

Background: Tiparp is an aryl hydrocarbon receptor (AHR) repressor, but its role in dioxin toxicity is unknown.
Results: Loss of Tiparp increases sensitivity to dioxin toxicity and lethality. Tiparp ADP-ribosylates AHR, which is reversed by the mono-ADP-ribosylase, MacroD1.
Conclusion: We identify new roles for Tiparp, MacroD1, and ADP-ribosylation in AHR signaling and dioxin toxicity.
Significance: These data reveal the importance of TIPARP in regulating AHR activity in mice.
Keywords: ADP-ribosylation, aryl hydrocarbon receptor (AhR) (AHR), dioxin, gene expression, toxicity
Abstract
The aryl hydrocarbon receptor (AHR) mediates the toxic effects of the environmental contaminant dioxin (2,3,7,8-tetrachlorodibenzo-p-dioxin; TCDD). Dioxin causes a range of toxic responses, including hepatic damage, steatohepatitis, and a lethal wasting syndrome; however, the mechanisms are still unknown. Here, we show that the loss of TCDD-inducible poly(ADP-ribose) polymerase (Tiparp), an ADP-ribosyltransferase and AHR repressor, increases sensitivity to dioxin-induced toxicity, steatohepatitis, and lethality. Tiparp−/− mice given a single injection of 100 μg/kg dioxin did not survive beyond day 5; all Tiparp+/+ mice survived the 30-day treatment. Dioxin-treated Tiparp−/− mice exhibited increased liver steatosis and hepatotoxicity. Tiparp ADP-ribosylated AHR but not its dimerization partner, the AHR nuclear translocator, and the repressive effects of TIPARP on AHR were reversed by the macrodomain containing mono-ADP-ribosylase MACROD1 but not MACROD2. These results reveal previously unidentified roles for Tiparp, MacroD1, and ADP-ribosylation in AHR-mediated steatohepatitis and lethality in response to dioxin.
Introduction
The aryl hydrocarbon receptor (AHR)2 is a ligand-activated transcription factor and member of the basic helix-loop-helix/Per-AHR nuclear translocator (ARNT)-Sim (PAS) protein family that mediates the toxicities of the environmental contaminant, dioxin (2,3,7,8-tetrachlorodibenzo-p-dioxin; TCDD) (1). In the absence of ligand, such as dioxin, the AHR exists in the cytoplasm bound to a chaperone protein complex, containing heat shock protein 90, AHR-interacting protein (AIP; ARA9), and a 23-kDa co-chaperone protein, p23 (2–4). After binding dioxin, the AHR translocates to the nucleus, dimerizes with ARNT, and regulates the transcription of target genes such as the drug-metabolizing enzymes CYP1A1 and CYP1B1 as well as AHR repressor (AHRR), a tissue- and context-specific repressor of AHR (5–7). Activated AHR is also regulated through proteasome-mediated degradation (8). In addition to its roles in xenobiotic metabolism, dioxin toxicity, and vascular development (9), AHR has been reported to have an important role in T-cell differentiation and in the defense against bacterial infections (10, 11).
Dioxin is a highly toxic environmental contaminant produced during waste incineration and other high temperature industrial processes, and it remains a global health concern. Dioxin causes diverse toxic effects in laboratory rodents, including immunosuppression, steatohepatitis, neurobehavioral alterations, teratogenesis, impaired reproduction, and a lethal wasting syndrome (12–14). Because dioxin is linked to appetite suppression, body weight, and energy homeostasis, understanding the mechanisms of the wasting syndrome could have important implications for obesity and metabolic diseases. Acute lethality to dioxin varies among species and within rodent strains. For example, the lethal dose 50 (LD50) for guinea pigs, one of the most sensitive species, is 1–2 μg/kg bw dioxin, whereas the LD50 for hamsters is >5000 μg/kg bw (12, 15). In most strains of mice, lethality occurs only 2 or 3 weeks after a single dose of 114–300 μg/kg of dioxin, and it is associated with inhibition of gluconeogenesis but also with feed intake refusal and body weight loss (12, 15). There is approximately a 10-fold difference in susceptibility between the high sensitive C57BL/6 and low sensitive DBA/2 strains, which is due to polymorphic variations in the ligand-binding domain and in the C-terminal transactivation domain of the Ahr in each strain (16). C57BL/6 mice harbor the high affinity Ahrb1 b1 allele, whereas DBA/2 and 129 substrains harbor the low affinity Ahrd allele (17, 18). The molecular mechanisms of the dioxin-induced wasting syndrome remain obscure, but Ahr-deficient (Ahr−/−) mice are resistant to this and other dioxin-dependent outcomes, indicating a key role for AHR in dioxin-induced toxicity (19).
TCDD-inducible poly(ADP-ribose) polymerase (Tiparp; Parp7; Artd14) is an Ahr target gene that is expressed in many different tissues, including liver, heart, spleen, brain, and reproductive organs (20). TIPARP is a member of the poly(ADP)-ribose polymerase (PARP) family that uses NAD+ as a substrate to transfer ADP-ribose onto target proteins. TIPARP contains a CCCH-type zinc finger, a tryptophan-tryptophan-glutamate (WWE) domain involved in protein-protein interactions and a C-terminal catalytic domain (21). The majority of PARP family members transfer mono-ADP-ribose, and not poly-ADP-ribose to their substrates (22). ADP-ribosylation is a post-translational modification involved in several biological processes, such as immune cell function, the regulation of transcription, and DNA repair (23). ADP-ribosylation is reversible with its removal being catalyzed by macrodomain-containing proteins, such as macrodomain 1 (MacroD1) and macrodomain 2 (MacroD2) (24).
We recently reported that TIPARP mono-ADP-ribosylates itself and core histones, and as part of a negative feedback loop it represses AHR activity by increasing its proteolytic degradation (25). However, the impact of TIPARP on AHR signaling and dioxin-induced toxicity in vivo has not been determined. Here, we show that the loss of Tiparp increases sensitivity to dioxin-induced steatohepatitis and lethality. We also provide evidence that Tiparp mono-ADP-ribosylates AHR, but not ARNT, and that the actions of TIPARP are reversed by the ADP-ribosylase, MACROD1. Overall, our findings reveal previously unidentified roles for Tiparp, MACROD1, and ADP-ribosylation in AHR signaling, dioxin toxicity, and lethality.
Experimental Procedures
Mice
TiparpGt(ROSA)79Sor (26) were purchased from The Jackson Laboratory (Bar Harbor, ME). These mutant mice were produced on a mixed C57BL/6;129S4 background (26), before being backcrossed 2 to 3 generations onto C57BL/6.3 The mice were then backcrossed one generation onto C57BL/6J (stock no. 000664) at The Jackson Laboratory prior to being received in our laboratory. The exact number of backcrosses onto C57BL/6 was not known when we initiated these studies; however, single nucleotide polymorphism analysis done by The Jackson Laboratory revealed the mice to be ∼90% C57BL/6 or that they have been backcrossed at least three generations onto C57BL/6. Congenic C57BL/6 and 129S mice used for Ahr genotyping were from Charles River (Montreal, Quebec, Canada). The mice we received from the cryopreserved TiparpGt(ROSA)79Sor strain were maintained by breeding heterozygous mice throughout this study. Eight-week-old mice were injected intraperitoneally with 30 μg/kg bw of dioxin (Wellington Laboratories) dissolved in corn oil (Sigma) and 10 or 100 μg/kg bw of dioxin dissolved in dimethyl sulfoxide (DMSO; Sigma). Control mice received an equivalent volume of corn oil or DMSO. For the mortality studies, mice were followed for up to 30 days after a single injection of 10 or 100 μg/kg of dioxin. For food intake studies, mice were housed individually and provided intact pellets of food that were weighed daily. A baseline was determined for each mouse by monitoring food intake for 1 week prior to treatment. Briefly, standard mouse pellets (Harlan Laboratories) that weighed ∼4–5 g each were placed on top of the wire mouse feeder and a single pellet placed in a dish on the cage floor. To obtain the amount of food a mouse ate, we weighed the total amount of food the day before. The next morning, the food pellets on the wire feeder, the single pellet in the dish, and any additional pieces were collected and weighed. This amount was subtracted from the measurement made the day before to give us the food intake value. Because there was only a single pellet placed on the floor of the cage, with the rest of the pellets in the wire rack, contamination from water or urine was assumed to be minimal in comparison with the total amount of food and similar among the different cages. Coprophagia could not be accounted for under these conditions. After treatment, food intake (g/g of body weight) was calculated daily until the end of the study. To determine serum glucose levels, whole blood was collected from mice treated for 1–3 days with 100 μg/kg bw dioxin. Mice were fasted for 4 h prior to blood collection. Serum was then isolated and analyzed using the Autokit glucose reagent (Wako Diagnostics) following the manufacturer's recommendations. Care and treatment of animals followed the guidelines set by the Canadian Council on Animal Care and was approved by the University of Toronto Animal Care Committee.
Genotyping for Tiparp, Ahrb1, and Ahrd
Genomic DNA was isolated from the tails of pups born from Tiparp+/− intercrosses. Tiparp genotypes were determined by PCR analysis of tail biopsies. The common forward primer was 5′-TGTCAGATCCCTCCTTCGTGAGGC-3′, the Tiparp+/+ reverse primer was 5′-GTATAGTACCTAGCACTGTTCACC-3′, and the Tiparp−/− reverse primer was 5′-GAAGCCTATAGAGTACGAGCCGTAGA-3′. Homozygous null mice were designated Tiparp−/−. A 20-μl reaction contained 1× Jumpstart Taq buffer and 0.2 units of Jumpstart Taq polymerase (Sigma), 1.75 mm MgCl2, 0.2 mm dNTPs, 0.2 μm of each primer, and 2 μl of DNA. PCR cycling conditions included an initial step of 3 min at 94 °C that was followed by 35 cycles of 30 s at 94 °C to denature DNA, 30 s at 59 °C for primer annealing, and 70 °C for 45 s for elongation. This was followed by a final step of 7 min at 70 °C. PCR products were resolved using 1% agarose gel electrophoresis followed by visualization with ethidium bromide.
The Ahr genotypes were determined using real time quantitative PCR (qPCR). Genomic DNA used to genotype Tiparp was purified using GenElute Mammalian Genomic DNA Miniprep kit (Sigma). Discrimination between the Ahrb1 and Ahrd alleles was based on common Ahr forward 5′-TCGACATAACGGACGAATCC-3′ and reverse 5′-CATACAGCTTAGGTGCTGAGTC-3′ primers and allele-specific probes designed to take advantage of differences between the Ahrb1 and Ahrd alleles in the exon 10 sequence of the Ahr gene (18, 27). Ahrb1 allele-specific probe was 5′6FAMTCAACTTTGZENCTGAACTCGGCTTGCIABkFQ-3′. Ahrd-allele specific probe was 5′-6FAMAACTTTGAaZENGAACTCCGCTTGCIABkFQ-3′. The underlined nucleotides distinguish the Ahr alleles, and the lowercase boldface nucleotide does not match the mouse genomic sequence and represents a T to A point mutation that was necessary for specificity of the Ahrd probe for the Ahrd rather than the Ahrb1 allele. 6FAM (6-carboxyfluorescein, reporter dye), ZEN (internal quencher), and IABkFQ (Iowa Black FQ, quencher dye) were included on the probes (Integrated DNA Technologies, Coralville, IA). A 10-μl reaction was set up using 25 ng of genomic DNA, 2× Kapa Probe reaction mix (Kapa Biosystems), 0.9 μm forward primer, 0.3 μm reverse primer, and 0.2 μm probe. qPCR conditions to detect the Ahrb1 allele using the Ahrb1 probe were 95 °C for 5 min followed by 40 cycles of 95 °C for 10 s and 64 °C for 40 s. For detection of the Ahrd allele using the Ahrd probe, the following qPCR conditions were used, 95 °C for 5 min followed by 40 cycles of 95 °C for 10 s and 59 °C for 40 s. All samples were also analyzed using Kapa SYBR FAST Green reaction mix, 0.9 μm forward primer, 0.3 μm reverse primer using the following qPCR conditions 95 °C for 5 min, followed by 40 cycles of 95 °C for 10 s and 59 °C for 40 s. Ahr probe-specific reactions were normalized to Ahr gene levels detected by Kapa SYBR FAST Green and analyzed using the comparative CT method.
