

Background: Caveolae are membrane domains that have been shown to enhance Gαq signals.
Results: Osmotic stress deforms caveolae, reduces the association of Gαq, and reduces Gαq-mediated Ca2+ release.
Conclusion: Mechanical deformation of caveolae weakens Ca2+ responses through changes in Gαq interactions.
Significance: These studies connect the mechanical and signaling functions of caveolae.
Keywords: calcium, caveolae, cell signaling, fluorescence, osmotic swelling, G proteins
Abstract
Caveolae are membrane invaginations that can sequester various signaling proteins. Caveolae have been shown to provide mechanical strength to cells by flattening to accommodate increased volume when cells are subjected to hypo-osmotic stress. We have previously found that caveolin, the main structural component of caveolae, specifically binds Gαq and stabilizes its activation state resulting in an enhanced Ca2+ signal upon activation. Here, we show that osmotic stress caused by decreasing the osmolarity in half reversibly changes the configuration of caveolae without releasing a significant portion of caveolin molecules. This change in configuration due to flattening leads to a loss in Cav1-Gαq association. This loss in Gαq/Cav1 association due to osmotic stress results in a significant reduction of Gαq/phospholipase Cβ-mediated Ca2+ signals. This reduced Ca2+ response is also seen when caveolae are reduced by treatment with siRNA(Cav1) or by dissolving them by methyl-β-cyclodextran. No change in Ca2+ release with osmotic swelling can be seen when growth factor pathways are activated. Taken together, these results connect the mechanical deformation of caveolae to Gαq-mediated Ca2+ signals.
Introduction
Caveolae are flask-shaped, protein-dense invaginations on the plasma membrane of many mammalian cells. Caveola domains are formed by the aggregation of caveolin-1 or caveolin-3 (Cav1 or Cav3), along with cholesterol and other proteins (1–3). The “U”-shaped and multiply palmitoylated Cav1/3 proteins reside in the inner leaflet of the plasma membrane, and the association of ∼144 Cav1/3 molecules (4), along with a cofactor called cavin (5) and cholesterol, induces inward curvature giving the domains their distinct shape.
The function(s) of caveolae have been the subject of numerous studies. Cav1 and Cav3 knock-out mice show cardiac hypertrophy (6–8), and caveola has been linked to some forms of muscular dystrophy (9). On the cellular level, caveolae are thought to play a role in endocytosis and trafficking (10), although this has been disputed (11). Caveolae can also serve as a platform to organize and corral signaling molecules such as the ones involved in nitric oxide production (8, 12). Early studies of caveolae postulated that they can play a role in mechanosensing, and the high density of caveolae in cells that undergo mechanical stress supported this idea. Recently, Sinha et al. (13) found that caveolae passively act as mechanical sensors by buffering stress induced by osmotic swelling independent of actin dynamics. Specifically, their studies show that increased osmotic stress results in the reversible flattening of caveolae to accommodate increased cell volume. Accompanying this flattening is a reduction in Cav1/cavin interactions and the release of Cav1 molecules into the plasma membrane.
Our laboratory has investigated the role played by caveolae in signaling through the Gαq-PLCβ2 (14–17). PLCβ is the main effector of Gαq, and its activation catalyzes the hydrolysis of phosphatidylinositol 4,5-bisphosphate to generate diacylglycerol and 1,4,5-trisphophate (18–20). This latter molecule diffuses to the endoplasmic reticulum to open Ca2+ stores resulting in an increase in intracellular Ca2. Unlike other types of G protein subunits, Gαq binds to the scaffold domain of Cav1 and Cav3, and this binding is strengthened when Gαq is activated (14). The association site between caveolin and Gαq differs from Gαq and PLCβ, and so Gαq continues to signal through PLCβ when bound to caveolae. The interaction between Cav proteins and Gαq has the effect of targeting Gαq and its associated receptors to caveola domains (16). Because the interactions between Cav1 and Gαq are strengthened upon activation, localization of Gαq to caveolae can stabilize its activated state and enhance Ca2+ signals (14–16).
The effect of caveola deformation on its ability to regulate Gαq signals is unclear. On the one hand, it is possible that caveola deformation better exposes the Gαq-binding site on Cav proteins, but on the other hand, it is possible that caveola deformation destabilizes Gαq-Cav contacts. Here, we first characterize the effect of relatively mild hypo-osmotic stress on the properties of caveolae in a cultured muscle cell line. Then, we show how this stress disrupts Gαq/Cav interactions and reduces the resulting Ca2+ signals. These studies show that mechanical stress can impact Ca2+ signals in cells by disrupting Gαq/Cav1 association.
Experimental Procedures
Materials
Rat aortic smooth muscle (A10) cells were purchased from the ATCC. Adult ventricular canine cardiomyocytes were gifts from Dr. Ira S. Cohen (Stony Brook University). mCherry-Cav1, eYFP-Cav1, and eCFP-Cav1 were constructed from canine Cav1-eGFP by excising it as a BamHI fragment and subcloning it into the same sites in pmCherry-C1, pEYFP-C1, and pECFP-C1 (Clontech) as described previously (16). eCFP-Gαq DNA was a gift from Dr. Catherine Berlot (Geisinger Clinic). Primary antibodies to PLCβ1 (mouse), PLCβ3 (rabbit), PLCβ1 (goat), Gαq (rabbit or goat), and Cav1 (rabbit) were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Anti-Cav3 (mouse) was from BD Biosciences. The YFP-tagged membrane marker construct was purchased from Clontech. Secondary antibodies Alexa-Fluor-488-labeled anti-rabbit or anti-mouse and Alexa-Fluor-647 anti-rabbit, anti-mouse, or anti-goat were purchased from Invitrogen.
