fig. 6, C and D). Also, although overexpressed NF-Q2C fusion proteins redistribute AnkG to the membrane efficiently (Fig. 2), the association is completely lost during immunoprecipitation experiments from HEK lysates (data not shown). Indeed, with or without phosphorylation by CK2, Nav1.2 binds to AnkG much more strongly than KCNQ3 and KCNQ2. The ratio of Nav and KCNQ peak conductance measured by voltage-clamp at rat sciatic nerve nodes of Ranvier (56) and L5 cortical pyramidal neuron AISs (5) is similar, ∼40:1. Our studies suggest that relatively weak KCNQ2/3-AnkG binding contributes to this conductance density ratio. This ratio is physiologically and energetically advantageous, due to the relatively hyperpolarized voltage dependence and slow kinetics of KCNQ2/3 channel activation. These parameters allow KCNQ2/3 channels to strongly influence excitability in the subthreshold range yet play a diminished role once threshold is passed (5, 56, 57).
Second, we mapped the AnkG subregions binding to the KCNQ2/3 and Nav1.2 anchor domains. We found that the strongest binding is contained within the first through seventh AnkG ankyrin repeats (R1–7), with weak but detectable additional binding elsewhere in MB. Work published by others while our paper was in preparation agrees with those findings (40, 58). However, our mutagenesis of hairpin tips beyond R7 did not reveal any additional critical binding residues (data not shown). In particular, we were unable to find any evidence for KCNQ2/3 or Nav1.2 interaction with hairpins 13, 14, or 15 of AnkG implicated in binding the major cardiac sodium channel, Nav1.5 (41).
By deletion and substitution mutagenesis within R1–7, we identified different regions and key residues needed for Nav and KCNQ2/3 binding. Alanine substitution of two arginine residues on the same face of the R1 front α-helix abolished KCNQ2/3 but not Nav channel binding (Fig. 7, B and C, and Fig. 10, B–E, site 2). Replacing two lysines at the third hairpin tip abolished Nav1.2 binding but only slightly diminished KCNQ2/3 (Fig. 10, B–E, site 3). R7 deletion much reduced KCNQ3 binding but had little effect on Nav1.2 (Fig. 10, B–E, site 5). Of note, Wang et al. (40) recently identified two phenylalanine residues on adjoining hairpin portions of repeats 4 and 5 (Fig. 10, B and E, sites 4) that are critical for binding an unphosphorylated Nav1.2 peptide and slightly reduced binding of an unphosphorylated KCNQ2 peptide (32). These in-solution biochemical studies are helpful guides for interpretation of already published and future structural studies (discussed below).
Third, we provide the first evidence for specific functional roles played by the alternatively spliced, non-ankyrin repeat, AnkG N terminus. We found that two of three alternatively spliced AnkG N termini strongly inhibit binding of KCNQ2/3 but not Nav1.2 anchors (Figs. 6E, 7, B and C, 8, and 9). This was unexpected, as these N termini have to our knowledge not previously attracted any study, although they are very highly conserved and, like KCNQ2/3, are found only in vertebrates (Fig. 9A). Our findings can be summarized in a gated binding pocket model (Fig. 10). It is notable that an ∼80-residue C-terminal domain in KCNQ2 and KCNQ3 is highly conserved across the vertebrates (3, 24). The Nav peptide domain, which is highly conserved and functionally of primary importance for AnkG binding, is much smaller, ∼30 residues (22–24, 34, 43). Although structural information regarding the channel ligands is currently limited (discussed further below), an economical model is that the larger, phosphorylated KCNQ2/3 anchor domain may access the R1–7 binding region less easily than the analogous but smaller phosphorylated Nav anchor.
Additional experiments will seek to further define mechanisms regulating the access of Nav and KCNQ2/3 ligands, their binding sites on AnkG, and whether such access might be regulated dynamically in vivo. Although the Nav and KCNQ2/3 anchor domains evolved in early vertebrate ancestors (basal chordates), the R1–7 repeats, including residues critical for binding, are much more ancient (39, 59). Our findings both support and extend the “evolved fit” model of ankyrin interaction proposed by Bennett and co-workers (11, 12) a quarter century ago. In this model, roles of ankyrin in binding multiple ligands that are already established (in both evolutionary and developmental terms) underlie the high sequence conservation of the MB domain. Nonetheless, innovations arise when new ligands gain anchor motifs that evolve toward optimal affinity for binding to pre-existing AnkG surfaces. Here, the Nav channel DII-III loop evolved the ability to occupy existing binding sites in the R1–6 region, probably in a basal, invertebrate chordate (24). The KCNQ2/3 ancestor gene evolved the ability to share this region 50–100 million years later, in a common ancestor of jawed vertebrates. Many invertebrate ankyrin N termini are short, 8–10 residues (Fig. 9A). The relatively lengthy (21–39 residue), highly conserved non-ankyrin repeat N termini of vertebrates (Fig. 9A) evolved in the same interval as KCNQ channel ankyrin binding. Growing the ankyrin gene in this way allowed for fine-tuning relative access of the two channels to their binding sites while conserving the actual binding surfaces they share.