RNA Extraction and Gene Expression Analysis
Livers were removed, washed in ice-cold PBS, weighed, and then flash-frozen in liquid nitrogen. Frozen livers were homogenized in TRIzol reagent (Life Technologies, Inc.), and total RNA was isolated using the Qiagen RNeasy Plus kit and reverse-transcribed as described previously (25). Primers used to amplify lipoprotein lipase (Lpl) were forward 5′-GGATGGACGGTAACGGGAAT-3′ and reverse 5′-GGCCCGATACAACCAGTCTA-3′; cluster of differentiation 36 (Cd36) were forward 5′-ACCCCTCCAGAATCCAGACA-3′ and reverse 5′-ACACAGGCTTTCCTTCTTTGC-3′; phosphoenolpyruvate carboxykinase (Pck1) were forward 5′-CCTAGTGCCTGTGGGAAGAC-3′ and reverse 5′-AAGTTGCCTTGGGCATCAAAC-3′; sterol regulatory element-binding transcription factor 1 (Srebp1) 5′-GCACACAAAAGCAAATCACTG-3′ and reverse 5′-TCTCCACCACTTCGGGTTTC-3′; fatty acid synthase (Fas) 5′-GCTGCGGAAACTTCAGGAAAT-3′and reverse 5′-AGAGACGTGTCACTCCTGGACTT-3′; Cyp1a1 5′-CGTTATGACCATGATGACCAAGA-3′ and reverse 5′-TCCCCAAACTCATTGCTCAGAT-3′; Cyp1b1 forward 5′-GTGCGGCAAAAGCATGTCTC and reverse 5′-GGGGAAAAGCAACGTTCTGAC-3′; Tiparp forward 5′-TCCCCGTGTCTGTGGAAAGCATG-3′ and reverse 5′-TTGACCGGAGGGGGCCTTCT-3′; tumor necrosis factor α (Tnfα) forward 5′-AGGGTCTGGGCCATAGAACT-3′ and reverse 5′-CCACCACGCTCTTCTGTCTAC-3′; interleukin 1 β (Il1β) forward 5′-GGACCCATATGAGCTGAAAGCT-3′ and reverse 5′-TGTCGTTGCTTGGTTCTCCTT-3′; and chemokine CXC motif ligand 2 (Cxcl2) forward 5′-AAGTTTGCCTTGACCCTGAA-3′ and reverse 5′-AGGCACATCAGGTACGATCC-3′. All genes were amplified using Kapa SYBR Green 2× mix, normalized to ribosomal 18S levels and analyzed using the comparative CT method. For the microarray studies, RNA was isolated from male liver tissues treated with 30 μg/kg dioxin. RNA was processed, labeled, and hybridized to the GeneChIP Mouse Exon 1.0 ST array (Affymetrix) at The Centre for Applied Genomics (Toronto, Ontario, Canada). Differentially regulated genes with an absolute fold change of ≥1.5 and p value ≤0.05 were identified using Affymetrix Transcriptome Analysis software. The average gene signal was converted to a log2 scale using a Tukey's bi-weight average for the gene signal intensity for all the samples in a treatment condition. Traditional unpaired one-way ANOVA was performed for dioxin-treated versus corn oil-treated Tiparp+/+ and dioxin-treated versus corn oil-treated Tiparp−/− samples. Microarray data have been deposited in the National Center for Biotechnology Information's Gene Expression Omnibus with accession number GSE61534.
Reporter Gene Assays
For luciferase reporter gene assays, HuH7 cells were transfected with pcDNA-TIPARP (25) and Cyp1a1 luciferase plasmid (pGudLuc; generously provided by Dr. Michael Denison, University of California at Davis) or pCMV-FLAG-MACROD1, pCMV-FLAG-MACROD1_G182E, pCMV-FLAG-MACROD1_D184A, or pCMV-FLAG-MACROD2 and treated with 10 nm dioxin for 24 h as described previously (25). Human MacroD1 and MacroD2 cDNAs were amplified by PCR using pCMV6 Myc-DDK MACROD1 or pCMV6 Myc-DDK MACROD2 variant 1 as templates, respectively (Origene, Rockville, MD). The PCR primers for MACROD1 were forward primer 5′-CAAAGAATTCATGTCTCTACAGAGCCGACTGTC-3′ and reverse 5′-CAAAGTCGACGGCCACGGGGAAGTAGTGGG-3′, and for MACROD2 were forward primer 5′-CAAAGAATTCATGTACCCCAGCAACAAGAAGAAAAA-3′ and reverse 5′-CAAACTCGAGTTTAGTTCCATTTCTTTGTTCCTTCG-3′. Following amplification, the MacroD1 sequence was digested with EcoRI and SalI, and the MacroD2 sequence was digested with EcoRI and XhoI. The digested sequences were cloned into EcoRI and XhoI sites of pCMV-2B (Invitrogen). The plasmids pCMV-FLAG-MACROD1_G182E and pCMV-FLAG-MACROD1_D184A were generated by PCR mutagenesis using the forward primer 5′-TCCCTGCTCGGAGGCGGTGAAGTGGACGGCTGCATTCATCGG-3′ and reverse primer 5′-CCGATGAATGCAGCCGTCCACTTCACCGCCTCCGAGCAGGGA-3′ for G182E, and forward primer 5′-CTGCTCGGAGGCGGTGGCGTGGCCGGCTGCATTCATCGG-3′ and reverse primer 5′-CCGATGAATGCAGCCGGCCACGCCACCGCCTCCGAGCAG-3′ for D184A. Nonsynonymous SNPs in the human TIPARP were selected from Exome Variant Server, NHLBI GO Exome Sequencing Project (ESP), Seattle, WA. The TIPARP plasmids containing the different SNPs were generated using PCR mutagenesis with the following PCR primer: F40S (rs369539588), 5′-CTCCATTGAAGACTTGTTCTAAGAAAAAGGATCAGAAA-3′, 5′-TTTCTGATCCTTTTTCTTAGAACAAGTCTTCAATGGAG-3′; K42R (rs144427222), 5′-CATTGAAGACTTGTTTTAAGAGAAAGGATCAGAAAAGATTG-3′, 5′-CAATCTTTTCTGATCCTTTCTCTTAAAACAAGTCTTCAATG-3′; N238D (rs140486151), 5′-TACCACACTCACCAAGAGGACGGAATTGAAATTTGCATG-3′, 5′-CATGCAAATTTCAATTCCGTCCTCTTGGTGAGTGTGGTA-3′; N338S (rs148252230), 5′-TCCACACCACCCTCTAGCAGTGTCAACTCTATTTACCAC-3′, 5′-GTGGTAAATAGAGTTGACACTGCTAGAGGGTGGTGTGGA-3′; P361A (rs141280180), 5′-TTTGGATGGAGAGAGTATGCCGAGTCTGTCATTCGATTG-3′, 5′-CA ATCGAATGACAGACTCGGCATACTCTCTCCATCCAAA-3′; E362K (rs150739414), 5′-GGATGGAGAGAGTATCCCAAGTCTGTCATTCGATTGATT-3′, 5′-AATCAATCGAATGACAGACTTGGGATACTCTCTCCATCC-3′; I365V (rs139609776), 5′-GAGTATCCCGAGTCTGTCGTTCGATTGATTGAAGAAGCC-3′, 5′-GGCTTCTTCAATCAATCGAACGACAGACTCGGGATACTC-3′; N372T (rs143409745), 5′-CGATTGATTGAAGAAGCCACCTCTCGGGGTCTGAAAGAG-3′, 5′-CTCTTTCAGACCCCGAGAGGTGGCTTCTTCAATCAATCG-3′; F381S (rs147143306), 5′-GGTCTGAAAGAGGTTCGATCTATGATGTGGAATAACCAC-3′, 5′-GTGGTTATTCCACATCATAGATCGAACCTCTTTCAGACC-3′; L412F (rs140142843), 5′-CGCTCCTGTTTTATACTGTTTCCATATTTACAGACACTT-3′, 5′-AAGTGTCTGTAAATATGGAAACAGTATAAAACAGGAGCG-3′; A425P (rs200460123), 5′-GGTGGGGTTCCCACACAACCTCCTCCACCTCTTGAAGCA-3′, 5′-TGCTTCAAGAGGTGGAGGAGGTTGTGTGGGAACCCCACC-3′; R475W (rs150068199), 5′-GCAGAGGATAAAAGTTATTGGATCATTTACAATCTTTTT-3′, 5′-AAAAAGATTGTAAATGATCCAATAACTTTTATCCTCTGC-3′; N479S (rs145315559), 5′-AGTTATCGGATCATTTACAGTCTTTTTCATAAGACTGTG-3′, 5′-CACAGTCTTATGAAAAAGACTGTAAATGATCCGATAACT-3′; E487D (rs370596394), 5′-TTTCATAAGACTGTGCCTGACTTTAAATACAGAATTTTG-3′, 5′-CAAAATTCTGTATTTAAAGTCAGGCACAGTCTTATGAAA-3′; R521C (rs146547620), 5′-GAACAGGAAAATGTTTGGCTGTGACAGGATAATAAATGAG-3′, 5′-CTCATTTATTATCCTGTCACAGCCAAACATTTTCCTGTTC-3′; N526S (rs150170944), 5′-GCCGTGACAGGATAATAAGTGAGAGACATTTATTTCA-3′, 5′-TGAAATAAATGTCTCTCACTTATTATCCTGTCACGGC-3′; M558T (rs368152871), 5′-TGTGGAAAGCATGCTACAACGTTTGGACAAGGCAGTTATT-3′, 5′-AATAACTGCCTTGTCCAAACGTTGTAGCATGCTTTCCACA-3′.
Histology
Liver tissues from 100 μg/kg dioxin-treated animals were collected on days 1, 3, or 6. Control tissue was from DMSO-treated animals on days 3 or 6 after dosing. Liver tissues from 10 μg/kg dioxin-treated Tiparp−/− animals were collected after the mice were euthanized, which ranged from days 5 to 13 after dosing. Tiparp+/+ animals were euthanized to day-match the Tiparp−/− animals, and their liver tissues were collected. Staining for β-galactosidase activity to visualize cells expressing the lacZ reporter (inserted in intron 1 of the Tiparp gene) was performed on optimal cutting temperature compound (OCT)-embedded tissue following standard methods. H&E staining was done following standard methods with representative images obtained using an AxioImager M2 microscope, AxioCam MRm camera, and AxioVision software (Zeiss). Oil-Red-O/hematoxylin staining was done on sections from OCT-embedded tissue using standard methods. Images were acquired using a Nikon Eclipse 80i microscope and Nikon Elements software (NIS-Elements Basic Research, version 3.10).
Tissue Lipid Analysis
Livers from mice treated with 100 μg/kg dioxin were flash-frozen and used to measure tissue triglyceride, free fatty acid, and cholesterol levels. Free fatty acids were quantified using a kit following the manufacturer's recommendations (MAK044, Sigma). Triglycerides and cholesterol were extracted from livers in chloroform/methanol (2:1, v/v) and quantified as described previously (29).
NAD Detection Assay
Total cellular NAD was determined using NAD/NADH quantification kit (MAK037; Sigma). Briefly, 20 mg of liver tissues was homogenized in NADH/NAD extraction buffer. The samples were clarified by centrifugation, and the supernatant was deproteinized by filtering it though a 10-kDa cutoff spin filter (Millipore). The assay was then performed according to the manufacturer's instructions. Values were corrected for dilutions and protein content of the samples.