Cell Studies
A10 cells were maintained in Dulbecco's modified Eagle's medium (DMEM, GIBCO catalog no. 11965, 320–355 mosm) supplemented with 1 mm sodium pyruvate,10% fetal bovine serum, 50 units/ml penicillin, and 50 μg/ml streptomycin sulfate. Canine left ventricular myocytes were maintained in M199 medium supplemented with 15% fetal bovine serum, 1% streptomycin, and 0.5% gentamicin. All cells were maintained at 37 °C in a humidified atmosphere of 5% CO2. Cells were transfected with eGFP-Cav1 and mCherry-Cav1 vectors (1 μg/0.5 × 106 cells in 35-mm dish) or other fluorescent-tagged proteins as indicated using LipofectamineTM 2000 transfection reagent (Invitrogen) according to the manufacturer's protocol. For measurements, cells were transferred to Leibovitz 15 media, 300 mosm, for viewing. Cells were treated by 150 mosm (1:1 dilution) hypo-osmotic shock for 3–5 min at room temperature or treated by 10 mm methyl-β-cyclodextran (mβCD) for 30 min at room temperature before measurements.
Endogenous Cav1 was down-regulated by treating the cells with siRNA(Cav1) from Dharmacon, Inc., according to the manufacturer's instructions. The efficiency of down-regulation was ∼62.5% as estimated by immunofluorescence using the anti-Cav1 antibody bound to Alexa-Fluor-647-conjugated secondary antibody (n = 96).
Colocalization Studies
A10 cells were seeded onto glass bottom dishes (MatTek Corp.) 1 day before immunostaining. The cells were washed and fixed with 4% paraformaldehyde for 1 h. The fixed cells were washed three times with PBS and incubated with 0.2% NP-40 in PBS for 5 min and then blocked in PBS containing 4% goat or horse serum for 1 h. The cells were incubated with primary antibody Gαq, Cav1, Cav3, PLCβ1, PLCβ2, and PLCβ1 at 1:200 to 1:500 dilution with 1% goat or horse serum in PBS for 1 h. Cells were then washed three times for 10 min with TBS (150 mm NaCl, 25 mm Tris, pH 7.6), followed by addition of Alexa-Fluor-488-labeled anti-rabbit or Alexa-Fluor-647 anti-mouse secondary antibody diluted to 1:1000 with 1% goat or horse serum in PBS and subsequent incubation at 37 °C for 1 h, and washed three times with TBS. Cells were viewed in TBS buffer on a laser-scanning confocal Olympus Fluoview1000 microscope equipped with a 488-nm argon ion laser for excitation for Alexa-Fluor-488 and a 633-nm HeNe laser for Alexa-Fluor-647. Background samples were treated identically but did not contain primary antibody. Colocalization analysis was performed using the MacBiophotonics version of ImageJ.
FRET Imaging
Fluorescence resonance energy transfer was performed with an Olympus Fluoview1000 instrument on A10 cells co-expressing eGFP- or mCherry-tagged Cav1 proteins as described (16). eGFP and mCherry were excited using 488 argon ion laser line and 543 nm HeNe(G) laser, respectively, and 480–495- and 543–660-nm bandpass filters to collect emission images, respectively. FRET imaging of eCFP-Gαq and eYFP-Cav1 was carried out as described previously (16). FRET efficiencies were calculated using the Olympus Fluoview software whose algorithm calculates FRET by sensitized emission after correcting for spectral bleed through using images of control cells expressing only donor or acceptor proteins with the same intensity distributions as the sample (16). We note that the FRET efficiency values did not change significantly over a 10-fold range of acceptor/donor intensity ratios.
FRAP Measurements
A10 cells were seeded on glass-bottom dishes (MatTek) for 24 h. Cav1 antibody (sc-897, Santa Cruz Biotechnology) was incubated with Alexa-Fluor-488 secondary antibody and then diluted with needle buffer (140 mm KCl and K2HPO4, pH 7.4) to 8 ng/μl. Anti-Cav1 reacted with Alexa-Fluor-488 secondary antibody was microinjected into A10 cells using an InjectMan NI2 with a FemtoJet pump from Eppendorf mounted on an Axiovert 200 M (Carl Zeiss) equipped with a long working distance 40× phase 2 objective. Samples were microinjected into the cytoplasm using an injection pressure of 45 hectopascals and a compensation pressure of 22 hectopascals. After microinjection, cells were allowed to recover before subjecting them to either normal or hypo-osmotic conditions.
FRAP studies were carried out by setting the focal plane on the membrane and selecting a circular region for bleaching (17). The region of interest was illuminated with a high intensity (100% transmittivity) 488-nm argon ion laser for 500 ms, and the recovery was observed for 225 s under low intensity illumination (1% transmittivity). Cells that exhibited cell movement or excessive photobleaching were not considered for the analysis. The corrected fluorescence recovery was fitted as described previously, using a one component exponential fit (17).
Ca2+ Measurements
A10 cells (1 × 107) were incubated with 1 μm Fura-2-AM in Hanks' balanced salt solution (HBSS, Gibco 14025) with 1% BSA and 5 mm glucose. After 30 min of incubation at room temperature, the cells were washed twice with PBS, harvested, and adjusted to 0.5 × 106/ml with HBSS2+, and fluorescence measurements were taken (16). Intracellular calcium release was measured under normal or swollen conditions or with 10 mm mβ-cyclodextran treatment for 30 min at room temperature. Cells were stimulated by 5 μm carbachol or a growth factor mixture consisting of hepatocyte growth factor (80 ng/ml), platelet-derived growth factor (80 ng/ml), and epidermal growth factor (20 nm). The osmolarity was lowered from 300 to 150 mosm by diluting the media by 50% with deionized water for 5 min at room temperature. Reversal of hypo-osmotic stress was carried out by changing the media to HBSS2+ and allowing recovery for 3–5 min until the fluorescence base line was stable and then stimulating the cells with 5 μm carbachol. Where indicated, 4 mm EGTA was added to the HBSS2+ medium immediately before stimulation. Calcium release in A10 cells treated with siRNA(Cav1) was measured on a single cell basis (15) on an Olympus Fluoview1000 microscope by labeling the cells with 5 μm Calcium Green, as described previously (16), to minimize the amount of reagents used. Briefly, untreated or siRNA(Cav1)-treated cells were loaded with 5 μm Calcium Green (Invitrogen) under different osmotic conditions, and the change in fluorescence was monitored on an Olympus confocal Fluoview1000 microscope. Changes in cell intensity were analyzed by ImageJ. The change in fluorescent ratio (∼1.25) was scaled to calcium release for wild type cell suspensions.