While our work was in final stages of preparation, Wang et al. (40) reported two crystal structures including the R1 through R7 regions of Ankyrin B fused to two ligands; one of the ligands studied was a portion of the Nav1.2 anchor peptide (32). This first glimpse at the main ankyrin KCNQ/Nav binding region is very informative, most notably revealing how repeats 5 and 6 (encoded by a single, conserved exon) depart from the ankyrin canonical fold while adhering closely to its 33 residue, solenoidal periodicity (Fig. 6, A and B). Our experiments implicate the same binding surface on AnkG shown to engage ligands by Wang et al. (40). However, our CK2 phosphorylation results indicate that portions of Nav1.2 and KCNQ2/3 up to 15–20 residues away from the Nav1.2 peptide studied by Wang et al. (40) are critical for ankyrin recognition. The completed work sets the stage for more refined mutational studies constrained by the known R1–7 structure. Future structural experiments should include the non-ankyrin repeat N terminus as well as larger and untethered portions of the channel anchor domains including CK2 phosphorylation sites. Phosphomimetic mutations at those sites may allow assessing the effect of CK2 phosphorylation on ankyrin-channel interactions.
Our studies highlight conserved vertebrate molecular mechanisms influencing neuronal excitability through precise control of the ratio of Nav and KCNQ2/3 channels on axons. The findings shed light on causes of epilepsy and other neuropsychiatric disease arising from mutations in genes for AnkG and its channel ligands. Identification of the channel binding regions makes possible targeted screening for agents capable of modulating relative channel affinity and, thus, neuronal responsiveness. Treatments that inhibit Nav binding or enhance KCNQ2/3 binding, thereby reducing the Nav/KCNQ ratio at AISs and nodes of Ranvier, are candidate therapies for acute seizures, epilepsy, and mood disorder.
Supplementary Material
Acknowledgments
We thank the members of the Cooper lab for helpful discussions, Angel Lopez for sharing unpublished results, and Baouyen Tran for critical reading of the manuscript. We thank the following investigators for generous gifts of cDNA clones: Vann Bennett (Duke University) for AnkG-270 and neurofascin-186-HA, Thomas Jentsch (Leibniz-Institute for Molecular Pharmacology) for KCNQ2 and KCNQ3, and Robert J. Dunn (McGill University) for Nav1.2.
Note Added in Proof
Supplemental Table S1 was missing from the version of this article that was published on May 21, 2015 as a Paper in Press. Supplemental Table 1 is now included.
Author Contributions—M. X. performed the experiments. E. C. C. and M. X. designed the study, analyzed the data, and wrote the paper.
This work was supported, in whole or in part, by National Institutes of Health Grant RO1 NS049119 (NINDS; to E. C. C. and M. X.) and T32NS043124 (to M. X.). The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Table S1.
- KCNQ
- voltage-gated KCNQ potassium channel
- AnkG
- Ankyrin-G
- Nav
- voltage-gated sodium channel
- Nav II-III
- intracellular loop of Nav1.2 between homology domains II and III
- AIS
- axon initial segment
- MB
- Membrane binding domain
- TBB
- 4,5,6,7-tetrabromobenzotriazole
- TBCA
- 4,5,6,7-tetrabromo-2-azabenzimidazole
- CK2
- protein kinase CK2.