ADP-ribosylation Assays
GST-tagged fusion proteins were purified according to standard procedures. ADP-ribosylation assays were carried out as described previously (25). Human PARP10 (ARTD10) cDNA was PCR-amplified using pEV-HA-RFO-ARTD10 (PARP10) as template (a gift from Professor Bernard Lüscher, IBMB Aachen, Germany). PCR-amplified sequence was digested with EcoRI and XhoI and cloned into the appropriately digested pGEX-4T1 plasmid. Briefly, ∼1 μg of GST-mouse Tiparp (mTiparp), human TIPARP (hTIPARP), or chicken (chTiparp) fusion protein was incubated with ∼4 μg of GST-AHR(1–434), GST-AHR(430–848), GST-ARNT(1–450), GST-ARNT(440–789), GST-AIP, GST-PCK1, GST-MACROD1, or GST-MACROD2. Note a C-terminal histidine tag was added to GST-AHR(430–848) and GST-ARNT(1–450), which helped reduce protein degradation during purification. For GST-PARP10 reactions, ∼1 μg of fusion protein was incubated with ∼4 μg of GST-AHR(430–848). ADP-ribosylation assays were done in 30-μl reactions and incubated at 30 °C for 20 min in reaction buffer (50 mm Tris base, pH 8.0, 4 mm MgCl2, 0.2 mm DTT) and 5 μCi o f32P-NAD+ (PerkinElmer Life Sciences) and/or 500 μm β-NAD+ (Sigma). Reactions were stopped by the addition of sample buffer, separated by SDS-PAGE, and visualized by autoradiography. Prior to autoradiography, PVDF membranes were stained with GelCode Blue to visualize the proteins and to ensure equal loading.
Western Blots and Co-immunoprecipitation Assays
For hepatic Ahr protein detection, whole cell extracts were prepared by homogenizing 100 mg of liver tissue in RIPA buffer. Ten micrograms of total protein was separated by SDS-PAGE and transferred to a nitrocellulose membrane. Membranes were incubated with anti-AHR antibody (SA-210; Enzo Scientific),stripped, and then incubated with anti-β-actin antibody (Sigma A-2228). For hepatic Cyp1a1 and Cyp1b1 protein detection, microsomes were prepared as described previously (30). Five micrograms of microsomal protein was separated by SDS-PAGE and transferred to a nitrocellulose membrane. The membranes were incubated with anti-Cyp1a1 (31), anti-Cyp1b1 (H105, Santa Cruz Biotechnology), or anti-ERp29 (Abcam ab11420). For transfection experiments, HuH7 cells were seeded in 6-well plates and transfected with pEGFP-TIPARP (WT) or pCMV-FLAG-MacroD1 with Lipofectamine 3000 (Life Technologies, Inc.). Six hours following transfection, cells were treated with 10 nm dioxin or DMSO for 18 h, and whole cell extracts were prepared. Cell extracts were separated by SDS-PAGE and transferred to a PVDF membrane. The membranes were incubated with anti-CYP1A1 (B4, Santa Cruz Biotechnology), anti-GFP (JL8, Clontech), anti-FLAG, or anti-β-actin. COS-1 cells were seeded in 6-well plates and transfected with pEGFP-TIPARP (WT or H532A), pRC-AHR, pCMV-FLAG-MACROD1, or pCMV-FLAG-MACROD2 with Lipofectamine 2000 (Life Technologies, Inc.). Six hours following transfection, cells were treated with 10 nm dioxin or DMSO for 18 h, and whole cell extracts were prepared. Two micrograms of anti-FLAG M2 (F1804, Sigma) antibody were incubated with 100 μg of whole cell extracts and protein G-agarose beads for 3 h at 4 °C. Beads were washed five times and eluted in sample buffer, separated by SDS-PAGE, and transferred to a nitrocellulose or PVDF membranes. Membranes were exposed to anti-GFP (Clontech) or anti-FLAG antibodies. Input samples were probed with anti-FLAG, anti-GFP, anti-AHR (H-211, Santa Cruz Biotechnology), or anti-β-actin. After washing, bands were visualized using SuperSignal substrate (Pierce). Western blots were quantified using ImageJ (National Institutes of Health).
Immunofluorescence
HuH7 cells were seeded onto coverslips and transfected with pcDNA-MacroD1. Twenty four hours after transfection, cells were incubated with 1 μm Mitotracker Deep Red 633 FM mitochondrial probe (Molecular Probes, M22426) in growth medium for 20 min and then fixed in 4% paraformaldehyde and permeabilized with 0.4% Triton X-100 in PBS for 20 min. Cells were incubated with anti-MacroD1 (Abcam, ab122688) followed by goat anti-rabbit-AlexaFluor® 568 (Life Technologies, Inc.) and mounted with Vectashield medium containing DAPI (Vector Laboratories). Images were acquired using an AxioImager M2 microscope, AxioCam MRm camera, and AxioVision software (Zeiss).
Statistical Analysis
All data were presented as means and S.E. Two-way or one-way analysis of variance (ANOVA) followed by Tukey's post hoc test was used to assess statistical significance (p < 0.05) using GraphPad Prism Software (San Diego).
Results
Characterization of Tiparp−/− Mice
To determine the role of Tiparp in dioxin toxicity, we established a colony of B6;129S4-TiparpGt(ROSA)79Sor (Tiparp−/−) mice and exposed them to dioxin. The characterization and creation of the Tiparp−/− mice have been described elsewhere (26). Because female Tiparp−/− mice are sterile (26), we intercrossed mice heterozygous for the Tiparp allele to generate the Tiparp+/+ (+/+), heterozygous (+/−), and homozygous null (−/−) animals (Fig. 1A). Because all the mice used in our study were generated from Tiparp+/− mice on a mixed C57BL/6;129S4 background that were only backcrossed at least three generations onto C57BL/6, we developed an Ahrb1 allele- and Ahrd allele-specific qPCR fluorescent probe-based Ahr genotyping assay. Genomic DNA isolated from 15 Tiparp+/+, 18 Tiparp−/−, 4 C57BL/6 (B6), and 4 129S mice were amplified using common Ahr forward and reverse primers and either an Ahrb1 allele- (Fig. 1B) or Ahrd allele-specific (Fig. 1C) fluorescent probe. The same genomic DNA samples were also amplified using the same common Ahr primers but in the presence of SYBR Green to normalize Ahr levels in all samples as described under “Experimental Procedures.” As expected, the Ahrb1 allele-specific probe only detected Ahr in genomic DNA isolated from B6, but not 129 mice, whereas the Ahrd allele-specific probe only detected Ahr in genomic DNA isolated from 129 mice but not B6 mice (Fig. 1, B and C). The Ahr gene amplified from all samples isolated from Tiparp+/+ and Tiparp−/− mice was only detected by the Ahrb1 allele- but not the Ahrd allele-specific probe, indicating that the mice harbored the Ahrb1 allele (Fig. 1, B and C). The lack of detection of the Ahr by the Ahrd allele-specific probe was not due to a lack of amplification of the Ahr gene sequence in the Tiparp+/+ (WT) and Tiparp−/− (KO), because the ratio of Ahr between WT/KO detected by SYBR Green was ∼1 (Fig. 1C). Dioxin-stimulated increases in hepatic Tiparp mRNA levels were observed in Tiparp+/+ but not in Tiparp−/− mice (Fig. 1D). Because the βgeo (a fusion of neo and lacZ) gene is inserted downstream of the Tiparp promoter, we used X-Gal staining of livers isolated from Tiparp+/+ or Tiparp−/− mice treated with a single intraperitoneal dose of 100 μg/kg dioxin. Negligible X-Gal staining was observed in livers from DMSO-treated mice. X-Gal staining was observed in livers from Tiparp−/− treated with dioxin for 24 h, and this was increased at 48 h of treatment. No X-Gal staining was observed in livers from similarly treated Tiparp+/+ mice (Fig. 1E).
FIGURE 1.
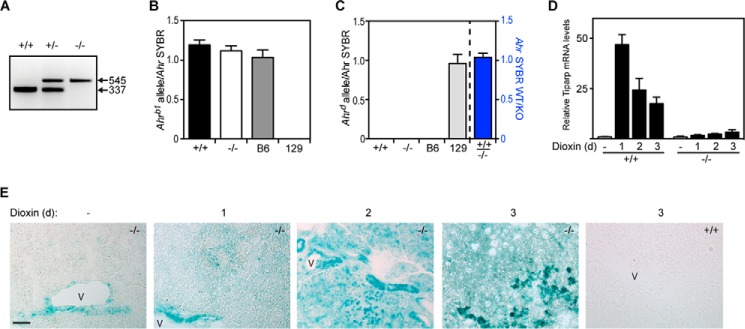
Characterization of Tiparp−/− mice. A, PCR genotyping showing bands of 337 and 545 bp indicating the wild type and mutant alleles, respectively. qPCR genotyping of the Ahr gene using an Ahrb1 allele-specific fluorescent probe (B) or Ahrd allele-specific probe (C). qPCR was performed on genomic DNA from n = 15 (Tiparp+/+ (+/+)), n = 18 Tiparp−/− (−/−), n = 4 C57BL/6 (B6), and n = 4 129 mice. The data were analyzed as described under “Experimental Procedures.” D, Dioxin- and time-dependent changes in Tiparp mRNA levels in male Tiparp+/+ (+/+) and Tiparp−/− (−/−) mice treated with DMSO or 100 μg/kg dioxin for 1–3 days (n = 4). E, representative hepatic β-galactosidase staining in mice euthanized 1–3 days after treatment. β-Galactosidase expression increased with time and was present throughout the liver with the highest levels seen in cells surrounding blood vessels (v). Control sections were from Tiparp+/+ livers treated with dioxin for 3 days. Scale bar represents 50 μm, and all images are to the same scale.
Loss of Tiparp Increases Dioxin-induced Gene and Protein Expression Levels
We next examined the effect of Tiparp deletion on hepatic AHR target gene expression in male mice treated for 6 and 24 h with corn oil or 30 μg/kg dioxin. Male Tiparp−/− mice treated for 6h exhibited increased hepatic expression of Cyp1a1 (Fig. 2, A and B) mRNA and protein levels compared with Tiparp+/+ animals. Despite increased Cyp1b1 mRNA levels, no protein was detected after 6 h of dioxin treatment (Fig. 2, C and D). The increased dioxin-induced hepatic Cyp1a1 and Cyp1b1 mRNA and protein levels observed in Tiparp−/− compared with Tiparp+/+ mice were also evident at 24 h (Fig. 2, E–H). A similar lag in the detection of dioxin-induced hepatic Cyp1b1 protein levels compared with mRNA levels has been previously reported (32, 33). Hepatic gene expression microarray studies supported the increased dioxin responsiveness of Tiparp−/− mice. We identified 766 genes with an absolute fold change of ≥1.5 (p < 0.05) in Tiparp−/− mice (supplemental Table 1) compared with 169 genes in Tiparp+/+ mice (supplemental Table 2), with 79 genes common to both datasets (Fig. 2I). Using Western blotting (Fig. 2J), we observed reduced dioxin-induced proteolytic degradation of Ahr protein levels in liver extracts from Tiparp−/− compared with Tiparp+/+ mice. This was in agreement with studies in Tiparp−/− mouse embryonic fibroblasts (25), and it further supports a role for Tiparp in the regulation of Ahr protein levels.
FIGURE 2.
