

Background: FNR proteins are O2-responsive bacterial transcription factors.
Results: Pseudomonas putida possesses three FNR proteins with iron-sulfur clusters that have different sensitivities to O2.
Conclusion: The mechanism of the iron-sulfur cluster reaction with O2 is conserved among FNR proteins.
Significance: Differential sensitivity of multiple FNR proteins extends the range of O2-responsive gene expression within a single bacterium.
Keywords: bacterial signal transduction, bacterial transcription, iron-sulfur protein, microbiology, transcription factor, transcription regulation
Abstract
The Escherichia coli fumarate-nitrate reduction regulator (FNR) protein is the paradigm for bacterial O2-sensing transcription factors. However, unlike E. coli, some bacterial species possess multiple FNR proteins that presumably have evolved to fulfill distinct roles. Here, three FNR proteins (ANR, PP_3233, and PP_3287) from a single bacterial species, Pseudomonas putida KT2440, have been analyzed. Under anaerobic conditions, all three proteins had spectral properties resembling those of [4Fe-4S] proteins. The reactivity of the ANR [4Fe-4S] cluster with O2 was similar to that of E. coli FNR, and during conversion to the apo-protein, via a [2Fe-2S] intermediate, cluster sulfur was retained. Like ANR, reconstituted PP_3233 and PP_3287 were converted to [2Fe-2S] forms when exposed to O2, but their [4Fe-4S] clusters reacted more slowly. Transcription from an FNR-dependent promoter with a consensus FNR-binding site in P. putida and E. coli strains expressing only one FNR protein was consistent with the in vitro responses to O2. Taken together, the experimental results suggest that the local environments of the iron-sulfur clusters in the different P. putida FNR proteins influence their reactivity with O2, such that ANR resembles E. coli FNR and is highly responsive to low concentrations of O2, whereas PP_3233 and PP_3287 have evolved to be less sensitive to O2.
Introduction
Fumarate-nitrate reduction regulator (FNR)2 proteins are a major subgroup of the cyclic-AMP receptor protein family of bacterial transcription regulators (1). The major function of FNR proteins is the reprogramming of gene expression to coordinate the switch from aerobic to anaerobic metabolism when facultative anaerobes like Escherichia coli are starved of O2 (2–7). The paradigm for O2-sensing transcription factors is the E. coli FNR protein. The N-terminal region of FNR contains four essential cysteine residues that coordinate an O2-sensitive [4Fe-4S] cluster (8, 9). In the absence of O2, the [4Fe-4S] cluster is stable, and FNR exists as a homodimer that is capable of high affinity, site-specific DNA binding to an FNR box (TTGATNNNNATCAA) (9, 10). When bound to target DNA, FNR activates the expression of genes encoding proteins required for anaerobic metabolism and represses those utilized under aerobic conditions (2, 4), such that when O2 is available, anaerobic metabolism is shutdown in favor of the more energetically efficient aerobic respiratory metabolism. Molecular oxygen reacts with the FNR [4Fe-4S] cluster in a series of steps that ultimately yields the apo form of the protein (Equations 1–3) (11–13).
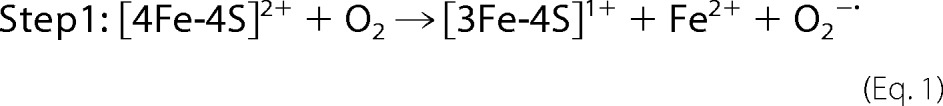 |
Recent work has shown that step 2 (Equation 2) is more complex than previously envisaged because it involves the conversion of the [3Fe-4S]1+ cluster to a persulfide-coordinated [2Fe-2S]2+ form. The [4Fe-4S] to [2Fe-2S] conversion can therefore be written as in Equation 4 (14).
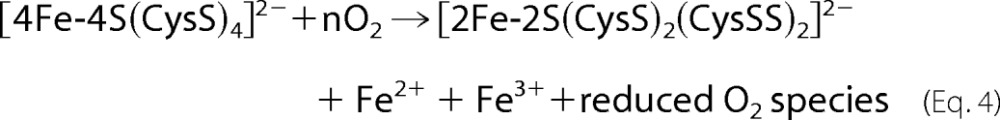 |
The retention of cluster sulfide (as CysSS) permits facile repair of the FNR [4Fe-4S] cluster in the presence of Fe2+ and a reducing agent (14). Molecular oxygen-dependent conversion of the [4Fe-4S] cluster to the persulfide-ligated [2Fe-2S] causes conformational rearrangements at the dimer interface that result in a charge-clash in the dimerization helices (15). Consequently, FNR monomerizes and is no longer able to bind DNA or to regulate transcription (10). Upon prolonged exposure to O2 in vitro and in vivo, the dominant FNR species is the monomeric apo form, which is capable of acquiring a [4Fe-4S] cluster via the action of the Isc (iron-sulfur cluster) biosynthetic machinery, such that FNR continually monitors the cytoplasm for the availability of O2 (16–18).
Unlike many bacteria, Pseudomonas putida and Burkholderia spp. possess multiple FNR family proteins that retain the characteristic cluster of cysteine residues in the N-terminal sensory domain. Transcriptomic analysis of the opportunistic pathogen Burkholderia cenocepacia revealed the presence of a 50-gene low oxygen-activated (lxa) locus that was associated with persistence of this obligate aerobe under anaerobic conditions (19). The lxa locus includes the FNR protein BCAM0287, which was induced 17-fold under low O2 (microaerobic) conditions. In addition, two other FNR protein encoding genes were induced during growth under a 6% O2 atmosphere, BCAM0049 (induced 77-fold, compared with aerobic conditions) and BCAM1483 (induced 3.5-fold) (19). Although an FNR box-like motif was associated with many genes induced under microaerobic conditions, the functions of the multiple FNR regulators in B. cenocepacia are poorly defined. Similarly, the properties and functions of the three FNR proteins of P. putida KT2440 are poorly understood. Here for the first time, three FNR proteins (PP_3233, PP_3287, and PP_4265, the last of which is also known as ANR) from a single bacterial species, P. putida KT2440, have been isolated, and their responses to O2 in vivo and in vitro have been assessed.
Experimental Procedures
Overproduction and Purification of Proteins
The ANR open reading frame was amplified from P. putida KT2440 genomic DNA using the primers MS87 (5′-TTTTTCTAGACATGTCCGAGCCAGTCAAACT-3′) and MS88 (5′-TTTTCTCGAGTCAGGCCTCGATTGCACCACA-3′) containing engineered XbaI and XhoI sites, respectively, and ligated into pGEX-KG to give construct pGS2268 (see Table 1). The resulting GST-ANR fusion protein was overproduced following IPTG induction in aerobic cultures of E. coli BL21 harboring pGS2268 for 2 h at 37 °C. The fusion protein was purified from cell-free extracts using glutathione-Sepharose 4B (GE Healthcare) equilibrated with 25 mm HEPES, 100 mm NaCl, 100 mm NaNO3, 2.5 mm CaCl2 at pH 7.5. The ANR protein was released from the fusion protein by on-column thrombin cleavage. The ANR, PP_3233 and PP_3287 open reading frames were amplified from P. putida KT2440 genomic DNA and ligated into the pCOLD-TF vector (Takara Clontech) (to give pGS2414, pGS2403, and pGS2413, respectively) using the following primers: ANR, MS140 (5′-TTTTCATATGTCCGAGCCAGTCAAACTGCG-3′) and MS150 (5′-TTTTTCTAGATCAGGCCTCGATTGCACCAC-3′) containing engineered NdeI and XbaI sites, respectively; PP_3233, MS141 (5′-TTTTCATATGTCAGGCTCTGCAGAAATGGG-3′) and MS92 (5′-TTTTCTCGAGTCAAGTGGGCTCCTCCAGGC-3′) containing engineered NdeI and XhoI sites, respectively; PP_3287, MS142 (5′-TTTTCATATGCCTGGCCAGCTGAAGGTCAC-3′) and MS151 (5′-TTTTTCTAGATCAGGGGCCCTTGGCCTCAC-3′) containing engineered NdeI and XbaI sites, respectively. The resulting His6-Tig-ANR, -PP_3233, and -PP_3287 fusion proteins were overproduced in aerobic cultures of E. coli BL21 harboring pGS2414, pGS2403, or pGS2413, respectively, for 24 h at 15 °C. The fusion proteins were purified from cell-free extracts on nickel-charged Hi-Trap chelating columns (GE Healthcare) equilibrated with 20 mm sodium phosphate, 0.5 m NaCl, pH 7.4, and were eluted by application of a linear imidazole gradient (0–1 m) followed by desalting into 25 mm HEPES, 100 mm NaCl, 100 mm NaNO3, pH 7.5. Protein concentration was determined by the Bio-Rad protein reagent (20).
TABLE 1.