References
- 1. Bean B. P. (2007) The action potential in mammalian central neurons. Nat. Rev. Neurosci. 8, 451–465 [DOI] [PubMed] [Google Scholar]
- 2. Devaux J. J., Kleopa K. A., Cooper E. C., Scherer S. S. (2004) KCNQ2 is a nodal K+ channel. J. Neurosci. 24, 1236–1244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pan Z., Kao T., Horvath Z., Lemos J., Sul J. Y., Cranstoun S. D., Bennett V., Scherer S. S., Cooper E. C. (2006) A common ankyrin-G-based mechanism retains KCNQ and NaV channels at electrically active domains of the axon. J. Neurosci. 26, 2599–2613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schwarz J. R., Glassmeier G., Cooper E. C., Kao T. C., Nodera H., Tabuena D., Kaji R., Bostock H. (2006) KCNQ channels mediate IKs, a slow K+ current regulating excitability in the rat node of Ranvier. J. Physiol. 573, 17–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Battefeld A., Tran B. T., Gavrilis J., Cooper E. C., Kole M. H. (2014) Heteromeric Kv7.2/7.3 channels differentially regulate action potential initiation and conduction in neocortical myelinated axons. J. Neurosci. 34, 3719–3732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhou D., Lambert S., Malen P. L., Carpenter S., Boland L. M., Bennett V. (1998) AnkyrinG is required for clustering of voltage-gated Na channels at axon initial segments and for normal action potential firing. J. Cell Biol. 143, 1295–1304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rasband M. N. (2010) The axon initial segment and the maintenance of neuronal polarity. Nat. Rev. Neurosci. 11, 552–562 [DOI] [PubMed] [Google Scholar]
- 8. Wimmer V. C., Reid C. A., So E. Y., Berkovic S. F., Petrou S. (2010) Axon initial segment dysfunction in epilepsy. J. Physiol. 588, 1829–1840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hedstrom K. L., Ogawa Y., Rasband M. N. (2008) AnkyrinG is required for maintenance of the axon initial segment and neuronal polarity. J. Cell Biol. 183, 635–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Galiano M. R., Jha S., Ho T. S., Zhang C., Ogawa Y., Chang K. J., Stankewich M. C., Mohler P. J., Rasband M. N. (2012) A distal axonal cytoskeleton forms an intra-axonal boundary that controls axon initial segment assembly. Cell 149, 1125–1139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Michaely P., Bennett V. (1995) Mechanism for binding site diversity on ankyrin: comparison of binding sites on ankyrin for neurofascin and the Cl−/HCO3a anion exchanger. J. Biol. Chem. 270, 31298–31302 [DOI] [PubMed] [Google Scholar]
- 12. Davis J. Q., Bennett V. (1990) The anion exchanger and Na+K+-ATPase interact with distinct sites on ankyrin in in vitro assays. J. Biol. Chem. 265, 17252–17256 [PubMed] [Google Scholar]
- 13. Li J., Mahajan A., Tsai M. D. (2006) Ankyrin repeat: a unique motif mediating protein-protein interactions. Biochemistry 45, 15168–15178 [DOI] [PubMed] [Google Scholar]
- 14. Michaely P., Tomchick D. R., Machius M., Anderson R. G. (2002) Crystal structure of a 12 ANK repeat stack from human ankyrinR. EMBO J. 21, 6387–6396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ferreira M. A., O'Donovan M. C., Meng Y. A., Jones I. R., Ruderfer D. M., Jones L., Fan J., Kirov G., Perlis R. H., Green E. K., Smoller J. W., Grozeva D., Stone J., Nikolov I., Chambert K., Hamshere M. L., Nimgaonkar V. L., Moskvina V., Thase M. E., Caesar S., Sachs G. S., Franklin J., Gordon-Smith K., Ardlie K. G., Gabriel S. B., Fraser C., Blumenstiel B., Defelice M., Breen G., Gill M., Morris D. W., Elkin A., Muir W. J., McGhee K. A., Williamson R., MacIntyre D. J., MacLean A. W., St C. D., Robinson M., Van Beck M., Pereira A. C., Kandaswamy R., McQuillin A., Collier D. A., Bass N. J., Young A. H., Lawrence J., Ferrier I. N., Anjorin A., Farmer A., Curtis D., Scolnick E. M., McGuffin P., Daly M. J., Corvin A. P., Holmans P. A., Blackwood D. H., Gurling H. M., Owen M. J., Purcell S. M., Sklar P., Craddock N., and Wellcome Trust Case Control, C. (2008) Collaborative genome-wide association analysis supports a role for ANK3 and CACNA1C in bipolar disorder. Nat. Genet. 40, 1056–1058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bi C., Wu J., Jiang T., Liu Q., Cai W., Yu P., Cai T., Zhao M., Jiang Y. H., Sun Z. S. (2012) Mutations of ANK3 identified by exome sequencing are associated with autism susceptibility. Hum. Mutat. 33, 1635–1638 [DOI] [PubMed] [Google Scholar]