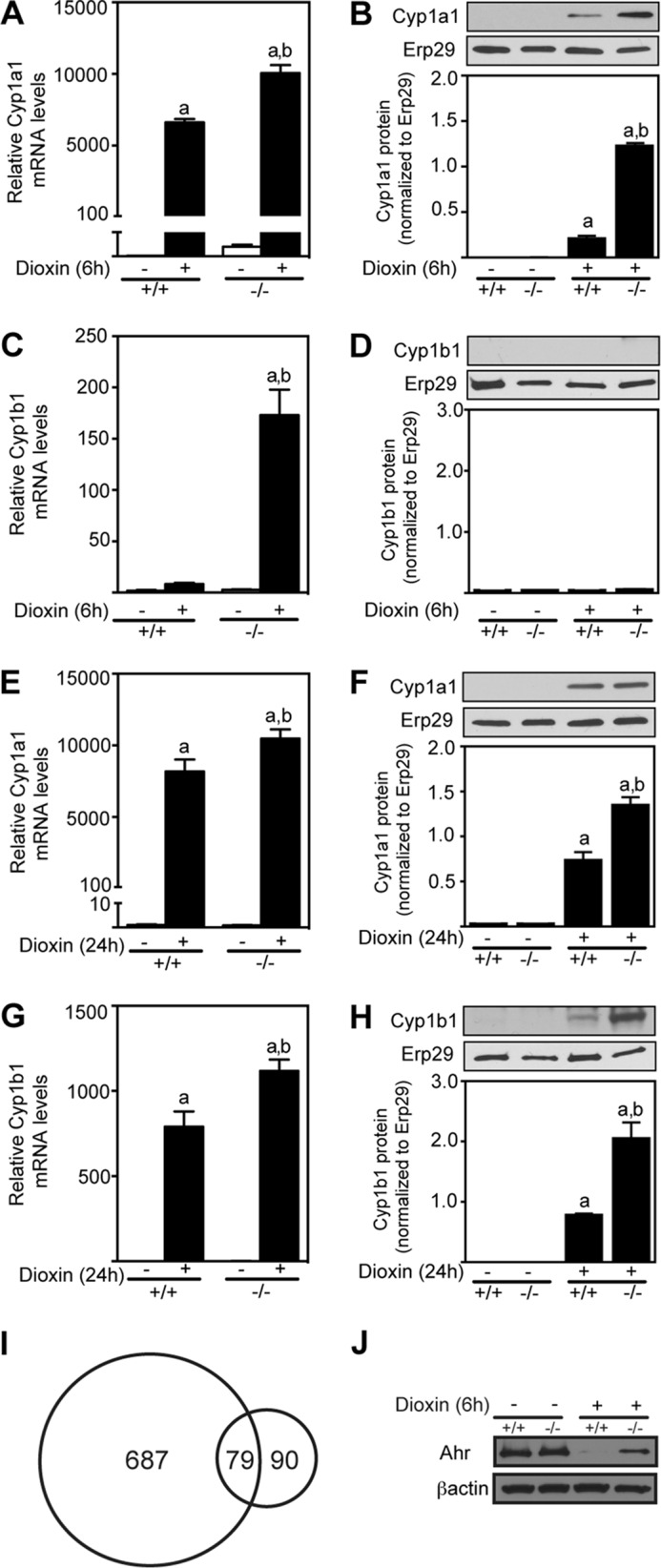
Dioxin-induced hepatic Cyp1a1 and Cyp1b1 mRNA and protein expression levels are increased in Tiparp−/− mice. Male Tiparp+/+ (+/+) and Tiparp−/− (−/−) mice were treated with either corn oil (−) or 30 μg/kg dioxin (+) for 6 h (A–D) or 24 h (E–H). The Cyp1a1 (A and E) and Cyp1b1 (C and G) mRNA levels are shown relative to time-matched control (no dioxin (−))-treated Tiparp+/+ (+/+) samples. Data represent the mean ± S.E. (n = 4). Representative Cyp1a1 (B and F), Cyp1b1 (D and H), and Erp29 microsomal protein levels were detected by Western blotting after 6 and 24 h. Cyp1a1 and Cyp1b1 proteins levels were normalized to Erp29 levels. Data are representative of three animals per treatment group. I, overlap of differentially regulated dioxin-induced hepatic genes in Tiparp+/+ and Tiparp−/− mice treated for 6 h. J, hepatic Ahr and β-actin protein levels were detected by Western blotting after 6 h of dioxin treatment. Data are representative of 3–4 animals per treatment group. a, p < 0.05 two-way ANOVA compared with control Tiparp+/+ mice; b, p < 0.05 two-way ANOVA compared with dioxin-treated Tiparp+/+ mice.
Tiparp−/− Mice Display Increased Sensitivity to Dioxin-induced Toxicity and Lethality
To determine whether the loss of Tiparp affects dioxin-induced toxicity, we injected male and female Tiparp+/+ and Tiparp−/− mice with a single nonlethal dose of 100 μg/kg dioxin and monitored the mice for 6 days as described by others (34). Although 100 μg/kg dioxin is a high dose, a dose 2-fold higher causes lethality between 22 and 26 days in C57BL/6 mice (35) and a dose 20-fold higher is not lethal in Ahr knock-out mice (19). No dioxin-treated male (Fig. 3A) or female (Fig. 4A) Tiparp−/− mice survived the 6-day experiment. All Tiparp−/− mice treated with a single injection of 100 μg/kg dioxin died between days 3 and 5. Dioxin-treated Tiparp−/− mice appeared weakened after 2 days and became moribund between 3 and 5 days and had to be euthanized. All Tiparp+/+ mice were normal in physical appearance at the end of the 30-day observation period (Figs. 3A and 4A).
FIGURE 3.

Loss of Tiparp increases dioxin-induced hepatotoxicity and lethal wasting syndrome in male mice. A, Kaplan-Meier survival curves for male Tiparp+/+ and Tiparp−/− mice treated with a single 100 μg/kg intraperitoneal injection of dioxin and monitored for 30 days. B, body weight; C, food intake; D, serum glucose levels; and E, hepatic Pck1 mRNA levels after dioxin treatment. Thymus (F) and liver weight (G) expressed as percentage of total body weight were measured from Tiparp+/+ and Tiparp−/− mice. H, serum alanine aminotransferase (ALT) activity was determined 3 and 6 days after treatment. Data shown are the mean ± S.E. n = 4–5 (A–E and H); n = 6 (F and G). a, p < 0.05, ANOVA followed by Tukey's post hoc test compared with genotype-matched control-treated mice. (H) b indicates p < 0.05, ANOVA followed by Tukey's post hoc test compared with day 6-treated Tiparp+/+ mice. I, representative H&E staining of livers (n = 3). Control (no dioxin (−)) animals were injected with DMSO and were euthanized on day 3. The asterisks indicate focal inflammatory infiltration; the arrowheads indicate microvesicular steatosis, and the arrow indicates cytoplasmic clearing. Scale bar represents 50 μm, and all images are to the same scale.
FIGURE 4.

Loss of Tiparp increases dioxin-induced hepatotoxicity and lethal wasting syndrome in female mice. A, Kaplan-Meier survival curves for Tiparp+/+ and Tiparp−/− mice. Female mice were treated with a single 100 μg/kg intraperitoneal injection of dioxin and monitored for up to 30 days. B, Tiparp−/− mice exhibit increased weight loss (% body weight relative to pretreatment). C, food intake of Tiparp+/+ and Tiparp−/− mice after dioxin treatment. Serum glucose levels were determined with a commercially available kit (D), and hepatic Pck1 mRNA levels were determined as described under “Experimental Procedures” (E). Thymus (F) and liver (G) weights expressed as percentage of total body weight were measured from Tiparp+/+ and Tiparp−/− mice. Serum alanine-aminotransferase (ALT) activity was determined after 3 and 6 days of treatment (H). Data shown are the mean ± S.E. n = 4–5 (A–E and H); n = 6 (F and G). a, p < 0.05, ANOVA followed by Tukey's post hoc test compared with genotype-matched control treated mice. E, b indicates p < 0.05, ANOVA followed by Tukey's post hoc test compared with day 3 dioxin-treated Tiparp+/+ mice. Mice were given a single injection of 100 μg/kg dioxin and euthanized after 1, 3, or 6 days (n = 3) (I). The control (no dioxin (−)) animals received a single injection of DMSO and were euthanized after 3 days. Liver sections were stained with H&E. The asterisks indicate focal inflammatory infiltration; the arrowheads indicate microvesicular steatosis 3 days post-injection (−/−), and the arrow indicates cytoplasmic clearing 6 days post-injection (+/+). Each image is representative of at least three animals. Scale bar represents 50 μm, and all images are to the same scale.
We then treated male and female Tiparp+/+ and Tiparp−/− mice with a single intraperitoneal injection of 100 μg/kg dioxin and analyzed the animals at days 1 and 3, as well as day 6 for Tiparp+/+ mice only. Tiparp−/− mice exhibited reduced body weight on day 3 (Figs. 3B and 4B) as a result of decreased food intake on days 1 and 2 (Figs. 3C and 4C). No changes in food intake or body weight were observed in Tiparp+/+ mice. Fasting serum glucose levels were reduced from day 1 to 3 in Tiparp−/− mice with no changes observed in Tiparp+/+ animals (Figs. 3D and 4D). However, no difference was observed in dioxin-induced repression of phosphoenolpyruvate carboxykinase (Pck1; Pepck) mRNA levels between genotypes (Figs. 3E and 4E). Dioxin-induced thymic involution and hepatomegaly, two end points associated with dioxin toxicity, responded as expected and were similar in both genotypes at day 3 (Fig. 3, F and G). Comparable findings were observed in female mice (Fig. 4, F and G). Serum alanine aminotransferase activity, a marker of hepatotoxicity, was markedly increased in male (Fig. 3H) and female (Fig. 4H) Tiparp−/− mice compared with Tiparp+/+ mice. We also analyzed liver sections by H&E staining for general pathology and additional evidence of hepatotoxicity (Fig. 3I). DMSO-treated Tiparp+/+ and Tiparp−/− mice displayed histologically normal liver architecture. On day 1, dioxin-treated Tiparp+/+ livers exhibited slight hepatocyte cytoplasmic clearing within periportal regions and inflammatory cell infiltration (Fig. 3I). In contrast, day 1 Tiparp−/− livers displayed increased degenerative changes, including focal inflammatory infiltration and increased cytoplasmic clearing of periportal hepatocytes. On day 3, the livers from Tiparp+/+ mice displayed regions of cytoplasmic clearing and hydropic degeneration, whereas Tiparp−/− mouse livers were characterized by a predominant microvesicular steatosis with regions of focal vacuolation. Six days after dioxin treatment, livers from Tiparp+/+ mice displayed distinct inflammatory cell infiltration and increased clearing of the cytoplasm with the appearance of large vacuoles within hepatocytes. Similar findings were observed in livers isolated from dioxin-treated female mice (Fig. 4, H and I).
We next determined the mRNA levels of Ahr-regulated cytokines (36, 37). Dioxin-induced hepatic tumor necrosis factor α (Tnfα) mRNA levels were similar in day 3-treated Tiparp+/+ and Tiparp−/− mice (Fig. 5A). However, dioxin-treated Tiparp−/− mice exhibited increased hepatic expression of interleukin 1β (Il1β) and chemokine (CXC motif) ligand 2 (Cxcl2) (Fig. 5, B and C), which for Cxcl2 was 100-fold higher when compared with Tiparp+/+ mice.
FIGURE 5.
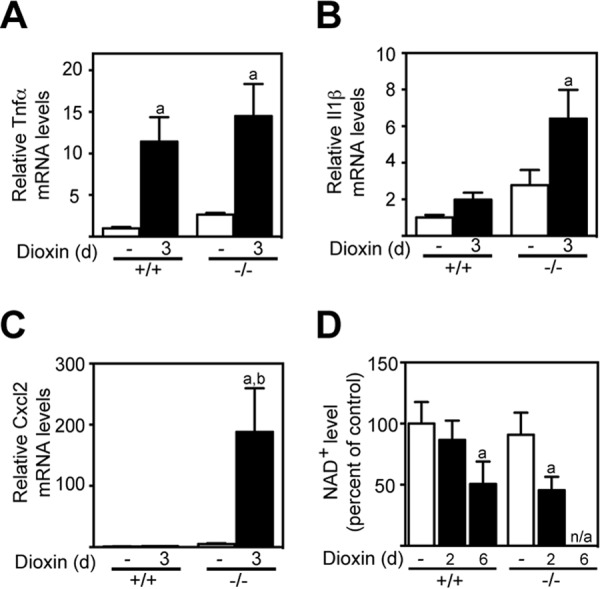
Dioxin increases hepatic cytokine levels and decreases NAD+ levels in Tiparp+/+ and Tiparp−/− mice. Hepatic Tnfα (A), Il1β (B), and Cxcl2 (C) mRNA levels were determined as described under “Experimental Procedures.” D, hepatic NAD+ levels in dioxin-treated Tiparp+/+ and Tiparp−/− mice. Data represent the mean ± S.E. (n = 4). a, p < 0.05 one-way ANOVA compared with control-treated Tiparp+/+. b, p < 0.05 one-way ANOVA compared with dioxin-treated Tiparp+/+.
Dioxin reduces hepatic cellular NAD+ levels, which in turn may alter the activity of NAD+-dependent proteins (38). The dioxin-dependent reduction in cellular NAD+ levels is thought to be due to increased expression of TIPARP and its consumption of NAD+ (39). Hepatic NAD+ levels were unchanged in 2-day dioxin-treated Tiparp+/+ mice but were significantly reduced in day 6-treated animals (Fig. 5D). A similar reduction in hepatic NAD+ levels was observed in day 2-treated Tiparp−/− mice, suggesting that Tiparp is not responsible for dioxin-induced reduction in cellular NAD+ levels.
