

Background: FK506-binding protein 51 (FKBP51) inhibits and FKBP52 stimulates transcription by various steroid receptors. Exchange of a single residue largely reverses this pattern of regulation.
Results: Unlike FKBP52, FKBP51 has two distinct coupled conformational transitions surrounding this mutation site.
Conclusion: Structural analysis of FKBP51 transient states can inform inhibitor design by conformational selection.
Significance: The differential plasticity of the FKBP domains may underlie their differential regulation.
Keywords: allosteric regulation, conformational change, nuclear magnetic resonance (NMR), prolyl isomerase, x-ray crystallography
Abstract
Interchanging Leu-119 for Pro-119 at the tip of the β4-β5 loop in the first FK506 binding domain (FK1) of the FKBP51 and FKBP52 proteins, respectively, has been reported to largely reverse the inhibitory (FKBP51) or stimulatory (FKBP52) effects of these co-chaperones on the transcriptional activity of glucocorticoid and androgen receptor-protein complexes. Previous NMR relaxation studies have identified exchange line broadening, indicative of submillisecond conformational motion, throughout the β4-β5 loop in the FK1 domain of FKBP51, which are suppressed by the FKBP52-like L119P substitution. This substitution also attenuates exchange line broadening in the underlying β2 and β3a strands that is centered near a bifurcated main chain hydrogen bond interaction between these two strands. The present study demonstrates that these exchange line broadening effects arise from two distinct coupled conformational transitions, and the transition within the β2 and β3a strands samples a transient conformation that resembles the crystal structures of the selectively inhibited FK1 domain of FKBP51 recently reported. Although the crystal structures for their series of inhibitors were interpreted as evidence for an induced fit mechanism of association, the presence of a similar conformation being significantly populated in the unliganded FKBP51 domain is more consistent with a conformational selection binding process. The contrastingly reduced conformational plasticity of the corresponding FK1 domain of FKBP52 is consistent with the current model in which FKBP51 binds to both the apo- and hormone-bound forms of the steroid receptor to modulate its affinity for ligand, whereas FKBP52 binds selectively to the latter state.
Introduction
The FK506-binding protein of 51-kDa (FKBP51)2 and FKBP52 are two closely homologous proteins, each containing a pair of FK506-binding domains (FK1 and FK2) followed by a tetratricopeptide repeat domain that mediates their interactions with Hsp90. FKBP52 was first characterized as a co-chaperone of Hsp90 in the activated hormone-bound steroid receptor complexes that are formed with the progesterone, androgen, or the glucocorticoid receptor proteins (1). FKBP52 enhances the binding of the glucocorticoid receptor complex to the dynein motor protein, thus facilitating microtubular transport of the receptor from the cytosol to the nucleus (2). FKBP51 is the predominant Hsp90 co-chaperone for the unliganded state of the glucocorticoid receptor (3, 4), which induces a lower binding affinity for glucocorticoids (5) and dynein (6). Steroid hormone binding to the FKBP51-bound glucocorticoid receptor is believed to induce the release of FKBP51 and the subsequent binding of FKBP52 (7).
The hormone-bound glucocorticoid receptor strongly induces the fkbp5 gene encoding FKBP51, yielding a direct negative feedback control loop (8). Presumably acting via the glucocorticoid receptor, single nucleotide polymorphisms in the fkbp5 gene exhibit a strong correlation with recurrence of depressive episodes, the rate of response to antidepressant therapies, and in psychological stress disorders (9, 10). As a necessary chaperone for the Akt-specific phosphatase PHLPP (11, 12), FKBP51 also provides indirect feedback regulation by inhibiting glucocorticoid receptor phosphorylation via the Akt-p38 kinase pathway (13, 14).