- 17. Weckhuysen S., Mandelstam S., Suls A., Audenaert D., Deconinck T., Claes L. R., Deprez L., Smets K., Hristova D., Yordanova I., Jordanova A., Ceulemans B., Jansen A., Hasaerts D., Roelens F., Lagae L., Yendle S., Stanley T., Heron S. E., Mulley J. C., Berkovic S. F., Scheffer I. E., de Jonghe P. (2012) KCNQ2 encephalopathy: emerging phenotype of a neonatal epileptic encephalopathy. Ann. Neurol. 71, 15–25 [DOI] [PubMed] [Google Scholar]
- 18. Weckhuysen S., Ivanovic V., Hendrickx R., Van Coster R., Hjalgrim H., Møller R. S., Grønborg S., Schoonjans A. S., Ceulemans B., Heavin S. B., Eltze C., Horvath R., Casara G., Pisano T., Giordano L., Rostasy K., Haberlandt E., Albrecht B., Bevot A., Benkel I., Syrbe S., Sheidley B., Guerrini R., Poduri A., Lemke J. R., Mandelstam S., Scheffer I., Angriman M., Striano P., Marini C., Suls A., De Jonghe P., and KCNQ2 Study Group (2013) Extending the KCNQ2 encephalopathy spectrum: clinical and neuroimaging findings in 17 patients. Neurology 81, 1697–1703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kato M., Yamagata T., Kubota M., Arai H., Yamashita S., Nakagawa T., Fujii T., Sugai K., Imai K., Uster T., Chitayat D., Weiss S., Kashii H., Kusano R., Matsumoto A., Nakamura K., Oyazato Y., Maeno M., Nishiyama K., Kodera H., Nakashima M., Tsurusaki Y., Miyake N., Saito K., Hayasaka K., Matsumoto N., Saitsu H. (2013) Clinical spectrum of early onset epileptic encephalopathies caused by KCNQ2 mutation. Epilepsia 54, 1282–1287 [DOI] [PubMed] [Google Scholar]
- 20. Cooper E. C., Jan L. Y. (2003) M-channels: neurological diseases, neuromodulation, and drug development. Arch. Neurol. 60, 496–500 [DOI] [PubMed] [Google Scholar]
- 21. Peters H. C., Hu H., Pongs O., Storm J. F., Isbrandt D. (2005) Conditional transgenic suppression of M channels in mouse brain reveals functions in neuronal excitability, resonance, and behavior. Nat. Neurosci. 8, 51–60 [DOI] [PubMed] [Google Scholar]
- 22. Garrido J. J., Giraud P., Carlier E., Fernandes F., Moussif A., Fache M. P., Debanne D., Dargent B. (2003) A targeting motif involved in sodium channel clustering at the axonal initial segment. Science 300, 2091–2094 [DOI] [PubMed] [Google Scholar]
- 23. Lemaillet G., Walker B., Lambert S. (2003) Identification of a conserved ankyrin-binding motif in the family of sodium channel α subunits. J. Biol. Chem. 278, 27333–27339 [DOI] [PubMed] [Google Scholar]
- 24. Hill A. S., Nishino A., Nakajo K., Zhang G., Fineman J. R., Selzer M. E., Okamura Y., Cooper E. C. (2008) Ion channel clustering at the axon initial segment and node of Ranvier evolved sequentially in early chordates. PLoS Genet. 4, e1000317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang X., Bennett V. (1998) Restriction of 480/270-kD ankyrin G to axon proximal segments requires multiple ankyrin G-specific domains. J. Cell Biol. 142, 1571–1581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Biervert C., Schroeder B. C., Kubisch C., Berkovic S. F., Propping P., Jentsch T. J., Steinlein O. K. (1998) A potassium channel mutation in neonatal human epilepsy. Science 279, 403–406 [DOI] [PubMed] [Google Scholar]
- 27. Schroeder B. C., Kubisch C., Stein V., Jentsch T. J. (1998) Moderate loss of function of cyclic-AMP-modulated KCNQ2/KCNQ3 K+ channels causes epilepsy. Nature 396, 687–690 [DOI] [PubMed] [Google Scholar]
- 28. Auld V. J., Goldin A. L., Krafte D. S., Marshall J., Dunn J. M., Catterall W. A., Lester H. A., Davidson N., Dunn R. J. (1988) A rat brain Na+ channel α subunit with novel gating properties. Neuron 1, 449–461 [DOI] [PubMed] [Google Scholar]