Deletion of Tiparp Increases Dioxin-induced Hepatosteatosis
Because dioxin causes intrahepatic lipid accumulation in WT but not Ahr-null mice (12, 35), we determined the intrahepatic lipid levels and qualitatively examined for the presence of lipids by Oil-Red-O staining. Macroscopically, DMSO-treated livers from male Tiparp+/+ mice were normal (Fig. 6A). Livers from day 6 dioxin-treated male Tiparp+/+ mice were enlarged and slightly pale in color (Fig. 6A). Livers from day 1 dioxin-treated Tiparp−/− mice were pale in color, and this discoloration was more apparent on day 3, indicative of lipid accumulation. Increases in intrahepatic triglycerides and cholesterol, but not free fatty acids, were observed in day 3 dioxin-treated Tiparp+/+ mice. However, significantly greater increases in intrahepatic free fatty acids, triglycerides, and cholesterol were observed in similarly treated male Tiparp−/− mice (Fig. 6, B–D) and female Tiparp−/− mice (Fig. 7, A–C). Livers isolated from male (Fig. 6E) and female (Fig. 7D) Tiparp+/+ mice 1 day after treatment with dioxin and control livers from Tiparp+/+ and Tiparp−/− mice were negative for Oil-Red-O staining. Only small droplets of lipid were detected in the livers of Tiparp+/+ mice euthanized 3 days after dioxin treatment. On day 6, the livers from Tiparp+/+ mice exhibited increased Oil-Red-O staining in a zonal pattern. Increased lipid accumulation was evident in the livers of dioxin-treated male (Fig. 6E) and female (Fig. 7D) Tiparp−/− mice after day 1, with large amounts of intracytoplasmic lipid accumulation observed at day 3.
FIGURE 6.

Dioxin-induced steatosis is increased in male Tiparp−/− mice. A, livers from male Tiparp+/+ mice given a single intraperitoneal injection of DMSO or 100 μg/kg dioxin and euthanized after 6 days. Similarly treated Tiparp−/− mice were euthanized after 1 or 3 days (n = 4). Intrahepatic free fatty acids (B), cholesterol (C), and triglycerides (D) from Tiparp+/+ and Tiparp−/− mice 3 days after dioxin treatment. Data represent the mean ± S.E. (n = 3). E, Oil-Red-O and hematoxylin-stained liver sections from Tiparp+/+ and Tiparp−/− mice. The control (no dioxin (−)) liver sections are from DMSO-treated mice euthanized 3 days (−/−) and 6 days (+/+) post-injection. The scale bar represents 50 μm, and all images are to the same scale. Hepatic mRNA levels of Lpl (F), Cd36 (G), Srebp1 (H), and Fas (I) were determined as described under “Experimental Procedures.” Data represent the mean ± S.E. (n = 4). For all data, p < 0.05 was determined by ANOVA followed by Tukey's post hoc test comparison. Significantly different compared with DMSO- (a) or dioxin-treated (b) Tiparp+/+ mice.
FIGURE 7.

Dioxin-induced steatosis is increased in female Tiparp−/− mice. Intrahepatic free fatty acids (A), cholesterol (B), and triglycerides (C) from Tiparp+/+ and Tiparp−/− mice after 3 days of treatment were determined with commercially available kits. Data represent the mean ± S.E. (n = 3). D, Oil-Red-O and hematoxylin-stained liver sections from Tiparp+/+ and Tiparp−/− mice treated with DMSO or 100 μg/kg dioxin and euthanized after 1, 3, or 6 days. Dioxin-treated livers had an accumulation of fat (stained red) after 1 day (−/−) or 3 days (+/+). The control (no dioxin (−)) liver sections are representative of livers from DMSO-treated mice euthanized 3 (−/−) and 6 days (+/+) post-injection. The scale bar represents 50 μm, and all images are to the same scale. E–H, hepatic lipid transport and lipogenic gene expression of 3-day dioxin-treated Tiparp+/+ and Tiparp−/− mice. Hepatic mRNA levels of Lpl (E), Cd36 (F), Srebp1 (G), and Fas (H) were determined as described under “Experimental Procedures.” Data represent the mean ± S.E. (n = 4). For all data, p < 0.05 was determined by ANOVA followed by Tukey's post hoc test comparison. Significantly different compared with DMSO-treated (a) or dioxin-treated (b) Tiparp+/+ mice.
We then analyzed the hepatic levels of transcripts encoding genes involved in lipid uptake and lipogenesis. We observed that Lpl mRNA levels were unaffected by dioxin in Tiparp+/+ mice but were increased 6-fold in male Tiparp−/− mice (Fig. 6F). The scavenger receptor encoded by cluster of differentiation 36 (Cd36) was increased 5-fold by dioxin treatment in Tiparp+/+ mice and increased to a greater extent (14-fold) in similarly treated Tiparp−/− mice (Fig. 6G). The dioxin-dependent increases in Cd36 mRNA levels were consistent with previous studies (40, 41). Hepatic expression of lipogenic genes, including sterol regulatory element-binding transcription factor 1 (Srebp1) and fatty acid synthase (Fas), was decreased in Tiparp+/+ mice after dioxin treatment (Fig. 6, H and I). Significantly greater decreases in Srebp1 and Fas mRNA levels were observed in Tiparp−/− mice. We observed similar results in dioxin-treated female mice (Fig. 7, E–H). Collectively, these data suggest that the increased sensitivity of Tiparp−/− mice to dioxin-induced hepatosteatosis was due to increased uptake of circulating lipids rather than increased hepatic lipogenesis.
Single Intraperitoneal Injection of 10 μg/kg bw Dioxin Induces Steatohepatosis and Causes Lethality in Tiparp−/− Mice
Because 100 μg/kg bw dioxin is a relatively high dose, we treated Tiparp+/+ and Tiparp−/− male mice with a single injection of 10 μg/kg dioxin and monitored them for 30 days. Dioxin-treated Tiparp−/− mice did not survive beyond day 8 with the day of death ranging from days 5 to 8 (n = 4) (Fig. 8A). Tiparp−/− mice exhibited reduced bodyweight on days 3 and 5 (Fig. 8B). No changes in body weight were observed in Tiparp+/+ mice. Livers from Tiparp−/− mice were excised after euthanasia and matched with Tiparp+/+ mice that were euthanized on the same day. We analyzed liver sections from Tiparp+/+ and Tiparp−/− mice 5–8 days after a single injection with 10 μg/kg dioxin by H&E (Fig. 8C) and Oil-Red-O (Fig. 8D) staining. These liver sections shown in Fig. 8 were from day 5 animals and were representative of the other three pairs of Tiparp+/+ and Tiparp−/− mice examined. Liver sections from treated Tiparp+/+ mice were normal in appearance, whereas livers from treated Tiparp−/− mice displayed increased focal inflammatory infiltration and increased cytoplasmic clearing of hepatocytes (Fig. 8C). The infiltration of inflammatory cells was more evident than that observed in livers from 100 μg/kg dioxin-treated animals (see Fig. 3 and 4). Oil-Red-O staining revealed small droplets of lipids in the livers of Tiparp+/+ mice euthanized 5 days after 10 μg/kg dioxin treatment (Fig. 8D). In contrast, livers from Tiparp−/− mice exhibited large amounts of intracytoplasmic lipid accumulation. These data further support the increased sensitivity of Tiparp−/− mice to dioxin-induced toxicity, hepatosteatosis, and lethality.
FIGURE 8.
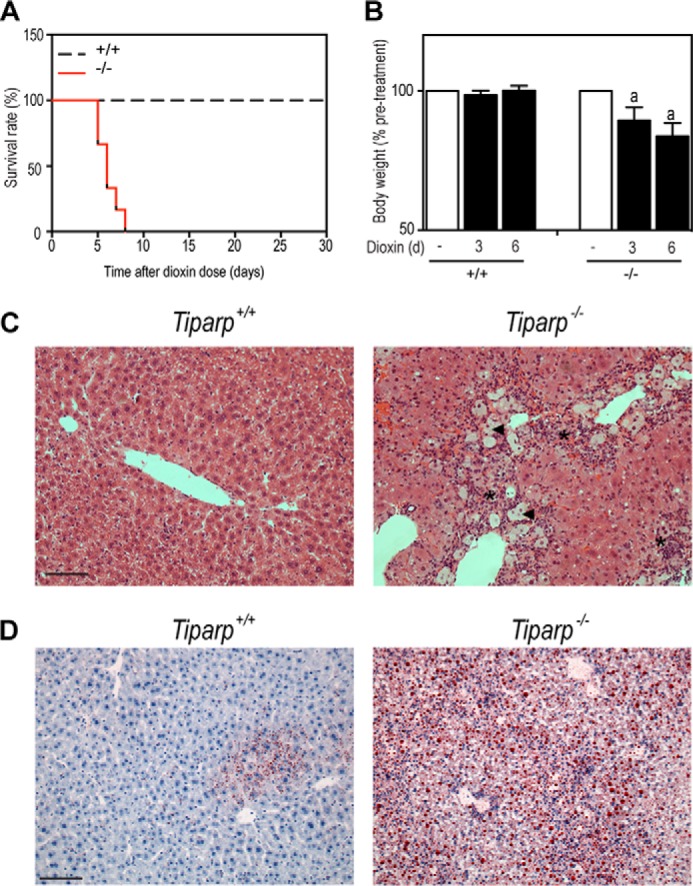
Single intraperitoneal injection of 10 μg/kg bw is lethal to Tiparp−/− mice. A, Kaplan-Meier survival curves for male Tiparp+/+ and Tiparp−/− mice treated with a single 10 μg/kg intraperitoneal injection of dioxin and monitored for 30 days. B, body weight at indicated days after dioxin treatment. C, representative H&E staining of livers (n = 4) from Tiparp+/+ and Tiparp−/− mice on day 5 after receiving a single injection 10 μg/kg dioxin. The asterisks indicate focal inflammatory infiltration, the arrowheads indicate microvesicular steatosis, and the arrow indicates cytoplasmic clearing. D, Oil-Red-O and hematoxylin-stained liver sections from Tiparp+/+ and Tiparp−/− mice 5 days after receiving a single injection of 10 μg/kg dioxin. The scale bar represents 50 μm, and all images are to the same scale. B, p < 0.05 was determined by ANOVA followed by Tukey's post hoc test comparison. Significantly different compared with same day matched (a) dioxin-treated Tiparp+/+ mice.
TIPARP and MACROD1 Specifically Regulate AHR Activity through Mono-ADP-ribosylation
To identify a molecular mechanism by which TIPARP may affect AHR activity, we tested whether AHR, ARNT, and AIP were substrates of TIPARP using an in vitro radioactive ADP-ribosylation assay. We incubated full-length GST-tagged mouse or human TIPARP with N- or C-terminal truncations of human AHR or ARNT, or with full-length AIP in the presence or absence of 32P-NAD+. Mono-ADP-ribosylation is indicated by the lack of shift in the apparent molecular weight following SDS-PAGE analysis (25). Murine (Fig. 9, A–C) and human (Fig. 9, D–F) TIPARP specifically mono-ADP-ribosylated the C-terminal transactivation domain of AHR (amino acids 430–848) but not ARNT, AIP, or the N-terminal region of AHR. Chicken Tiparp, but not human PARP10 (ARTD10), also ADP-ribosylated the AHR transactivation domain (Fig. 9, G and H). Because we observed a marked reduction of serum glucose levels in dioxin-treated Tiparp−/− mice, we tested whether PCK1 (PEPCK) is a substrate of TIPARP. Human PCK1 was mono-ADP-ribosylated by murine Tiparp (Fig. 9I) and human TIPARP (Fig. 9J).
FIGURE 9.

Tiparp selectively mono-ADP-ribosylates AHR. GST-murine Tiparp (mTiparp) was incubated with GST-AHR(1–434) or GST-AHR(430–848) (A), GST-ARNT(1–450) or GST-ARNT (440–789) (B), and GST-AIP in the presence of 32P-NAD+ (C) under the conditions shown. GST-TIPARP (human) was incubated with GST-AHR(1–434) or GST-AHR(430–848) (D), GST-ARNT(1–450) or GST-ARNT (440–789) (E), and GST-AIP in the presence of 32P-NAD+ (F) under the conditions shown. GST-chicken Tiparp (chTiparp) was incubated with GST-AHR(1–434) or GST-AHR(430–848) (G), GST-PARP10 was incubated GST-AHR(430–848) (H), and GST-mTiparp (I) or GST-TIPARP (J) was incubated with His-PCK1 (I) in the presence of 32P-NAD+ under the conditions shown. Prior to autoradiography, PVDF membranes were stained with Gel Code Blue (GCB) to visualize the proteins to ensure equal loading. The data shown are representative of three independent experiments.