Bacterial strains and plasmids used in this work
AmpR, ampicillin resistance; CmR, chloramphenicol resistance; GmR, gentamicin resistance; KanR, kanamycin resistance; TetR, tetracycline resistance.
| Relevant characteristics | Source | |
|---|---|---|
| Bacterial strain | ||
| JRG6348 | A derivative of E. coli JRG1728 lac, fnr carrying a chromosomal FNR-dependent promoter-lacZ fusion; CmR | Dr. David Lee (University of Birmingham, Birmingham, UK) |
| JRG6725 | P. putida KT2440 parent strain; source of genomic DNA for amplification and cloning anr, PP_3233, and PP_3287 genes for protein expression and construction of JRG6721, JRG6722, and JRG6723 | Laboratory collection |
| JRG6721 | P. putida KT2440 PP_3233, anr (PP_3287+) | This work |
| JRG6722 | P. putida KT2440 PP_3287, anr (PP_3233+) | This work |
| JRG6723 | P. putida KT2440 PP_3233, PP_3287 (ANR+) | This work |
| Plasmid | ||
| pAS12 | pEX18Ap containing gene replacement cassette for PP_4265, AmpR, GmR | This study |
| pBBR1-MCS-5 | Broad host range vector, KanR. | Ref. 35 |
| pDelta_PP_3233 | pEMG containing replacement cassette for PP3233, KanR | This study |
| pDelta_PP_3287 | pEMG containing replacement cassette for PP3287, KanR | This study |
| pEMG | Gene replacement vector with two flanking I-SceI sites, KanR | Ref. 23 |
| pEX18Ap | Gene replacement vector, sacB, AmpR | Ref. 22 |
| pFLP2 | Broad host range vector with FLP recombinase, AmpR | Ref. 22 |
| pGS422 | pUC13 containing the FF-41.5 promoter (EcoRI-HindIII) | Ref. 36 |
| pGS652 | pBluescript containing the NN-41.5 promoter (EcoRI-HindIII) | Ref. 37 |
| pGS810 (pFF-41.5) | pRW50 derivative with lacZ under the control of a class II FNR-dependent promoter, TetR | Ref. 38 |
| pGS2268 | pGEX-KG (39) containing ANR for expression as a GST fusion protein; AmpR | This work |
| pGS2350 | pBAD-HisB (Invitrogen) derivative for expression of E. coli fnr, AmpR | This work |
| pGS2351 | pBAD-HisB derivative for expression of P. putida anr; AmpR | This work |
| pGS2352 | pBAD-HisB derivative for expression of P. putida PP_3233, AmpR | This work |
| pGS2353 | pBAD-HisB derivative for expression of P. putida PP_3287, AmpR | This work |
| pGS2403 | As for pGS2414 but containing PP_3233 | This work |
| pGS2413 | As for pGS2414 but containing PP_3287 | This work |
| pGS2414 | pCOLD-TF (Takara Clontech) containing anr for expression as a His6-Tig fusion protein, AmpR | This work |
| pGS2508 | PP_3233 ligated into pBB1MSC-5 (EcoRI-XhoI) for expression of PP_3233 in P. putida, GmR | This work |
| pGS2509 | PP_3287 ligated into pBB1MSC-5 (EcoRI-XhoI) for expression of PP_3287 in P. putida; GmR | This work |
| pPS858 | Source of gentamicin resistance cassette; AmpR, GmR | Ref. 22 |
| pSWI | Broad host range vector with I-SceI endonuclease, AmpR | Ref. 40 |
Gel Filtration, Protein Reconstitution, and Total Amino Acid Analysis
The oligomeric state of ANR was estimated from the elution volume of a sample (50 μl of 250 μm protein) applied to a calibrated Superdex 200 column. The column was equilibrated with 25 mm Tris-HCl, pH 7.5, containing 500 mm NaCl and 2 mm EDTA, and the standards used to calibrate the column were blue dextran, hemoglobin, ovalbumin, cytochrome c, and aprotinin.
The ANR, PP_3233, and PP_3287 proteins were reconstituted overnight under anaerobic conditions at 25 °C in 25 mm HEPES, 100 mm NaCl, 100 mm NaNO3, pH 7.5, to which 0.5 mm l-cysteine, 12.5 mm DTT, an 8-fold molar excess of (NH4)2Fe(SO4)2 and 0.2 μm NifS cysteine desulfurase were added. Reconstituted proteins were purified on a heparin column (GE Healthcare) and eluted in 25 mm Tris-HCl containing 500 mm NaCl, pH 7.5 (21). Iron content was determined as previously described (21). Total amino acid analysis was carried out by Alta Bioscience (University of Birmingham, Birmingham, UK) following complete acid hydrolysis of ANR protein samples that had previously had the protein content estimated by the Bio-Rad protein assay (20).
UV-visible and CD Spectroscopy
Sealed anaerobic cuvettes containing reconstituted FNR proteins were injected with increasing amounts of air-saturated buffer as indicated and were incubated at 25 °C for 10 min followed by spectroscopic analysis. Absorbance measurements were made with a Cary UV-visible spectrophotometer. Changes in absorbance at 405 nm were used to monitor the conversion of the clusters. The extinction coefficient for the E. coli FNR iron-sulfur cluster (ϵ406 nm = 16,200 m−1 cm−1) was used to calculate the amount of [4Fe-4S]2+ cluster in the reconstituted proteins. The spectra shown are typical of at least three experiments. CD measurements were made with a Jasco J-810 spectropolarimeter. Aliquots of ANR (680 μl) were diluted to 29.8 μm iron-sulfur cluster under anaerobic conditions for initial measurements before treating with oxygenated buffer (∼220 μm O2) to give ∼2-fold molar excess O2 and then incubated for 15 min at room temperature prior to further measurements.
Kinetic Measurements
Reactions were initiated by the injection of air-saturated buffer (final concentration, ∼100 μm O2) into sealed anaerobic cuvettes containing reconstituted ANR, PP_3233, or PP_3287 proteins (final concentration, ∼6–9 μm [4Fe-4S]) at 25 °C. The dead time of mixing was ∼5 s. Changes in absorbance at 420 nm were used to monitor the conversion of the clusters. The A420 nm decay data were fitted to a single or double exponential function (as appropriate) using the program Origin (version 8; OriginLab). Where a double exponential function was fitted, the higher rate constant was assumed to correspond to the initial reaction with O2. Reported rate constants are mean values with standard errors from three repeats.
Liquid Chromatography-Mass Spectrometry of ANR
For LC-MS, an aliquot of ANR (20 μl, 80 μm [4Fe-4S]) was combined with an equal volume of oxygenated buffer (∼220 μm O2) or anaerobic buffer and allowed to react for 15 min. Samples were diluted to 2.9 μm final concentration, with an aqueous mixture of 1% (v/v) acetonitrile, 0.3% (v/v) formic acid, sealed, removed from the anaerobic cabinet, and loaded (5 μl) onto a ProSwift RP-1S column (4.6 × 50 mm) (Thermo Scientific) on a Ultimate 3000 UHLPC system (Dionex, Leeds, UK). Bound protein was eluted (0.2 ml/min) using a linear gradient (15 min) from 1% to 100% (v/v) acetonitrile, 0.1% (v/v) formic acid. The eluent was continuously infused into a Bruker microQTOF-QIII mass spectrometer, running Hystar (Bruker Daltonics, Coventry, UK), using positive mode electrospray ionization. Compass Data Analysis with Maximum Entropy v1.3 (Bruker Daltonics, Coventry) was used for processing of spectra under LC peak. The mass spectrometer was calibrated with ESI-L tuning mix (Agilent Technologies).
Construction of Plasmids and Bacterial Strains
To investigate the responses of the three P. putida FNR proteins in vivo, it was necessary to create P. putida KT2440 strains that only expressed one of the three FNR proteins encoded by the genome. Two different strategies were used to create unmarked deletion mutants. The P. putida gene PP_4265 encoding ANR was deleted using sacB counter selection and FLP recombinase excision as described by Hoang et al. (22). The primer pairs oAS23 (5′-GGAATTCAGCCAGATCGGCGACCTGTA-3′), oAS24 (5′-CGGGATCCTGTAGGCCAGTGTGCGCGAT-3′), oAS25 (5′-CGGGATCCACCTTGGCCTGGCGGTAGAA-3′), and oAS26 (5′-GCTCTAGACTGTCGGCATGCACTTCCAG-3′) containing engineered EcoRI, BamHI, and XbaI restrictions sites (as indicated by underlining) were used to amplify 511- and 533-bp DNA fragments flanking the PP_4265 gene. The fragments were cloned into the suicide vector pEX18Ap flanking the gentamicin resistance cassette from plasmid pPS858 and used to generate the unmarked P. putida PP_4265 mutant strain (22).
