

Background: Rifampin is a representative inducer of P-gp, and the only known mechanism is binding between rifampin and PXR.
Results: Rifampin directly activates CK2, which phosphorylates Hsp90β. Consequently, PXR increases, and P-gp expression is induced.
Conclusion: A mechanism for inducing P-gp expression by rifampin exposure is newly identified.
Significance: A strategy for overcoming P-gp-derived drug resistance is suggested.
Keywords: ABC transporter, colorectal cancer, drug resistance, heat shock protein 90 (Hsp90), tuberculosis, casein kinase 2 (CK2), heat shock protein 90 β (Hsp90β), permeability glycoprotein (P-gp), pregnane X receptor (PXR), rifampin
Abstract
The P-glycoprotein (P-gp) encoded by the MDR1 gene is a drug-exporting transporter located in the cellular membrane. P-gp induction is regarded as one of the main mechanisms underlying drug-induced resistance. Although there is great interest in the regulation of P-gp expression, little is known about its underlying regulatory mechanisms. In this study, we demonstrate that casein kinase 2 (CK2)-mediated phosphorylation of heat shock protein 90β (Hsp90β) and subsequent stabilization of PXR is a key mechanism in the regulation of MDR1 expression. Furthermore, we show that CK2 is directly activated by rifampin. Upon exposure to rifampin, CK2 catalyzes the phosphorylation of Hsp90β at the Ser-225/254 residues. Phosphorylated Hsp90β then interacts with PXR, causing a subsequent increase in its stability, leading to the induction of P-gp expression. In addition, inhibition of CK2 and Hsp90β enhances the down-regulation of PXR and P-gp expression. The results of this study may facilitate the development of new strategies to prevent multidrug resistance and provide a plausible mechanism for acquired drug resistance by CK2-mediated regulation of P-gp expression.
Introduction
Primary and/or acquired drug resistance remains one of the major limitations of the clinical efficacy of cancer treatment. Cancer cells can become resistant to chemotherapy in many ways. One such type of resistance, ATP-binding cassette (ABC)3 transporter superfamily-derived resistance, is becoming an increasingly important field of research because ABC transporters comprise 46 members, each of which has multiple substrates (1). Effective solutions against ABC transporter-driven drug resistance have yet to be developed, although efforts to overcome this phenomenon have persisted and are important issues in cancer prevention and/or treatment (2).
P-glycoprotein (P-gp) is a member of the ABC transporter family (3) that is expressed in tumor cells as well as various organs, including the liver, kidney, intestine, brain, etc. P-gp facilitates the export of potentially toxic substances or metabolites from the body (4, 5). Thus, P-gps are believed to be involved in the defense against various types of xenobiotic stresses. One major transcription factor that regulates P-gp expression in the intestine is PXR (6).
PXR-regulating drugs have been identified as a (poorly understood) P-gp-inducing mechanism. Rifampin, an anti-tuberculosis agent, is a representative PXR inducer (7) that affects the bioavailability of several drugs that are substrates of CYP3A4 and/or P-gp (8). The drugs that interact with rifampin include not only anticancer drugs but also calcium channel blockers, antiepileptic drugs, and antiretroviral agents, among others. The list is continuously updated because this phenomenon is inhibiting treatment progress (9).
The interaction between rifampin and PXR was already discovered using structural determinants (10). However, because PXR regulates molecules such as heat shock protein 90β (Hsp90β) and cytoplasmic constitutive active/androstane receptor retention protein (CCRP), additional P-gp-inducing pathways are gradually being discovered (11–13). Using a phosphoproteomic approach, we focused on Hsp90β, a highly conserved 90-kDa molecular chaperone/heat shock protein, as a key regulator of P-gp expression. Hsp90β is phosphorylated by casein kinase 2 (CK2) and forms a complex with PXR in the cytoplasm (14, 15). In the study described herein, we reveal that this pathway can be induced by rifampin.
CK2 is a nuclear matrix-associated, ubiquitously expressed, and evolutionally conserved serine/threonine protein kinase consisting of two α or α′ catalytic and two β regulatory subunits (16). CK2 phosphorylates a wide range of cellular proteins and is involved in a variety of biological functions, such as cell cycle progression (17), signal transduction (18), circadian rhythms (19), and gene expression (20). Importantly, CK2 levels are elevated in tumors compared with normal tissue, where overexpression of its catalytic subunit leads to the development of T-cell lymphoma and mammary tumorigenesis (21, 22), indicating the significance of CK2 in proliferation as well as in the formation of neoplasias (23). Similar to Hsp90β, CK2 is also related to drug resistance. CK2 regulates MRP1 (multidrug resistance-related protein 1) function via putative phosphorylation and is believed to function in MRP1-dependent drug resistance (24). Herein, we show that CK2 plays an important role at the beginning of the rifampin-mediated P-gp induction pathway and is the driving force behind its direct interaction with rifampin.
This study aimed to address the mechanism of drug-induced P-gp expression as a part of multidrug resistance. Our results suggest that CK2-mediated Hsp90β phosphorylation is essential for PXR accumulation and subsequent regulation of P-gp expression in response to rifampin, based on the direct interaction between rifampin and CK2 at initial onset.
Experimental Procedures
Chemicals, Cell Culture, and Transfection
LS174T and HEK293 cell lines were obtained from the American Type Culture Collection (ATCC) and maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 120 μg/ml penicillin, and 200 μg/ml streptomycin. Transfection was performed using Lipofectamine® 2000 according to the manufacturer's instructions (11668-019, Invitrogen). 5,6-Dichloro-1-β-d-ribofuranosylbenzimidazole (DRB) and 4,5,6,7-tetrabromotriazole (TBB) were purchased from Sigma-Aldrich. CK2α siRNA was designed as described previously (25) and synthesized by Invitrogen. Negative control siRNA (SilencerTM) was purchased from Ambion. Luciferase assays were performed using a Dual-Luciferase assay kit (Promega). BEP-800 (S14198) was purchased from Selleckchem.
cDNA and Mutagenesis Preparation
Vector of CSNK2A1 human cDNA clone (SC107027, OriGene) was changed to pcDNA3.1+ using BamHI enzyme. His-160/Asp-175 was changed to Ala-160/Ala-175 in one clone using site-directed mutagenesis. Insertion of HSP90AB1 human cDNA clone (SC108085, OriGene), was moved into pcDNA3.1+ using HindIII and ExoRI enzymes. Then Ser-225/Ser-254 was changed to Asp-225/Asp-254 and Ala-225/Ala-254 using site-directed mutagenesis. Cloning and mutagenesis quality was checked at the end of each process using direct sequencing. All processes were performed by the cloning team of CosmoGenetech. PXR siRNA was custom-built as sense (cguuuguucgcuuccugagtt) and antisense (cucaggaagcgaacaaacgtg) by Genolution.