Macrodomain-containing proteins bind to ADP-ribosylated proteins and can act as mono-ADP-ribosylases, providing evidence that this is a reversible modification (42). To determine whether macrodomain proteins release ADP-ribose from modified TIPARP, we incubated MACROD1 (LRP16) or MACROD2 with in vitro radiolabeled full-length mouse or human TIPARP. MACROD1 and MACROD2 effectively hydrolyzed the mono-ADP-ribosyl linkage from mTiparp (Fig. 10A) as evidenced by reduced Tiparp auto-ribosylation in the presence of either protein. Using co-immunoprecipitation (co-IP) experiments, we found that AHR interacted with FLAG-MACROD1 and not FLAG-MACROD2 (Fig. 10B). Notably, this interaction was increased by dioxin and occurred in a TIPARP-dependent manner. This interaction was also dependent on its catalytic activity, as evidenced by co-IP studies done in the presence of the TIPARP H532A catalytic mutant (Fig. 10C). Because the ability of TIPARP to repress AHR activity is also dependent on its catalytic activity (25), we determined whether MACROD1 or MACROD2 reversed TIPARP-mediated repression of AHR-regulated reporter gene activity. The presence of MACROD1 or FLAG-MACROD1, but not FLAG-MACROD2, reversed the TIPARP-dependent inhibition of reporter gene activity (Fig. 10D). Western blotting confirmed similar protein expression levels of FLAG-MACROD1 and FLAG-MACROD2 (Fig. 10E). MACROD1 has been reported to localize to the mitochondria (43), but we observed both mitochondrial and nuclear localization (Fig. 10F). Moreover, MACROD1 has also been reported to be a cofactor for androgen receptor and estrogen receptor α (44). We next determined whether the ability of MACROD1 to rescue TIPARP-mediated repression of AHR was dependent on its macrodomain by mutating glycine at position 182 to a glutamate (G182E) or aspartate at position 184 to an alanine (D184A). Equivalent mutations in MACROD2 block its ability to hydrolyze ADP-ribosylated residues on PARP10 (42). The MacroD1 mutants exhibited a reduced ability to hydrolyze ADP-ribosylated TIPARP under our assay conditions (Fig. 10G). Expression of MACROD1G182E or MACROD1D184A did not reverse the TIPARP-mediated repression of AHR-dependent reporter gene activity (Fig. 10H), confirming the importance of the macrodomain in counteracting the actions of TIPARP. Western blotting confirmed that MACROD1, MACROD1G182E, and MACROD1D184A were expressed at similar levels (Fig. 10I). In agreement with our previous study (25), overexpression of TIPARP in HuH7 cells repressed dioxin-induced CYP1A1 mRNA (Fig. 10J) and protein levels (Fig. 10, K and L). This reduction was prevented by overexpression of MACROD1, showing that MACROD1 reverses the inhibitory action of TIPARP at the CYP1A1 gene.
FIGURE 10.

TIPARP and MacroD1 regulate AHR activity through reversible mono-ADP-ribosylation. A, MacroD1 more effectively removes mono-ADP-ribose from modified GST-mTiparp compared with MacroD2. B, MACROD1 but not MACROD2 forms a complex with AHR that is dependent on TIPARP. C, interactions between MACROD1 and AHR were reduced in the presence of the TIPARP catalytic mutant (H532A) compared with TIPARP (WT). D, MACROD1 but not MacroD2 prevents TIPARP-dependent repression of AHR regulated reporter gene activity in transiently transfected HuH7 cells, n = 4. E, Western blots of FLAG-MACROD1 and FLAG-MACRoD2 in HuH7 cells. F, subcellular localization of MACROD1. MACROD1 was located predominantly in the mitochondria, but it was also found in the cytoplasm and nucleus. HuH7 cells were transfected with pcDNA-MACROD1, fixed, immunostained for MACROD1, and mounted using Vectashield containing DAPI to visualize DNA. G, two point mutants in MACROD1 prevent its ability to effectively remove mono-ADP-ribose from TIPARP. H, expression of MACROD1G182E or MACROD1D184A in HuH7 cells does not prevent TIPARP-dependent repression of AHR-regulated reporter gene activity, n = 4. I, Western blots of FLAG-MACROD1 (WT), FLAG-MACROD1G182E, and FLAG-MACROD1D184A mutants in transfected HuH7 cells. A–C, E–G, and I, data are representative of three independent experiments. A and G, PVDF membranes were stained with GelCode Blue (GCB) prior to autoradiography to visualize the proteins to ensure equal loading. J, dioxin-induced CYP1A1 mRNA levels were determined in extracts from HuH7 cells transfected with GFP-TIPARP and/or FLAG-MACROD1. K, Western blots of CYP1A1, GFP-TIPARP, and FLAG-MACROD1 in extracts isolated from transfected HuH7 cells. L, quantification of CYP1A1 proteins levels using ImageJ, n = 3. a, significant differences (p < 0.05) compared with dioxin in the absence of TIPARP or macrodomain proteins as determined by ANOVA of the mean followed by Tukey's post hoc test.
SNPs in Human TIPARP Differentially Affect the Ability of TIPARP to Repress AHR Activity
We were interested to determine whether some of the identified SNPs in human TIPARP affected its ability to repress AHR. We investigated 17 nonsynonymous SNPs that were identified by NHLBI GO Exome Sequencing Project. The SNPs spanned the length of the protein coding sequence and, with the exception of F40S (rs369539588) and K42R (rs144427222), were located within the zinc finger, WWE, and catalytic domains (Fig. 11A). Plasmids containing the TIPARP SNPs were created by PCR mutagenesis, and their ability to repress AHR-dependent CYP1A1-regulated reporter gene activity was determined as described under “Experimental Procedures.” We observed small but significant differences in the ability of some of the SNPs to affect the ability of TIPARP to repress (Fig. 11B). The F40S resulted in increased repression of AHR activity, whereas N338S (rs148252230), P361A (rs141280180), and E362K (rs150739414) in the WWE domain and M558T (rs368152871) located in the catalytic domain slightly, but significantly, reduced the ability of TIPARP to repress AHR. The expression of each TIPARP SNP was confirmed by Western blotting (Fig. 11C).
FIGURE 11.
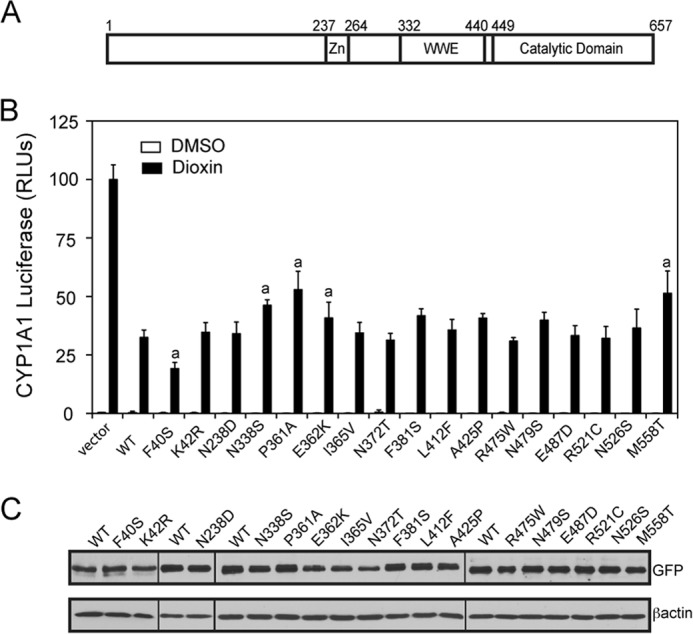
SNPs in human TIPARP differentially affect its ability to repress AHR-dependent CYP1A1-regulated reporter gene activity. A, schematic of TIPARP with its known domains. Zn, zinc finger; WWE, tryptophan-tryptophan-glutamate. The numbers above the domains refer to amino acid positions. B, effect of overexpression of GFP-TIPARP and various SNPs in HuH7 on the TIPARP-dependent repression of AHR-regulated reporter gene activity. C, Western blots of GFP-TIPARP and various SNPs and βACTIN in extracts isolated from transfected HuH7 cells. The data are representative of three independent experiments. a, significant differences (p < 0.05) compared with dioxin in the presence of WT TIPARP. RLU, relative light unit.
Discussion
Despite numerous studies, the mechanisms and downstream target genes regulating dioxin toxicity remain unknown. Here, we show that the product of Tiparp, a well known Ahr target gene, protects against dioxin-induced toxicity and lethality in mice. We also identify previously unknown roles for Tiparp, Macrod1, and mono-ADP-ribosylation in Ahr signaling (Fig. 12).
FIGURE 12.

Schematic representation of TIPARP, MACROD1, and mono-ADP-ribosylation in AHR signaling. Dioxin binds and activates AHR resulting in its nuclear translocation, heterodimerization with ARNT, and increase in TIPARP expression levels. TIPARP catalyzes the mono-ADP-ribosylation of AHR and/or other target proteins causing reduced AHR activity and increased AHR proteolytic degradation (25). MACROD1 binds and hydrolyzes ADP-ribose reversing the actions of TIPARP. The actions of TIPARP and MACROD1 regulate the AHR activity with the loss of TIPARP leading to increased AHR activity and dioxin-induced toxicity.
Lethality in Tiparp−/− mice occurs through an accelerated wasting syndrome in which the mice succumb within days after a single intraperitoneal injection of dioxin. We propose that under normal conditions, Ahr induces the expression of Tiparp, which in concert with MacroD1 dynamically regulates AHR activity through mono-ADP-ribosylation. In the absence of Tiparp, Ahr activity is increased due to reduced mono-ADP-ribosylation resulting in increased sensitivity to dioxin-induced toxicities. Although the increased dioxin sensitivity may be due to reduced mono-ADP-ribosylation of Ahr, it remains possible that the loss of mono-ADP-ribosylation of other yet to be identified Tiparp substrates exacerbates dioxin toxicity in the Tiparp−/− animals.
Dioxin causes and sensitizes mice to diet-induced steatohepatitis, which is characterized by hepatic steatosis accompanied with inflammation that may progress to cirrhosis and hepatocellular carcinoma (36, 39, 45). The observed steatohepatitis in Tiparp−/− mice is of significance because it is related to dioxin-induced chronic toxicity, including metabolic disease and liver cancer. Interestingly, loss of TIPARP is associated with the development of ovarian cancer (46), suggesting an anti-cancer role for this enzyme. The increase in intrahepatic lipid levels in dioxin-treated Tiparp−/− mice suggests that this enzyme plays a role in lipid homeostasis and fatty liver disease. TIPARP knockdown in immortalized human hepatoma, breast, and gastric carcinoma cell lines also results in increased AHR target gene expression in response to dioxin (25), and its overexpression represses AHR activity (Fig. 11) (25). Because of the known differences in affinities of AHR agonists for human AHR compared with mouse Ahrb1 and the differential gene expression profiles observed between mouse and human hepatocytes in response to dioxin (47), it will be important to determine whether TIPARP influences lipid uptake and AHR signaling in human hepatocytes or other human primary cells. Moreover, it will be necessary to determine whether some of the SNPs in TIPARP, particularly those that were found to increase or decrease the ability of TIPARP to repress AHR in a reporter gene assay can affect other AHR-dependent outcomes.
All of our studies were done using mice on a mixed C57BL/6;129 background rather than on the more commonly used C57BL/6 background. SNP analysis revealed that the C57BL/6;129 Tiparp+/+ and Tiparp−/− mice are 90% C57BL/6, and Ahr genotyping confirmed that they express the Ahrb1 that is present in C57BL/6 rather than the Ahrd that is present in 129 substrains. However, other genetic factors may influence Ahr activity, and it will be necessary to complete similar studies in congenic C57BL/6 mice as well as to develop tissue-specific Tiparp null models to fully understand its role in Ahr biology and toxicity.
