

Background: Full-length PTEN-induced putative kinase 1 (PINK1) is an important regulator of mitophagy, but the function of its highly labile cytosolic counterpart is often overlooked.
Results: Cytosolic PINK1 is stabilized by TRAF6/NF-κB activation via Lys-63-linked ubiquitination and promotes the removal of apparently healthy mitochondria.
Conclusion: Cytosolic PINK1 can be stabilized to bring about non-selective mitophagy.
Significance: The phenomenon may represent a cytoprotective response to counteract oxidative stress.
Keywords: mitophagy, parkin, Parkinson disease, PTEN-induced putative kinase 1 (PINK1), ubiquitylation (ubiquitination)
Abstract
The potential cellular function of the 53-kDa cytosolic form of PINK1 (PINK1-53) is often overlooked because of its rapid degradation by the proteasome upon its production. Although a number of recent studies have suggested various roles for PINK1-53, how this labile PINK1 species attains an adequate expression level to fulfil these roles remains unclear. Here we demonstrated that PINK1-53 is stabilized in the presence of enhanced Lys-63-linked ubiquitination and identified TRAF6-related NF-κB activation as a novel pathway involved in this. We further showed that a mimetic of PINK1-53 promotes mitophagy but, curiously, in apparently healthy mitochondria. We speculate that this “non-selective” form of mitophagy may potentially help to counteract the build-up of reactive oxygen species in cells undergoing oxidative stress and, as such, represent a cytoprotective response.
Introduction
Far from being solitary and static structures, mitochondria are now recognized to be dynamic organelles that constantly undergo membrane remodeling through repeated cycles of fusion and fission as well as regulated turnover via a specialized lysosome-mediated degradation pathway known as “mitophagy.” Collectively, these processes help to maintain the quality and, thereby, optimal function of mitochondria and allow the organelle to respond rapidly to changes in cellular energy status. Recent studies have revealed that two genes, whose mutations are linked to familial parkinsonism, i.e. parkin (encoding a ubiquitin ligase) and PINK1 (encoding a serine/threonine kinase), are important for mitophagy (1). According to the proposed model (2), a key initial event that occurs upon mitochondrial depolarization is the selective accumulation of PINK1 on the outer membrane of the damaged organelle. This accumulation allows PINK1 to recruit parkin (3), whose latent ubiquitin ligase activity becomes unmasked along the way, in part because of its phosphorylation by PINK1 (4, 5). PINK1 also phosphorylates ubiquitin, which binds and activates parkin (6, 7). Activated parkin then promotes the ubiquitination and subsequent degradation of many outer mitochondrial membrane proteins (8, 9). During the process, parkin-decorated mitochondria progressively cluster toward the perinucleus region to form mitoaggresomes, which, by virtue of their association with lysosomal components, are removed over time in an autophagy-dependent manner.
Selective mitophagy as described above does not occur in healthy mitochondria because the PINK1 protein containing a mitochondrion-targeting signal at its N terminus is normally imported rapidly into the inner mitochondrial membrane through the sequential actions of the translocase of outer mitochondrial membrane complex and the translocase of inner mitochondrial membrane complex. During the importation process, the full-length PINK1 is progressively modified by a series of mitochondrial proteases to a 53-kDa cleaved form (hereafter designated PINK1-53) that is degraded rapidly by the proteasome (10, 11). Current evidence suggests that PINK1-53 is retrotranslocated to the cytosol and degraded through the N-end rule pathway via the actions of the ubiquitin protein ligase E3 component n-recognin (UBR) family of E3 ligases (12). Although the above proteolytic events would ensure that PINK1 is kept at low levels under normal conditions (and, as such, to prevent unintended mitophagy from occurring), it is intriguing to note that the cell has to go through such an elaborate process (i.e. to synthesize, import, cleave it twice, and degrade the protein) just to restrict its expression. It is attractive, therefore, to speculate that the 53-kDa PINK1 protein may be stabilized under certain conditions to subserve some cellular roles. Supporting this, accumulation of PINK1-53 has been reported in the brains of both idiopathic and PINK1-related Parkinson disease patients (13, 14). Moreover, recent studies have also implicated several functional roles for PINK1-53 (including being a neuroprotectant and a promoter of neurite outgrowth as well as an inhibitor of mitophagy) (15–17), although none of these studies describe how this labile species of PINK1 may attain an adequate expression level in the first place to fulfil the suggested roles.
Here we show that PINK1-53 is stabilized in the presence of Lys-63-linked ubiquitination. Importantly, we identified TRAF6 as an E3 ligase responsible for the phenomenon and demonstrated that TRAF6-related NF-κB activation promotes the stability of PINK1-53. Contrary to a recent report (16), we found that PINK1-53 does not inhibit parkin-mediated mitophagy. Instead, it appears to trigger parkin translocation to normal mitochondria, leading to their clearance. Taken together, our study elucidated a novel mechanism by which the otherwise highly labile PINK1-53 may be stabilized and, at the same time, expanded the role of PINK1 to include its participation in “non-selective” mitophagy.
Materials and Methods
Antibodies and Reagents
Antibodies used were as follows: mouse anti-β-actin and anti-FLAG-peroxidase (Sigma); mouse anti-GFP, anti-[c-myc]-peroxidase, and anti-HA-peroxidase (Roche Diagnostics); mouse anti-Tim23 and rabbit anti-Tom20 (Santa Cruz Biotechnology); mouse anti-Lys-63 (clone HWA4C4) (Enzo Life Science); rabbit anti-PINK1 (BC100-494, Novus Biologicals); rabbit mAB anti-p65, anti-IκB kinase β (IKKβ), anti-phospho-IκBα, and rabbit anti-AKT (Cell Signaling Technology), Rhodamine-Red-conjugated anti-mouse and anti-rabbit IgG, Alexa Fluor 488-conjugated anti-mouse, Alexa Fluor 647-conjugated anti-mouse and anti-rabbit IgG, and Alexa Fluor 405-conjugated anti-rabbit and anti-mouse IgG (Molecular Probes); FITC-conjugated anti-rabbit IgG (BD Biosciences), and anti-mouse and anti-rabbit peroxidase (GE Healthcare). Chemicals/reagents used were as follows: MitoTracker Red CMXROS (Molecular Probes); 3-methyladenine, dimethyl sulfoxide (DMSO),5 TNFα, phorbol 12-myristate 13-acetate (PMA), carbonyl cyanide m-chlorophenylhydrazone (CCCP), and valinomycin (Sigma); clasto-lactacystin-β-lactone (Enzo Life Science), MG132 (A. G. Scientific), Z-IIe-Glu (OtBu)-Ala-Leu-H (aldehyde) (PSI) (Peptides International); Bafilomycin A1 (A. G. Scientific); puromycin (Clontech); BMS-34551 (Sigma); and SC-514 (Calbiochem). Plasmids used were as follows. Untagged PINK1 was used because tagged versions of PINK1 have been reported to alter its localization (5). Untagged PINK1 was cloned from HEK293 cDNA into pCDNA3 at the BamHI and EcoRI sites. The PINK1 truncated mutant PINK1Δ104 (untagged) was subcloned from untagged PINK1 with residue 1–103 amino acid deleted and phenylalanine at position 104 replaced with methionine and inserted into pCDNA3 via the BamHI and EcoRI sites. The primers used in the PCR reaction were 5′-ATAAGGATCCGCCACCATGGGGCTAGGGCTGGGCCTCATC-3′ (forward) and 5′-ATAAGAATTCTCACAGGGCTGCCCTCCATG-3′ (reverse). PINK1Δ104-IRES-GFP was cloned by PCR amplification from the pcDNA3-PINK1Δ104 construct using the following forward and reverse primers: forward, 5′ ATGATATCCGCCACCATGGGGCTAG 3′; reverse, 5′ ATGGATCCTCACAGGGCTGCCCTC 3′. The amplified fragment was digested with EcoRV/BamHI and inserted into the EcoRV and BamHI sites of an intermediate lentivector, pL6mCWIRESEGFP. Subsequently, the PINK1Δ104-IRES-GFP cassette was isolated from this vector by EcoRV/BsrGI digestion and inserted into the HpaI/BsrGI-digested FUGW vector, replacing an original tdTomato insert. FLAG-mcherry-parkin was cloned into a BstBI site of the FUGW vector, replacing an original tdTomato insert. TRAF6 was cloned into pCMV-myc at the EcoRI and XhoI sites. Plasmid constructs expressing HA-tagged ubiquitin and its mutants, FLAG-tagged parkin, myc-tagged CHIP, UbcH7, and Ubc13 and HA-tagged HHARI, have been described previously (18). HA-tagged ubiquitin C-terminal hydrolase L1 (UCHL1) and N-terminal GFP-tagged Parkin were gifts from Ted Dawson (John Hopkins University) and Tso-Pang Yao (Duke University), respectively. The following plasmids were obtained from Addgene: GFP-tagged ataxin3 (catalog no. 22122, Henry Paulson), GFP-tagged A20 (catalog no. 22141, Yihong Ye), FLAG-tagged wild-type IKKβ (catalog no. 11103) and its mutants K44M (catalog no. 11104) and S177E/S181E (catalog no. 11105, Anjana Rao), HA-tagged wild-type IκBα (catalog no. 21985), IκBα S32A/S36A (catalog no. 24143, Warner Greene), and HA-tagged TRAF3 (catalog no. 44032, Shao-Cong Sun). The following plasmids were purchased: Myc, His6-tagged Mulan, RNF11, RNF144B, March5, and RNF185 (Abgent); HA-tagged SENP8 (GeneCopoeia); and EGFP-N3 (Clontech). p65 shRNA (ID TRCN0000014685) with a mature antisense sequence of AACTCATCATAGTTGATGGTG, IKKβ shRNA (ID TRCN0000018917) with a mature antisense sequence of ATGTTCAAGATATGAACCAGC, and the RNAi consortium (TRC) lentiviral non-targeting shRNA control were obtained from Dharmacon GE.