- 29. Cunha S. R., Mohler P. J. (2008) Obscurin targets ankyrin-B and protein phosphatase 2A to the cardiac M-line. J. Biol. Chem. 283, 31968–31980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang X., Davis J. Q., Carpenter S., Bennett V. (1998) Structural requirements for association of neurofascin with ankyrin. J. Biol. Chem. 273, 30785–30794 [DOI] [PubMed] [Google Scholar]
- 31. Dunn K. W., Kamocka M. M., McDonald J. H. (2011) A practical guide to evaluating colocalization in biological microscopy. Am. J. Physiol. Cell Physiol. 300, C723–C742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Barlow A. L., Macleod A., Noppen S., Sanderson J., Guérin C. J. (2010) Colocalization analysis in fluorescence micrographs: verification of a more accurate calculation of pearson's correlation coefficient. Microsc. Microanal. 16, 710–724 [DOI] [PubMed] [Google Scholar]
- 33. Adler J., Parmryd I. (2010) Quantifying colocalization by correlation: the Pearson correlation coefficient is superior to the Mander's overlap coefficient. Cytometry A 77, 733–742 [DOI] [PubMed] [Google Scholar]
- 34. Bréchet A., Fache M. P., Brachet A., Ferracci G., Baude A., Irondelle M., Pereira S., Leterrier C., Dargent B. (2008) Protein kinase CK2 contributes to the organization of sodium channels in axonal membranes by regulating their interactions with ankyrin G. J. Cell Biol. 183, 1101–1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hien Y. E., Montersino A., Castets F., Leterrier C., Filhol O., Vacher H., Dargent B. (2014) CK2 accumulation at the axon initial segment depends on sodium channel Nav1. FEBS Lett. 588, 3403–3408 [DOI] [PubMed] [Google Scholar]
- 36. Howard R. J., Clark K. A., Holton J. M., Minor D. L., Jr. (2007) Structural insight into KCNQ (Kv7) channel assembly and channelopathy. Neuron 53, 663–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kang S., Xu M., Cooper E. C., Hoshi N. (2014) Channel-anchored protein kinase CK2 and protein phosphatase 1 reciprocally regulate KCNQ2-containing M-channels via phosphorylation of calmodulin. J. Biol. Chem. 289, 11536–11544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pagano M. A., Poletto G., Di Maira G., Cozza G., Ruzzene M., Sarno S., Bain J., Elliott M., Moro S., Zagotto G., Meggio F., Pinna L. A. (2007) Tetrabromocinnamic acid (TBCA) and related compounds represent a new class of specific protein kinase CK2 inhibitors. Chembiochem 8, 129–139 [DOI] [PubMed] [Google Scholar]
- 39. Cai X., Zhang Y. (2006) Molecular evolution of the ankyrin gene family. Mol. Biol. Evol. 23, 550–558 [DOI] [PubMed] [Google Scholar]
- 40. Wang C., Wei Z., Chen K., Ye F., Yu C., Bennett V., Zhang M. (2014) Structural basis of diverse membrane target recognitions by ankyrins. Elife 10.7554/eLife.04353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lowe J. S., Palygin O., Bhasin N., Hund T. J., Boyden P. A., Shibata E., Anderson M. E., Mohler P. J. (2008) Voltage-gated Nav channel targeting in the heart requires an ankyrin-G dependent cellular pathway. J. Cell Biol. 180, 173–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Abdi K. M., Mohler P. J., Davis J. Q., Bennett V. (2006) Isoform specificity of ankyrin-B: a site in the divergent C-terminal domain is required for intramolecular association. J. Biol. Chem. 281, 5741–5749 [DOI] [PubMed] [Google Scholar]
- 43. Gasser A., Ho T. S., Cheng X., Chang K. J., Waxman S. G., Rasband M. N., Dib-Hajj S. D. (2012) An ankyrinG-binding motif is necessary and sufficient for targeting Nav1.6 sodium channels to axon initial segments and nodes of Ranvier. J. Neurosci. 32, 7232–7243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ho T. S., Zollinger D. R., Chang K. J., Xu M., Cooper E. C., Stankewich M. C., Bennett V., Rasband M. N. (2014) A hierarchy of ankyrin-spectrin complexes clusters sodium channels at nodes of Ranvier. Nat. Neurosci. 