Results
Caveola Stabilizes Cells from Increased Membrane Tension due to Hypo-osmotic Osmotic Pressure
Although Sinha et al. (13) used mainly the endothelial cells, for these studies, we used rat aortic smooth muscle (A10) cells that have a robust Gαq/PLCβ signaling system and are rich in caveolae. We first subjected A10 cells to osmotic swelling by stepwise addition of water to lower the osmolarity from 300 to 150 mosm. This stepwise addition allowed for cell recovery mechanisms that involve ion movement to relieve osmotic pressure (21). In Fig. 1, we show sample images of untreated A10 cells along with box plots showing the ratio of cell diameters under osmotic stress versus nonstressed conditions. We found that we could dilute the medium until the osmolarity reached 90 mosm before the cells showed a significant increase in size as measured by phase contrast microscopy. In many cases, further dilution to 30 mosm resulted in cell rupture. Treating the cells with siRNA(Cav1) for 24 h reduced the amount of dilution where increased volume is observed to 140 mosm compared with 90 mosm in control cells (Fig. 1). This stability of cell size with reduced osmolarity is consistent with flattening of caveolae to accommodate a larger cell volume.
FIGURE 1.

Changes in cell volume with dilution. Images of cells placed in Leibovitz's medium (L-15) and then diluted with deionized water (1:9 dilution for 30 mosm, 1:2.3 dilution for 90 mosm, and 1:1 for 150 mosm hypo-osmotic shock) (left) are shown. Cells were viewed after 5 min at room temperature using a ×40 objective. Right, box plots showing changes in the ratio of cells at different osmotic strength divided by those under hypo-osmotic conditions where n = 15–32.
We repeated this study using A10 cells that have been treated with methyl-β-cyclodextran (mβCD). mβCD will chelate cholesterol that is needed to maintain the integrity of caveola domains, and treatment with mβCD results in caveola flattening and dissolution. We found that cells treated with mβCD did not show an increased volume (Fig. 1). This behavior suggests that even though mβCD perturbs the structure of caveolae, it still provides some mechanical strength to the cells.
Osmotic Pressure Disrupts Caveola Domains
Previous studies suggested that the flattening of caveolae brought about by hypo-osmotic conditions is accompanied by the release of Cav1 molecules (13). We tested whether this is the case for A10 cells by measuring the degree of Förster resonance energy transfer (FRET) between eGFP-Cav1 donors and mCherry-Cav1 acceptors when cells are subjected to hypo-osmotic conditions. As found previously (16), the expressed proteins had a cytosolic population that presumably was from Golgi- and plasma membrane-localized molecules (Fig. 2A). The fluorescent-Cav1 molecules gave significantly higher whole cell FRET values as compared with negative controls (0.124 ± 0.008, n = 36, versus 0.001 ± 0.002, n = 24). Addition of water to reduce the osmolarity to 150 mosm primarily reduced the FRET from the plasma membrane region to reduce the overall FRET by ∼15% (0.094 ± 0.006, n = 36). This reduction in FRET suggests some degree of disassembly of caveolae under osmotic pressure and is analogous to the behavior seen for Cav1 in mouse lung epithelial cells (13).
FIGURE 2.
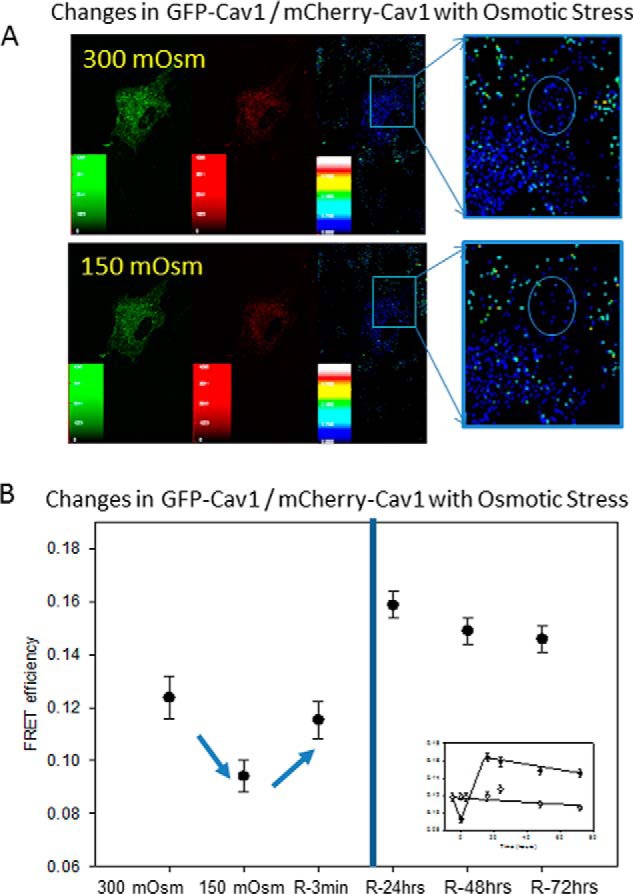
Hypo-osmotic stress reduces FRET between Cav1 molecules. A, FRET between GFP-Cav1 and mCherry-Cav1 in A10 cells under iso-osmotic conditions (upper panels) and 5 min after the osmolarity is reduced to 150 mosm (lower panels). B, changes in FRET before (control) and 5 min after a reduction of osmolarity from 300 to 150 mosm (swollen). Values designated by R refer to FRET measurements taken after the cells were returned to 300 mosm for 3 min and 24, 48, and 72 h (R-3min, R-24 h, R-48 hrs, and R-72 hrs, respectively). The inset shows a comparison of the FRET values for cells at 150 mosm (●) and controls at 300 mosm (○) over the 72-h period. The data are an average of over 36 cells, and S.D. is shown.