For the generation of unmarked gene deletion mutants of the genes encoding PP_3233 and PP_3287, the I-SceI endonuclease based knock-out strategy for P. putida described by Martínez-García and de Lorenzo was used (23). The following primers were used to amplify upstream and downstream regions of PP_3233 and join both fragments by sewing PCR: PP_3233 Upstream-Fwd (5′-GAATTCAAGGCTTTTTCGCGTTCTC-3′, engineered EcoRI site underlined), PP_3233 Upstream-Rev (5′-GAGACCTGCATGGACGAAGGACGATGCCTCCGCTTTTTTC-3′), PP_3233 Downstream-Fwd (5′-CTTCGTCCATGCAGGTCTC-3′), and PP_3233 Downstream-Rev (5′-AAGCTTATTTATCGTCAGCACCCAGAGT-3′, engineered HindIII site underlined). For PP_3287 the following primers: PP_3287 Upstream-Fwd (5′-GAATTCTGCGATACGTAGGTAGAGCATC-3′, engineered EcoRI site underlined), PP_3287 Upstream1-Rev (5′-AGACATCCGCAACATGAAGCTTTCAGGCCTCCTTCGCATTACG-3′), PP_3287 Downstream-Fwd (5′-GCTTCATGTTGCGGATGTCT-3′), and PP_3287 Downstream-Rev (5′-GGATCCCCACGTTGCATGATCTTGAG-3′, engineered BamHI site underlined) were used. The PCR products were ligated into the suicide vector pEMG and used to generate double knock-out mutants P. putida PP_3233 PP_4265 (JRG6721) and P. putida PP_3287 PP_4265 (JRG6722), as well as P. putida PP_3233 PP_3287 (JRG6723) following the protocol described by Martínez-García and de Lorenzo (23).
The PP_3233 and PP_3287 genes including their promoter regions were amplified by PCR from P. putida KT2440 genomic DNA using the primer pairs PP_3233 (5′-TTTTGAATTCGGCCTGATCAACACGTGAAC-3′ and 5′-TTTTCTCGAGTCGTCAGCACCCAGAGTGC-3′) and PP_3287 (5′-TTTTGAATTCGCCAGCTACACGTTGCGAA-3′ and 5′-TTTTCTCGAGATGATCTTGAGGCGGGCGA-3′) containing engineered EcoRI and XhoI sites (underlined) for ligation into pBBR1MCS-5 to give pGS2508 and pGS2509, respectively (see Table 1).
For the heterologous reporter system, expression plasmids for use in E. coli JRG6348 as well as an equivalent E. coli fnr expression plasmid to act as a control were created (see Table 1). The open reading frames corresponding to FNR, ANR, PP_3233, and PP_3287 were amplified by PCR to incorporate a XhoI restriction site downstream of the open reading frames: MS125 (5′-ATCCCGGAAAAGCGAATTAT-3′) and MS126 (5′-TTTTCTCGAGTCAGGCAACGTTACGCGTAT-3′) for fnr; MS122 (5′-TCCGAGCCAGTCAAACTGCG-3′) and MS88 (5′-TTTTCTCGAGTCAGGCCTCGATTGCACCACA-3′) for anr; MS124 (5′-TCAGGCTCTGCAGAAATGGG-3′) and MS92 (5′-TTTTCTCGAGTCAAGTGGGCTCCTCCAGGC-3′) for PP_3233; MS123 (5′-CCTGGCCAGCTGAAGGTCAC-3′) and MS90 (5′-TTTTCTCGAGTCAGGGGCCCTTGGCCTCAC-3′) for PP_3287. After digestion with XhoI, the products were ligated into pBADHisB (Invitrogen) following NcoI and XhoI digestion and filling in the NcoI site so that the ATG start codon was provided by the vector and the encoded proteins lacked His tags. The authenticity of all constructs was confirmed by DNA sequencing.
In Vivo Transcription Assays
P. putida KT2440 mutants, JRG6721, JRG6722, and JRG6723 with deletions of two of the three FNR encoding genes, i.e. capable of expressing either PP_3287, PP_3233, or anr only, were transformed with the FNR-dependent reporter plasmid pGS810 (pFF-41.5; see Table 1). Where indicated, JRG6721 and JRG6722 were transformed with pGS810 and either pGS2508 or pGS2509 (expressing PP_3233 or PP_3287 under the control of their respective native promoters: see Table 1). Cultures were grown in L-broth supplemented with appropriate antibiotics—tetracycline (35 μg ml−1) and gentamicin (20 μg ml−1)—in 50 ml of shaking (200 rpm) conical flasks containing 10, 20, 30, 40, or 50 ml of medium at 30 °C for 3 h. To test the effects of nitric oxide on ANR, PP_3233, and PP_3287 activities, 1-hydroxy-2-oxo-3-(N-methyl-3-aminopropyl)-3-methyl-1-triazene (NOC-7, 20 μm; 40 μm nitric oxide) was added to anaerobic cultures grown in mineral medium 154 (1.4 g KH2PO4, 5.7 g Na2HPO4, 0.6 g NaCl, 1.7 g K2SO4, 0.55 mg MnSO4·4H2O, 50 mg MgSO4·7H2O, 3 mg/liter FeCl3) supplemented with 0.4% (w/v) yeast extract and 30 mm l-arginine and appropriate antibiotics. The effects of oxidative stress were tested in aerobic L-broth cultures supplemented with paraquat (0.2 mm). All P. putida cultures were incubated at 30 °C for 3 h. β-Galactosidase activities were measured as described by Miller (24).
RNA Isolation and qRT-PCR
Cultures of E. coli JRG6348 transformed with the pBAD-HisB-derivatives pGS2350, pGS2351, pGS2352, or pGS2353 (encoding FNR, PP_4265 (ANR), PP_3233, and PP_3287) all expressed under the control of the pBAD promoter to eliminate any differential transcriptional control over the production of the regulators (see Table 1) were grown under anaerobic conditions (sealed tubes filled to the neck) in M9 minimal medium supplemented with L-broth (5%, v/v), glycerol (0.4%, v/v), trimethylamine N-oxide (20 mm), sodium fumarate (20 mm), and ampicillin (100 μg ml−1) at 37 °C until the A600 reached ∼0.2 (2). Aliquots were removed, and mRNA was stabilized by the addition of 0.4 volume of ice-cold ethanol-phenol (95%:5%) at pH 4.5. The cultures were then exposed to air by shaking, and incubation was continued for 20 min before taking further samples for total RNA preparation using the RNeasy RNA purification kit (Qiagen) according to the manufacturer's instructions. RNA was quantified using a NanoDrop 1000 spectrophotometer (Thermo Scientific). Relative lacZ RNA quantities were determined for triplicate cultures as previously described (25).
To determine the abundances of the anr, PP_3233 and PP_3287 transcripts in P. putida strains, qRT-PCR was used with RNA samples isolated as described above. The genes for normalization were gyrA and gyrB. The primers used were: anr, 5′-TCTTTCGCTGAACCTGGAAG-3′ and 5′-AGCCAAAACTGTCACCCTG-3′; gyrA, 5′-GTCAACGGTTCCAGCGGTA-3′ and 5′-TTCCGGGTTGTCGATGAGC-3′; gyrB, 5′-GCAGCCGAGGTCATCATGA-3′ and 5′-GCGTTCACAACCGACACAC-3′; PP_3233, 5′-ACGAAGTGGACAAACTGGAG-3′ and 5′-GAAAATTCTTGATCGCCCCAG-3′; and PP_3287, 5′-GAATTTCTACCAACCTGCCATG-3′ and 5′-TTGCGGATGTCTCGTGAAG-3′.
Results and Discussion
P. putida
KT2440 possesses three FNR proteins: PP_3233, PP_3287 and PP_4265 (hereafter ANR). Compared with the E. coli FNR protein, ANR is 53% identical (76% similar over 226 amino acid residues), PP_3233 is 46% identical (67% similar over 225 amino acid residues), and PP_3287 is 41% identical (58% similar over 224 amino acid residues). The four cysteine residues that coordinate the [4Fe-4S] cluster that is essential for the function of E. coli FNR are conserved, and thus all three P. putida FNR proteins were predicted to contain cysteine-ligated [4Fe-4S] clusters; however, the amino acid residues in the vicinity of the clusters differ (Figs. 1 and 2). Previous studies have shown that replacement of amino acid residues adjacent to cluster coordinating cysteine residues can have profound effects on the reactivity of the E. coli FNR iron-sulfur cluster with O2 (12, 26). These observations suggested that the three P. putida FNR proteins might have evolved different sensitivities to O2.
FIGURE 1.