17, 1664–1672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rueckert E. H., Barker D., Ruderfer D., Bergen S. E., O'Dushlaine C., Luce C. J., Sheridan S. D., Theriault K. M., Chambert K., Moran J., Purcell S. M., Madison J. M., Haggarty S. J., Sklar P. (2013) Cis-acting regulation of brain-specific ANK3 gene expression by a genetic variant associated with bipolar disorder. Mol. Psychiatry 18, 922–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cunningham F., Amode M. R., Barrell D., Beal K., Billis K., Brent S., Carvalho-Silva D., Clapham P., Coates G., Fitzgerald S., Gil L., Girón C. G., Gordon L., Hourlier T., Hunt S. E., Janacek S. H., Johnson N., Juettemann T., Kähäri A. K., Keenan S., Martin F. J., Maurel T., McLaren W., Murphy D. N., Nag R., Overduin B., Parker A., Patricio M., Perry E., Pignatelli M., Riat H. S., Sheppard D., Taylor K., Thormann A., Vullo A., Wilder S. P., Zadissa A., Aken B. L., Birney E., Harrow J., Kinsella R., Muffato M., Ruffier M., Searle S. M., Spudich G., Trevanion S. J., Yates A., Zerbino D. R., Flicek P. (2015) Ensembl 2015. Nucleic Acids Res. 43, D662–D669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cole C., Barber J. D., Barton G. J. (2008) The Jpred 3 secondary structure prediction server. Nucleic Acids Res. 36, W197–W201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Buchan D. W., Minneci F., Nugent T. C., Bryson K., Jones D. T. (2013) Scalable web services for the PSIPRED Protein Analysis Workbench. Nucleic Acids Res. 41, W349–W357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zakon H. H. (2012) Adaptive evolution of voltage-gated sodium channels: the first 800 million years. Proc. Natl. Acad. Sci. U.S.A. 109, 10619–10625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hallermann S., de Kock C. P., Stuart G. J., Kole M. H. (2012) State and location dependence of action potential metabolic cost in cortical pyramidal neurons. Nat. Neurosci. 15, 1007–1014 [DOI] [PubMed] [Google Scholar]
- 51. Chang K. J., Zollinger D. R., Susuki K., Sherman D. L., Makara M. A., Brophy P. J., Cooper E. C., Bennett V., Mohler P. J., Rasband M. N. (2014) Glial ankyrins facilitate paranodal axoglial junction assembly. Nat. Neurosci. 17, 1673–1681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Xu K., Zhong G., Zhuang X. (2013) Actin, spectrin, and associated proteins form a periodic cytoskeletal structure in axons. Science 339, 452–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sanchez-Ponce D., Muñoz A., Garrido J. J. (2011) Casein kinase 2 and microtubules control axon initial segment formation. Mol. Cell. Neurosci. 46, 222–234 [DOI] [PubMed] [Google Scholar]
- 54. Brachet A., Leterrier C., Irondelle M., Fache M. P., Racine V., Sibarita J. B., Choquet D., Dargent B. (2010) Ankyrin G restricts ion channel diffusion at the axonal initial segment before the establishment of the diffusion barrier. J. Cell Biol. 191, 383–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Nakada C., Ritchie K., Oba Y., Nakamura M., Hotta Y., Iino R., Kasai R. S., Yamaguchi K., Fujiwara T., Kusumi A. (2003) Accumulation of anchored proteins forms membrane diffusion barriers during neuronal polarization. Nat. Cell Biol. 5, 626–632 [DOI] [PubMed] [Google Scholar]
- 56. Röper J., Schwarz J. R. (1989) Heterogeneous distribution of fast and slow potassium channels in myelinated rat nerve fibres. J. Physiol. 416, 93–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Shah M. M., Migliore M., Valencia I., Cooper E. C., Brown D. A. (2008) Functional significance of axonal Kv7 channels in hippocampal pyramidal neurons. Proc. Natl. Acad. Sci. U.S.A. 105, 7869–7874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Barry J., Gu Y., Jukkola P., O'Neill B., Gu H., Mohler P. J., Rajamani K. T., Gu C. (2014) Ankyrin-G directly binds to kinesin-1 to transport voltage-gated na channels into axons. Dev. Cell 28, 117–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bennett V., Baines A. J. (2001) Spectrin and ankyrin-based pathways: metazoan inventions for integrating cells into tissues. Physiol. Rev. 81, 1353–1392 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.