We then determined whether the value of Cav1 FRET returns to iso-osmotic values by swelling the cells to 150 mosm for 5 min and then transferring to medium at 300 mosm. The results are shown in Fig. 2B. We found that soon after return to basal conditions, the value of FRET recovered to its original value within error (0.115 ± 0.007, n = 36). However, over the next 24 h, the values of FRET for the samples subjected to hypo-osmotic condition pressure showed a significant increase that was stable over the next 2 days, whereas the FRET values of control samples were unchanged (Fig. 2B). This “recoil” effect shows enhanced Cav1-Cav1 association and suggests a restructuring of caveolae after deformation.
We reasoned that the disruption and disaggregation of caveolae might result in increased mobility as caveolin molecules move out of the relatively immobile caveola domains. We followed changes in caveolae with osmotic pressure by fluorescence recovery after photobleaching (FRAP), which measures diffusion of fluorescent molecules into a region that was bleached by a high intensity laser. To this end, we transfected A10 cells with eGFP-Cav1 and monitored the recovery of fluorescence of control and osmotically swollen cells. First, we monitored the recovery of cells expressing only a small tri-palmitoylated peptide that localizes to the plasma membrane (YFP-MEM, Clontech) under control and osmotic stress conditions. The recoveries under both conditions fit best to a single exponential and were identical to each other suggesting that the fluidity of the membrane was not greatly affected by osmotic stress (Fig. 3A, inset). We then monitored the diffusion of eGFP-Cav1 at 300, 150, and 30 mosm and could not detect movement under any condition (see Fig. 3A). This result suggests that either the flattening of the caveola does not result in large scale dissolution of caveola domains and/or that the dissociated Cav1 molecules are not fully released into the plasma membrane.
FIGURE 3.

Changes in diffusion of Cav1 molecules under osmotic stress. A, FRAP measurements of A10 cells transfected GFP-Cav1 molecules under iso-osmotic (300 mosm) (●) and hypo-osmotic (150 mosm) (○) conditions. We note that the iso-osmotic results are the same as those reported previously (17) and that repeating the studies at 30 mosm gave identical results as the 150 mosm recovery shown in this figure. The inset shows recoveries for YFP-MEM expressing in A10 cells and viewed under identical conditions where the medium was at 300 mosm (●) and 150 mosm (○). B, FRAP studies following the recovery of anti-Cav1 + Alexa-Fluor-488 secondary antibody microinjected into A10 cells at 300 mosm (●) and 150 mosm (○).
To better understand the effect of osmotic stress on caveola structure, we used an alternative approach in which we microinjected anti-Cav1 with bound Alexa-Fluor-488 secondary antibody into the cells and followed its diffusion with osmotic swelling. If the antibody was not bound to its caveola epitope, then it would be freely diffusing in the cytoplasm. However, when the antibody binds to Cav1, its diffusion would be extremely limited.
We microinjected the fluorescent-labeled Cav1 antibody complex into the cytoplasm of A10 cells. At 300 mosm, we found a relatively fast recovery (Fig. 3B). Because the size of the bleach spot encompasses several caveolae, it is unlikely that this diffusion corresponds to movement of the antibody within a single domains. Rather, it is more likely that it is a combination of cytosolic movement of the unbound antibody and movement of individual Cav1 molecules. Subjecting the cells to increased osmotic stress at 150 or 30 mosm causes a significant reduction in antibody diffusion at both osmolarities. We interpret this loss in mobility of the antibody to be due to association of the antibody to Cav1 epitopes that become exposed under osmotic stress. These results correlate well with the idea that swelling causes a restructuring of caveola domains.
Osmotic Pressure Disrupts Caveola Gαq/Cav Interactions
We have previously found that a significant population of Gαq resides in caveola domains where they interact specifically with the conserved scaffold domain of Cav1 and Cav3 (16). Therefore, it is possible that Gαq molecules are released from caveolae under osmotic stress. We tested this possibility by monitoring changes in colocalization between Cav1 and Gαq under hypo-osmotic stress. We found a significant reduction in colocalization between the proteins (Fig. 4A). We note that we found a similar reduction in colocalization in adult canine cardiac ventricular myocytes (data not shown) which is a primary muscle cell line where we have shown that Gαq-Cav3 impacts Ca2+ (15).
FIGURE 4.

Osmotic stress reduces association between Gαq and Cav1. A, coimmunofluorescence studies showing the colocalization between Cav1 (green) and Gαq (red) at iso-osmotic (300 mosm) and hypo-osmotic (150 mosm) conditions in A10 cells (left). The compiled data were from three separate experiments where n = 17–33 for each experiment (right). B, FRET between eCFP-Gαq and eYFP-Cav1 with osmotic stress where n = 30.
We supported the colocalization studies by FRET in A10 cells transfected with eCFP-Gαq and eYFP-Cav1. We found that reducing the osmolarity from 300 to 150 mosm significantly (i.e. p ≤ 0.001) reduces the amount of FRET from 0.078 ± 0.005 S.E. (n = 30 cells with 3 points sampled in each) to 0.054 ± 0.004 S.E. (n = 30 with 3 points sampled in each) (Fig. 4B). Together with the colocalization studies, these results suggest that Gαq/Cav1 interactions are altered under osmotic stress.