The FK1 domain mediates the selectivity of interaction for steroid receptor binding exchange (15, 16), dynein binding (6), and inhibition of the Akt kinase (12). The peptidylprolyl isomerase activity of this domain is not required for receptor binding exchange (16) or Akt kinase inhibition (12). Using both yeast heterologous expression and murine FKBP52 knock-out experiments, Smith and colleagues (16) demonstrated that a FKBP52-like L119P mutation near the tip of the β4-β5 loop in the FK1 domain of human FKBP51 increases reporter gene expression by ∼3.5-fold for the human glucocorticoid and androgen receptors. The complementary P119L mutation in FKBP52 yielded a ∼2-fold decrease in reporter gene expression, indicating that the transcriptional activity of these steroid receptors can be substantially reversed by a single point mutation.
The conformational plasticity of the steroid receptor protein offers the opportunity for its various interaction partners to couple to these conformational transitions in the process of regulation. Although the conformational states of unliganded steroid receptor proteins remain poorly characterized, crystal structures of various ligand-bound states demonstrate that the conformational transitions of the ligand binding domain which are induced by steroid antagonists generally differ from those induced by steroid agonists, and these distinct conformations can differentially interact with co-regulators (17). In addition, a concurrent conformational transition in the Hsp90 subunits has been proposed to serve as a component of a larger scale allosteric response (18).
Arguably most relevant to the present study, the binding of Hsp70 to the isolated glucocorticoid receptor-ligand binding domain induces a conformational transition that affects the residues neighboring the ligand binding pocket, and not only markedly reduces the intrinsic hormone binding affinity of apo-glucocorticoid receptor-ligand binding domain but also stimulates the release of hormone from the liganded receptor (19). Based on this Hsp70-mediated modulation of glucocorticoid receptor-ligand binding domain hormone affinity, Agard and colleagues (19) have proposed an ATP-dependent regulatory mechanism for altering receptor activity in response to the intracellular hormone concentration.
The markedly differing conformational dynamics of the FK1 domains of FKBP51 and FKBP52 provide a potential mechanism for differentially coupling to the regulatory transitions of the steroid receptor proteins as well as providing a potential basis for selective drug design (20). Unlike FKBP52, FKBP51 exhibits elevated 15N R2 relaxation rates throughout much of the β4-β5 loop with a magnetic field strength dependence that indicates conformational dynamics in the submillisecond time frame (21). This conformational exchange line broadening is quenched in the L119P variant of FKBP51. The complementary P119L variant of FKBP52 induces a FKBP51-like pattern of exchange line broadening for the residues along the β4-β5 loop, indicative of a similar underlying conformational transition (20).
The FKBP52-like L119P variant of FKBP51 also suppresses exchange line broadening of residues in the β2 and β3a strands near Phe-67, which has its structurally buried phenyl ring packed directly under the tip of the β4-β5 loop in the unliganded crystal structures (22). The bifurcated main chain hydrogen bonding of the amides of Phe-67 and Asp-68 with the carbonyl oxygen of Gly-59 disrupts the standard antiparallel hydrogen bonding pattern between the β2 and β3a strands. We earlier proposed that a transition in this bifurcated hydrogen bonding interaction could provide the structural basis of the conformational line broadening observed for those strands (20). Interestingly, the analogous P119L mutation in FKBP52 has no effect on the line broadening of the β2 and β3a strands, indicating an additional nonequivalence in the conformational plasticity of FKBP51 and FKBP52.
It has remained unresolved whether the exchange line broadening in the β4-β5 loop and in the β2 and β3a strands of FKBP51 arise from a single concerted conformational transition or from two distinct coupled transitions. In the year following our initial characterization of the conformational exchange line broadening dynamics surrounding the bifurcated main chain hydrogen bonding of the amides of Phe-67 and Asp-68 with the carbonyl oxygen of Gly-59, Hausch and colleagues (23) reported the crystal structures of two so-called iFit inhibitor-bound forms of the FK1 domain from FKBP51 in which the conformation of the β3a strand surrounding Phe-67 is altered so as to disrupt this bifurcated main chain hydrogen bonding interaction. They interpreted these structures to demonstrate an induced-fit mechanism of binding, despite the fact that the structure of the final ligand-bound state cannot discriminate between an induced-fit or a conformational selection-based mechanism (24). Particularly germane to these issues is whether FKBP51 significantly populates a similar conformation in the absence of the inhibitor. NMR relaxation analysis of systematically designed FK1 domain variants can provide a fruitful approach to characterizing such transient conformational substates (25, 26).