Multiple sequence alignment of P. putida FNR proteins and the E. coli FNR protein. Clustal Omega alignment of E. coli FNR and P. putida ANR, PP_3233, and PP_3287 proteins (41). The cysteine residues that coordinate the FNR iron-sulfur cluster (bold type), residues adjacent to cluster ligating cysteine residues that are substituted compared with FNR (white type on black), the DNA recognition helix (shaded gray), Asp-154 of FNR (bold black type on gray), residues that are identical in all four proteins (*), residues with strongly similar properties (:), and residues with weakly similar properties (.) are indicated.
FIGURE 2.
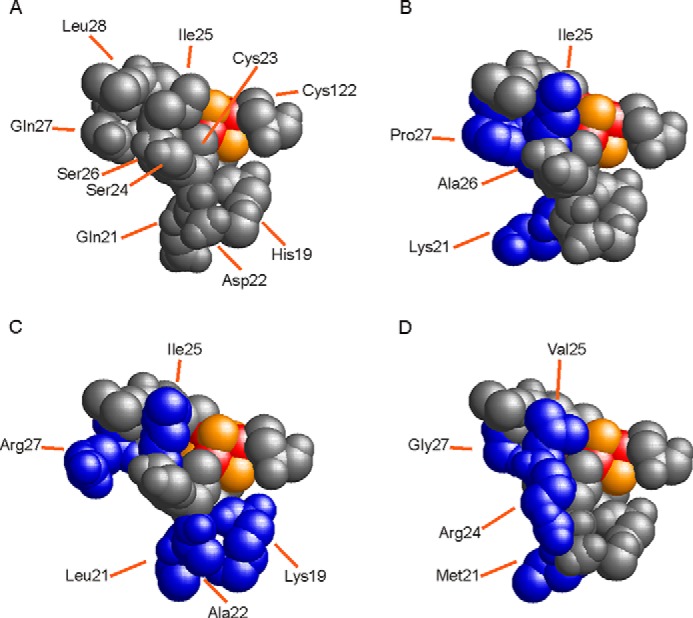
Models of the N-terminal [4Fe-4S] cluster binding regions of four FNR proteins. The models were constructed in SWISS-Model (42) using endonuclease III (Protein Data Bank code 2abk) as the template as described by Jervis et al. (12). The images were produced as space-filled representations in RasWin (43). The iron (red) and sulfide (gold) atoms of the [4Fe-4S] cluster are shown. Amino acids are labeled and numbered according to E. coli FNR. Conserved amino acids are colored gray, and those of the P. putida FNR proteins that differ from those present in E. coli FNR are colored blue. A, E. coli FNR. B, P. putida ANR. C, P. putida 3233. D, P. putida 3287.
The Reaction of the P. putida ANR Iron-Sulfur Cluster with O2 Resembles That of E. coli FNR
ANR was released from a GST-ANR fusion by “on-column” treatment with the protease thrombin. Application of the resulting apo-ANR protein to a calibrated gel filtration column indicated that unlike apo-FNR, which is monomeric (10), apo-ANR was dimeric, despite retaining Asp-154 (FNR numbering) that is proposed to cause a charge clash preventing dimerization of apo-FNR (Fig. 1 and Ref. 15). This suggests that additional residues in the dimer interface also contribute to determine the oligomeric state of ANR and FNR. After anaerobic iron-sulfur cluster reconstitution, the iron content of ANR was 4.1 ± 0.3 iron atoms per subunit (n = 3), based on protein estimation by total amino acid analysis. The anaerobic UV-visible spectrum of ANR was characteristic of a [4Fe-4S] protein (ϵ405 nm = ∼18,000 m−1 cm−1), and upon addition of O2 the spectrum changed to resemble that of a [2Fe-2S] protein, with broad absorbance bands at 320, 420, and 550 nm (Fig. 3A). Upon prolonged (16 h) exposure to air, the [2Fe-2S] form was degraded to the apo-ANR protein. Titration of reconstituted ANR with aerobic buffer revealed a progressive decrease in absorbance in the 400–420-nm region associated with conversion of the [4Fe-4S] form to the [2Fe-2S] form (Fig. 3A). The CD spectrum of reconstituted [4Fe-4S] ANR exhibited positive bands at 296, 325, 375, and 420 nm, reminiscent of [4Fe-4S] FNR (21). Following exposure to O2 (∼2-fold molar excess), these bands were replaced by a broad spectrum with two positive bands at 325 and 450 nm and one negative band at 375 nm, similar to the [2Fe-2S] form of FNR (Fig. 3B) (21). Treatment of the [4Fe-4S] form of ANR with 2 molar equivalents of O2 for 15 min followed by analysis of the resulting [2Fe-2S] form by LC-MS revealed the presence of up to five sulfur adducts, with one and two additional sulfurs as the major species (Fig. 3C). Thus, it was concluded that the reaction of the ANR [4Fe-4S] cluster with O2 proceeds via the same mechanism as that described for FNR, including the retention of cluster sulfide (14). The retention of cluster sulfide as S0 has implications for the repair of [4Fe-4S] clusters (14). Anaerobic incubation of [2Fe-2S] ANR with a 4-fold molar excess of ferrous ions in the presence of the reducing agent DTT regenerated the [4Fe-4S] form, as judged by the UV-visible spectrum of the protein (Fig. 3D). Thus, the mechanism of [4Fe-4S] repair proposed for E. coli FNR is likely to be a common feature of this family of regulators and probably other iron-sulfur proteins (14).
FIGURE 3.

Oxidation and repair of the P. putida ANR protein. A, UV-visible spectrum of reconstituted ANR containing ∼15 μm [4Fe-4S] cluster under anaerobic conditions (thick line). The changes in the ANR spectrum upon successive additions of aerobic buffer (25 mm Tris-HCl containing 500 mm NaCl, pH 7.5) (thin lines) are presented along with the final spectrum ([2Fe-2S] form) shown in gray. B, CD spectra of [4Fe-4S] ANR (29.8 μm) before (solid line) and after (dashed line) exposure to O2 (∼2-fold molar excess). The arrow indicates the movement of spectral features in response to O2. The buffer was 9 mm Tris, 17 mm HEPES, 1.7 mm CaCl2, 236 mm NaCl, 66 mm NaNO3, pH 7.5. C, detection of persulfide forms of apo-ANR after exposure of [4Fe-4S] ANR to O2. Mixtures of ANR reconstituted under anaerobic conditions (initially 80 μm [4Fe-4S]2+ cluster) were analyzed by LC-MS after incubation with anaerobic buffer for 15 min (gray line) and after treatment with 2 molar equivalents of O2 for 15 min (black line). The peak at 28,343 Da corresponds to the peak ANR monomer (mass, 28,347 Da) with two disulfide bonds. The peaks labeled S0–5S0 correspond to successive S0 additions (+32 Da). D, restoration of the ANR [4Fe-4S] cluster by treatment of purified [2Fe-2S] ANR (∼40 μm cluster) with ferrous ions (4-fold molar excess) and DTT (3 mm). The gray line shows the initial spectrum of [2Fe-2S] ANR, the dashed and solid black lines show the spectra obtained 50 and 160 min after the addition of ferrous ions and DTT.
Reactions of the [4Fe-4S] Clusters of PP_3233 and PP_3287 with O2 Result in Conversion to [2Fe-2S] Forms
Several attempts were made to overproduce the P. putida PP_3233 and PP_3287 proteins, but they were consistently found as insoluble aggregates when expressed at high levels, except when fused to the C terminus of the chaperone Trigger factor (Tig). Therefore, PP_3233 and PP_3287 were isolated as Tig fusions, and a Tig fusion of ANR was also generated to permit direct comparisons.
Anaerobic reconstitution of the iron-sulfur clusters of the three Tig fusion proteins resulted in UV-visible spectra characteristic of [4Fe-4S] proteins, with a broad absorbance at 400–420 nm (Fig. 4, A–C). Titration with aerobic buffer resulted in spectral changes that were consistent with conversion from [4Fe-4S] to [2Fe-2S] forms (Fig. 4, A–C). The response of the Tig-ANR fusion (Fig. 4A) was similar to that of the untagged ANR protein (Fig. 3A), suggesting that the Tig tag did not impair cluster acquisition or O2-mediated cluster conversion. Thus, it was concluded that all the P. putida FNR proteins acquired [4Fe-4S] clusters that underwent conversion to [2Fe-2S] clusters in the presence of O2.
FIGURE 4.

Absorbance spectra of Tig-tagged P. putida FNR proteins after treatment with increasing amounts of O2. Absorbance spectra obtained by titration of anaerobic solutions of proteins with air-saturated (220 μm O2 at 25 °C) buffer (25 mm Tris-HCl containing 500 mm NaCl, pH 7.5). The initial and final spectra are shown as bold lines. After each O2 addition, the sample was incubated for 10 min at 25 °C before obtaining the spectrum shown. The arrows indicate the direction of spectral change during the titration. A, ANR. B, PP_3233. C, PP_3287. The insets show the spectral changes in the visible region with an expanded ordinate (absorbance) scale. The spectra shown are typical of at least three measurements.