Osmotic Pressure Reversibility Attenuates Calcium Signals
Because caveolae have been found to stabilize the activated state of Gαq and enhance Ca2+ signals, we determined whether these signals are diminished under osmotic swelling because the studies above indicate a reduction in Cav and Gαq association. We measured the release of Ca2+ with carbachol stimulation of A10 cell suspensions loaded with the fluorescent-calcium indicator, Fura-2. Under normal osmolarity (300 mosm), these cells showed a rapid and large increase in intracellular calcium (∼220 nm). This curve can be fit to a skewed Gaussian with an initial slope of 20 nm/s (Fig. 5A). However, when the osmolarity is reduced to 150 mosm, the net release of Ca2+ with stimulation is significantly reduced from 220 to ∼140 nm (Fig. 5). Additionally, the initial slope of the curve is lowered by almost a factor of 4 making the shape of the curve more symmetric and delaying the maximum release of Ca2+ over 20 s. When the cells were returned to normal osmolarity, there was a partial recovery (Fig. 5A); the amount of Ca2+ released was somewhat lower (∼190 nm) and occurred ∼10 s slower than controls.
FIGURE 5.

Osmotic stress reduces Ca2+ release upon Gαq stimulation. A, A10 cells in suspension (0.5 × 106 cells/ml) were loaded with Fura-2-AM, and the change in signal in response to 5 μm carbachol was measured at 300 mosm (○), 150 mosm (■), and 5 min after hypo-osmotic cells were returned to 300 mosm for 3 min (gray diamonds), where the data are an average of 9–16 independent experiments and S.D. is shown. These studies carried out at initial physiological Ca2+ levels. Ca2+ levels were determined following the procedure of Tsien and co-workers (30). B, calcium response to carbachol for A10 cells at 300 and 150 mosm taken in the absence of extracellular Ca2+ where n = 15 and S.D. is shown. C, comparison of the Ca2+ increase with stimulation by 5 μm carbachol of control cells (○) and cells treated with mβCD (●) at 300 mosm, where n = 9 and S.D. is shown. D, A10 cells at 300 and 150 mosm stimulated by a mixture containing 80 ng/ml human growth factor, 80 ng/ml PDGF, and 20 nm EGF. Data are an average of 7–9 independent experiments, and S.D. is shown. The inset shows the time range where the calcium increase with carbachol addition occurs. E, single cell calcium release taken by fluorescence microscopy of untreated and siRNA(Cav1)-treated A10 cells loaded with Calcium Green and stimulated with carbachol at physiological and hypo-osmotic conditions where the calcium concentrations were estimated by scaling the ratio of the fluorescence response to solution calcium measurements, and where n = 15–19 and S.D. is shown.
The above studies were carried in medium at normal physiological Ca2+ concentrations. Thus, the increase in Ca2+ upon Gαq/PLCβ stimulation is a combination of Ca2+ release from intracellular stores and movement of extracellular Ca2+ into the cytosol from opening of Ca2+-activated Ca2+ channels. To distinguish between these possibilities, we repeated the studies in the absence of extracellular Ca2+. Because the cells are not stable for extended periods without extracellular Ca2+, we carried out measurements immediately after removing the Ca2+. We found an identical decrease in Ca2+ release with reduced osmolarity as under physiological Ca2+ concentrations (Fig. 5B). However, we note that the delay in Ca2+ release is no longer present suggesting that the delay seen with osmotic stress may be due to changes in downstream extracellular Ca2+ entry.
To support the idea that the mechanical deformation of caveolae due to hypo-osmotic conditions reduces Gαq-mediated Ca2+ signals, we measured the Ca2+ release with carbachol stimulation of untreated A10 cells and cells whose caveolae have been deformed by mβCD treatment (22, 23). These cells showed a greatly reduced calcium response that was similar but weaker than the responses seen for hypo-osmotic stress (Fig. 5C). Although the trend and the shape of the mβCD-treated cells was similar to cells under hypo-osmotic stress, the reduction of Ca2+ release was more pronounced suggesting that mβCD treatment may disrupt additional steps in the Gαq/PLCβ-mediated Ca2+ release pathway.
To determine whether the effect of osmotic stress on Ca2+ release was specific to the Gαq/PLCβ signaling pathway, we repeated the measurements by stimulating the cells with a mixture of growth factors (hepatocyte growth factor, platelet-derived growth factor, and epidermal growth factor) that will increase calcium through receptor kinase-phospholipase Cγ pathways. At 300 mosm, growth factor stimulation results in a delayed and much lower Ca2+ response as compared with Gαq stimulation (Fig. 5D). When the osmolarity was reduced to 150 mosm and the cells were stimulated with growth factors, the basal level of Ca2+ was elevated as before, but both the amount of Ca2+ released and the shift in the time of release are within error of control samples.
To better connect caveolae with calcium signaling, we treated cells with siRNA(Cav1) and measured the change in response to osmotic stress. These studies were done viewing single cells to minimize the amount of siRNA used. The compiled data are shown in Fig. 5E. We found that untreated cells show a similar behavior as reported previously (16), which is similar to that seen for cell suspensions. When cells are treated with siRNA(Cav1) to achieve an ∼65% knockdown, the amount of Ca2+ released is lower, and the duration of the peak is shorter, mimicking the behavior seen for untreated cells under osmotic stress. When these cells were subjected to mild osmotic stress in the range where cell rupture does not occur (210 mosm), we found that Ca2+ release is no longer sensitive to osmotic stress and that release was identical to the behavior at normal osmotic pressure. Taken together, these studies support the idea that deformation of caveolae affects Ca2+ signals mediated through Gαq.
Effect of Osmotic Stress on Membrane-bound PLCβ/Gαq
It is possible that some of the observed changes in Ca2+ responses could be potentiated by interactions between PLCβ and its binding partners. Typically, a significant population of PLCβ localizes to the plasma membrane where it is bound to Gαq (24), and it is possible that hypo-osmotic stress could disrupt Gαq/PLCβ interactions. We investigated this possibility by monitoring the change in colocalization between PLCβ1 and Gαq in A10 cells. Although no significant change in the mean value of colocalization is observed, the distribution of the values was much broader (Fig. 6). These results suggest that although Gαq/PLCβ interactions were largely preserved under increased osmotic pressure, some population might be destabilized.