Cell Culture, Western Blot Analysis, and Proteasome Assay
HEK293 cells were grown in DMEM containing 10% FBS in a 5% CO2 atmosphere. Stably transfected N-terminal GFP-tagged Parkin HeLa cells, a gift from Keiji Tanaka and Noriyuki Matsuda (Tokyo Metropolitan Institute of Medical Science), were maintained in the abovementioned medium supplemented with 5 μg/ml puromycin. Transfections of cells were carried out using Lipofectamine PLUS (Invitrogen) or X-tremeGENE HP DNA (Roche) transfection reagent according to the instructions of the manufacturer. Transfected cells were lysed with PBS containing 1% Triton X-100 and analyzed by Western blot as described previously (19). The proteasome assay was performed with fresh Triton X-100-soluble fractions as described previously (20).
Mouse Primary Neuron Preparation and Transfection
Mouse studies were approved by and conformed to the guidelines of the Institutional Animal Care Committee of Tan Tock Seng Hospital-National Neuroscience Institute. Embryonic day 17.5 mouse fetuses from C57BL/6 were obtained, and their meninges were removed. The cortex was isolated and dissociated with 0.25% trypsin for 20 min, followed by the addition of trypsin inhibitor. Cells were washed, titrated, and resuspended in neurobasal medium supplemented with B27 and GlutaMAX (Gibco). Cells were transfected with the nucleofector kit for mouse neurons (Lonza) according to the instructions of the manufacturer. Briefly, 106 cells were pooled and electroporated with 10 μg each of FLAG-mcherry-parkin with the FUGW-IRES-GFP or FUGW-PINK1Δ104-IRES-GFP plasmids. The O-0005 program was used. 200,000 cells were then plated onto 13-mm coverslips coated with 1 μg/ml poly-l-lysine. 4 days later, cells were fixed, stained, and analyzed.
Immunoprecipitation and Immunocytochemistry
HEK293 cells were harvested and lysed in radioimmune precipitation assay buffer (containing 150 mm NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 1% Nonidet P-40, 10 μg/ml aprotinin, 1 mm PMSF, 1% (v/v) phosphatase inhibitor mixture II and III, and 0.5 mm N-ethylmaleimide) and centrifuged at 13,500 rpm for 15 min at 4 °C. The supernatant was subjected to immunoprecipitation as described previously (21) with anti-PINK1. PINK1 was detected with Clean-Blot immunoprecipitation detection reagent HRP (Thermo Scientific) which specifically recognizes the native primary antibody and as such prevents the interference of the denatured heavy immunoglobulin chain that migrates at similar position as PINK1–53 on SDS-PAGE. For immunocytochemical analysis, HEK293 or GFP-Parkin HeLa cells were treated 24 h post-transfection with the indicated pharmacological reagents. Cells were processed and imaged as described previously (19). Quantitative results reported are an average of at least three experiments.
NF-κB Inhibitors, shRNA Treatment, and Luciferase Assay
HEK293 cells were transfected with untagged PINK1 alone or with either control, p65, or IKKβ shRNA. For NF-κB inhibitor experiments, 36 h after transfection, the cells were washed with fresh medium and treated with 20 μm BMS-34551 (Sigma) or 20 μm SC-514 (Calbiochem) for 1 h and, subsequently, with 10 ng/ml TNFα for 4 h. For the shRNA experiment, 24 h after transfection, cells were treated with 5 μg/ml puromycin for 20 h to select for transfected cells. 20 h later, cells were washed with fresh medium and treated with 10 ng/ml TNFα for 4 h. For the NF-κB luciferase assay, HEK 293 cells were also transfected with the pSEAP2-control vector and the pNFκB-MetLuc2 reporter vector (Clontech). Media was collected after TNFα treatment, and the luciferase assay was performed on 10× diluted media with the Ready-To-GlowTM dual secreted reporter assay kit (Clontech) in accordance with the instructions of the manufacturer. Technical triplicates and experimental triplicates were performed. Relative luciferase activity was calculated by normalizing the luciferase values with the pSEAP values. Luminescence (-fold difference) was computed by normalizing the relative luciferase activity to untreated cells or cells transfected with control shRNA.
Statistical Analysis
Statistical significance for all quantitative data obtained was analyzed using Student's t test (*, p < 0.05; **, p < 0.001).
Results
Lys-63-linked Ubiquitination Stabilizes PINK1-53
Under normal conditions, PINK1-53 protein is highly unstable, even when its precursor full-length protein is overexpressed in cells (Fig. 1A). Appreciable amounts of PINK1-53 could only be observed in the presence of proteasome inhibitors (e.g. MG132, PSI, lactacystin) but not autophagy inhibitors (e.g. 3-methyladenine, bafilomycin), suggesting that the cleaved PINK1 species is rapidly and selectively degraded by the proteasome (Fig. 1A). Conversely, in the presence of CCCP, a chemical uncoupler that collapses the mitochondrial membrane potential (ΔΨm) and prevents the importation of PINK1 into the mitochondria, full-length PINK1 but not PINK1-53 accumulates selectively (Fig. 1A). Together, these results demonstrate the highly labile nature of the PINK1-53 protein under normal growth conditions and support the current model of PINK1 processing.
FIGURE 1.

PINK1-53 expression is enhanced in the presence of Lys-63-linked ubiquitination. A, anti-PINK1 immunoblots of lysates prepared from control or PINK1-transfected HEK293 cells that were either untreated or treated with 1 μm MG132, 1 μm PSI, 5 μm Lactacytsin, 10 mm 3-methyladenine (3MA), and 200 nm Bafilomycin A1 for 16 h or 10 μm CCCP for 4 h, as indicated. The bands corresponding to full-length PINK1 (63 kDa) and PINK1-53 (53 kDa) are indicated. The blots above were stripped and reprobed with anti-actin as a loading control. This experiment was repeated at least three times. B, representative immunoblots of at least three experimental sets showing the expression level of full-length PINK1 (63 kDa) and PINK1-53 (53 kDa) in the presence of 1 μm MG132 (16 h) or various forms of HA-ubiquitin (Ub). The numbers indicate the average -fold (Avg. fold) change in the densitometric level of PINK1-53. C, same as B, except that HA-ubiquitin was replaced by myc-Ubc13, myc-UbcH7, FLAG-parkin, or HA-HHARI. The asterisk denotes cells treated with 1 μm MG132. This experiment was duplicated. D, representative immunoblots of at least three experimental sets showing the expression level of full-length (63 kDa) and PINK1-53 (53 kDa) in the presence of Ubc13 coexpression alone or with GFP-Ataxin-3, HA-UCHL-1, HA-SENP8, or GFP. (DUB, Deubiquitinating enzyme). E, same as C, except that PINK1 is expressed in some cases in the presence of both myc-Ubc13 and FLAG-parkin or HA-HHARI. The blots above were stripped and reprobed with anti-actin antibody to reflect loading variations. This experiment was duplicated. F, the relative chymotrypsin-like proteasome activities of at least three sets of lysates prepared from PINK1 overexpressing cells cotransfected with various E2s and E3s as indicated. **, p < 0.001 compared with the first column; Student's t test.