PP_3233 and PP_3287 React More Slowly than ANR with O2 in Vitro
Under pseudo-first order reaction conditions (O2:[4Fe-4S] ratio of ∼13), the A420 nm decays for ANR (both ANR and the Tig-ANR fusion), Tig-PP_3233, and Tig-PP_3287 were measured (Fig. 5). For ANR and Tig-ANR, the data were best fitted to a double-exponential function with observed rate constants (kobs) for the first reaction of 0.034 ± 0.007 s−1 for ANR and 0.028 ± 0.0015 s−1 for the Tig-ANR fusion (Fig. 5A). This again indicates that fusion to Tig did not significantly affect the reactivity of the ANR iron-sulfur cluster, and thus it was assumed that a Tig tag would not affect the reactivity of the PP_3233 and PP_3287 clusters. For the Tig-PP_3233 and Tig-PP_3287 fusion proteins, the data were best fitted to a single-exponential function yielding kobs values of 0.0038 ± 0.0002 s−1 for Tig-PP_3233 and 0.0055 ± 0.0001 s−1 for Tig-PP_3287 (Fig. 5, B and C). Division of the observed rate constants by the O2 concentration provides an estimate of the apparent second order rate constants for the fusion proteins: ANR, 280 m−1 s−1 (cf. 309 m−1 s−1 for the untagged ANR); PP_3233, 38 m−1 s−1; and PP_3287, 55 m−1 s−1. These values indicate that the [4Fe-4S] cluster of ANR displays similar sensitivity to O2 as previously reported for E. coli FNR (278 m−1 s−1), but the iron-sulfur clusters of PP_3233 and PP_3287 were significantly less reactive with O2 in vitro, more closely resembling the previously characterized variant FNR-S24F (80 m−1 s−1), which is also less responsive to O2 in vivo (11, 12).
FIGURE 5.

Kinetics of O2-mediated [4Fe-4S] cluster conversion. Samples of reconstituted Tig fusions of ANR (A, inset untagged ANR), PP_3233 (B), and PP_3287 containing ∼8 μm [4Fe-4S] cluster (C) were mixed with a 13-fold molar excess of O2 as aerobic buffer at 25 °C. The buffer was 25 mm HEPES, 100 mm NaCl, 100 mm NaNO3, pH 7.5. Loss of the [4Fe-4S] clusters was monitored at 420 nm as a function of time (gray lines). Data were fitted to exponential functions as described in the text (black lines). The upper data set (not fitted) in each panel shows the response when anaerobic buffer was used in place of aerobic buffer. The rate constants reported in the text from these experiments are mean values with standard errors from three repeats.
The Responses of PP_3233 and PP_3287 to Increased Culture Aeration Are Weaker than That of ANR
To determine whether ANR, PP_3233, and PP_3287 act as O2 sensors in vivo, three double mutant strains of P. putida were created in which two of the three genes encoding FNR proteins were deleted (Table 1). Cultures of these strains were grown in 50-ml conical flasks at 30 °C with shaking (200 rpm). For aerobic cultures, the flasks contained 10 ml of medium; for O2-limited cultures, the flasks contained 40 ml of medium. Strains that lacked anr exhibited impaired growth under O2-limited conditions, and strains that lacked either PP_3233 or PP_3287 were impaired under aerobic conditions (Fig. 6). This is consistent with relative O2 sensitivities of the ANR, PP_3233, and PP_3287 iron-sulfur clusters.
FIGURE 6.

Growth of P. putida under aerobic and O2-limited conditions. P. putida mutants that lack two of the three fnr genes present in the wild-type strain were grown under aerobic conditions (A, 10 ml of medium in a 50-ml conical flask with shaking at 200 rpm, 30 °C) or O2-limiting conditions (B, 40 ml of medium in a 50-ml conical flask with shaking at 200 rpm, 30 °C). Growth was monitored by measuring the optical density of the cultures (A600 nm). Wild-type (ANR+, PP_3233+, PP_3287+; filled diamonds); JRG6723 (ANR+; filled squares); JRG6722 (PP_3233+; filled triangles); JRG6721 (PP_3287+; open squares). The data points show the means and standard deviations (n = 6). *, significant difference between: wild-type (A) and all mutant strains and strains possessing the anr gene (i.e. wild-type and JRG6723) and those lacking anr (JRG6721 and JRG6722) (p ≤ 0.05) (B).
For E. coli FNR, it has been shown that Glu-209, Ser-212, and Arg-213 in the DNA recognition helix make the major interactions with the FNR box: TTGATCTAGATCAA (FF site). The amino acid sequences of the DNA recognition helices of the P. putida FNR proteins are very similar to those of E. coli FNR (PP_3287 has Cys in place of Ser), suggesting that ANR, PP_3233, and PP_3287 will recognize an FNR box (Fig. 1). Preliminary electromobility shift assays indicated that all three P. putida FNR proteins could bind at the FF site under anaerobic conditions. Therefore, the P. putida mutants were transformed with plasmid pGS810, which carries the FNR-dependent FF-41.5 (pFF-41.5) promoter fused to lacZ (Table 1). Cultures were grown under O2-limited conditions (50 ml of medium in a 50-ml shaking conical flask) and aerobic conditions (10 ml of medium in a 50-ml shaking conical flask). Measurement of β-galactosidase activity for P. putida JRG6723 (expresses only anr) cultures showed that ANR activity decreased ∼5-fold in response to enhanced aeration (Fig. 7, A and B). Measurement of the anr transcript by qRT-PCR and ANR protein by Western blotting with anti-serum raised against E. coli FNR for cultures grown in 50-ml shake flasks containing 50, 40, 30, 20, or 10 ml of medium, resulting in increasing O2 transfer to the cultures as the volume of medium decreased, showed that the amounts of anr transcript and ANR protein were similar in all the cultures (Fig. 7B). However, measurement of ANR-dependent transcription in cultures grown under these same conditions showed that increased aeration progressively lowered FF-41.5 promoter activity (Fig. 7B). Thus, it was concluded that ANR has properties similar to E. coli FNR and is a bona fide O2-responsive gene regulator in P. putida. However, β-galactosidase activities for the P. putida strains lacking the anr gene (i.e. P. putida JRG6722 expresses only PP_3233, and P. putida JRG6721 expresses only PP_3287) showed only a small decrease in response to increased aeration and low levels of β-galactosidase activity (128 ± 6 and 184 ± 3 Miller units, respectively, under O2-limited conditions compared with 8550 ± 54 Miller units for ANR) (Fig. 7A). This observation could result from poor expression of the PP_3233 and PP_3287 proteins. Therefore qRT-PCR was used to determine whether PP_3233 and PP_3287 were expressed in JRG6722 and JRG6721 under the conditions tested. The data indicated that the level of expression of PP_3233 was ∼10-fold lower, and PP_3287 was ∼5-fold lower than anr. Low levels of PP_3233 and PP_3287 mRNA were consistent with the hypothesis that expression of the three P. putida FNR proteins is likely to be temporally and/or spatially distinct. Therefore, to increase expression of PP_3233 and PP_3287, these genes and their respective promoter regions were ligated into the broad host range vector pBBR1-MCS-5 (Table 1). The resulting expression plasmids were used to transform P. putida JRG6722 and JRG6721 carrying the pGS810 reporter plasmid creating strains that only expressed PP_3233 or PP_3287. Cultures expressing PP_3233 from pGS2508 (ANR−, PP_3233++, PP_3287−) or PP_3287 from pGS2509 (ANR−, PP_3233−, PP_3287++) were grown under O2-limited and aerobic conditions, and qRT-PCR showed that the level of PP_3233 and PP_3287 expression was increased by ∼10- and ∼5-fold compared with the expression of chromosomal anr. Unfortunately, the corresponding increase in PP_3233 and PP_3287 proteins could not be determined because the E. coli FNR anti-serum did not cross-react with these proteins. Nevertheless, for both overexpression strains, FNR-dependent β-galactosidase activitydecreased significantly with increased aeration (Fig. 7A). Thus, it was concluded that transcription activation by PP_3233 and PP_3287 was inhibited by O2.
FIGURE 7.