Immobilized Metal Ion Affinity Chromatography, Two-dimensional Gel Electrophoresis, and Mass Spectrometry
Immobilized metal ion affinity chromatography (IMAC) was performed using a phosphoprotein enrichment kit according to the manufacturer's instructions (BD Biosciences). IMAC-isolated phosphoproteins were then precipitated with 10% trichloroacetic acid (TCA) and resuspended in rehydration buffer consisting of 8 m urea, 2% CHAPS, and 2% immobilized pH gradient buffer (pH 3–10). Samples were soaked in an immobilized pH gradient strip (pH 3–10) and isoelectrofocused using an IPGphor isoelectric focusing system (Amersham Biosciences). Proteins were further resolved by 4–12% gradient SDS-PAGE and visualized by silver staining. Pro-QD staining (Molecular Probe) was used to specifically detect phosphoproteins. Gels were scanned using a Typhoon 8600 laser imager (Amersham Biosciences). Spot detection and determination of densitometric intensity were performed using Image Master 2D Elite software.
Gel pieces containing proteins of interest were excised, destained, and washed prior to in-gel digestion with trypsin. Briefly, gel pieces were incubated with 10 ng/μl sequencing grade modified trypsin (Promega) in 50 mm NH4HCO4 buffer, pH 8.0, at 37 °C for 15 h, as described previously (26). Digested peptides were analyzed by nano-liquid chromatography tandem mass spectrometry (LC-MS/MS) on a fused silica microcapillary C18 column (particle size, 5 μm; inner diameter, 75 μm; length, 100 mm). LC separation was conducted using a linear gradient of buffer B (0.1% formic acid in acetonitrile) in buffer A (0.1% formic acid in H2O). The percentages of buffer B at specific time points were as follows: 0 min, 0%; 5 min, 0%; 23 min, 8%; 40 min, 16%; 76 min, 48%; 80 min, 80%. After each column run, the column was washed with 80% buffer B for 20 min and then re-equilibrated with the initial solvent (0% buffer B). The flow rate was set to 250 ml/min.
Separated peptides were subsequently analyzed on an LTQ linear ion trap mass spectrometer (ThermoFinnigan, San Jose, CA), with the electrospray voltage set to 2.0 kV and the threshold for switching from MS to MS/MS set to 1,000. The normalized collision energy for MS/MS was 35% of the main radio frequency amplitude, and the duration of activation was 30 ms. All spectra were acquired in the data-dependent mode. Each complete MS scan was followed by MS/MS scans of the nine most intense peaks from the initial scan. The MS/MS parameters used were as follows: microscan number, 1; maximum injection time, 100 ms; peak repeat count for dynamic exclusion, 1; repeat duration, 30 s; dynamic exclusion duration, 180 s; exclusion mass width, ±1.5 Da; dynamic exclusion list size, 50.
Identification of the Hsp90β Phosphorylation Site
To identify the phosphorylation site on Hsp90, LS174T colon cancer cells were treated with rifampin for 24 h, and Hsp90 was purified from protein extracts by immunoprecipitation. Immunoprecipitated Hsp90 was subjected to 4–12% gradient SDS-PAGE and silver-stained, and Hsp90 bands were subsequently isolated and digested into peptides as described above. Identification of the exact phosphorylation site was achieved by MS/MS spectroscopy of the phosphopeptides using an LTQ linear ion-trap mass spectrometer (ThermoFinnigan). SEQUEST (ThermoFinnigan) was used to analyze the MS data, and results were validated manually.
Western Blot Analysis
Proteins were separated using 4–12% SDS-PAGE and transferred to nitrocellulose membranes. Membranes were blocked with 5% nonfat milk and probed with an Omni-Phos phosphorylation assay kit (Chemicon) or with antibodies specific for P-gp, CK2, and Hsp90 (all from Santa Cruz Biotechnology, Inc.), or actin (control) (Cell Signaling). Anti-phospho-Hsp90β antibodies were generated using synthetic phosphopeptides as immunogens (Peptron) and validated using an enzyme-linked immunosorbent assay (ELISA). Membranes were then incubated with horseradish peroxidase-conjugated anti-mouse IgG or anti-rabbit IgG (Santa Cruz Biotechnology) and visualized using an enhanced chemiluminescent (ECL) system (Santa Cruz Biotechnology).
In Vitro Kinase Assay
Recombinant Hsp90β was incubated with bacterially expressed CK2α at 30 °C for 30 min in kinase assay buffer (25 mm Tris-Cl, pH 7.5, 150 mm NaCl, 1 mm MgCl2, 1 mm dithiothreitol (DTT)) with 40 mm (10 μCi) [γ-32P]ATP. Proteins were subjected to SDS-PAGE, autoradiographed, and transferred onto nitrocellulose membranes. Transferred proteins were analyzed by Western blotting with an anti-phospho-Hsp90β antibody.
Immunofluorescence Analysis
HEK293 cells were cultured on glass chamber slides and treated with rifampin and DRB, a specific inhibitor of CK2, for 24 h. After treatment, cells were washed with phosphate-buffered saline (PBS), fixed with 4% formaldehyde, permeabilized in 0.3% Triton X-100, and blocked in 4% bovine serum albumin (BSA) for 1 h. Cells were then stained with an anti-P-gp antibody and analyzed by confocal microscopy using a Zeiss LSM510 Meta microscope.
Intracellular Calcein Accumulation Assay
The intracellular accumulation of calcein was measured using the cell marker reagent calcein acetoxymethyl ester. Cells were incubated with calcein acetoxymethyl ester (0.5 μm) at 37 °C for 10 min, and accumulation was halted after 10 min by rapidly washing the cells three times with ice-cold PBS. Cells were then lysed by incubation with 250 μl of 1% Triton X-100 in PBS for 1 h. Cell lysates (200 μl) were transferred into a 96-well plate, and fluorescence, which represents intracellular calcein, was measured using a Victor3 plate reader (PerkinElmer Life Sciences) with excitation and emission wavelengths of 485 and 535 nm, respectively. A Bradford protein assay kit was used to determine the amount of cellular protein in each lysate (Bio-Rad).
GST Pull-down Assay
Purified Hsp90β-glutathione S-transferase (GST) was mixed with PXR in 500 μl of ADB II (20 mm MOPS, pH 7.2, 2.5 mm β-glycerol phosphate, 1 mm Na3VO4, 1 mm DTT, and 1 mm CaCl2) containing pre-equilibrated glutathione-Sepharose 4B beads, followed by incubation at 4 °C with gentle rotation. After 1 h, CK2α was added to the mixture and incubated for an additional 2 h at 4 °C. Beads were then centrifuged at 600 × g for 1 min and washed five times with 1 ml of PBS. Bound proteins were resuspended in 50 μl of 2× lithium dodecyl sulfate loading buffer and analyzed by Western blotting for PXR, GST, and Hsp90β.
Measurement of the -Fold Change in Rifampin-induced PXR mRNA Expression by Real-time PCR
Total RNA was extracted from rifampin-treated and -untreated cells using TRIzol® reagent (15596-018, Invitrogen). The RNA was quantified by NanoDrop technology (Thermo Scientific NanoDrop 2000 spectrophotometer, Thermo Fisher Scientific), and the purity was checked by measuring the absorbance of 260 and 280 nm. First strand cDNA was synthesized using 1 μg of RNA in a 50-μl reaction. The reaction mix was composed of 10× TaqMan RT buffer (5 μl), 25 mm MgCl2 (11 μl), dNTPs (10 μl), random hexamers (2.5 μl), RNase inhibitor (1 μl), reverse transcriptase (1.25 μl), RNA (1 μg), and nuclease-free water (up to 50 μl). Real-time PCR was carried out using TaqMan® Universal PCR Master Mix (4304437, Applied Biosystems (Foster City, CA)). PXR mRNA was quantified by real-time PCR using a 7900HT fast real-time PCR system (Applied Biosystems) using human PXR as a probe (4331182, Applied Biosystems). The relative mRNA expression level of PXR was calculated by the ΔΔCt method. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (4331182, Applied Biosystems) was used as an internal control for normalization.