Given that the stability of a protein may be enhanced by Lys-63-linked ubiquitination, we wondered whether this form of ubiquitin modification that is typically uncoupled from the proteasome could influence the stability of the otherwise short-lived PINK1-53 protein, which is presumably normally ubiquitinated via linkages associated with proteasomal degradation. To examine this, we coexpressed full-length untagged PINK1 in HEK293 cells in the absence or presence of HA-tagged wild-type or mutant ubiquitin species that we have reported previously to either support (i.e. Lys-63, K48R) or prevent (i.e. Lys-48, Lys-63R) Lys-63 ubiquitin chain formation (20). MG132-treated PINK1-transfected cells were included as a positive control. Consistent with the proteasome-independent role of Lys-63-linked ubiquitination, we found that the level of PINK1-53 is increased in the presence of Lys-63 HA-ubiquitin coexpression and, to a comparable extent, to that found in MG132-treated PINK1-transfected cells (Fig. 1B). This increase in PINK1–53 level was also observed with Lys-48-Arg HA-ubiquitin coexpression. Comparatively, PINK1-53 expression is reduced dramatically in transfected cells coexpressing wild-type, Lys-48, or Lys-63-Arg HA-ubiquitin (Fig. 1B). Our results therefore suggest that enhanced Lys-63-linked ubiquitination can stabilize PINK1-53. To complement these findings, we repeated our experiments with Ubc13 (which specifies for Lys-63-linked ubiquitination) and UbcH7 (for which no specificity for the Lys-63-linked chain has been reported) in place of the ubiquitin mutants. In agreement with our results above, we found that the level of PINK1-53 is enhanced in the presence of Ubc13 but not UbcH7 coexpression (Fig. 1C), suggesting that endogenously promoted Lys-63-linked ubiquitination can similarly stabilize the PINK1-53 species. Given this, we surmised that the removal of Lys-63-linked chains by deubiquitinating enzymes specific for this ubiquitin topology will counteract the stabilizing effect of Ubc13 overexpression on PINK1-53. For this purpose, we coexpressed PINK1 and Ubc13 together with Ataxin 3, which has been reported to cleave Lys-63-linked chains (22), and included UCHL1 (a related ubiquitin hydrolase), SENP8 (a NEDD8-specific deneddylase), and GFP as controls. As expected, Ubc13-mediated enhancement of PINK1-53 level is significantly and selectively mitigated by Ataxin 3 (Fig. 1D).
Because parkin is known to be capable of mediating Lys-63-linked ubiquitination (23) and is furthermore intimately associated with PINK1 function, we wondered whether overexpression of parkin can affect the processing of PINK1 under normal culture conditions (i.e. in the absence of CCCP). Interestingly, we found that ectopically expressed parkin enhances the level of full-length PINK1 and, in the process, appears to retard the cleavage of PINK1 into the 53-kD form (Fig. 1, C and E). The stabilization of the longer PINK1 form by parkin seems to be specific to E3 because a related member, HHARI, fails to elicit the phenomenon (Fig. 1C). Given the observed effect of parkin on PINK1, we next examined whether parkin overexpression might affect Ubc13-mediated enhancement of PINK1-53. We found that overexpressed parkin, but not HHARI, effectively prevented the Ubc13-mediated phenomenon (Fig. 1E), presumably because PINK1-53 is not generated in the first place in the presence of overexpressed parkin. Notably, none of the PINK1 stabilization events (other than those occurring in MG132-treated cells) are associated with proteasomal dysfunction because the activity of the 20S proteasome is not significantly affected in the variously transfected cells (Fig. 1F).
TRAF6 Overexpression Stabilizes PINK1-53 via Lys-63-linked Ubiquitination
Because parkin fails to promote the stabilization of PINK1-53, we were naturally curious to find out the E3 that could do the job. We selected a list of candidate E3 members whose activity is associated with Lys-63-linked ubiquitination and/or mitochondrial function, including TRAF6, UCHL1, CHIP, Mulan, March5, RNF-11, RNF-144B, and RNF-185 (24). Among these, we found that TRAF6 overexpression could appreciably enhance the level of PINK1-53 (Fig. 2, A–D). Interestingly, Murata et al. (25) have demonstrated recently that TRAF6 interacts with and stabilizes full-length PINK1 (25). Here we found that TRAF6 also dramatically promotes the level of PINK1-53. We further found that, in the presence of A20, a deubiquitinating enzyme known to disrupt the formation of Lys-63-linked chains by TRAF6 (26), the stabilization of PINK1 by TRAF6 is reduced in a dose-dependent manner (Fig. 2E), suggesting that the phenomenon is dependent on Lys-63-linked ubiquitination. Notably, A20 coexpression can also overcome Ubc13-mediated stabilization of PINK1-53 (Fig. 2F).
FIGURE 2.
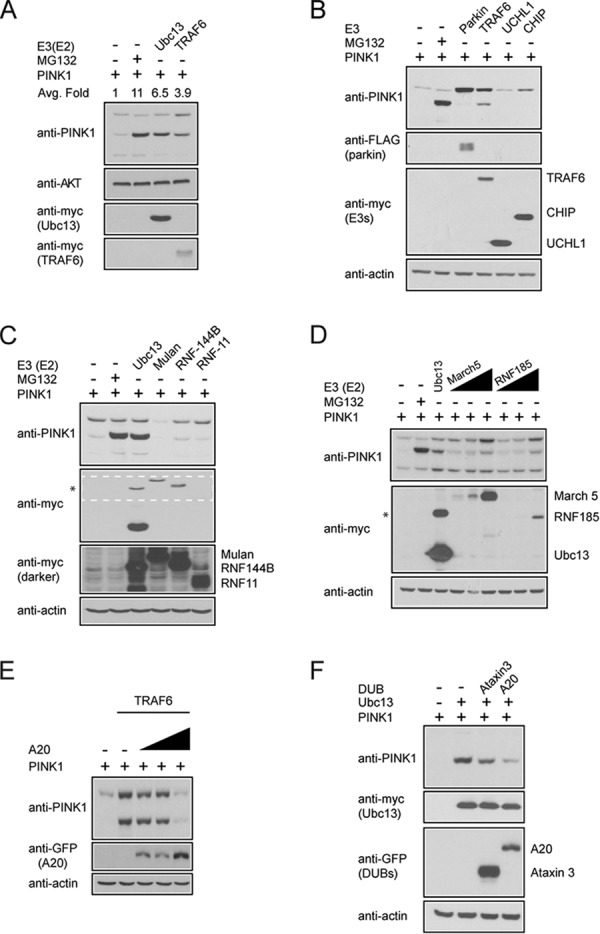
TRAF6 promotes the stability of PINK1-53 via Lys-63-linked ubiquitination. A, representative immunoblots of at least three experimental sets showing the expression level of full-length PINK1 and PINK1-53 in the absence or presence of myc-TRAF6 or myc-Ubc13. The numbers indicate the average -fold (Avg. Fold) change in the densitometric level of PINK1-53. Anti-AKT blot served as a loading control. B and C, representative anti-PINK1, anti-FLAG, and anti-myc immunoblots of three sets of cell lysates prepared from PINK1 overexpressed HEK293 cells cotransfected with various E3 ligases as indicated. CHIP, carboxyl terminus of Hsc70-interacting protein. As a control, PINK1 overexpressed cells were either treated with 1 μm MG132 or cotransfected with myc-Ubc13. The asterisk denotes a nonspecific band in the Ubc13-containing lane. A darker exposure of the region outlined in white dotted lines in C is presented below to show the expression of myc-RNF11 in particular, along with myc-Mulan and myc-RNF144B. D, the same as above, except that increasing concentrations of March5 and RNF185 (at ratios of 1:2, 1:4, and 1:10) were used because of their poor expression in transfected cells. This experiment was duplicated. The asterisk denotes a nonspecific band in the Ubc13-containing lane. E, representative anti-PINK1 and anti-GFP immunoblots of cell lysates prepared from cells transfected with myc-TRAF6 and increasing amounts of GFP-A20 at ratios of 1:2, 1:4, and 1:8. The blots above were stripped and reprobed with actin as a loading control. This experiment was duplicated. F, representative anti-PINK1 immunoblot showing the expression of PINK1-53 in the presence or absence of myc-Ubc13 alone or in combination with GFP-tagged Ataxin 3 and A20. The above blots were stripped and reprobed with actin as a loading control. This experiment was duplicated. DUB, deubiquitinating enzyme.