Responses of P. putida FNR proteins to O2in vivo. A, the output from an FNR-dependent promoter decreases in response to enhanced aeration of P. putida cultures expressing only one of the three FNR proteins. All the strains were transformed with the FF-41.5-lacZ reporter plasmid pGS810. The rate of culture aeration was increased by decreasing the volume of medium in the shaking conical flasks (50 ml of medium for O2-limited cultures and 10 ml of medium for aerobic cultures). Cultures were grown at 30 °C for 3 h, at which point samples were taken for measurement of β-galactosidase activity. The β-galactosidase activities of the aerobic cultures were divided by those of the O2-limited cultures. The error bars are the standard deviation from the mean values of the aerobic:O2-limited ratios (n = 4). ANR, PP_3233, and PP_3287 indicate chromosomal expression of the corresponding genes; PP_3233++ and PP_3287++ indicate expression of the corresponding genes from a multicopy plasmid. B, concentration of cytoplasmic ANR does not respond to changes in culture aeration. Shown are Western blots developed with anti-serum raised against E. coli FNR for cell samples from P. putida cultures that express only anr grown in shaking 50-ml conical flasks containing 10, 20, 30, 40, or 50 ml of medium (lanes 1–5) to impose an increasing degree of O2 limitation on the cultures. The equivalent region of a Coomassie Blue-stained gel is shown as a loading control (M indicates protein standard markers: 37, 25, and 20 kDa, top to bottom). The outputs from the pFF-41.5 reporter (pGS810) for cultures grown as described above are shown below each lane (mean values ± standard deviation, n = 3). C, inactivation of FNR proteins upon exposure of anaerobic cultures to air. Cultures of E. coli JRG6348 expressing either no FNR (vector), E. coli FNR, P. putida ANR, PP_3233, or PP_3287, as indicated, were grown under anaerobic conditions and the abundance of FNR-protein-dependent lacZ transcription was measured by qRT-PCR. The cultures were exposed to air for 20 min, and then the abundance of the lacZ transcript was measured again. The relative abundance of lacZ mRNA after transfer to aerobic conditions is shown. The error bars are the standard deviation from the mean (n = 3).
To confirm the in vivo O2 responsiveness of the P. putida FNR proteins, a heterologous reporter system consisting of an E. coli fnr, lac mutant (JRG6348) with a chromosomal copy of the FNR-dependent FF-41.5 promoter fused to lacZ was transformed with plasmids expressing FNR, ANR, PP_3233, or PP_3287 under the control of the pBAD promoter (Table 1). Measurement of the decrease in lacZ transcript abundance by qRT-PCR after exposure of anaerobic cultures to O2 for 20 min showed that the activities of E. coli FNR and all three P. putida regulators decreased, with FNR- and ANR-dependent transcription showing the greatest responses (Fig. 7C). The weaker responses of PP_3233 and PP_3287 suggested that these proteins were less sensitive to O2 compared with FNR and ANR, consistent with the in vitro data presented above.
Signal Specificity
Transcription factors that utilize iron-sulfur clusters as sensory modules have been shown to respond to O2 (e.g. FNR), redox state (e.g. SoxR), nitric oxide (e.g. NsrR), and iron-sulfur cluster/iron homeostasis (e.g. IscR) (27, 28). Some of these transcription factors respond to more than one of these signals. Hence, the E. coli FNR and SoxR proteins respond to nitric oxide in addition to their primary signals, O2 and redox cycling, respectively. In vitro kinetic measurements with the [4Fe-4S] form of FNR indicated that it is much more sensitive to nitric oxide than it is to O2. However, in vivo, FNR is only nitrosylated when the major nitric oxide sensors (e.g. NsrR and NorR) and detoxification systems (e.g. NorVW, NrfA, and Hmp) are overwhelmed. Thus, FNR serves primarily as an O2 sensor with a secondary nitric oxide sensing role (25). By contrast, the iron-sulfur clusters of regulators that are primarily nitric oxide sensors (e.g. NsrR and Wbl proteins) or redox sensors (e.g. SoxR) are generally stable for several hours in the presence of O2 (29–31). The data described above show that the three P. putida FNR proteins respond to O2 in vitro and in vivo, suggesting that they are primarily O2 sensors. To determine whether they also share the nitric oxide- or redox-responsive characteristics of E. coli FNR and SoxR, respectively, anaerobic cultures of P. putida expressing only one of the three FNR proteins and carrying the FNR-dependent pFF-41.5 fused to lacZ were supplemented with the nitric oxide donor NOC-7; in addition, aerobic cultures were exposed to the redox cycling agent paraquat. The responses of PP_3233 and PP_3287 were similar, nitric oxide had little or no effect under anaerobic conditions, and paraquat had no effect under aerobic conditions (Fig. 8). However, for ANR, nitric oxide significantly inactivated anaerobic reporter gene expression, whereas paraquat again had no effect under aerobic conditions (Fig. 8). Thus, the response of ANR was similar to that reported previously for E. coli FNR, further confirming the similarities between these two proteins, but PP_3233 and PP_3287 were less responsive with both O2 and nitric oxide compared with ANR (25). Nevertheless, in all cases the greatest responses were provoked by culture aeration, and therefore, it was concluded that O2 is the major modulator of the activity of all three P. putida FNR proteins.
FIGURE 8.
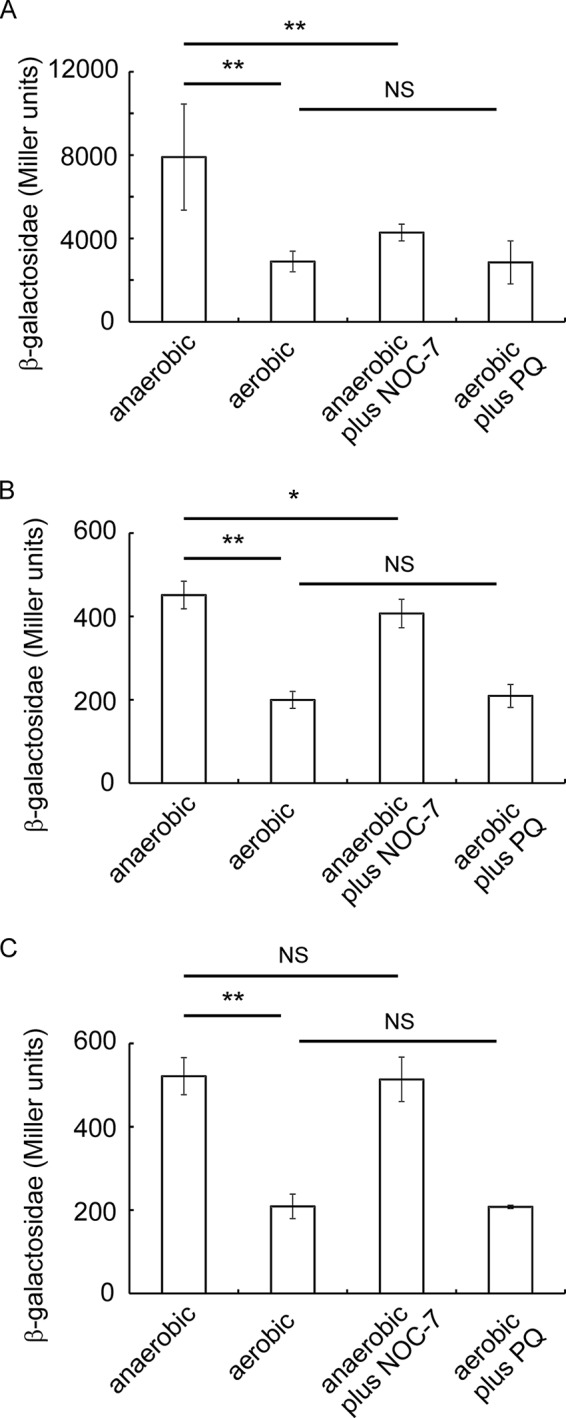
Responses of P. putida FNR proteins to nitric oxide and oxidative stress in vivo. P. putida strains capable of expressing only anr from the chromosome (ANR) or only PP_3233 from a multicopy plasmid (PP_3233++) or only PP_3287 from a plasmid (PP_3287++) were transformed with the FF-41.5-lacZ reporter plasmid pGS810. Cultures were grown at 30 °C for 3 h under anaerobic (50 ml of anaerobic minimal medium 154 supplemented with 0.4% (w/v) yeast extract and 30 mm l-arginine in a sealed 50-ml conical flask) or aerobic (10 ml of L-broth in a 50-ml conical flask, 200 rpm shaking) conditions in the presence and absence of NOC-7 (20 μm) or paraquat (PQ; 200 μm) as indicated. Samples were taken for measurement of β-galactosidase activity as a proxy for FNR protein activity: ANR (A), PP_3233++ (B), and PP_3287++ (C). The error bars are the standard deviation from the mean values (n = 3).). p values were determined by Student's t test. **, p < 0.01; *, p < 0.1; NS, p > 0.5.