Proteasome Inhibition Assay
LS174T cells were incubated in 60-mm plates and treated with 0–2.5 μm MG132 (474790, Calbiochem) for 8 h or with 2.5 μm MG132 for 0–8 h. The cells were lysed with radioimmune precipitation assay buffer after two washes with PBS and centrifuged at 13,000 rpm for 10 min. The supernatants were subjected to Western blotting with anti-PXR antibodies (sc-48403, Santa Cruz Biotechnology).
Cell Fractionation Assay
The nuclear localization of several proteins was evaluated by a cell fractionation assay using a Qproteome cell compartment kit (37502, Qiagen GmbH (Hilden, Germany)). Briefly, LS174T cells were subcultured in 6-well plates to 50% confluence. After 24 h, the cells were treated with rifampin (80 μm) or DMSO as a negative control. After 24 h of treatment, the cells were collected, a lysate was prepared, and cytosolic and nuclear fractions were prepared according to the manufacturer's protocol. Protein quantification was done by the Bradford assay using Bio-Rad protein assay dye reagent (500-0006, Bio-Rad). In total, 25 μg of protein was used for Western blotting with protein separation performed on a precast 4–12% polyacrylamide gel (Invitrogen). The separated proteins were transferred to an Amersham Biosciences PVDF membrane (GE Healthcare), and nonspecific proteins were blocked using 5% skim milk in Tris-buffered saline containing 0.1% Tween 20 (TBST). The membrane was then incubated with specific primary antibodies overnight at 4 °C. After a single wash with TBST, the membrane was incubated with horseradish peroxidase-conjugated secondary antibodies for 1 h at room temperature. The proteins were detected with Western blotting Luminol reagent (sc-2048, Santa Cruz Biotechnology) and enhanced chemiluminescence reagent (RPN-2235, GE Healthcare). The detected proteins were exposed to Eastman Kodak Co. medical x-ray film.
Analysis of the Molecular Docking of CK2 and Rifampin
A molecular docking simulation was performed using SYBYL-X version 2.1 (Tripos International, St. Louis, MO). The crystal structure of CK2 (Protein Data Bank code 4FBX) was used to determine the best docking position for rifampin at the binding site (27). The structure of rifampin was obtained from another Protein Data Bank structure (entry 2HW2) (28). Docking site residues were selected as the range within 10 Å of TBB. The considered residue numbers are 44–55, 65–73, 94–98, 110–124, 161–165, and 171–178. The protein and ligand were minimized using the Tripos force field with Gasteiger-Hückel charges until the root mean square gradient was <0.05. Surflex docking was performed using the following settings. Hydrogen and heavy atoms were incorporated to allow protein movement, the covalent force field weighting was set at full for the “Ligand” option and 0.3 for the “Protein” value, the maximum number of poses per ligand was set to 30, and the minimum root mean square deviation between final poses was set to 0.05. The Surflex-Dock scoring function was applied to generate docking results, and complex structures that matched as closely as possible to the productive bound form of the ligand were selected (i.e. complexes with the highest matching score).
In Vitro Kinase Assay for CK2 and Rifampin
CK2 kinase activity was measured using a casein kinase 2 assay kit (17-132, Millipore). Briefly, TBB, a specific inhibitor of CK2 (T0826, Sigma), and rifampin (R3501, Sigma), were tested in a phosphorylation reaction mixture with [γ-32P]ATP (3,000 Ci/mmol; NEG002A, PerkinElmer Life Sciences), CK2 (14-197, Millipore), or CK2α (14-445, Millipore). Reaction mixtures were incubated for 15 min at 30 °C according to the manufacturer's instructions. Reactions were stopped by the addition of 40% TCA (T6399, Sigma), and 32P-labeled substrates were detected by scintillation counting.
Direct Binding Assay
A direct binding assay was performed according to the manufacturer's instructions using a MultiScreen HTS 96-well filtration plate and vacuum manifold (MSHVN4B10 and MSVMHTS00, respectively (Millipore)). [3H]Emodin (2.0 Ci/mmol) and [3H]rifampin (7.3 Ci/mmol) were purchased from Moravek Biochemicals (MT-1759 and MT-1626, respectively). TBB (T0826), rifampin (R3501), and emodin (E7881) were purchased from Sigma-Aldrich. [3H]Emodin or [3H]rifampin was mixed with purified CK2α protein, which was bound to beads in ADB I buffer (20 mm MOPS, pH 7.2, 25 mm β-glycerol phosphate, 5 mm EGTA, 1 mm sodium orthovanadate, and 1 mm dithiothreitol). TBB, emodin, or rifampin was for inhibition assay. The plates were incubated at room temperature for 40 min. The assay reactions from the plates were filtered and washed twice with 200 μl of ice-cold 50 mm Tris-HCl (pH 7.8). Finally, the remaining [3H]rifampin or [3H]emodin was quantified after adding 20 μl of scintillation mixture to each well.
Results
Identification of Phosphoproteins during P-gp Induction by Rifampin
Because phosphorylation can functionally activate many proteins in cellular processes, proteins whose phosphorylation status is modulated during the induction of MDR1 (multidrug resistance 1) gene expression were first identified. The phosphoproteomics-based approach was utilized in the study. We found that P-gp and MDR1 expression levels increased following rifampin treatment in LS174T colon cancer cells in a dose- and time-dependent manner (data not shown), which is consistent with a previous report (6). To isolate phosphoproteins, protein extracts from LS174T cells treated with 40 μm rifampin or dimethyl sulfoxide (DMSO; vehicle) for 24 h were loaded onto a Fe3+-IMAC column and separated by two-dimensional gel electrophoresis. After visualization of the separated proteins using the phosphoprotein-specific Pro-QD stain, two-dimensional profiles of phosphoproteins were compared between those exposed to DMSO and rifampin (Fig. 1A). Eight phosphoproteins whose intensities were significantly different between the two treatments were identified using trypsin digest and nano-LC/MS/MS of spots (data not shown). Of these eight proteins, Hsp90β showed the highest differential staining intensity between treatment groups (Fig. 1, A and B). Hsp90β protein levels were also confirmed using Western blot analysis by comparing Hsp90β expression before and after IMAC elution. Hsp90β expression did not differ among the basal extracts of LS174T cells treated with rifampin or DMSO. However, the level of Hsp90β in the IMAC elute was much higher in rifampin-treated cells than in control (Fig. 1C). This result suggests that rifampin treatment increases the phosphorylation of Hsp90β rather than its protein level per se.
FIGURE 1.

Phosphorylation of Hsp90β is induced by rifampin. A, two-dimensional PAGE of purified phosphorylated proteins from control and rifampin-treated LS174T cells. Phosphorylated Hsp90β is indicated by a red circle. B, representative mass spectra of Hsp90β. C, IMAC eluates were subjected to Western blotting with an anti-Hsp90 antibody.