To examine whether PINK1-53 is ubiquitinated by TRAF6 without the confounding presence of ectopic full-length PINK1, we generated a truncation mutant of PINK1 that is deleted of the N terminus 104 residues (i.e. PINK1Δ104) (Fig. 3A). The PINK1Δ104 mutant shares sequence identity with PINK1-53, except for its first amino acid residue, which is methionine, from the start codon instead of phenylalanine, a modification that allows PINK1Δ104 to escape the N-rule degradation pathway (12). Consistent with this, we found that PINK1Δ104 is robustly expressed in the absence of proteasome inhibition (Fig. 3A). Notably, overexpressed PINK1Δ104 resides predominantly in the cytosol without appreciable localization (if at all) to the mitochondria (Fig. 3B). This is in agreement with the observations reported by others (12). As an initial investigation, we checked whether PINK1Δ104 may be directly ubiquitinated by overexpressed HA-tagged Lys-63 ubiquitin, especially given our earlier finding that PINK1-53 is stabilized in the presence of Lys-63 ubiquitin overexpression (Fig. 1B). For this purpose, we immunoprecipitated PINK1Δ104 from transfected cells coexpressing HA-UbLys-63 and observed robust PINK1 ubiquitination in the presence but not absence of the ubiquitin mutant (Fig. 3C), suggesting that PINK1-53 can be modified via Lys-63-linked ubiquitin chains. Next we examined the role of TRAF6 in promoting the ubiquitination of PINK1Δ104. We used exogenous wild-type ubiquitin because we wished to know whether TRAF6 would assemble Lys-63-linked chains on PINK1Δ104 when given the choice. In the presence (but not absence) of TRAF6 and wild-type HA-tagged ubiquitin, PINK1Δ104 immunoprecipitates display a PINK1-positive high molecular weight laddering pattern that is immunoreactive for anti-HA (Fig. 3, D and E), indicating that PINK1Δ104 is ubiquitinated by TRAF6. The specificity of this TRAF6-mediated reaction is illustrated by the absence of the clear laddering pattern when E3 is substituted by a related member, TRAF3 (Fig. 3D). When probed with an antibody specific for Lys-63-linked ubiquitin topology, the PINK1- and HA-positive high molecular weight bands that are generated in the presence of TRAF6 exhibit robust staining for the ubiquitin chain (Fig. 3E). These results suggest that Δ104 PINK1 is ubiquitinated by TRAF6 and via Lys-63-linked ubiquitination. Therefore, it appears that the fate of the labile PINK1-53 cytosolic species may be altered en route to its degradation by TRAF6-mediated Lys-63-linked ubiquitination in a manner that would result in its stabilization.
FIGURE 3.

PINK1-53 is ubiquitinated by TRAF6 via Lys-63-linked chains. A, left panel, schematic showing the domains present in WT PINK1 and PINK1Δ104. Right panel, anti-PINK1 immunoblot of cell lysates prepared from HEK293 cells transfected with either vector, untagged PINK1, or PINK1Δ104. The anti-AKT blot served as a loading control. MTS, mitochondrial-targeting sequence. B, representative confocal images of HEK293 cells overexpressing untagged PINK1Δ104 immunostained with anti-PINK1 (green) and anti-Tim23 (red). Note that PINK1Δ104 does not colocalize with Tim23. C, anti-PINK1 and anti-HA (UbK63) immunoblots of PINK1Δ104 immunoprecipitates from cells expressing HA-tagged Lys-63 ubiquitin or PINK1Δ104 alone or in combination as indicated. This experiment was duplicated. D, representative anti-PINK1 and anti-HA immunoblots showing the extent of ubiquitination (Ub) in PINK1 immunoprecipitates (IP-PINK1) from control or PINK1Δ104-transfected cells expressing HA-ubiquitin in the absence or presence of myc-TRAF6 or HA-TRAF3. The expression of PINK1 and HA-ubiquitin is shown in the INPUT blots. E, the same as D, except that TRAF3 is omitted and the blots include anti-UbLys-63 staining. These experiments were repeated at least three times.
NF-κB Pathway Activation Stabilizes PINK1-53
The involvement of TRAF6, a transducer of the NF-κB pathway, as a regulator of PINK1 stability suggests that NF-κB signaling activation might be relevant here. To address this, we treated PINK1-expressing cells with TNFα and PMA, which are known pharmacological activators of the pathway. We found that both TNFα and PMA treatment, like TRAF6 overexpression, result in the stabilization of PINK1-53 without exerting any apparent effects on proteasome function (Fig. 4A). Furthermore, we found that the stability of PINK1-53 is reduced significantly in TNFα-treated PINK1-expressing cells in the presence of A20 coexpression (Fig. 4B), suggesting again that Lys-63-linked ubiquitination is important for PINK1-53 stabilization.
FIGURE 4.
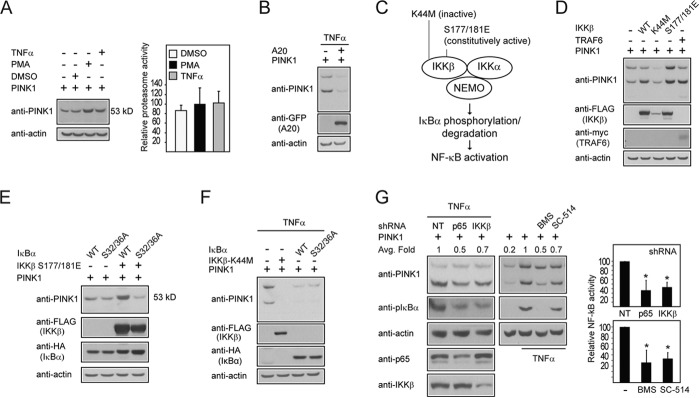
The NF-κB pathway activation stabilizes PINK1-53. A, representative immunoblots of at least three experimental sets showing the expression level of PINK1-53 in untreated PINK1-transfected HEK293 cells or those treated with vehicle (DMSO), 10 ng/ml TNFα, or 10 ng/ml PMA for 4 h. Shown are the chymotrypsin-like proteasome activities of lysates prepared from these cells. B, representative anti-PINK1 and anti-GFP immunoblots of lysates prepared from TNFα-treated cells expressing PINK1 alone or with GFP-A20. C, schematic depicting the components of the NF-κB signaling pathway. NEMO, NF-κB essential modulator. D, representative immunoblots of at least three experimental sets showing the expression level of full-length PINK1 and PINK1-53 in PINK1-transfected HEK293 coexpressing FLAG-WT, inactive (K44M), or a constitutively active (S177E/S181E) IKKβ or myc-TRAF6, as indicated. E, representative immunoblots of at least three experimental sets showing the expression level of PINK1–53 in PINK1-transfected HEK293 coexpressing HA-WT, or dominant negative S32/36A IκBα either alone or in combination with FLAG-(S177E/S181E) IKKβ, as indicated. F, representative anti-PINK1 and anti-FLAG immunoblots of cell lysates prepared from TNFα-treated HEK293 cells overexpressing untagged PINK1 with FLAG-K44M IKKβ, HA-WT IκBα, or mutant IκBα. This experiment was duplicated. G, representative immunoblots showing the levels of PINK1-53 in the presence or absence of p65, IKKβ shRNA, or non-targeting control shRNA (NT) or a pharmacological inhibitor of IKK, i.e. BMS-34551 (BMS) or SC-514. The blots were stripped and reprobed with actin as a loading control. Portions of cell lysates were also immunoblotted for NF-κB-related components. Numbers indicate the average -fold (Avg. Fold) change in the densitometric level of PINK1-53. The bar graph shows the activity of NF-κB in a separate luciferase-based assay in the absence or presence of p65 and IKKβ shRNA or a pharmacological inhibitor of IKK, i.e. BMS-34551 or SC-514 (*, p < 0.05). These experiments were repeated three times.