Conclusions
The research described here suggests that the three FNR proteins of P. putida have evolved to fulfill distinct but overlapping roles. All three regulators, ANR, PP_3233, and PP_3287, acquired [4Fe-4S] clusters under anaerobic conditions and were converted to [2Fe-2S] forms upon exposure to O2 in vitro. ANR has the least number of nonconservative amino acid substitutions in the vicinity of the cluster-ligating cysteine residues compared with E. coli FNR and hence was expected to exhibit similar cluster reactivity to FNR (Fig. 1). The double-exponential nature of the ANR [4Fe-4S] cluster reaction with O2, the detection of sulfur adducts after conversion of [4Fe-4S]2+ ANR to the [2Fe-2S] form, and the capacity to repair the [4Fe-4S] cluster by simply providing ferrous ions under reducing conditions are consistent with the reaction scheme for E. coli FNR and O2 proposed by Zhang et al. (14) in which the [2Fe-2S]2+ cluster is ligated by one (Equations 5 and 6) or two (Equations 5 and 7) cysteine persulfides (CysSS).
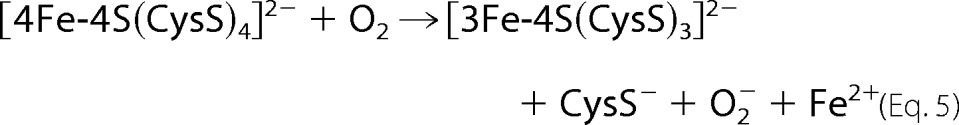 |
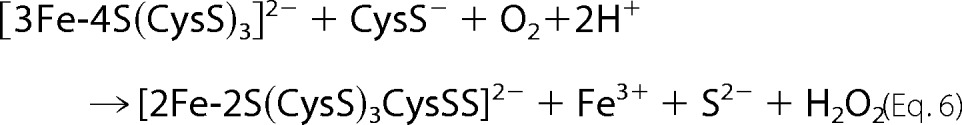 |
 |
Furthermore, ANR resembled FNR in exhibiting a secondary response when cultures were exposed to micromolar levels of nitric oxide (Fig. 8). Thus, the observations reported here are consistent with P. putida ANR acting as an E. coli-type O2 sensor regulator, in accordance with its ability to regulate the expression of multiple terminal oxidases of the P. putida respiratory chain and the ability of the closely related P. aeruginosa anr gene (encoded protein 88% identical, 94% similar over 244 amino acids) to complement the anaerobic growth phenotype of an E. coli fnr mutant (32, 33).
The [4Fe-4S] clusters of PP_3233 and PP_3287 also underwent conversion to [2Fe-2S] clusters upon exposure to O2, but these reactions were slower than that of ANR, and the responses of these proteins when cultures were exposed to nitric oxide were weaker than that observed for ANR (Fig. 8). The kinetic data for the reaction of the PP_3233 and PP_3287 [4Fe-4S] clusters with O2 fitted well to a single-exponential function rather than a double-exponential function, implying that, unlike FNR and ANR, the initial cluster oxidation step to generate the [3Fe-4S]1+ intermediate (Equation 5) was much slower than the subsequent decay of the [3Fe-4S]1+ to the [2Fe-2S]2+ form (Equations 6 and 7). Thus, it is suggested that the mechanism for [4Fe-4S] to [2Fe-2S] cluster conversion in PP_3233 and PP_3287 was similar to that described for FNR (11) and ANR, but the PP_3233 and PP_3287 [4Fe-4S] clusters appear to be more stable when bacteria are exposed to air or nitric oxide. The relative rates of ANR, PP_3233, and PP_3287 cluster reactions with O2 results in differential responses to O2 availability.
The in vivo properties of ANR, PP_3233, and PP_3287 were consistent with the observed reactivities of the [4Fe-4S] clusters with O2. Previous work with E. coli FNR showed that replacement of Ser-24, which is located immediately adjacent to the cluster ligand Cys-23, by Arg resulted in significant aerobic FNR activity, indicative of stabilization of the FNR-S24R iron-sulfur cluster (12). Interestingly, PP_3287 has Arg in the position equivalent to Ser-24 in FNR (Figs. 1 and 2), and thus this amino acid substitution could at least partially account for the lower reactivity of PP_3287 with O2. On the other hand, PP_3233 resembles E. coli FNR by retaining a Ser residue at the equivalent of position 24 (Figs. 1 and 2); however, amino acid substitutions in other locations are known to influence the reactivity of the E. coli FNR iron-sulfur cluster with O2 (26, 34). Like S24R, another amino acid substitution that promoted aerobic FNR activity was also located immediately adjacent to Cys-23, but this time on the other flank (D22G) (34). The equivalent position in PP_3233 is occupied by Ala (Figs. 1 and 2), and thus by analogy, replacement of the acidic Asp residue might alter the redox properties of the PP_3233 iron-sulfur cluster, such that it is less O2 reactive.
Although the observations reported here resolve several aspects of the properties of the three FNR proteins possessed by P. putida KT2440, many questions remain, including: (i) What are the conditions encountered by P. putida that induce PP_3233 and PP_3287 target gene expression? (ii) Do the three P. putida FNR proteins control distinct but overlapping regulons, perhaps by making productive interactions with additional transcription factors or alternative sigma factors? (iii) What is the imperative for employing multiple FNR proteins to extend the range of O2-responsive gene expression? Further detailed biochemical and physiological studies are now required to address these questions and in so doing discern the mechanism of the observed differential sensitivities to O2 of these closely related proteins and the broader implications for the control of gene expression in P. putida.
Acknowledgments
We thank Victor de Lorenzo for the plasmids pEMG and pSWI, David Lee for E. coli JRG6348, Jon Shaw for plasmid pBBR1-MCS-5, and Annika Steen for creating the P. putida PP_4265 mutant strain.
This work was supported by Grants BB/G018960/1, BB/G019347/1, and BB/L008114/1 from the Biotechnology and Biological Sciences Research Council UK, a Ph.D. scholarship from Higher Committee for Education and Development in Iraq (to S. I.), and Deutsche Forschungsgemeinschaft Grant SCHO 888/4–1 (to M. S.). The authors declare that they have no conflicts of interest with the contents of this article.
- FNR
- fumarate-nitrate reduction regulator
- ANR
- arginine deiminase nitrate reductase regulator
- NOC-7
- 1-hydroxy-2-oxo-3-(N-methyl-3-aminopropyl)-3-methyl-1-triazene
- qRT-PCR
- quantitative RT-PCR.
References
- 1. Körner H., Sofia H. J., Zumft W. G. (2003) Phylogeny of the bacterial superfamily of Crp-Fnr transcription regulators: exploiting the metabolic spectrum by controlling alternative gene programs. FEMS Microbiol. Rev. 27, 559–592 [DOI] [PubMed] [Google Scholar]
- 2. Constantinidou C., Hobman J. L., Griffiths L., Patel M. D., Penn C. W., Cole J. A., Overton T. W. (2006) A reassessment of the FNR regulon and transcriptomic analysis of the effects of nitrate, nitrite, NarXL, and NarQP as Escherichia coli K12 adapts from aerobic to anaerobic growth. J. Biol. Chem. 281, 4802–4815 [DOI] [PubMed] [Google Scholar]
- 3. Kang Y., Weber K. D., Qiu Y., Kiley P. J., Blattner F. R. (2005) Genome-wide expression analysis indicates that FNR of Escherichia coli K-12 regulates a large number of genes of unknown function. J. Bacteriol. 187, 1135–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Myers K. S., Yan H., Ong I. M., Chung D., Liang K., Tran F., Keleş S., Landick R., Kiley P. J. (2013) Genome-scale analysis of Escherichia coli FNR reveals complex features of transcription factor binding. PLoS Genet. 9, e1003565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Partridge J. D., Scott C., Tang Y., Poole R. K., Green J. (2006) Escherichia coli transcriptome dynamics during transition from anaerobic to aerobic conditions. J. Biol. Chem. 281, 27806–27815 [DOI] [PubMed] [Google Scholar]
- 6. Partridge J. D., Sanguinetti G., Dibden D. P., Roberts R. E., Poole R. K., Green J. (2007) Transition of Escherichia coli from aerobic to micro-aerobic conditions involves fast and slow reacting components. J. Biol. Chem. 282, 11230–11237 [DOI] [PubMed] [Google Scholar]
- 7. Rolfe M. D., Ocone A., Stapleton M. R., Hall S., Trotter E. W., Poole R. K., Sanguinetti G., Green J. (2012) Systems analysis of transcription factor activities in environments with stable and dynamic oxygen concentrations. Open Biol. 2, 120091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sharrocks A. D., Green J., Guest J. R. (1990) In vivo and in vitro mutants of FNR the anaerobic transcriptional regulator of E. coli. FEBS Lett. 270, 119–122 [DOI] [PubMed] [Google Scholar]