FIGURE 6.
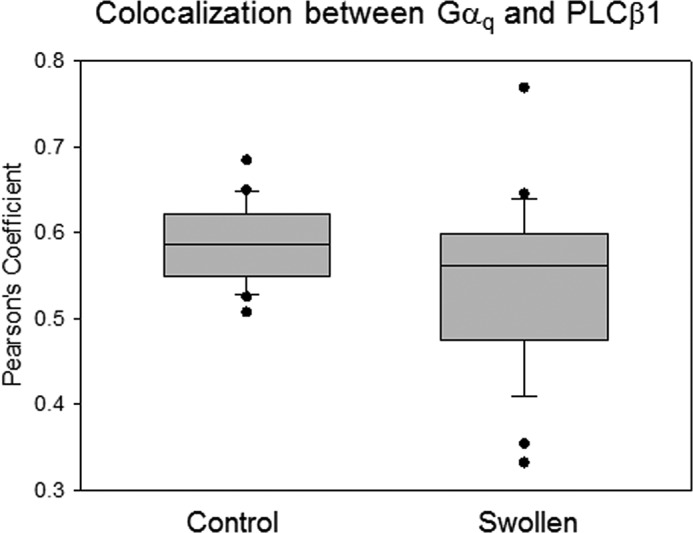
Osmotic stress does not significantly reduce Gαq and PLCβ1 colocalization. Tabulation of coimmunofluorescence studies between Gαq and PLCβ1 at iso-osmotic (300 mosm) and hypo-osmotic (150 mosm) conditions in A10 cells where the data are a compilation from three separate experiments, and where n = 30 for each experiment.
In contrast to the G-protein-regulated PLCβ enzymes, PLCδ enzymes are activated by increased cellular calcium levels. Association of PLCβ will inhibit PLCδ activity, although G protein activation will displace PLCβ from PLCδ resulting in a synergistic calcium response (25). To determine whether changes in PLCβ-PLCδ association contribute to the observed calcium behavior, we monitored FRET between the labeled proteins under basal and osmotically swollen conditions. No differences were observed (Fig. 7). We note that identical results were obtained when monitoring the colocalization of the two proteins under normal and hypo-osmotic conditions (data not shown).
FIGURE 7.
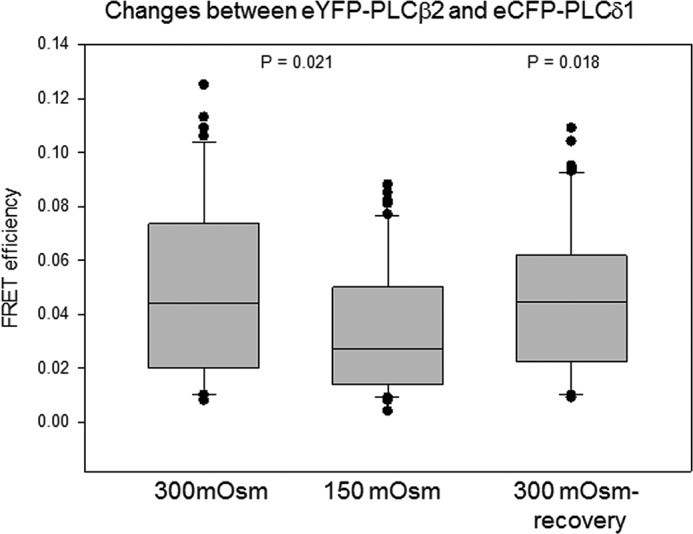
PLCβ and PLCδ association is not affected by osmotic stress. A10 cells were transfected with eYFP-PLCβ2 and eCFP-PLCδ1, and FRET was monitored at 300 and 150 mosm and after recovery back to 300 mosm, and where n = 15 for all samples.
Discussion
In this study, we show that mechanical deformation of caveolae by osmotic swelling can directly affect Ca2+ signals by disrupting caveola/Gαq interactions, thus linking the mechanical and signaling properties of caveolae. Caveolae have long been known to give mechanical strength to membranes and are prominent in contractile cells. The connection between mechanosensing and cell signaling has been mainly attributed to the ability of caveolae to serve as platforms that organize signaling domains (26). Additionally, mechanosensitivity has been shown to trigger caveolin phosphorylation that in turn modulates Src family kinases (27). Although it is clear that caveola deformation may have many indirect effects on signaling pathways, we show here that these effects can be direct.
We subjected A10 rat aortic smooth muscle cells to osmotic stress by lowering the osmolarity from 300 to 150 mosm to deform caveolae. Although previous studies subjected epithelial cells to a larger drop in osmolarity over a short duration, we found that A10 cells do not tolerate high osmotic stress even for a relatively short amount of time (5 min). Our data that compare the stability of cells treated with mβCD or with siRNA(Cav1) to mock-treated controls support the idea that the presence of caveolae gives mechanical strength to cells and give us a working range of osmolarity to carry out our signaling studies.
Consistent with previous work (13), our studies suggest that subjecting A10 cells to hypo-osmotic stress reduces the interaction of Cav1 molecules as seen by a reduction in intermolecular FRET between GFP-Cav1 and mCherry-Cav1, which is consistent disruption of caveolae. We note that although the reduction in FRET is significant (∼25%), a considerable amount of FRET remains suggesting that most Cav molecules remain in close proximity. Although these results may correlate to a partial dissolution of domains, it is also possible that they instead reflect a change in Cav1 configuration that causes the position of the fluorescent tags to be less optimal for energy transfer. It is notable that our FRAP studies detect little movement of eGFP-Cav1 molecules at 300, 150, and 30 mosm suggesting that almost all of the tagged Cav1 molecules remain in domains under our experimental conditions. By correlating these results to our studies monitoring the exposure of a Cav1 epitope with swelling, we conclude that the major result of osmotic stress is to change the configuration of Cav molecules rather than to release major populations into the plasma membrane. This idea correlates well with the flattening of caveolae by osmotic stress observed by Sinha et al. (13). It is important to note that the diffusion of GFP-Cav1 as followed by FRAP was identical at both 150 and 30 mosm showing that changes in ionic strength do not play a significant role in the diffusion behavior of Cav1 or the association between Cav1 and its antibody. Taken together, we conclude that the caveola domains remain intact, although restructured, with reduced osmolarity.