Phosphorylation of Hsp90β at Ser-225/Ser-254 Correlates with Rifampin-induced P-gp Expression
We next identified the specific amino acid residues of Hsp90β that are phosphorylated during rifampin-mediated P-gp induction. Hsp90β protein was purified from DMSO- or rifampin-treated LS174T cells by immunoaffinity chromatography. Purified proteins were subjected to SDS-PAGE, and bands corresponding to Hsp90β were excised, digested with trypsin (Fig. 2A), and analyzed by MS/MS. The MS/MS peaks, corresponding to phospho-Ser, -Thr, or -Tyr, exhibited a 79.97-Da mass shift compared with the corresponding unmodified residues. Induction of neutral loss of phosphoric acid (97.98 Da) on phospho-Ser/Thr-containing peptides has been reported to occur upon low energy collisional activation in positive ion mode. As shown in Fig. 2B, phosphopeptides were detected specifically in the Hsp90β from rifampin-treated LS174T cells. From the MS/MS sequence analysis of these phosphopeptides, both Ser-225 and Ser-254 were identified as phosphorylated residues (Fig. 2B). In contrast to rifampin-treated cells, neither Ser-225- nor Ser-254-containing phosphopeptides were detected in the Hsp90β band from DMSO-treated LS174T cells by MS/MS sequence analysis (data not shown). In addition, Hsp90β protein expression was unchanged in rifampin-treated LS174T cells; however, the expression of phosphorylated Ser-225 or Ser-254 of Hsp90β increased upon treatment with rifampin (Fig. 2C). In addition, a 2-aminothieno[2,3-d]pyrimidine ATP competitive Hsp90 inhibitor, BEP-800, was applied to confirm the possibility of P-gp regulation by Hsp90β phosphorylation. Upon rifampin treatment, the expression of P-gp, PXR, and phosphorylated Ser-225 or Ser-254 of Hsp90β increased. However, the rifampin effect was canceled out when BEP-800 was added in combination with rifampin. Hsp90β expression was unaffected by BEP-800 and/or rifampin treatment (Fig. 2D). These findings indicate that phosphorylation of Hsp90β at Ser-225/Ser-254 is required for the rifampin-mediated induction of P-gp expression.
FIGURE 2.

Identification of Hsp90β phosphorylation sites. A, protein extracts were prepared from LS174T cells treated with either vehicle (DMSO) or rifampin (40 μm) and immunoprecipitated (IP) with an anti-Hsp90β antibody. Recovered proteins were separated by SDS-PAGE and visualized by silver staining. B, MS/MS spectra of Hsp90β phosphopeptides in which the phosphorylation sites were localized to Ser-225 (top) and Ser-254 (bottom). Bands containing Hsp90β were excised from the gel, digested with trypsin, and analyzed by LC-MS/MS, as described under “Experimental Procedures.” C, LS174T cells were incubated with vehicle (DMSO) or rifampin (40 μm) for 24 h. Cell lysates were subjected to Western blot analysis with antisera raised against phosphorylated Hsp90β Ser-225 and Ser-254. Blots were reprobed an anti-Hsp90β antibody. D, P-gp, PXR, Hsp90β, and phosphorylated Hsp90β expression was determined by Western blotting with or without BEP-800 and rifampin.
CK2 Is the Kinase Responsible for Ser-225/254 Phosphorylation of Hsp90β
To develop several possible strategies for preventing multidrug resistance, it is necessary to determine the kinase upstream of Hsp90β responsible for rifampin-induced P-gp expression. Amino acid sequences flanking Ser-225 and Ser-254 in Hsp90β contain a consensus phosphorylation site for the Ser/Thr protein kinase CK2, which has acidic residues at position n + 3 (16). As shown in Fig. 3A, CK2 readily phosphorylated recombinant Hsp90β, which was markedly suppressed by the presence of DRB, a specific pharmacological inhibitor of CK2. To further determine whether CK2 activity is responsible for the observed phosphorylation of Ser-225 and Ser-254, phosphorylated Hsp90β was analyzed by Western blot using an anti-Hsp90β-phospho-Ser-225/254 antibody. CK2 consistently catalyzed the phosphorylation of Hsp90β at Ser-225 and Ser-254. Furthermore, this effect was suppressed by the presence of DRB in vitro (Fig. 3B). Moreover, peptides containing Ser-225 or Ser-254 of Hsp90β were phosphorylated by CK2, and DRB abolished CK2-mediated peptide phosphorylation, as monitored by the consumption of ATP using the Kinase-GloTM luminescent assay (Fig. 3C). To confirm the effect of rifampin on CK2, CK2α protein expression was measured by Western blot analysis in rifampin-treated LS174T cells. As shown in Fig. 3D, incubation with rifampin increased the expression of CK2α. Moreover, rifampin treatment consistently and significantly stimulated the kinase activity of CK2 on Hsp90β Ser-225/254 residues according to the Kinase-GloTM luminescent assay; this effect was sensitive to treatment with DRB (Fig. 3E). In addition, rifampin treatment increased the phosphorylation of Hsp90β Ser-225/Ser-254 in LS174T cells, which was abrogated by incubation with CK2 inhibitors DRB and TBB (Fig. 3F). Finally, an siRNA specific to CK2α was applied to HEK293 cells. Treatment of rifampin also increased Ser-225/Ser-254 phosphorylation of Hsp90β, and knockdown of CK2α by the siRNA markedly suppressed the induction of rifampin-induced Ser-225/Ser-254 phosphorylation (Fig. 3G). Taken together, these results indicate that Hsp90β is efficiently phosphorylated by CK2, and Ser-225/254 residues are the major CK2 acceptor sites; this relationship between CK2 and Hsp90β constitutes a part of rifampin-induced P-gp induction. Additionally, the phosphorylation effect was confirmed using the phosphomimetic mutant and control mutant (Ser-225/Ser-254 to Asp-225/Asp-254 and Ala-225/Ala-254, respectively) of Hsp90β cDNA. Expression of P-gp was higher at Asp-225/Asp-254 than that of Ala-225/Ala-254. Hsp90β protein levels were equal among transfections of WT, Ala-225/Ala-254, and Asp-225/Asp-254 (Fig. 3H).
FIGURE 3.

CK2 phosphorylates Ser-225 and Ser-254 of Hsp90β for P-gp induction. A and B, recombinant Hsp90β was incubated with purified CK2α (8 ng) and/or DRB (40 μm). Samples were analyzed by autoradiography (A) or Western blotting with anti-phospho-p225-Hsp90β and anti-phospho-p254-Hsp90β antibodies (B). C, peptides containing Ser-225 or Ser-254 of Hsp90β were incubated with purified CK2α and/or DRB (40 μm). The amount of ATP in the reaction was determined using a Kinase-GloTM luminescent assay kit (Promega). D, protein extracts from LS174T cells treated with or without rifampin (40 μm) were subjected to Western blotting with anti-P-gp and anti-CK2α antibodies. E, peptides containing Ser-225 or Ser-254 of Hsp90β were incubated with protein extracts from LS174T cells treated with rifampin (40 μm) and/or DRB (40 μm). Kinase activity was determined using a Kinase-GloTM Luminescent assay kit. F, protein extracts from LS174T cells treated with 40 μm rifampin and DRB (40 μm) or TBB (80 μm) were subjected to Western blotting with anti-phospho-p225-Hsp90β, anti-phospho-p254-Hsp90β, and Hsp90β antibodies. G, HEK293 cells were transfected with a control siRNA (40 nm) or CK2α siRNA (40 nm) for 48 h in the presence of rifampin (40 μm). Cell lysates were analyzed by Western blotting with antibodies specific for CK2α, phospho-p225-Hsp90β, and phospho-p254-Hsp90β. H, LS174 cells were transfected with a wild type (Ser-225/Ser-254), control mutated (Ala-225/Ala-254), or phosphomimetically mutated (Asp-225/Asp-254) Hsp90β cDNA. Protein levels of P-gp were compared by Western blotting. RLU, relative luminescence units. Error bars, S.D.