- 9. Khoroshilova N., Popescu C., Münck E., Beinert H., Kiley P. J. (1997) Iron-sulfur cluster disassembly in the FNR protein of Escherichia coli by O2: [4Fe-4S] to [2Fe-2S] conversion with loss of biological activity. Proc. Natl. Acad. Sci. U.S.A. 94, 6087–6092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lazazzera B. A., Beinert H., Khoroshilova N., Kennedy M. C., Kiley P. J. (1996) DNA binding and dimerization of the Fe-S-containing FNR protein from Escherichia coli are regulated by oxygen. J. Biol. Chem. 271, 2762–2768 [DOI] [PubMed] [Google Scholar]
- 11. Crack J. C., Green J., Cheesman M. R., Le Brun N. E., Thomson A. J. (2007) Superoxide-mediated amplification of the oxygen-induced switch from [4Fe-4S] to [2Fe-2S] clusters in the transcriptional regulator FNR. Proc. Natl. Acad. Sci. U.S.A. 104, 2092–2097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jervis A. J., Crack J. C., White G., Artymiuk P. J., Cheesman M. R., Thomson A. J., Le Brun N. E., Green J. (2009) The O2 sensitivity of the transcription factor FNR is controlled by Ser24 modulating the kinetics of the [4Fe-4S] to [2Fe-2S] cluster conversion. Proc. Natl. Acad. Sci. U.S.A. 106, 4659–4664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Reinhart F., Achebach S., Koch T., Unden G. (2008) Reduced apo-fumarate nitrate reductase regulator (apoFNR) as the major form of FNR in aerobically growing Escherichia coli. J. Bacteriol. 190, 879–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang B., Crack J. C., Subramanian S., Green J., Thomson A. J., Le Brun N. E., Johnson M. K. (2012) Reversible cycling between cysteine persulfide-ligated [2Fe-2S] and cysteine-ligated [4Fe-4S] clusters in the FNR regulatory protein. Proc. Natl. Acad. Sci. U.S.A. 109, 15734–15739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Moore L. J., Mettert E. L., Kiley P. J. (2006) Regulation of FNR dimerization by subunit charge repulsion. J. Biol. Chem. 281, 33268–33275 [DOI] [PubMed] [Google Scholar]
- 16. Dibden D. P., Green J. (2005) In vivo cycling of the Escherichia coli transcription factor FNR between active and inactive states. Microbiology 151, 4063–4070 [DOI] [PubMed] [Google Scholar]
- 17. Engel P., Trageser M., Unden G. (1991) Reversible interconversion of the functional state of the gene regulator FNR from Escherichia coli in vivo by O2 and iron availability. Arch. Microbiol. 156, 463–470 [DOI] [PubMed] [Google Scholar]
- 18. Mettert E. L., Kiley P. J. (2005) ClpXP-dependent proteolysis of FNR upon loss of its O2-sensing [4Fe-4S] cluster. J. Mol. Biol. 354, 220–232 [DOI] [PubMed] [Google Scholar]
- 19. Sass A. M., Schmerk C., Agnoli K., Norville P. J., Eberl L., Valvano M. A., Mahenthiralingam E. (2013) The unexpected discovery of a novel low-oxygen-activated locus for the anoxic persistence of Burkholderia cenocepacia. ISME J. 7, 1568–1581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 21. Crack J. C., Le Brun N. E., Thomson A. J., Green J., Jervis A. J. (2008) Reactions of nitric oxide and oxygen with the regulator of fumarate and nitrate reduction, a global transcriptional regulator, during anaerobic growth of Escherichia coli. Methods Enzymol. 437, 191–209 [DOI] [PubMed] [Google Scholar]
- 22. Hoang T. T., Karkhoff-Schweizer R. R., Kutchma A. J., Schweizer H. P. (1998) A broad host range Flp-FRT recombination system for site-specific excision of chromosomally located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212, 77–86 [DOI] [PubMed] [Google Scholar]
- 23. Martínez-García E., de Lorenzo V. (2011) Engineering multiple genomic deletions in Gram-negative bacteria: analysis of the multi-resistant antibiotic profile of Pseudomonas putida KT2440. Environ. Microbiol. 13, 2702–2716 [DOI] [PubMed] [Google Scholar]
- 24. Miller J. H. (1972) Experiments in Molecular Genetics, pp. 352–355, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 25. Crack J. C., Stapleton M. R., Green J., Thomson A. J., Le Brun N. E. (2013) Mechanism of [4Fe-4S](Cys)4 cluster nitrosylation is conserved among NO-responsive regulators. J. Biol. Chem. 288, 11492–11502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bates D. M., Popescu C. V., Khoroshilova N., Vogt K., Beinert H., Münck E., Kiley P. J. (2000) Substitution of leucine 28 with histidine in the Escherichia coli transcription factor FNR results in increased stability of the [4Fe-4S]2+ cluster to oxygen. J. Biol. Chem. 275, 6234–6240 [DOI] [PubMed] [Google Scholar]
- 27. Crack J. C., Green J., Thomson A. J., Le Brun N. E. (2012) Iron-sulfur cluster sensor-regulators. Curr. Opin. Chem. Biol. 16, 35–44 [DOI] [PubMed] [Google Scholar]
- 28. Crack J. C., Green J., Hutchings M. I., Thomson A. J., Le Brun N. E. (2012) Bacterial iron-sulfur regulatory proteins as biological sensor-switches. Antioxid. Redox Signal. 17, 1215–1231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Smith L. J., Stapleton M. R., Fullstone G. J., Crack J. C., Thomson A. J., Le Brun N. E., Hunt D. M., Harvey E., Adinolfi S., Buxton R. S., Green J. (2010) Mycobacterium tuberculosis WhiB1 is an essential DNA-binding protein with a nitric oxide-sensitive iron-sulfur cluster. Biochem. J. 432, 417–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Crack J. C., Smith L. J., Stapleton M. R., Peck J., Watmough N. J., Buttner M. J., Buxton R. S., Green J., Oganesyan V. S., Thomson A. J., Le Brun N. E. (2011) Mechanistic insight into the nitrosylation of the [4Fe-4S] cluster of WhiB-like proteins. J. Am. Chem. Soc. 133, 1112–1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hidalgo E., Bollinger J. M., Jr., Bradley T. M., Walsh C. T., Demple B. (1995) Binuclear [2Fe-2S] clusters in the Escherichia coli SoxR protein and role of the metal centers in transcription. J. Biol. Chem. 270, 20908–20914 [DOI] [PubMed] [Google Scholar]
- 32. Ugidos A., Morales G., Rial E., Williams H. D., Rojo F. (2008) The coordinate regulation of multiple terminal oxidases by the Pseudomonas putida ANR global regulator. Environ. Microbiol. 10, 1690–1702 [DOI] [PubMed] [Google Scholar]
- 33. Zimmermann A., Reimmann C., Galimand M., Haas D. (1991) Anaerobic growth and cyanide synthesis of Pseudomonas aeruginosa depend on anr, a regulatory gene homologous with fnr of Escherichia coli. Mol. Microbiol. 5, 1483–1490 [DOI] [PubMed] [Google Scholar]
- 34. Kiley P. J., Reznikoff W. S. (1991) Fnr mutants that activate gene expression in the presence of oxygen. J. Bacteriol. 173, 16–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kovach M. E., Elzer P. H., Hill D. S., Robertson G. T., Farris M. A., Roop R. M., 2nd, Peterson K. M. (1995) Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistance cassettes. Gene 166, 175–176 [DOI] [PubMed] [Google Scholar]
- 36. Sharrocks A. D., Green J., Guest J. R. (1991) FNR activates and represses transcription in vitro. Proc. Biol. Sci. 245, 219–226 [DOI] [PubMed] [Google Scholar]
- 37. Gostick D. O., Green J., Irvine A. S., Gasson M. J., Guest J. R. (1998) A novel regulatory switch mediated by the FNR-like protein of Lactobacillus casei. Microbiology 144, 705–717 [DOI] [PubMed] [Google Scholar]
- 38. Wing H. J., Williams S. M., Busby S. J. (1995) Spacing requirements for transcription activation by Escherichia coli FNR protein. J. Bacteriol. 177, 6704–6710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Guan K. L., Dixon J. E. (1991) Eukaryotic proteins expressed in Escherichia coli: an improved thrombin cleavage and purification procedure of fusion proteins with glutathione S-transferase. Anal. Biochem. 192, 262–267 [DOI] [PubMed] [Google Scholar]
- 40. Wong S. M., Mekalanos J. J. (2000) Genetic footprinting with mariner-based transposition in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U.S.A. 97, 10191–10196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sievers F., Wilm A., Dineen D., Gibson T. J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Söding J., Thompson J. D., Higgins D. G. (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kiefer F., Arnold K., Künzli M., Bordoli L., Schwede T. (2009) The SWISS-MODEL repository and associated resources. Nucleic Acids Res. 37, D387-D392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sayle R. A., Milner-White E. J. (1995) RasMol: Biomolecular graphics for all. Trends Biochem. Sci. 20, 374–376 [DOI] [PubMed] [Google Scholar]