In the canonical pathway, the NF-κB-related transcription factors are bound and inhibited by the IκB proteins. Activators of the pathway induce the activation of the IKK complex (comprising three members, i.e. IKKα, IKKβ, and NF-κB essential modulator (NEMO)) that is responsible for the phosphorylation of IκBα and its subsequent proteasomal degradation via ubiquitination (Fig. 4C). To dissect the role of the pathway in stabilizing PINK1-53 further, PINK1 was coexpressed in the presence of wild-type IKKβ, which is expected to promote NF-κB signaling. Alongside this, an inactive IKKβ mutant (K44M) and a constitutively active counterpart (S177E/S181E) were included as controls. In the presence of wild-type or S177E/S181E IKKβ coexpression, PINK1-53 is stabilized to a comparable extent to that brought about by TRAF6 (Fig. 4D). This stabilization effect is abolished when wild-type IKKβ is substituted by the inactive K44M mutant (Fig. 4D). Along the same line of investigation, we coexpressed PINK1 with wild-type IκBα along with its dominant-negative (S32A/S36A) mutant, both of which were expected to repress NF-κB activation. As anticipated, neither of the IκBα species leads to appreciable stabilization of PINK1-53 (Fig. 4E). Moreover, both wild-type and mutant IκBα (along with the IKKβ K44M mutant) are able to block TNFα-mediated enhancement of the PINK1-53 level (Fig. 4F). Therefore, the negative effects of IκBα need to be repressed for PINK1-53 stabilization to occur in the presence of NF-κB activation. The importance of this is illustrated further by the ability of the constitutively active S177E/S181E IKKβ mutant to promote the expression of PINK1-53 in the presence of wild-type IκBα but not the dominant-negative species (Fig. 4E). Again, none of the genetic components of the NF-κB pathway that were tested here has any effects on 20S proteasome activity (data not shown), suggesting that their respective effect on PINK1-53 is independent of the proteasome. As an alternative approach, we repeated our experiments in the presence of shRNA directed at silencing p65 (a subunit of NF-κB) or IKKβ expression. Both strategies led to a significant reduction in the level of PINK1-53 in TNFα-treated PINK1-expressing cells relative to control cells transfected with non-targeting shRNA (Fig. 4G). The efficacy of the p65 and IKKβ shRNAs in reducing their expression and, thereby, NF-κB activation was ascertained by means of phospho-IκBα immunoblotting and a NF-κB-linked luciferase assay (Fig. 4G). Additionally, we also tried to pharmacologically inhibit the pathway via treatment of cells with BMS-34551 or SC-514, which are commercially available selective inhibitors of IKK. Similarly, we saw a reduction in the level of PINK1-53 in TNFα-treated PINK1-expressing cells in the presence of these inhibitors (Fig. 4G), the effects of which on NF-κB signaling are again confirmed by the abovementioned assays. Taken together, our results strongly suggest that the NF-κB pathway is a major regulator of the stability of PINK1-53, which may occur at various levels of the pathway upstream or downstream of TRAF6.
The PINK1-53 Mimetic Promotes Parkin-mediated Mitophagy in the Apparent Absence of Mitochondrial Depolarization
Interestingly, Fedorowicz et al. (16) recently reported that cytosolic cleaved PINK1 (i.e. PINK1-53) represses parkin translocation to mitochondria and, consequently, inhibits mitophagy (16). Given this finding and our results above, it is attractive to speculate that TRAF6/NF-κB activation may potentially counteract mitophagy through the promotion of PINK1-53 expression. As an initial approach to examine this, we overexpressed PINK1Δ104 (which mimics PINK1-53) in GFP-parkin-expressing HeLa cells and subjected them to CCCP treatment to see whether mitophagy is retarded. Two time points (i.e. 4 h and 24 h) post-CCCP treatment were chosen to reflect the two key aspects of the mitophagy process, i.e. coaggregation of parkin with mitochondria (visualized by means of Tim23 staining) and clearance of parkin-decorated mitochondria, respectively. Surprisingly, in the presence of PINK1Δ104 expression, neither the coaggregation of parkin with mitochondria nor the subsequent clearance of these structures was affected (Fig. 5A). Moreover, unlike wild-type PINK1 that is colocalized with parkin/Tim23-positive mitochondria in CCCP-treated cells, PINK1Δ104 remains in the cytosol in the midst of juxtanuclear parkin-mitochondrion clustering (Fig. 5B). Similar observations were made when we repeated the experiment in HEK293 cells cotransfected with parkin and PINK1Δ104 (Fig. 5C). Therefore, PINK1Δ104 expression does not appear to retard mitophagy in our hands. We were unsure about the discrepancy between our finding and that reported by Fedorowicz et al. (16). However, we noted that Fedorowicz et al. (16) used valinomycin instead of CCCP as a mitochondrial depolarizer in their experimental paradigm. We therefore repeated our experiments by treating PINK1Δ104-transfected GFP-parkin-expressing HeLa cells with 1 μm valinomycin for 1.5 and 16 h to replicate the conditions described by Fedorowicz et al. (16). Interestingly, using this paradigm, we found that, at 1.5 h post-valinomycin treatment, HeLa-GFP Parkin cells expressing PINK1Δ104 show a significant reduction in the percentage of parkin-mitochondrion colocalization compared with control-treated cells (Fig. 6, A and B). This is similar to what Fedorowicz et al. (16) have reported. However, at 16 h post-valinomycin treatment, we again found that the expression of PINK1Δ104 did not compromise the clearance of damaged mitochondria (Fig. 6, A and C). Taken together, our results do not support a role for PINK1-53 as an inhibitor of parkin-mediated mitophagy in the presence of CCCP. In the presence of valinomycin, PINK1Δ104 appears to moderately retard parkin translocation to the mitochondria, although it does not provide strong inhibition to parkin-mediated mitophagy.
FIGURE 5.

PINK1Δ104 expression fails to retard parkin-mediated mitophagy in the presence of CCCP. A, representative confocal images of GFP-parkin HeLa cells transiently transfected with untagged PINK1Δ104 treated with 10 μm CCCP for 4 or 24 h. The cells were immunostained with anti-PINK1 (red) and anti-Tim23, a mitochondrial marker (white). The outline indicates PINK1Δ104-transfected cells with mitochondria clearance. Right panel, the percentage of cells with parkin-mitochondria colocalization at 4 h of CCCP treatment and mitochondrial clearance at 24 h of CCCP treatment. The total number of cells analyzed (n) is indicated. These experiments were repeated at least three times. B, representative confocal images of GFP-parkin-expressing HeLa cells transiently transfected with untagged wild-type PINK1 or PINK1Δ104 (red) that are treated with 10 μm CCCP for 4 h. Notice that although wild-type PINK1 is colocalized with the mitochondrial cluster around the perinucleus region, PINK1Δ104 remains cytosolic. This experiment was duplicated. C, representative confocal images of HEK293 cells transiently transfected with untagged PINK1Δ104 and GFP-tagged parkin treated with 10 μm CCCP for 4 or 24 h. The outlines denote cotransfected cells with mitochondrial clearance. This experiment was duplicated.
FIGURE 6.
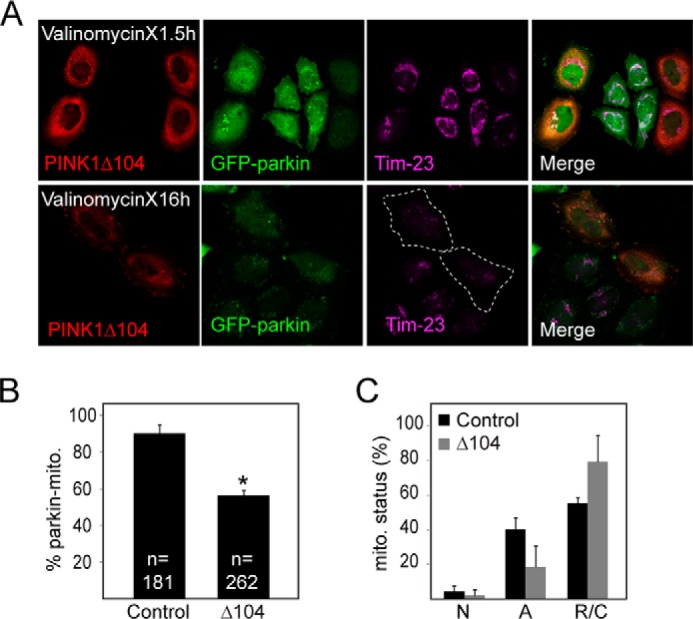
PINK1Δ104 expression fails to retard parkin-mediated mitophagy in the presence of valinomycin. A, representative confocal images of GFP-parkin HeLa cells transiently transfected with untagged PINK1Δ104 treated with 1 μm valinomycin for 1.5 or 16 h as indicated. The cells were immunostained with anti-PINK1 (red) and anti-Tim23, a mitochondrial marker (magenta). The outline indicates PINK1Δ104-transfected cells with mitochondrial clearance. B and C, the percentage of cells with parkin-mitochondria colocalization at 1.5 h of valinomycin treatment (with the total number indicated) (*, p < 0.05) and (C) mitochondrial clearance at 16 h of valinomycin treatment. N, normal; A, aggregated (parkin-positive); R/C, reduced/cleared) with at least 50 cells analyzed in each set. These experiments were repeated three times.