Effects of CK2 Inhibition on MDR1 Transcript Level, P-gp Expression, and Cellular Localization
The effect of CK2 inhibition on MDR1 transcript and P-gp protein levels was evaluated using semiquantitative reverse transcription polymerase chain reaction (RT-PCR) and Western blot analysis, respectively. Rifampin treatment consistently led to the accumulation of MDR1 mRNA and P-gp protein, whereas DRB abrogated rifampin-induced P-gp expression (Fig. 4A). The expression of P-gp induced by rifampin was identical to that of β-catenin, which binds to the cytoplasmic domain of the plasma membrane protein cadherin. In addition, P-gp expression was diminished in the presence of DRB (Fig. 4B). Furthermore, the function of P-gp expression in rifampin-treated HEK293 cells was assessed by examining the intracellular accumulation of calcein, a substrate probe of P-gp, in the presence and absence of DRB. As shown in Fig. 4C, treatment with rifampin significantly decreased intracellular calcein accumulation, which was completely restored by DRB. In addition, siRNA-mediated knockdown of CK2α clearly nullified the rifampin-induced up-regulation of P-gp expression (Fig. 4D). These results suggest that CK2 activity, which leads to the phosphorylation of Hsp90β at Ser-225/254, is closely associated with the drug-induced up-regulation of P-gp expression.
FIGURE 4.

CK2 activity is required for P-gp expression. A, protein (top) and mRNA (bottom) were prepared from HEK293 cells treated with vehicle (DMSO) or DRB (40 μm) in the presence of rifampin (40 μm) and analyzed by Western blotting with an anti-P-gp antibody and semiquantitative RT-PCR for MDR1 mRNA. B, immunofluorescence analysis of P-gp expression in HEK293 cells. After fixation, cells were stained with anti-P-gp or β-catenin antibodies and observed at ×400 magnification. C, HEK293 cells treated with vehicle (DMSO) or DRB (40 μm) in the presence or absence of rifampin (40 μm) were incubated with calcein acetoxymethyl ester (0.5 μm) for 10 min. Intracellular calcein fluorescence was measured and normalized to the protein concentration. D, HEK293 cells were transfected with control siRNA (40 nm) and CK2α siRNA (40 nm) for 48 h in the presence or absence of rifampin (40 μm). Cell lysates were analyzed by Western blotting with anti-CK2α and anti-P-gp antibodies. Error bars, S.D.
CK2-mediated Hsp90β Phosphorylation Increases PXR Protein Stability
PXR, a nuclear receptor protein that regulates MDR1 gene expression, forms a heteromeric complex with Hsp90β and is related to the stability of its client protein (15, 29). Therefore, we next determined whether CK2-mediated Hsp90β phosphorylation influences the extent of the Hsp90β-PXR interaction by immunoprecipitation. Rifampin treatment increased the interaction between PXR and Hsp90β in LS174T cells. Moreover, this effect was suppressed by the presence of DRB (Fig. 5A). Further in vitro experiments showed that PXR precipitated with Hsp90β in the presence of CK2 and ATP, suggesting that the Hsp90β-PXR interaction is increased by CK2-mediated Hsp90β phosphorylation (Fig. 5B). The effect of rifampin treatment on PXR expression was also evaluated by Western blot and RT-PCR. Rifampin treatment resulted in the up-regulation of PXR protein, but not mRNA (Fig. 5C). The PXR mRNA level was quantitatively confirmed using real-time PCR. The relative mRNA level did not change significantly according to rifampin treatment, consistent with RT-PCR results (Fig. 5D). To determine whether CK2 inhibition also affects PXR expression, LS174T cells were treated with rifampin in the presence or absence of DRB. In accordance with previous results, DRB treatment diminished the induction by rifampin of PXR protein expression (Fig. 5E). In an attempt to evaluate the influence of CK2 activity on PXR protein stability, the proteasome inhibitor, MG132, in combination with rifampin and/or DRB, was utilized after confirming the effect of MG132 on PXR protein expression. We found that PXR expression increased in a dose- and time-dependent manner (Fig. 5F). The inhibitory effect of DRB on PXR protein expression was also abrogated by the addition of MG132, showing that proper CK2 activity is necessary for PXR protein stability (Fig. 5G). When treating PXR siRNA, expression of PXR and P-gp was decreased. Even when PXR siRNA was treated with rifampin, expression of PXR and P-gp was decreased compared with only rifampin treatment. However, expression levels of CK2α and Hsp90β were not affected with or without PXR siRNA transfection (Fig. 5H). Together, these results indicate that Hsp90β, which is phosphorylated by CK2, interacts with PXR and leads to an increase in its stability via inhibition of proteasome-mediated degradation.
FIGURE 5.

Phosphorylation of Hsp90β is required for its interaction with PXR. A, Hsp90β was immunoprecipitated (IP) from LS174T cells treated with rifampin (40 μm) or DRB (40 μm). Precipitated proteins were subjected to Western blotting with an anti-PXR antibody. B, recombinant GST or GST-Hsp90β was incubated with purified CK2 (8 ng) for 30 min and then incubated with in vitro-translated PXR for GST pull-down assays. Precipitated proteins were then subjected to Western blotting with antibodies specific for Hsp90β, PXR, and GST. C, proteins (top) or mRNA (bottom) were prepared from LS174T cells treated with either DMSO or rifampin (40 μm) and analyzed by Western blotting with an anti-PXR antibody or semiquantitative RT-PCR for PXR mRNA. D, LS174T cells were treated with DMSO or rifampin for 24 h, after which mRNA was isolated and converted to cDNA. A quantitative analysis was performed using real-time PCR with normalization against GAPDH. The results were obtained in three independent experiments. E and G, protein extracts were prepared from HEK293 cells treated with vehicle (DMSO) or DRB (40 μm) in the presence of rifampin (40 μm), exposed to MG-132 (20 μm) for 8 h, and subjected to Western blotting with an anti-PXR antibody. F, PXR protein quantities were compared according to different concentrations of MG132 in LS174T cells treated for 8 h (top). The amounts of PXR in LS174T cells treated with 2.5 μm MG132 for different time periods were compared (bottom). H, using PXR siRNA and rifampin, the protein levels of CK2α, Hsp90β, P-gp, and PXR were confirmed by Western blotting. C, E, F, G, and H, blots were reprobed with an anti-β-actin antibody, which served as a loading control. Error bars, S.D.