AHR influences many signaling pathways, including stem cell expansion and T-cell differentiation (48). T-cell function is regulated via ADP-ribosylation, and Tiparp expression is induced in response to viral infection (49). Tiparp has also been reported to safeguard the pluripotency of ES cells (50). It will be important to determine the extent to which other Ahr-regulated pathways are influenced by the induction of Tiparp and/or its activity. Tiparp expression is also induced by nuclear hormone receptors (51) and platelet-derived growth factor (52), and its loss impacts several platelet-derived growth factor target tissues (26, 53). Therefore, it is likely that Tiparp exerts many effects that are independent of Ahr.
AHRR was first described as an AHR target gene and repressor of AHR activity (7). AHRR is a very effective inhibitor of AHR activity in vitro (54) but exhibits gene- and tissue-specific inhibition of Ahr action in vivo (55). Moreover, AHRR expression levels do not correlate with CYP1 activity in human skin fibroblasts nor do they correlate with CYP1A1 responsiveness in mice exposed to the AHR agonist, benzo[a]pyrene) (56, 57). Treatment of Ahrr−/− mice with 3-methylcholanthrene results in tissue-specific rather than whole body increases in Cyp1a1 mRNA levels. There are also no increases in hepatic Cyp1a1 mRNA levels compared with WT animals (55). These data suggest that Ahrr does not regulate Ahr in liver, which is central in dioxin toxicity, because Ahr loss in hepatocytes protects against many dioxin-associated toxicities (58). To the best of our knowledge, the impact of Ahrr loss on dioxin-induced wasting syndrome has not been reported. The increased sensitivity of Tiparp−/− mice to dioxin-induced hepatic gene expression and toxicity is consistent with its role as a negative auto regulator of Ahr activity and support the view that Tiparp is a key regulator of hepatic Ahr activity (25). It will be necessary to determine whether Ahr-dependent gene expression profiles and other Ahr-regulated end points are increased in all Ahr-responsive tissues in the absence of Tiparp.
Tiparp has been reported to mediate dioxin-dependent suppression of gluconeogenesis in chicken (Gallus gallus) embryo hepatocytes (38). Species differences between chTiparp and mTiparp may account for the differences seen in our study. ChTiparp, however, exhibits the same mono-ADP-ribosyltransferase activity and ability to ADP-ribosylate AHR compared with the mouse and human homologs (Fig. 10). The repressive effects of chTiparp on AHR activity have not been reported. Tiparp has also been proposed to mediate dioxin-induced hepatosteatosis by lowering cellular NAD+ levels and consequently reducing the activity of NAD+-dependent enzymes (39). We observed dioxin-induced reduction in hepatic NAD+ levels in Tiparp−/− mice, suggesting that this is not mediated by Tiparp. We cannot exclude the possibility that local and/or small cellular decreases in NAD+ levels may influence the activity of NAD+-dependent enzymes, such as sirtuin 1 and 3. Other PARP family members might be responsible for dioxin-dependent decreases in cellular NAD+ levels. PARP1 is responsible for 80–90% of the cellular PARP activity, and continuous activation can reduce intracellular NAD+ levels by 80% (59). In addition to its role in the DNA damage response, PARP1 is enriched at 90% of RNA polymerase II-bound regions near the transcription start sites of actively expressed genes (60). However, whether PARP1 influences AHR activity and whether it is recruited to AHR target genes in a dioxin-dependent manner have not been reported.
We are only beginning to identify the cellular and physiological functions of TIPARP and other mono-ADP-ribosylating members of the PARP family (22). Most studies have focused on the auto-ribosylation activity of these enzymes, and it will be important to identify the protein targets of TIPARP to fully characterize its physiological role and the impact of mono-ADP-ribosylation on protein function. The selective mono-ADP-ribosylation of AHR by TIPARP, and not by PARP10, and its specific reversal by MACROD1, but not MACROD2, show the unique and selective cellular pathways regulated by mono-ADP-ribosylating PARPs and macrodomain proteins. Our findings also add mono-ADP-ribose to the list of post-translational modifications regulating AHR action and provide new insight into AHR signaling, revealing a novel regulatory mechanism for an ancient receptor pathway. Interestingly, deletion and insertion mutants within the transactivation domain of rat Ahr are thought to be responsible for the marked differences in resistance to dioxin-induced toxicity between strains (28).
One important question is whether the specific targeting of Ahr by Tiparp provides protection against dioxin-induced toxicity or whether other protein targets of Tiparp are involved. The mono-ADP-ribosylation of PCK1 shows that there are TIPARP substrates that are beyond the canonical AHR signaling pathway. Important future experiments will be to fully characterize the mono-ADP-ribose modifications of TIPARP and AHR, as well as to identify other TIPARP substrates.
In summary, we provide evidence that the loss of the Ahr target gene Tiparp in mice increases their sensitivity to dioxin toxicity, steatohepatitis, and lethality. We also show that AHR is mono-ADP-ribosylated by TIPARP and that the inhibitory action of TIPARP on AHR activity is reversed by the ADP-ribosylase, MACROD1. These findings support our previous work showing that TIPARP is a negative regulator of AHR activity (25) and reveal the importance of ADP-ribosylation in the regulation of AHR function.
Supplementary Material
Acknowledgments
We thank David Riddick (University of Toronto) and Tim Zacharewski (Michigan State University) and lab members for critically reading the manuscript.
Note Added in Proof
In support of the mono-ADP-ribosylation of PCK1 by TIPARP reported in our study, Tiparp was shown to ADP-ribosylate both Pck1 and Pck2 in a chicken embryo model (Diani-Moore, S., Zhang, S., Ram, P., and Rifkind, A. B. (2013) Aryl hydrocarbon receptor activation by dioxin targets phosphoenolpyruvate carboxykinase (PEPCK) for ADP-ribosylation via 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)-induced poly(ADP-ribose) polymerase (TiPARP). J. Biol. Chem. 288, 21514–21525).
This work was supported by Canadian Institutes of Health Research Operating Grant MOP-125919, an unrestricted research grant from DOW Chemical, an Early Researcher Award from the Ontario Ministry of Innovation (to J. M.), and an Ontario Graduate Scholarship (to L. M.). The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Tables S1 and S2.
The amino acid sequence of this protein can be accessed through NCBI Protein Database under NCBI accession number GSE61534.
P. Soriano, personal communication.
- AHR
- aryl hydrocarbon receptor
- dioxin (TCDD)
- 2,3,7,8-tetrachlorodibenzo-p-dioxin
- TIPARP
- 2,3,7,8-tetrachlorodibenzo-p-dioxin inducible poly(ADP-ribose) polymerase
- ARNT
- AHR nuclear translocator
- AIP (ARA9)
- AHR interacting protein
- AHRR
- AHR repressor
- PARP
- poly(adenosine diphosphate (ADP)-ribose) polymerase
- PCK (PEPCK)
- phosphoenolpyruvate carboxykinase
- Lpl
- lipoprotein lipase
- Fas
- fatty acid synthase
- ANOVA
- analysis of variance
- qPCR
- quantitative PCR
- bw
- body weight
- SNP
- single nucleotide polymorphism.
References
- 1. Nebert D. W., Puga A., Vasiliou V. (1993) Role of the Ah receptor and the dioxin-inducible [Ah] gene battery in toxicity, cancer, and signal transduction. Ann. N.Y. Acad. Sci. 685, 624–640 [DOI] [PubMed] [Google Scholar]
- 2. Denis M., Cuthill S., Wikström A. C., Poellinger L., Gustafsson J. A. (1988) Association of the dioxin receptor with the Mr 90,000 heat shock protein: a structural kinship with the glucocorticoid receptor. Biochem. Biophys. Res. Commun. 155, 801–807 [DOI] [PubMed] [Google Scholar]
- 3. Perdew G. H. (1988) Association of the Ah receptor with the 90-kDa heat shock protein. J. Biol. Chem. 263, 13802–13805 [PubMed] [Google Scholar]
- 4. Carver L. A., Bradfield C. A. (1997) Ligand-dependent interaction of the aryl hydrocarbon receptor with a novel immunophilin homolog in vivo. J. Biol. Chem. 272, 11452–11456 [DOI] [PubMed] [Google Scholar]
- 5. Hahn M. E., Allan L. L., Sherr D. H. (2009) Regulation of constitutive and inducible AHR signaling: complex interactions involving the AHR repressor. Biochem. Pharmacol. 77, 485–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hankinson O. (1995) The aryl hydrocarbon receptor complex. Annu. Rev. Pharmacol. Toxicol. 35, 307–340 [DOI] [PubMed] [Google Scholar]
- 7. Mimura J., Ema M., Sogawa K., Fujii-Kuriyama Y. (1999) Identification of a novel mechanism of regulation of Ah (dioxin) receptor function. Genes Dev. 13, 20–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pollenz R. S. (2002) The mechanism of AH receptor protein down-regulation (degradation) and its impact on AH receptor-mediated gene regulation. Chem. Biol. Interact. 141, 41–61 [DOI] [PubMed] [Google Scholar]
- 9. Stevens E. A., Mezrich J. D., Bradfield C. A. (2009) The aryl hydrocarbon receptor: a perspective on potential roles in the immune system. Immunology 127, 299–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Quintana F. J., Basso A. S., Iglesias A. H., Korn T., Farez M. F., Bettelli E., Caccamo M., Oukka M., Weiner H. L. (2008) Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature 453, 65–71 [DOI] [PubMed] [Google Scholar]
- 11. Moura-Alves P., Faé K., Houthuys E., Dorhoi A., Kreuchwig A., Furkert J., Barison N., Diehl A., Munder A., Constant P., Skrahina T., Guhlich-Bornhof U., Klemm M., Koehler A. B., Bandermann S., et al. (2014) AhR sensing of bacterial pigments regulates antibacterial defence. Nature 512, 387–392 [DOI] [PubMed] [Google Scholar]
- 12. Poland A., Knutson J. C. (1982) 2,3,7,8-Tetrachlorodibenzo-p-dioxin and related halogenated aromatic hydrocarbons: examination of the mechanism of toxicity. Annu. Rev. Pharmacol. Toxicol. 22, 517–554 [DOI] [PubMed] [Google Scholar]
- 13. Birnbaum L. S. (1995) Developmental effects of dioxins and related endocrine disrupting chemicals. Toxicol. Lett. 82–83, 743–750 [DOI] [PubMed] [Google Scholar]
- 14. Birnbaum L. S. (1994) Endocrine effects of prenatal exposure to PCBs, dioxins, and other xenobiotics: implications for policy and future research. Environ. Health Perspect. 102, 676–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pohjanvirta R., Tuomisto J. (1994) Short-term toxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin in laboratory animals: effects, mechanisms, and animal models. Pharmacol. Rev. 46, 483–549 [PubMed] [Google Scholar]
- 16. Poland A., Palen D., Glover E. (1994) Analysis of the four alleles of the murine aryl hydrocarbon receptor. Mol. Pharmacol. 46, 915–921 [PubMed] [Google Scholar]
- 17. Poland A., Glover E. (1990) Characterization and strain distribution pattern of the murine Ah receptor specified by the Ahd and Ahb-3 alleles. Mol. Pharmacol. 38, 306–312 [PubMed] [Google Scholar]
- 18. Yu Z., Loehr C. V., Fischer K. A., Louderback M. A., Krueger S. K., Dashwood R. H., Kerkvliet N. I., Pereira C. B., Jennings-Gee J. E., Dance S. T., Miller M. S., Bailey G. S., Williams D. E. (2006) In utero exposure of mice to dibenzo[a,l]pyrene produces lymphoma in the offspring: role of the aryl hydrocarbon receptor. Cancer Res. 66, 755–762 [DOI] [PubMed] [Google Scholar]