Curiously, during the course of our investigations, we noticed that even in vehicle (DMSO)-treated GFP-parkin-expressing HeLa cells, the presence of PINK1Δ104 tends to promote the clustering of parkin with mitochondria at the perinucleus region (Fig. 7A), which is widely taken to be a preamble to mitophagy. Quantification analysis revealed that about 80% GFP-parkin cells transfected with PINK1Δ104 exhibit this feature of parkin-mitochondria coaggregation (Fig. 7A). Again, a similar observation was made in HEK293 cells cotransfected with parkin and PINK1Δ104 (Fig. 7B). Importantly, after a prolonged period, a significant percentage of these PINK1Δ104-transfected GFP-parkin-expressing HeLa cells exhibit a reduced level or clearance of mitochondria (Fig. 7A). To exclude the trivial possibility that PINK1Δ104 expression may result in mitochondrial depolarization that leads to parkin-mediated mitophagy, we repeated the experiment in the presence of MitoTracker Red, a red fluorescent dye that stains mitochondria in live cells, and its accumulation is dependent upon mitochondrial membrane potential. Interestingly, GFP-parkin coaggregated with MitoTracker Red-positive mitochondria in the presence of PINK1Δ104 coexpression (Fig. 7C), suggesting that cytosolic PINK1Δ104 (and, thereby, PINK1-53) can promote the translocation of parkin to apparently normal mitochondria to result in their subsequent clearance. Finally, given our demonstration that NF-κB activation can stabilize PINK1-53, we wondered whether it could also result in the removal of mitochondria. To examine this, we treated cells expressing wild-type PINK1 with PMA. As anticipated, we found that pharmacological activation of NF-κB significantly increases the clearance of mitochondria in these cells (Fig. 8). Therefore, the stabilization of PINK1-53 expression appears to promote mitophagy in a non-selective manner.
FIGURE 7.

PINK1Δ104 expression promotes removal of apparently healthy mitochondria. A, representative confocal images of GFP-parkin HeLa cells transiently transfected with untagged PINK1Δ104 treated with DMSO for 4 or 24 h. The cells were immunostained with anti-PINK1 (red) and anti-Tim23 (white). The outline indicates PINK1Δ104-transfected cells with mitochondrial clearance. Right panel, top, the percentage of cells with parkin-mitochondria colocalization. Right panel, bottom, the different status of mitochondria. N, normal; A, aggregated (parkin-positive); R, reduced; C, cleared (n > 150 for each group). This experiment was repeated three times. (*, p < 0.05). B, representative confocal images of HEK293 cells transfected with GFP-parkin and vector or untagged PINK1Δ104 (white). Parkin-mitochondria aggregation is observed in the presence of PINK1Δ104 coexpression despite PINK1Δ104 localization to the cytosol. C, representative confocal images of GFP-parkin HeLa cells transiently transfected with vector or untagged PINK1Δ104 treated with DMSO for 4 h. 100 nm MitoTracker Red was added 2 h into DMSO treatment. Cells were stained with anti-Tim23 (magenta). Note that GFP-parkin colocalization with mitoaggresome was only observed with PINK1Δ104 expression. Importantly, these mitoaggresomes are MitoTracker-positive, suggesting that the mitochondria are not depolarized. These experiments were duplicated.
FIGURE 8.
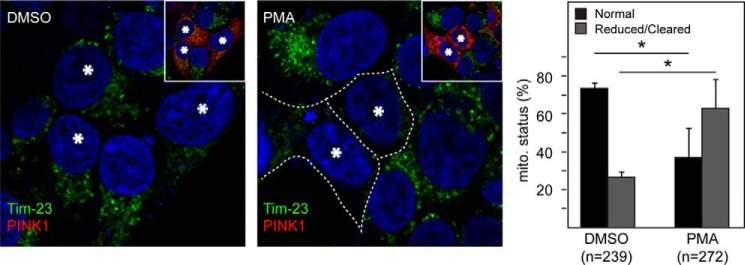
Pharmacological NF-κB activation promotes mitochondrial clearance in PINK1-expressing cells. Representative confocal images of HEK293 cells transiently transfected with untagged PINK1 treated with DMSO or 10 ng/ml PMA for 24 h (PINK1-transfected cells are denoted by asterisks and revealed by anti-PINK1 staining shown in the inset). The outline indicates the reduced mitochondrial network in PINK1-transfected cells treated with PMA. The percentage of cells showing normal or reduced/cleared mitochondria is shown in the right panel. The total number of cells analyzed (n) is indicated. This experiment was repeated three times (*, p < 0.05).
As an additional line of investigation, we wondered whether PINK1-53 could influence parkin-mediated mitophagy in neurons. Although controversial, a number of groups have reported parkin accumulation on mitochondria in CCCP-treated primary neurons as well as in induced pluripotent stem cell-derived human neurons (27–29). Using this paradigm, we examined whether PINK1Δ104 could promote the accumulation of parkin on mitochondria in the absence of CCCP treatment. For this purpose, we transfected primary mouse cortical neurons with PINK1Δ104 linked to an IRES-driven GFP (PINK1Δ104-IRES-GFP) in the presence of mcherry-tagged parkin cotransfection. As a control, we substituted PINK1Δ104-IRES-GFP with IRES-GFP. We found that the percentage of parkin-mitochondria colocalization is increased significantly in primary neurons coexpressing PINK1Δ104 and parkin (35.3%) relative to the control (6.7%) (Fig. 9), suggesting the potential ability of PINK1-53 to influence mitophagy in neurons.
FIGURE 9.
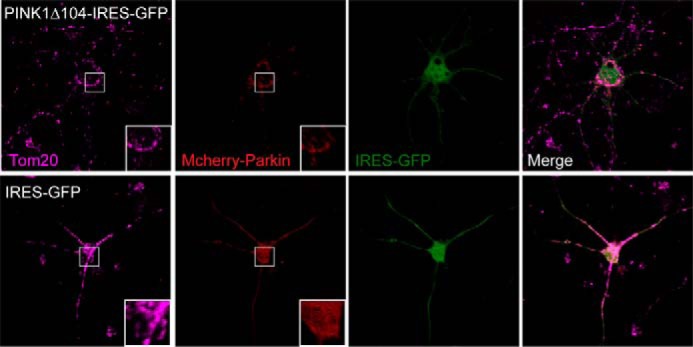
PINK1Δ104 expression promotes parkin colocalization with mitochondria in primary mouse cortical neurons. Representative confocal images of primary neurons cotransfected with PINK1Δ104-IRES-GFP or IRES-GFP and mCherry-parkin. The cells were immunostained with anti-Tom20 (magenta). Inset, magnified image of the boxed region. Note that mCherry parkin adopts a punctate appearance that colocalizes with Tom20 in the presence of PINK1Δ104-IRES-GFP (top panels) and a more uniform appearance in control cells expressing GFP alone (bottom panels). This experiment was triplicated with at least 20 parkin/PINK1-expressing neurons counted in each set.
Discussion
Taken together, we demonstrated here that Lys-63-linked ubiquitination stabilizes the otherwise rapidly degraded PINK1-53 and, at the same time, identified TRAF6-related NF-κB activation as a key pathway involved in this. We also showed that PINK1-53 may trigger parkin translocation to apparently normal mitochondria, leading to non-selective mitophagy. In essence, our findings have expanded our understanding regarding the biology of cytosolic PINK1.