Rifampin Directly Activates CK2-mediated Phosphorylation
The potential for rifampin to directly stimulate CK2 was verified. Following treatment with ADB I, CK2 or CK2α, the CK2 substrate peptide, and [γ-32P]ATP were incubated with TBB or rifampin. CK2 and CK2α activities both decreased following TBB treatment. IC50 concentrations were 10−6 to 10−5 m (Fig. 6). In contrast, CK2 and CK2α levels increased following rifampin treatment (Fig. 6). CK2α and [3H]rifampin binding was then confirmed in vitro. [3H]Emodin, a known CK2 binding partner (30), served as a positive control (Fig. 7, A and B). The amount of binding increased, depending on the concentrations of both drugs, and was inhibited with the addition of non-tritium-labeled drugs and TBB. Inhibition concentrations of rifampin, emodin, and TBB were determined using a prebinding assay (data not shown). Binding affinity of [3H]rifampin to Ala-160/Ala-175 mutated CK2α was compared with wild type CK2α. Affinity to mutated CK2α was lower than that of wild type CK2α (Fig. 7C). Fig. 8 shows the result from the molecular docking prediction between CK2 and rifampin. Binding energies (ΔGb) from the docking simulation with CK2 were −1.97 kcal/mol and −5.06 kcal/mol for rifampin and TBB, respectively. Log p values, which indicate hydrophobicity, were −18.719 and 2.877 for the CK2 binding pocket and rifampin, respectively. The binding site volume was 584.47 Å3. At first glance, it appeared that the ring structure of rifampin might bind inside the CK2 binding pocket, compared with TBB. However, due to the steric hindrance of piperazine (C4H10N2), the carbon chain group bound inside of the binding pocket. This aspect of the binding complex is similar to that by which rifampin binds to Arr enzymes (28). Rifampin showed interactions with 10 residues of CK2, including Arg-47, Val-53, Val-66, Lys-68, Phe-113, Val-116, Asn-118, His-160, Met-163, and Asp-175. Among them, His-160 and Asp-175 showed a hydrogen bond with ligand (Fig. 8C). In silico mutation with Ala showed an increase of binding energy (H160A, 2.95 kcal/mol; D175A, 2.80 kcal/mol), indicating a decrease in binding affinity.
FIGURE 6.

In vitro kinase assay. CK2 (A) or CK2α (B), a CK2 substrate peptide, [γ-32P]ATP, and various concentrations of TBB or rifampin were incubated for 10 min.
FIGURE 7.

Direct binding assay. The binding of CK2α to [3H]emodin (A) or [3H]rifampin (B) was quantified. The respective non-tritium-labeled drug and TBB were used for competition. C, the binding of [3H]rifampin to wild type CK2α or Ala-160/Ala-175 mutated CK2α was quantified.
FIGURE 8.

Molecular docking analysis of CK2 and rifampin. A, overview of CK2 and rifampin binding. B, close-up view of the binding site in CK2. C, hydrogen bonds between CK2 and rifampin. Green, CK2; cyan, binding site pocket; yellow, TBB; magenta, rifampin.
Hsp90β Location Confirmation
PXR and Hsp90β quantities were compared at the cytoplasm and nucleus according to the rifampin treatment (Fig. 9). PXR was increased at both the cytoplasm and nucleus after rifampin treatment. However, the increase was much higher at the nucleus than at the cytoplasm. Hsp90β was same with or without rifampin treatment at the cytoplasm. No signal of Hsp90β was found in the nucleus.
FIGURE 9.
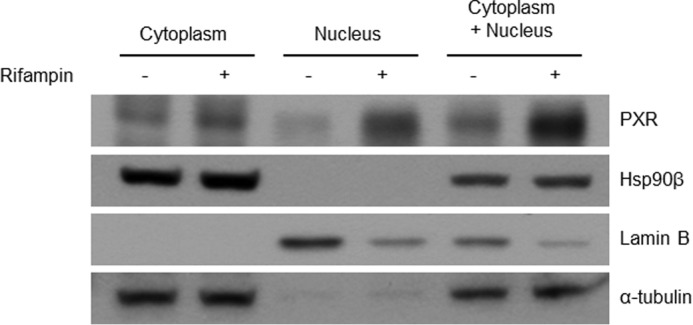
Hsp90β location confirming. Fractions of cytosol and nucleus were divided after 24-h rifampin or DMSO treatment of the LS174 cell line. The fifth and sixth lines were the sum of the 50% cytosolic and 50% nuclear fraction. Lamin-B and α-tubulin served as nuclear and cytosolic marker, respectively.
Graphic Presentation
The contents of this study are summarized in Fig. 10. As already reported in several papers, each step of Fig. 10, B–D, is again confirmed for solidifying our hypothesis of the rifampin-induced MDR1 overexpression mechanism. Rifampin activity of binding to CK2 and activating CK2 (Fig. 10A) has been added to this paper. To date, the mechanism of rifampin-induced MDR1 overexpression has only been known for binding between rifampin and PXR. However, the mechanisms shown in Fig. 10, B–D, are caused by Fig. 10A, and all of these steps constitute a new mechanism (Fig. 10E) of rifampin-induced MDR1 overexpression.
FIGURE 10.
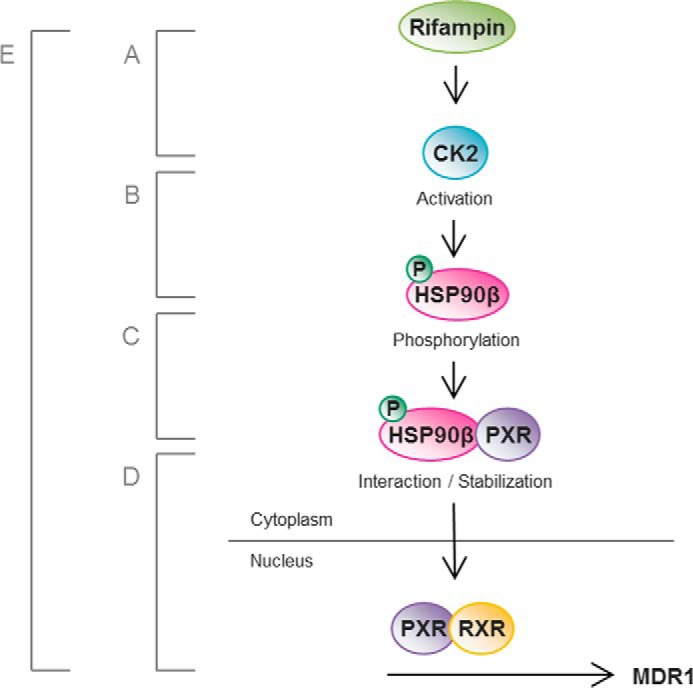
Graphic presentation. The hypothesis of this study is summarized as a simple illustration. A, rifampin binds directly to and activates CK2. B, CK2 phosphorylates Hsp90β at Ser-225 and Ser-254. C, Hsp90β binds to and stabilizes PXR in the cytoplasm. D, PXR is translocated into the nucleus in the absence of Hsp90β. E, the proposed pathway of MDR1 overexpression with rifampin treatment comprising steps A–D.