We monitored the recovery of caveola domains by FRET upon return to basal osmotic strength. We found that the value of FRET soon after recovery is the same within error as the control. However, over the next 24 h, FRET is significantly higher than untreated controls and remains high for 2 more days. The cause of this increase is not clear, but it could result from the irreversible release of specific caveola components, such as the membrane curvature sensing protein Pascin 2 (28) or changes in actin interaction. Alternatively, the higher degree of FRET could correspond to a different orientation of the Cav1 molecules that allow for more efficient transfer. In either case, these results show some irreversibility of the native caveola composition/structure after osmotically induced deformation.
Our results show a significant reduction in Gαq-Cav1 colocalization and loss in FRET with hypo-osmotic stress suggesting dissociation of Gαq proteins from the deformed caveola domains. The FRET response is less robust than expected from the colocalization results, most likely due to the need to overexpress both proteins that in turn promotes protein association. We note that the same reduction in colocalization is seen using cardiac myocytes (data not shown), which are rich in caveolae. Thus, mechanical deformation of caveolae either places Cav1 in an orientation that perturbs the Gαq association site or changes the membrane orientation of Gαq. As a control, we followed the colocalization of Gαq and PLCβ whose association is highly dependent on membrane orientation but independent of Cav1 association (14). As expected, we could not detect significant changes in colocalization either in A10 cells or cardiac myocytes, suggesting that the changes in Ca2+ signals that accompany hypo-osmotic stress are not due to changes in the membrane orientation of Gαq or large scale disruption of its association with PLCβ.
Increased osmotic stress greatly changed the release of Ca2+ with Gαq/PLCβ stimulation (12, 29). In unstimulated A10 cells, we found that lowering the osmolarity does not affect the basal Ca2+ levels and in contrast to previous reports showed that osmotic stress increases external Ca2+ leakage (29), suggesting that leakage may be cell-specific. When Gαq is stimulated, the increase in Ca2+ in cells experiencing osmotic stress is greatly reduced compared with controls, and the timing of the signal is delayed. Our studies in the absence of extracellular Ca2+ suggest that the reduction in signal can be directly traced to Gαq/PLCβ activity, although the delay in signaling results from downstream events, such as the opening of the Ca2+ channel on the plasma membrane. This reduction in signal correlates well with a loss in Gαq-Cav1 association due to mechanical deformation. Although many factors may contribute to the large changes in Ca2+ response with osmolarity, our results here suggest the underlying cause may be traced to Gαq-caveola association. Support for this idea comes from studies in which cells that were treated with siRNA(Cav1) or with mβCD showed a large reduction in Ca2+ release suggesting that the change in Ca2+ response is caveola-mediated. Additionally, slower Ca2+ signals generated by growth factors are not significantly affected by osmotic stress in accordance with the idea that caveola deformation specifically affects Gαq-related pathways.
Although rare, the changes in osmolarity studied here can occur under some natural conditions, such as hyponatremia, and these changes may be associated with mechanical deformation. Sinha et al. (13) showed that the membrane tension induced by a reduction in osmolarity gives a similar deformation of caveolae as using mechanical force. The oscillation of membrane tension that leads to caveola deformation is thought to occur in contractile cells, leading to the idea that the connection between caveola flattening and Ca2+ signals may play a role in cell function. It is also interesting to note that although the absence of caveolae is associated with cardiac hypertrophy (9), cardiac hypertrophy in turn is associated with malfunction of Gαq-signaling pathways leading to the idea that the reduced calcium output in cells without caveolae compensates for these lower levels by enhancing Gαq signals. This idea is presently being explored.
Acknowledgments
We thank Dr. Urszula Golebiewska and an anonymous reviewer for their helpful comments.
This work was supported, in whole or in part, by National Institutes of Health Grant GM053132. The authors declare that they have no conflicts of interest with the contents of this article.
- PLCβ
- phospholipase β
- mβCD
- methyl-β-cyclodextran
- eGFP
- enhanced GFP
- HBSS
- Hanks' buffered salt solution.