To date, research on PINK1 has largely focused on understanding the function of the full-length protein, which has been demonstrated recently by several groups to be an important regulator of mitophagy (28, 30). Comparatively fewer studies have looked into the role of cytosolic PINK1, perhaps because of its labile nature. Besides mitophagy, PINK1 is also known to confer cytoprotection against a wide spectrum of insults, including pro-oxidant compounds and proteasome toxins (31, 32). Although it remains unclear whether mitochondrially localized or extramitochondrial PINK1 is mediating the protective effects, studies that utilized a cytosolic PINK1 mimetic have revealed its ability to confer cytoprotection. For example, Haque et al. (33) showed previously that PINK1 devoid of its mitochondrion-targeting signal motif could still protect neurons against the toxic effects of 1-methyl-4-phenylpyridine (33). More recently, using a PINK1 F104A mutant that favors the production of PINK1-53 and a PINK1 P95A mutant that predominantly expresses the full-length protein, Deas et al. (17) arrived at the conclusion that N-terminally cleaved (i.e. not full-length) PINK1 protects cells against the build-up of oxidative stress, although the mechanism underlying this protection was not well defined. That cytosolic PINK1 is mediating the cytoprotection of PINK1 is a reasonable proposition, considering that the full-length species is usually imported rapidly into the mitochondria, where it is cleaved rapidly and that its stabilization is taken to be a specific preamble to mitophagy. However, the first argument holds for cytosolic PINK1, which, for the most part is a highly labile species that is acted upon by the proteasome, presumably as soon as it re-enters the cytosol from the mitochondria, where it is produced (12). Therefore, for whatever function PINK1-53 is expected to fulfil outside the mitochondria, it first has to be around long enough to perform the function. Notably, a number of studies have indicated that cytosolic PINK1 may be stabilized by chaperones such as Hsp90 and Bag5 (34, 35). Here we provide an alternate mechanism for cytosolic PINK1 stabilization through its ubiquitination via Lys-63-linkages. We also identified TRAF6 as an E3 ligase responsible for this, which recruited our interest in examining the role of the NF-κB pathway in promoting the stability of PINK1-53. We found that pharmacological or genetic activation of the NF-κB pathway resulted in PINK1-53 stabilization. Interestingly, and in a reciprocal fashion, PINK1 has been demonstrated by others to promote IL-1β-mediated inflammatory signaling via the positive regulation of TRAF6 and the associated NF-κB pathway (36). Taking this into consideration, we are tempted to think that PINK1-53 may have a role to play in inflammation. Whether this involves its effects on mitophagy remains to be investigated.
As mentioned earlier, Fedorowicz et al. (16) reported recently that cytosolic cleaved PINK1 (i.e. PINK1-53) represses parkin translocation to mitochondria and, consequently, inhibits mitophagy. This is an attractive mechanism because it provides an additional layer of regulation to ensure that mitophagy does not occur under normal conditions. However, using virtually the same PINK1-53 mimetic, we were unable to see a strong retardation of parkin-mediated mitophagy by PINK1-53 in cells subjected to CCCP or valinomycin treatment, although the PINK1-53 mimetic does appear to moderately retard the translocation of parkin to the mitochondria in cells treated with valinomycin. We used similar conditions as those reported by Fedorowicz et al. (16) and are therefore unsure about the discrepancy. However, because cleaved PINK1 is known to be degraded rapidly under normal conditions, even in systems that overexpress the full-length protein, we find it difficult to envision that a rapidly degraded PINK1 species would normally act to inhibit mitophagy through its binding to parkin. If it were to be the case, the interaction of cleaved PINK1 with parkin should preclude its degradation by the proteasome and allow its appreciable detection under normal conditions. Moreover, overexpressed parkin should, in theory, be able to overcome the inhibition by endogenous cleaved PINK1 through mass effect to result in mitophagy, but this is not the case, according to the observations by Fedorowicz et al. and others (2, 16).
Instead of retarding mitophagy, we observed the reverse taking place, i.e. PINK1-53 promotes mitophagy under conditions where mitochondria are not overtly depolarized. Why a cell would mediate the clearance of apparently normal mitochondria is bewildering. However, this is not unprecedented. For example, cancer cells within the tumor mass growing under hypoxic conditions are known to rid themselves of mitochondria to facilitate energy generation via glycolysis. Furthermore, recent reports also demonstrated that, in cells undergoing prolonged hypoxia, mitophagy may help to reduce the level of reactive oxygen species and, thereby, cell death and may represent an adaptive metabolic response (37). On the basis of these results and the recognition that the NF-κB signaling pathway, which stabilizes PINK1-53, is a stress response mechanism that is largely protective in nature, we propose that the removal of normal mitochondria via a PINK1-53-mediated mechanism could serve as a protective mechanism under conditions of stress. It is well known that the mitochondria, being the site of the electron transport chain, are one of the main sources of oxidative stress. Therefore, it is conceivable that the removal of mitochondria under conditions where reactive oxygen species are elevated might serve as an attempt by the cell to mitigate the stress.
Finally, pertaining to our findings above, we are cognizant that the majority of our studies were carried out in cultured cell lines, i.e. whether our results are relevant in the neuronal context awaits further characterization. Although we provided a piece of evidence in our study above, suggesting that PINK1-53 may potentially influence parkin-mediated neuronal mitophagy, we feel that this result, although interesting, is preliminary in nature, even though we conducted the study according to the experimental paradigm reported previously by others (27–29). A comprehensive characterization in primary neurons (involving different time points, autophagy markers, and NF-κB activation) needs to be carried out before we can confirm the role of PINK1-53 in neuronal mitochondrial physiology. This is pertinent particularly in view of the controversy surrounding parkin-mediated mitophagy in neurons (38). Furthermore, we utilized ectopically expressed PINK1 in our studies largely because of the notorious lack of specificity of current commercially available PINK1 antibodies in recognizing the endogenous protein. For this purpose, we used an untagged version of PINK1 to avoid artifacts arising from epitope tagging, which is another common problem when working with PINK1. Notwithstanding these caveats, our current findings elucidate a mechanism by which a short-lived PINK1 species may be stabilized and support a functional role for this protein in normal cellular physiology. Together with others who have investigated the function of cytosolic PINK1, our report emphasizes the need to understand this often overlooked PINK1 species better, and we hope that it will pave the way for more studies.
Acknowledgments
We thank Marc Fivaz and Willcyn Tang for technical assistance.
This work was supported by grants from the National Medical Research Council-TCR and CBRG, A*STAR Biomedical Research Council-TCRP, and the National University of Singapore.
- DMSO
- dimethyl sulfoxide
- PMA
- phorbol 12-myristate 13-acetate
- CCCP
- carbonyl cyanide m-chlorophenylhydrazone
- IKK
- IκB kinase
- IRES
- internal ribosome entry site
- HHARI
- human homolog of Ariadne
- 3MA
- 3-methyladenine.