Discussion
The induction of P-gp expression has long been acknowledged for decreasing the bioavailability of drugs co-administered with rifampin (31). Rifampin acts as a ligand and direct activator of nuclear receptor PXR, and its role in P-gp induction has been unknown until now. PXR is believed to be retained in the cytoplasm by binding to Hsp90 and CCRP (15). Upon activation by xenobiotics, such as rifampin, PXR dissociates and translocates into the nucleus, where it interacts with RXR to induce the transcriptional activation of several genes involved in drug metabolism (12, 32, 33). In addition to PXR, various transcription factors, including Sp1, YB-1, NF-Y, p53, NF-κB, HSF, and β-catenin/TCF-4, have been shown to regulate P-gp expression in vitro, but their effects have yet to be verified in vivo (5). The present study aimed to determine the mechanism underlying PXR regulation of P-gp induction by rifampin stimulation.
Using a phosphoproteomic approach, eight phosphorylated proteins were identified in colorectal carcinoma LS174T cells after rifampin exposure (data not shown). Of the eight phosphorylated proteins, Hsp90β exhibited the highest signal (Hsp90α was also included). In concordance with previous reports, our results strongly suggest that Hsp90 forms a complex with PXR in the cytoplasm before its translocation into the nucleus (12). Dissociation of Hsp90β and PXR in the cytoplasm prior to PXR translocation was verified in this study. Following treatment with rifampin, cytoplasmic PXR increased, and PXR in the nucleus was much more increased than that in the cytoplasm. However, Hsp90β was localized only to the cytoplasm regardless of rifampin treatment (data not shown). However, the functions of the remaining six proteins related to PXR or P-gp have yet to be determined. Therefore, multiple as yet unknown mechanisms probably regulate rifampin-induced P-gp induction, although each may not contribute equally.
The present study confirmed the ability of CK2 to phosphorylate Hsp90β, which acts upstream of the PXR pathway, as well as P-gp stimulation by rifampin treatment. The Hsp90β-phosphorylating function of CK2 per se was identified previously (14, 16, 34). Other possibilities for P-gp regulation were also confirmed. Under the same experimental conditions as Fig. 4A, treatment with inhibitors for kinases other than CK2, such as SB203580 (MAPK), PD98059 (MEK), and wortmannin (PI3K), showed no such suppression of P-gp expression (data not shown).
The ability of rifampin to independently activate CK2 has yet to be described. This pathway was evaluated in two experiments in this study. In a direct binding assay, the Vmax and Km values could not be determined due to the experimental conditions. To reach saturation status, the drug dose would have to increase, or the protein quantity would have to decrease. When the protein concentration was lower than the minimum recommended by the manufacturer, the experiment failed. Therefore, the binding assay was performed in a non-saturated state.
Because CK2 shows activity against more than 300 substrates based on a 2003 report (35) and is associated with several pathways, CK2 inhibitors have been used to improve the efficacy of many drugs. To date, combination therapy of CK2 inhibitors with anticancer agents has been investigated based on the known roles of CK2, including in apoptosis, the PI3K-Akt-mTOR pathway, angiogenesis, NF-κB, Wnt signaling, the epithelial-mesenchymal transition, DNA damage repair, and Hsp90 machinery (36). Hsp90 inhibitors are also being investigated as a combination partner with anticancer drugs. A recent report found that combination therapy with an Hsp90 inhibitor and paclitaxel increased the efficacy of paclitaxel in a non-small cell lung cancer system (37). The synergistic effect of the Hsp90 inhibitor was assumed to be due to the role of Hsp90 in cell survival, such as prosurvival signaling, and effects on the cell cycle (36). If the P-gp-inducing pathway identified in this study was also active in a lung cancer environment, then inhibition of P-gp may also contribute to the increased efficacy of paclitaxel.
CK2 and Hsp90β have been developed as targets of combination cancer therapy based on enhancing mechanisms of cancer drugs. In this regard, this study provides a new understanding of combination therapy by suppressing additional P-gp expression. In summary, we propose a mechanism for rifampin-induced up-regulation of P-gp. CK2-mediated phosphorylation of Hsp90β constitutes a crucial upstream molecular mechanism that regulates P-gp expression. Because increased P-gp expression is a major issue in rifampin-induced drug resistance, our findings may facilitate the development of new strategies to prevent P-gp-mediated multidrug resistance and provide mechanistic insight into the regulation of this drug efflux transporter.
This work was supported by National Research Foundation of Korea (NRF) Grant R13-2007-023-00000-0 funded by the Korean government (MSIP) and Korean Health Technology R&D Project, Ministry of Health and Welfare, Republic of Korea, Grant HI14C0067. The authors declare that they have no conflicts of interest with the contents of this article.
- ABC
- ATP-binding cassette
- P-gp
- permeability glycoprotein
- CK
- casein kinase
- Hsp
- heat shock protein
- PXR
- pregnane X receptor
- CCRP
- cytoplasmic constitutive active/androstane receptor retention protein
- DRB
- 5,6-dichloro-1-β-d-ribofuranosylbenzimidazole
- TBB
- 4,5,6,7-tetrabromotriazole
- IMAC
- immobilized metal ion affinity chromatography.
References
- 1. Gottesman M. M. (2002) Mechanisms of cancer drug resistance. Annu. Rev. Med. 53, 615–627 [DOI] [PubMed] [Google Scholar]
- 2. Persidis A. (1999) Cancer multidrug resistance. Nat. Biotechnol. 17, 94–95 [DOI] [PubMed] [Google Scholar]
- 3. Gant T. W., Silverman J. A., Thorgeirsson S. S. (1992) Regulation of P-glycoprotein gene expression in hepatocyte cultures and liver cell lines by a trans-acting transcriptional repressor. Nucleic Acids Res. 20, 2841–2846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Labialle S., Gayet L., Marthinet E., Rigal D., Baggetto L. G. (2002) Transcriptional regulators of the human multidrug resistance 1 gene: recent views. Biochem. Pharmacol. 64, 943–948 [DOI] [PubMed] [Google Scholar]
- 5. Scotto K. W. (2003) Transcriptional regulation of ABC drug transporters. Oncogene 22, 7496–7511 [DOI] [PubMed] [Google Scholar]
- 6. Geick A., Eichelbaum M., Burk O. (2001) Nuclear receptor response elements mediate induction of intestinal MDR1 by rifampin. J. Biol. Chem. 276, 14581–14587 [DOI] [PubMed] [Google Scholar]