References
- 1. Anderson R. G. (1998) The caveolae membrane system. Annu. Rev. Biochem. 67, 199–225 [DOI] [PubMed] [Google Scholar]
- 2. Marx J. (2001) Caveolae: a once-elusive structure gets some respect. Science 294, 1862–1865 [DOI] [PubMed] [Google Scholar]
- 3. Parton R. G., Simons K. (2007) The multiple faces of caveolae. Nat. Rev. Mol. Cell Biol. 8, 185–194 [DOI] [PubMed] [Google Scholar]
- 4. Parton R. G., Hanzal-Bayer M., Hancock J. F. (2006) Biogenesis of caveolae: a structural model for caveolin-induced domain formation. J. Cell Sci. 119, 787–796 [DOI] [PubMed] [Google Scholar]
- 5. Hill M. M., Bastiani M., Luetterforst R., Kirkham M., Kirkham A., Nixon S. J., Walser P., Abankwa D., Oorschot V. M., Martin S., Hancock J. F., Parton R. G. (2008) PTRF-Cavin, a conserved cytoplasmic protein required for caveola formation and function. Cell 132, 113–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Woodman S. E., Park D. S., Cohen A. W., Cheung M. W., Chandra M., Shirani J., Tang B., Jelicks L. A., Kitsis R. N., Christ G. J., Factor S. M., Tanowitz H. B., Lisanti M. P. (2002) Caveolin-3 knock-out mice develop a progressive cardiomyopathy and show hyperactivation of the p42/44 MAPK cascade. J. Biol. Chem. 277, 38988–38997 [DOI] [PubMed] [Google Scholar]
- 7. Schwencke C., Braun-Dullaeus R. C., Wunderlich C., Strasser R. H. (2006) Caveolae and caveolin in transmembrane signaling: Implications for human disease. Cardiovasc. Res. 70, 42–49 [DOI] [PubMed] [Google Scholar]
- 8. Stary C. M., Tsutsumi Y. M., Patel P. M., Head B. P., Patel H. H., Roth D. M. (2012) Caveolins: targeting pro-survival signaling in the heart and brain. Front. Physiol. 3, 393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Galbiati F., Razani B., Lisanti M. P. (2001) Caveolae and caveolin-3 in muscular dystrophy. Trends Mol. Med. 7, 435–441 [DOI] [PubMed] [Google Scholar]
- 10. Pelkmans L., Helenius A. (2002) Endocytosis via caveolae. Traffic 3, 311–320 [DOI] [PubMed] [Google Scholar]
- 11. Thomsen P., Roepstorff K., Stahlhut M., van Deurs B. (2002) Caveolae are highly immobile plasma membrane microdomains, which are not involved in constitutive endocytic trafficking. Mol. Biol. Cell 13, 238–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Harvey R. D., Calaghan S. C. (2012) Caveolae create local signalling domains through their distinct protein content, lipid profile, and morphology. J. Mol. Cell. Cardiol. 52, 366–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sinha B., Köster D., Ruez R., Gonnord P., Bastiani M., Abankwa D., Stan R. V., Butler-Browne G., Vedie B., Johannes L., Morone N., Parton R. G., Raposo G., Sens P., Lamaze C., Nassoy P. (2011) Cells respond to mechanical stress by rapid disassembly of caveolae. Cell 144, 402–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sengupta P., Philip F., Scarlata S. (2008) Caveolin-1 alters Ca2+ signal duration through specific interaction with the Gαq family of G proteins. J. Cell Sci. 121, 1363–1372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Guo Y., Golebiewska U., Scarlata S. (2011) Modulation of Ca2+ activity in cardiomyocytes through caveolae-Gαq interactions. Biophys. J. 100, 1599–1607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Calizo R. C., Scarlata S. (2012) A role for G-proteins in directing G-protein-coupled receptor–caveolae localization. Biochemistry 51, 9513–9523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Calizo R. C., Scarlata S. (2013) Discrepancy between fluorescence correlation spectroscopy and fluorescence recovery after photobleaching diffusion measurements of G-protein-coupled receptors. Anal. Biochem. 440, 40–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Exton J. H. (1996) Regulation of phosphoinositide phospholipases by hormones, neurotransmitters, and other agonists linked to G proteins. Annu. Rev. Pharmacol. Toxicol. 36, 481–509 [DOI] [PubMed] [Google Scholar]
- 19. Rebecchi M. J., Pentyala S. N. (2000) Structure, function and control of phosphoinositide-specific phospholipase C. Physiol. Rev. 80, 1291–1335 [DOI] [PubMed] [Google Scholar]
- 20. Suh P. G., Park J. I., Manzoli L., Cocco L., Peak J. C., Katan M., Fukami K., Kataoka T., Yun S., Ryu S. H. (2008) Multiple roles of phosphoinositide-specific phospholipase C isozymes. BMB Rep. 41, 415–434 [DOI] [PubMed] [Google Scholar]
- 21. D'Alessandro M., Russell D., Morley S. M., Davies A. M., Lane E. B. (2002) Keratin mutations of epidermolysis bullosa simplex alter the kinetics of stress response to osmotic shock. J. Cell Sci. 115, 4341–4351 [DOI] [PubMed] [Google Scholar]
- 22. Rothberg K. G., Heuser J. E., Donzell W. C., Ying Y. S., Glenney J. R., Anderson R. G. (1992) Caveolin, a protein component of caveolae membrane coats. Cell 68, 673–682 [DOI] [PubMed] [Google Scholar]
- 23. Rothberg K. G., Ying Y. S., Kamen B. A., Anderson R. G. (1990) Cholesterol controls the clustering of the glycophospholipid-anchored membrane receptor for 5-methyltetrahydrofolate. J. Cell Biol. 111, 2931–2938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dowal L., Provitera P., Scarlata S. (2006) Stable association between Gαq and phospholipase Cβ1 in living cells. J. Biol. Chem. 281, 23999–24014 [DOI] [PubMed] [Google Scholar]
- 25. Guo Y., Rebecchi M., Scarlata S. (2005) Phospholipase Cβ2 binds to and inhibits phospholipase Cδ1. J. Biol. Chem. 280, 1438–1447 [DOI] [PubMed] [Google Scholar]
- 26. Taggart M. J. (2001) Smooth muscle excitation-contraction coupling: a role for caveolae and caveolins? News Physiol. Sci. 16, 61–65 [DOI] [PubMed] [Google Scholar]
- 27. Radel C., Rizzo V. (2005) Integrin mechanotransduction stimulates caveolin-1 phosphorylation and recruitment of Csk to mediate actin reorganization. Am. J. Physiol. Heart Circ. Physiol. 288, H936–H945 [DOI] [PubMed] [Google Scholar]
- 28. Shvets E., Ludwig A., Nichols B. J. (2014) News from the caves: update on the structure and function of caveolae. Curr. Opin. Cell Biol. 29, 99–106 [DOI] [PubMed] [Google Scholar]
- 29. Tjondrokoesoemo A., Park K. H., Ferrante C., Komazaki S., Lesniak S., Brotto M., Ko J.-K., Zhou J., Weisleder N., Ma J. (2011) Disrupted membrane structure and intracellular Ca2+ signaling in adult skeletal muscle with acute knockdown of Bin1. PLoS ONE 6, e25740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Grynkiewicz G., Poenie M., Tsien R. Y. (1985) A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 260, 3440–3450 [PubMed] [Google Scholar]