References
- 1. Lim K. L., Ng X. H., Grace L. G., Yao T. P. (2012) Mitochondrial dynamics and Parkinson's disease: focus on parkin. Antioxid. Redox Signal. 16, 935–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Youle R. J., Narendra D. P. (2011) Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 12, 9–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Okatsu K., Oka T., Iguchi M., Imamura K., Kosako H., Tani N., Kimura M., Go E., Koyano F., Funayama M., Shiba-Fukushima K., Sato S., Shimizu H., Fukunaga Y., Taniguchi H., Komatsu M., Hattori N., Mihara K., Tanaka K., Matsuda N. (2012) PINK1 autophosphorylation upon membrane potential dissipation is essential for Parkin recruitment to damaged mitochondria. Nat. Commun. 3, 1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kondapalli C., Kazlauskaite A., Zhang N., Woodroof H. I., Campbell D. G., Gourlay R., Burchell L., Walden H., Macartney T. J., Deak M. (2012) PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2, 120080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Matsuda N., Sato S., Shiba K., Okatsu K., Saisho K., Gautier C. A., Sou Y. S., Saiki S., Kawajiri S., Sato F., Kimura M., Komatsu M., Hattori N., Tanaka K. (2010) PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 189, 211–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Koyano F., Okatsu K., Kosako H., Tamura Y., Go E., Kimura M., Kimura Y., Tsuchiya H., Yoshihara H., Hirokawa T., Endo T., Fon E. A., Trempe J. F., Saeki Y., Tanaka K., Matsuda N. (2014) Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 510, 162–166 [DOI] [PubMed] [Google Scholar]
- 7. Kane L. A., Lazarou M., Fogel A. I., Li Y., Yamano K., Sarraf S. A., Banerjee S., Youle R. J. (2014) PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell Biol. 205, 143–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Poole A. C., Thomas R. E., Yu S., Vincow E. S., Pallanck L. (2010) The mitochondrial fusion-promoting factor mitofusin is a substrate of the PINK1/parkin pathway. PLoS ONE 5, e10054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ziviani E., Tao R. N., Whitworth A. J. (2010) Drosophila parkin requires PINK1 for mitochondrial translocation and ubiquitinates mitofusin. Proc. Natl. Acad. Sci. U.S.A. 107, 5018–5023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Greene A. W., Grenier K., Aguileta M. A., Muise S., Farazifard R., Haque M. E., McBride H. M., Park D. S., Fon E. A. (2012) Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep. 13, 378–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jin S. M., Lazarou M., Wang C., Kane L. A., Narendra D. P., Youle R. J. (2010) Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 191, 933–942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yamano K., Youle R. J. (2013) PINK1 is degraded through the N-end rule pathway. Autophagy 9, 1758–1769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gandhi S., Muqit M. M., Stanyer L., Healy D. G., Abou-Sleiman P. M., Hargreaves I., Heales S., Ganguly M., Parsons L., Lees A. J., Latchman D. S., Holton J. L., Wood N. W., Revesz T. (2006) PINK1 protein in normal human brain and Parkinson's disease. Brain 129, 1720–1731 [DOI] [PubMed] [Google Scholar]
- 14. Muqit M. M., Abou-Sleiman P. M., Saurin A. T., Harvey K., Gandhi S., Deas E., Eaton S., Payne Smith M. D., Venner K., Matilla A., Healy D. G., Gilks W. P., Lees A. J., Holton J., Revesz T., Parker P. J., Harvey R. J., Wood N. W., Latchman D. S. (2006) Altered cleavage and localization of PINK1 to aggresomes in the presence of proteasomal stress. J. Neurochem. 98, 156–169 [DOI] [PubMed] [Google Scholar]
- 15. Dagda R. K., Pien I., Wang R., Zhu J., Wang K. Z., Callio J., Banerjee T. D., Dagda R. Y., Chu C. T. (2014) Beyond the mitochondrion: cytosolic PINK1 remodels dendrites through protein kinase A. J. Neurochem. 128, 864–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fedorowicz M. A., de Vries-Schneider R. L., Rüb C., Becker D., Huang Y., Zhou C., Alessi Wolken D. M., Voos W., Liu Y., Przedborski S. (2014) Cytosolic cleaved PINK1 represses Parkin translocation to mitochondria and mitophagy. EMBO Rep. 15, 86–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Deas E., Plun-Favreau H., Gandhi S., Desmond H., Kjaer S., Loh S. H., Renton A. E., Harvey R. J., Whitworth A. J., Martins L. M., Abramov A. Y., Wood N. W. (2011) PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum. Mol. Genet. 20, 867–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lim K. L., Chew K. C., Tan J. M., Wang C., Chung K. K., Zhang Y., Tanaka Y., Smith W., Engelender S., Ross C. A., Dawson V. L., Dawson T. M. (2005) Parkin mediates nonclassical, proteasomal-independent ubiquitination of synphilin-1: implications for Lewy body formation. J. Neurosci. 25, 2002–2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang C., Tan J. M., Ho M. W., Zaiden N., Wong S. H., Chew C. L., Eng P. W., Lim T. M., Dawson T. M., Lim K. L. (2005) Alterations in the solubility and intracellular localization of parkin by several familial Parkinson's disease-linked point mutations. J. Neurochem. 93, 422–431 [DOI] [PubMed] [Google Scholar]
- 20. Tan J. M., Wong E. S., Kirkpatrick D. S., Pletnikova O., Ko H. S., Tay S. P., Ho M. W., Troncoso J., Gygi S. P., Lee M. K., Dawson V. L., Dawson T. M., Lim K. L. (2008) Lysine 63-linked ubiquitination promotes the formation and autophagic clearance of protein inclusions associated with neurodegenerative diseases. Hum. Mol. Genet. 17, 431–439 [DOI] [PubMed] [Google Scholar]
- 21. Lim G. G., Chew K. C., Ng X. H., Henry-Basil A., Sim R. W., Tan J. M., Chai C., Lim K. L. (2013) Proteasome inhibition promotes Parkin-Ubc13 interaction and lysine 63-linked ubiquitination. PLoS ONE 8, e73235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Todi S. V., Winborn B. J., Scaglione K. M., Blount J. R., Travis S. M., Paulson H. L. (2009) Ubiquitination directly enhances activity of the deubiquitinating enzyme ataxin-3. EMBO J. 28, 372–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lim K. L., Dawson V. L., Dawson T. M. (2006) Parkin-mediated lysine 63-linked polyubiquitination: a link to protein inclusions formation in Parkinson's and other conformational diseases? Neurobiol. Aging 27, 524–529 [DOI] [PubMed] [Google Scholar]
- 24. Lim K. L., Lim G. G. (2011) Lys-63-linked ubiquitination and neurodegeneration. Neurobiol. Dis. 43, 9–16 [DOI] [PubMed] [Google Scholar]
- 25. Murata H., Sakaguchi M., Kataoka K., Huh N. H. (2013) SARM1 and TRAF6 bind to and stabilize PINK1 on depolarized mitochondria. Mol. Biol. Cell 24, 2772–2784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shembade N., Ma A., Harhaj E. W. (2010) Inhibition of NF-κB signaling by A20 through disruption of ubiquitin enzyme complexes. Science 327, 1135–1139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Narendra D., Tanaka A., Suen D. F., Youle R. J. (2008) Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 183, 795–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Vives-Bauza C., Zhou C., Huang Y., Cui M., de Vries R. L., Kim J., May J., Tocilescu M. A., Liu W., Ko H. S., Magrané J., Moore D. J., Dawson V. L., Grailhe R., Dawson T. M., Li C., Tieu K., Przedborski S. (2010) PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. U.S.A. 107, 378–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Seibler P., Graziotto J., Jeong H., Simunovic F., Klein C., Krainc D. (2011) Mitochondrial Parkin Recruitment is impaired in neurons derived from mutant PINK1 induced pluripotent stem cells. J. Neurosci. 31, 5970–5976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Narendra D. P., Jin S. M., Tanaka A., Suen D. F., Gautier C. A., Shen J., Cookson M. R., Youle R. J. (2010) PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 8, e1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Petit A., Kawarai T., Paitel E., Sanjo N., Maj M., Scheid M., Chen F., Gu Y., Hasegawa H., Salehi-Rad S., Wang L., Rogaeva E., Fraser P., Robinson B., St George-Hyslop P., Tandon A. (2005) Wild-type PINK1 prevents basal and induced neuronal apoptosis, a protective effect abrogated by Parkinson disease-related mutations. J. Biol. Chem. 280, 34025–34032 [DOI] [PubMed] [Google Scholar]
- 32. Dagda R. K., Chu C. T. (2009) Mitochondrial quality control: insights on how Parkinson's disease related genes PINK1, parkin, and Omi/HtrA2 interact to maintain mitochondrial homeostasis. J. Bioenerg. Biomembr. 41, 473–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Haque M. E., Thomas K. J., D'Souza C., Callaghan S., Kitada T., Slack R. S., Fraser P., Cookson M. R., Tandon A., Park D. S. (2008) Cytoplasmic Pink1 activity protects neurons from dopaminergic neurotoxin MPTP. Proc. Natl. Acad. Sci. U.S.A. 105, 1716–1721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lin W., Kang U. J. (2008) Characterization of PINK1 processing, stability, and subcellular localization. J. Neurochem. 106, 464–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang X., Guo J., Fei E., Mu Y., He S., Che X., Tan J., Xia K., Zhang Z., Wang G., Tang B. (2014) BAG5 protects against mitochondrial oxidative damage through regulating PINK1 degradation. PLoS ONE 9, e86276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lee H. J., Jang S. H., Kim H., Yoon J. H., Chung K. C. (2012) PINK1 stimulates interleukin-1β-mediated inflammatory signaling via the positive regulation of TRAF6 and TAK1. Cell Mol. Life Sci. 69, 3301–3315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhang H., Bosch-Marce M., Shimoda L. A., Tan Y. S., Baek J. H., Wesley J. B., Gonzalez F. J., Semenza G. L. (2008) Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J. Biol. Chem. 283, 10892–10903 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 38. Grenier K., McLelland G. L., Fon E. A. (2013) Parkin- and PINK1-dependent mitophagy in neurons: will the real pathway please stand up? Front. Neurol. 4, 100. [DOI] [PMC free article] [PubMed] [Google Scholar]