- 7. Greiner B., Eichelbaum M., Fritz P., Kreichgauer H. P., von Richter O., Zundler J., Kroemer H. K. (1999) The role of intestinal P-glycoprotein in the interaction of digoxin and rifampin. J. Clin. Invest. 104, 147–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Niemi M., Backman J. T., Fromm M. F., Neuvonen P. J., Kivistö K. T. (2003) Pharmacokinetic interactions with rifampicin: clinical relevance. Clin. Pharmacokinet. 42, 819–850 [DOI] [PubMed] [Google Scholar]
- 9. Baciewicz A. M., Chrisman C. R., Finch C. K., Self T. H. (2013) Update on rifampin, rifabutin, and rifapentine drug interactions. Curr. Med. Res. Opin. 29, 1–12 [DOI] [PubMed] [Google Scholar]
- 10. Watkins R. E., Wisely G. B., Moore L. B., Collins J. L., Lambert M. H., Williams S. P., Willson T. M., Kliewer S. A., Redinbo M. R. (2001) The human nuclear xenobiotic receptor PXR: structural determinants of directed promiscuity. Science 292, 2329–2333 [DOI] [PubMed] [Google Scholar]
- 11. Cowen L. E., Carpenter A. E., Matangkasombut O., Fink G. R., Lindquist S. (2006) Genetic architecture of Hsp90-dependent drug resistance. Eukaryot. Cell 5, 2184–2188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kakizaki S., Takizawa D., Tojima H., Horiguchi N., Yamazaki Y., Mori M. (2011) Nuclear receptors CAR and PXR: therapeutic targets for cholestatic liver disease. Front. Biosci. 16, 2988–3005 [DOI] [PubMed] [Google Scholar]
- 13. Lu X., Xiao L., Wang L., Ruden D. M. (2012) Hsp90 inhibitors and drug resistance in cancer: the potential benefits of combination therapies of Hsp90 inhibitors and other anti-cancer drugs. Biochem. Pharmacol. 83, 995–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Miyata Y., Yahara I. (1992) The 90-kDa heat shock protein, HSP90, binds and protects casein kinase II from self-aggregation and enhances its kinase activity. J. Biol. Chem. 267, 7042–7047 [PubMed] [Google Scholar]
- 15. Squires E. J., Sueyoshi T., Negishi M. (2004) Cytoplasmic localization of pregnane X receptor and ligand-dependent nuclear translocation in mouse liver. J. Biol. Chem. 279, 49307–49314 [DOI] [PubMed] [Google Scholar]
- 16. Pinna L. A. (2002) Protein kinase CK2: a challenge to canons. J. Cell Sci. 115, 3873–3878 [DOI] [PubMed] [Google Scholar]
- 17. Pinna L. A., Meggio F. (1997) Protein kinase CK2 (“casein kinase-2”) and its implication in cell division and proliferation. Prog. Cell Cycle Res. 3, 77–97 [DOI] [PubMed] [Google Scholar]
- 18. Hériché J. K., Chambaz E. M. (1998) Protein kinase CK2α is a target for the Abl and Bcr-Abl tyrosine kinases. Oncogene 17, 13–18 [DOI] [PubMed] [Google Scholar]
- 19. Akten B., Tangredi M. M., Jauch E., Roberts M. A., Ng F., Raabe T., Jackson F. R. (2009) Ribosomal s6 kinase cooperates with casein kinase 2 to modulate the Drosophila circadian molecular oscillator. J. Neurosci. 29, 466–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ahmed K., Gerber D. A., Cochet C. (2002) Joining the cell survival squad: an emerging role for protein kinase CK2. Trends Cell Biol. 12, 226–230 [DOI] [PubMed] [Google Scholar]
- 21. Seldin D. C., Leder P. (1995) Casein kinase II α transgene-induced murine lymphoma: relation to theileriosis in cattle. Science 267, 894–897 [DOI] [PubMed] [Google Scholar]
- 22. Landesman-Bollag E., Romieu-Mourez R., Song D. H., Sonenshein G. E., Cardiff R. D., Seldin D. C. (2001) Protein kinase CK2 in mammary gland tumorigenesis. Oncogene 20, 3247–3257 [DOI] [PubMed] [Google Scholar]
- 23. Guo C., Yu S., Davis A. T., Wang H., Green J. E., Ahmed K. (2001) A potential role of nuclear matrix-associated protein kinase CK2 in protection against drug-induced apoptosis in cancer cells. J. Biol. Chem. 276, 5992–5999 [DOI] [PubMed] [Google Scholar]
- 24. Stolarczyk E. I., Reiling C. J., Pickin K. A., Coppage R., Knecht M. R., Paumi C. M. (2012) Casein kinase 2α regulates multidrug resistance-associated protein 1 function via phosphorylation of Thr249. Mol. Pharmacol. 82, 488–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yamane K., Kinsella T. J. (2005) CK2 inhibits apoptosis and changes its cellular localization following ionizing radiation. Cancer Res. 65, 4362–4367 [DOI] [PubMed] [Google Scholar]
- 26. Feng S., Pan C., Jiang X., Xu S., Zhou H., Ye M., Zou H. (2007) Fe3+ immobilized metal affinity chromatography with silica monolithic capillary column for phosphoproteome analysis. Proteomics 7, 351–360 [DOI] [PubMed] [Google Scholar]
- 27. Enkvist E., Viht K., Bischoff N., Vahter J., Saaver S., Raidaru G., Issinger O. G., Niefind K., Uri A. (2012) A subnanomolar fluorescent probe for protein kinase CK2 interaction studies. Org. Biomol. Chem. 10, 8645–8653 [DOI] [PubMed] [Google Scholar]
- 28. Baysarowich J., Koteva K., Hughes D. W., Ejim L., Griffiths E., Zhang K., Junop M., Wright G. D. (2008) Rifamycin antibiotic resistance by ADP-ribosylation: structure and diversity of Arr. Proc. Natl. Acad. Sci. U.S.A. 105, 4886–4891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Huang H., Wang H., Sinz M., Zoeckler M., Staudinger J., Redinbo M. R., Teotico D. G., Locker J., Kalpana G. V., Mani S. (2007) Inhibition of drug metabolism by blocking the activation of nuclear receptors by ketoconazole. Oncogene 26, 258–268 [DOI] [PubMed] [Google Scholar]
- 30. Battistutta R., Sarno S., De Moliner E., Papinutto E., Zanotti G., Pinna L. A. (2000) The replacement of ATP by the competitive inhibitor emodin induces conformational modifications in the catalytic site of protein kinase CK2. J. Biol. Chem. 275, 29618–29622 [DOI] [PubMed] [Google Scholar]
- 31. Schuetz E. G., Beck W. T., Schuetz J. D. (1996) Modulators and substrates of P-glycoprotein and cytochrome P4503A coordinately up-regulate these proteins in human colon carcinoma cells. Mol. Pharmacol. 49, 311–318 [PubMed] [Google Scholar]
- 32. Lehmann J. M., McKee D. D., Watson M. A., Willson T. M., Moore J. T., Kliewer S. A. (1998) The human orphan nuclear receptor PXR is activated by compounds that regulate CYP3A4 gene expression and cause drug interactions. J. Clin. Invest. 102, 1016–1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Blumberg B., Sabbagh W., Jr, Juguilon H., Bolado J., Jr, van Meter C. M., Ong E. S., Evans R. M. (1998) SXR, a novel steroid and xenobiotic-sensing nuclear receptor. Genes Dev. 12, 3195–3205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Miyata Y. (2009) Protein kinase CK2 in health and disease: CK2: the kinase controlling the Hsp90 chaperone machinery. Cell Mol. Life Sci. 66, 1840–1849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Meggio F., Pinna L. A. (2003) One-thousand-and-one substrates of protein kinase CK2? FASEB J. 17, 349–368 [DOI] [PubMed] [Google Scholar]
- 36. Pinna L. A. (2012) Protein kinase CK2, John Wiley & Sons, Inc., New York [Google Scholar]
- 37. Proia D. A., Sang J., He S., Smith D. L., Sequeira M., Zhang C., Liu Y., Ye S., Zhou D., Blackman R. K., Foley K. P., Koya K., Wada Y. (2012) Synergistic activity of the Hsp90 inhibitor ganetespib with taxanes in non-small cell lung cancer models. Invest. New Drugs 30, 2201–2209 [DOI] [PMC free article] [PubMed] [Google Scholar]