- 19. Fernandez-Salguero P. M., Hilbert D. M., Rudikoff S., Ward J. M., Gonzalez F. J. (1996) Aryl-hydrocarbon receptor-deficient mice are resistant to 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced toxicity. Toxicol. Appl. Pharmacol. 140, 173–179 [DOI] [PubMed] [Google Scholar]
- 20. Ma Q., Baldwin K. T., Renzelli A. J., McDaniel A., Dong L. (2001) TCDD-inducible poly(ADP-ribose) polymerase: a novel response to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Biochem. Biophys. Res. Commun. 289, 499–506 [DOI] [PubMed] [Google Scholar]
- 21. Hottiger M. O., Hassa P. O., Lüscher B., Schüler H., Koch-Nolte F. (2010) Toward a unified nomenclature for mammalian ADP-ribosyltransferases. Trends Biochem. Sci. 35, 208–219 [DOI] [PubMed] [Google Scholar]
- 22. Vyas S., Matic I., Uchima L., Rood J., Zaja R., Hay R. T., Ahel I., Chang P. (2014) Family-wide analysis of poly(ADP-ribose) polymerase activity. Nat. Commun. 5, 4426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kraus W. L., Hottiger M. O. (2013) PARP-1 and gene regulation: progress and puzzles. Mol. Aspects Med. 34, 1109–1123 [DOI] [PubMed] [Google Scholar]
- 24. Feijs K. L., Forst A. H., Verheugd P., Lüscher B. (2013) Macrodomain-containing proteins: regulating new intracellular functions of mono(ADP-ribosyl)ation. Nat. Rev. Mol. Cell Biol. 14, 443–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. MacPherson L., Tamblyn L., Rajendra S., Bralha F., McPherson J. P., Matthews J. (2013) 2,3,7,8-Tetrachlorodibenzo-p-dioxin poly(ADP-ribose) polymerase (TiPARP, ARTD14) is a mono-ADP-ribosyltransferase and repressor of aryl hydrocarbon receptor transactivation. Nucleic Acid Res. 41, 1604–1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schmahl J., Raymond C. S., Soriano P. (2007) PDGF signaling specificity is mediated through multiple immediate early genes. Nat. Genet. 39, 52–60 [DOI] [PubMed] [Google Scholar]
- 27. Chang C., Smith D. R., Prasad V. S., Sidman C. L., Nebert D. W., Puga A. (1993) Ten nucleotide differences, five of which cause amino acid changes, are associated with the Ah receptor locus polymorphism of C57BL/6 and DBA/2 mice. Pharmacogenetics 3, 312–321 [DOI] [PubMed] [Google Scholar]
- 28. Pohjanvirta R., Sankari S., Kulju T., Naukkarinen A., Ylinen M., Tuomisto J. (1990) Studies on the role of lipid peroxidation in the acute toxicity of TCDD in rats. Pharmacol. Toxicol. 66, 399–408 [DOI] [PubMed] [Google Scholar]
- 29. Patel R., Patel M., Tsai R., Lin V., Bookout A. L., Zhang Y., Magomedova L., Li T., Chan J. F., Budd C., Mangelsdorf D. J., Cummins C. L. (2011) LXRβ is required for glucocorticoid-induced hyperglycemia and hepatosteatosis in mice. J. Clin. Invest. 121, 431–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lee C., Ding X., Riddick D. S. (2013) The role of cytochrome P450-dependent metabolism in the regulation of mouse hepatic growth hormone signaling components and target genes by 3-methylcholanthrene. Drug Metab. Dispos. 41, 457–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Park S. S., Fujino T., West D., Guengerich F. P., Gelboin H. V. (1982) Monoclonal antibodies that inhibit enzyme activity of 3-methylcholanthrene-induced cytochrome P-450. Cancer Res. 42, 1798–1808 [PubMed] [Google Scholar]
- 32. Santostefano M. J., Ross D. G., Savas U., Jefcoate C. R., Birnbaum L. S. (1997) Differential time-course and dose-response relationships of TCDD-induced CYP1B1, CYP1A1, and CYP1A2 proteins in rats. Biochem. Biophys. Res. Commun. 233, 20–24 [DOI] [PubMed] [Google Scholar]
- 33. Walker N. J., Portier C. J., Lax S. F., Crofts F. G., Li Y., Lucier G. W., Sutter T. R. (1999) Characterization of the dose-response of CYP1B1, CYP1A1, and CYP1A2 in the liver of female Sprague-Dawley rats following chronic exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol. Appl. Pharmacol. 154, 279–286 [DOI] [PubMed] [Google Scholar]
- 34. Bunger M. K., Moran S. M., Glover E., Thomae T. L., Lahvis G. P., Lin B. C., Bradfield C. A. (2003) Resistance to 2,3,7,8-tetrachlorodibenzo-p-dioxin toxicity and abnormal liver development in mice carrying a mutation in the nuclear localization sequence of the aryl hydrocarbon receptor. J. Biol. Chem. 278, 17767–17774 [DOI] [PubMed] [Google Scholar]
- 35. Uno S., Dalton T. P., Sinclair P. R., Gorman N., Wang B., Smith A. G., Miller M. L., Shertzer H. G., Nebert D. W. (2004) Cyp1a1(−/−) male mice: protection against high-dose TCDD-induced lethality and wasting syndrome, and resistance to intrahepatocyte lipid accumulation and uroporphyria. Toxicol. Appl. Pharmacol. 196, 410–421 [DOI] [PubMed] [Google Scholar]
- 36. Matsubara T., Tanaka N., Krausz K. W., Manna S. K., Kang D. W., Anderson E. R., Luecke H., Patterson A. D., Shah Y. M., Gonzalez F. J. (2012) Metabolomics identifies an inflammatory cascade involved in dioxin- and diet-induced steatohepatitis. Cell Metab. 16, 634–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Casado F. L., Singh K. P., Gasiewicz T. A. (2011) Aryl hydrocarbon receptor activation in hematopoietic stem/progenitor cells alters cell function and pathway-specific gene modulation reflecting changes in cellular trafficking and migration. Mol. Pharmacol. 80, 673–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Diani-Moore S., Ram P., Li X., Mondal P., Youn D. Y., Sauve A. A., Rifkind A. B. (2010) Identification of the aryl hydrocarbon receptor target gene TiPARP as a mediator of suppression of hepatic gluconeogenesis by 2,3,7,8-tetrachlorodibenzo-p-dioxin and of nicotinamide as a corrective agent for this effect. J. Biol. Chem. 285, 38801–38810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. He J., Hu B., Shi X., Weidert E. R., Lu P., Xu M., Huang M., Kelley E. E., Xie W. (2013) Activation of the aryl hydrocarbon receptor sensitizes mice to nonalcoholic steatohepatitis by deactivating mitochondrial sirtuin deacetylase Sirt3. Mol. Cell. Biol. 33, 2047–2055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lee J. H., Wada T., Febbraio M., He J., Matsubara T., Lee M. J., Gonzalez F. J., Xie W. (2010) A novel role for the dioxin receptor in fatty acid metabolism and hepatic steatosis. Gastroenterology 139, 653–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lu H., Cui W., Klaassen C. D. (2011) Nrf2 protects against 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)-induced oxidative injury and steatohepatitis. Toxicol. Appl. Pharmacol. 256, 122–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rosenthal F., Feijs K. L., Frugier E., Bonalli M., Forst A. H., Imhof R., Winkler H. C., Fischer D., Caflisch A., Hassa P. O., Lüscher B., Hottiger M. O. (2013) Macrodomain-containing proteins are new mono-ADP-ribosylhydrolases. Nat. Struct. Mol. Biol. 20, 502–507 [DOI] [PubMed] [Google Scholar]
- 43. Neuvonen M., Ahola T. (2009) Differential activities of cellular and viral macro domain proteins in binding of ADP-ribose metabolites. J. Mol. Biol. 385, 212–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yang J., Zhao Y. L., Wu Z. Q., Si Y. L., Meng Y. G., Fu X. B., Mu Y. M., Han W. D. (2009) The single-macro domain protein LRP16 is an essential cofactor of androgen receptor. Endocr. Relat. Cancer 16, 139–153 [DOI] [PubMed] [Google Scholar]
- 45. Marra F., Gastaldelli A., Svegliati Baroni G., Tell G., Tiribelli C. (2008) Molecular basis and mechanisms of progression of non-alcoholic steatohepatitis. Trends Mol. Med. 14, 72–81 [DOI] [PubMed] [Google Scholar]
- 46. Goode E. L., Chenevix-Trench G., Song H., Ramus S. J., Notaridou M., Lawrenson K., Widschwendter M., Vierkant R. A., Larson M. C., Kjaer S. K., Birrer M. J., Berchuck A., Schildkraut J., Tomlinson I., Kiemeney L. A., et al. (2010) A genome-wide association study identifies susceptibility loci for ovarian cancer at 2q31 and 8q24. Nat. Genet. 42, 874–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Forgacs A. L., Dere E., Angrish M. M., Zacharewski T. R. (2013) Comparative analysis of temporal and dose-dependent TCDD-elicited gene expression in human, mouse, and rat primary hepatocytes. Toxicol. Sci. 133, 54–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Esser C., Rannug A., Stockinger B. (2009) The aryl hydrocarbon receptor in immunity. Trends Immunol. 30, 447–454 [DOI] [PubMed] [Google Scholar]
- 49. Atasheva S., Akhrymuk M., Frolova E. I., Frolov I. (2012) New PARP gene with an anti-alphavirus function. J. Virol. 86, 8147–8160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Roper S. J., Chrysanthou S., Senner C. E., Sienerth A., Gnan S., Murray A., Masutani M., Latos P., Hemberger M. (2014) ADP-ribosyltransferases Parp1 and Parp7 safeguard pluripotency of ES cells. Nucleic Acid Res. 42, 8914–8927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sasse S. K., Mailloux C. M., Barczak A. J., Wang Q., Altonsy M. O., Jain M. K., Haldar S. M., Gerber A. N. (2013) The glucocorticoid receptor and KLF15 regulate gene expression dynamics and integrate signals through feed-forward circuitry. Mol. Cell. Biol. 33, 2104–2115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chen W. V., Delrow J., Corrin P. D., Frazier J. P., Soriano P. (2004) Identification and validation of PDGF transcriptional targets by microarray-coupled gene-trap mutagenesis. Nat. Genet. 36, 304–312 [DOI] [PubMed] [Google Scholar]
- 53. Schmahl J., Rizzolo K., Soriano P. (2008) The PDGF signaling pathway controls multiple steroid-producing lineages. Genes Dev. 22, 3255–3267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Karchner S. I., Jenny M. J., Tarrant A. M., Evans B. R., Kang H. J., Bae I., Sherr D. H., Hahn M. E. (2009) The active form of human aryl hydrocarbon receptor (AHR) repressor lacks exon 8, and its Pro 185 and Ala 185 variants repress both AHR and hypoxia-inducible factor. Mol. Cell. Biol. 29, 3465–3477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hosoya T., Harada N., Mimura J., Motohashi H., Takahashi S., Nakajima O., Morita M., Kawauchi S., Yamamoto M., Fujii-Kuriyama Y. (2008) Inducibility of cytochrome P450 1A1 and chemical carcinogenesis by benzo[a]pyrene in AhR repressor-deficient mice. Biochem. Biophys. Res. Commun. 365, 562–567 [DOI] [PubMed] [Google Scholar]
- 56. Bernshausen T., Jux B., Esser C., Abel J., Fritsche E. (2006) Tissue distribution and function of the Aryl hydrocarbon receptor repressor (AhRR) in C57BL/6 and aryl hydrocarbon receptor deficient mice. Arch. Toxicol. 80, 206–211 [DOI] [PubMed] [Google Scholar]
- 57. Tigges J., Weighardt H., Wolff S., Götz C., Förster I., Kohne Z., Huebenthal U., Merk H. F., Abel J., Haarmann-Stemmann T., Krutmann J., Fritsche E. (2013) Aryl hydrocarbon receptor repressor (AhRR) function revisited: repression of CYP1 activity in human skin fibroblasts is not related to AhRR expression. J. Invest. Dermatol. 133, 87–96 [DOI] [PubMed] [Google Scholar]
- 58. Walisser J. A., Glover E., Pande K., Liss A. L., Bradfield C. A. (2005) Aryl hydrocarbon receptor-dependent liver development and hepatotoxicity are mediated by different cell types. Proc. Natl. Acad. Sci. U.S.A. 102, 17858–17863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bai P., Cantó C. (2012) The role of PARP-1 and PARP-2 enzymes in metabolic regulation and disease. Cell Metab. 16, 290–295 [DOI] [PubMed] [Google Scholar]
- 60. Krishnakumar R., Kraus W. L. (2010) PARP-1 regulates chromatin structure and transcription through a KDM5B-dependent pathway. Mol. Cell 39, 736–749 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.