Experimental Procedures
Protein Preparation
Gene sequences for the wild type and sequence variants of the FK1 domain of human FK506-binding protein FKBP51 (Glu20-Glu140) were chemically synthesized (Genscript), with codon optimization for expression in Escherichia coli. The gene sequences were cloned into the expression vector pET11a and the plasmids were transformed into the BL21Star(DE3) strain (Invitrogen) for expression. The protein expression and purification procedure for the FKBP51 domain was carried out as previously described (20). All isotopically labeled samples were prepared via protein expression in minimal medium containing 0.1% 15NH4Cl as nitrogen source. All protein samples were concentrated via centrifugal ultrafiltration and then equilibrated into a pH 6.50 buffer containing 25 mm sodium phosphate, 2 mm dithiothreitol, and 2 mm tris(2-carboxyethyl)phosphine by a series of centrifugal concentration steps to a final concentration of 0.5 mm protein.
NMR Spectroscopy
NMR relaxation data were collected on a Bruker Avance III 600 MHz spectrometer and a Bruker Avance II 900 MHz spectrometer at 25 °C. HSQC-based T1, T1ρ, and heteronuclear NOE experiments were carried out as described by Lakomek et al. (27) using delays and spin lock field strengths for the T1ρ experiments as previously described for the wild type and L119P variants of the FK1 domain for FKBP51 (20). T1ρ data sets were collected at multiple frequency offsets (four at 60.8 MHz 15N and five at 91.2 MHz 15N).
To maximize the sensitivity for detecting variations in the conformational exchange line broadening effects between the wild type and the mutational variants, the transverse relaxation rates were analyzed in differential mode. For residues that do not exhibit conformational exchange line broadening, the backbone 15N R2 values are generally dominated by the dynamics of global molecular tumbling, which depends upon sample viscosity. The median R2 values were used to normalize the relaxation effects arising from slightly differing sample temperatures and protein concentrations.
The robustness of this normalization approach is directly indicated by the ΔR2 values for the residues not exhibiting substantial differential line broadening because these ΔR2 values incorporate the experimental errors of the individual relaxation measurements as well as the discrepancies that arise from a lack of equivalence for the conformational/orientational dynamics of the protein samples (20). As a means of further enhancing the robustness of the differential R2 analysis, except where explicitly noted, R2 relaxation values less than 25 s−1 were excluded from the ΔR2 calculations if the root mean square deviation fit to the optimal decay constant was larger than 1.5% of the initial intensity.
X-ray Crystallography
Initial crystallization conditions for the FK1 domain of the K121G variant was established using the Hampton Research Crystal Screen I and II. Large crystals were grown at room temperature in hanging drops. 2 μl of protein solution at 65 mg/ml concentration was mixed with an equal volume of reservoir solution containing 26% PEG 3350, 0.1 m HEPES, pH 7.5, 0.2 m ammonium acetate, and 5% isopropyl alcohol, and equilibrated overnight. The crystals belong to space group C2 with cell parameters: a = 69.435 Å, b = 31.990 Å, c = 57.435 Å, β = 119.03°. There is one molecule per asymmetric unit, with a crystal solvent content of 42%. Prior to data collection, crystals were transferred to a reservoir solution containing crystallization buffer supplemented with 25% glycerol, and then flash-cooled under a nitrogen stream at 100 K, and stored in liquid nitrogen. Diffraction data were collected at 100 K using beamline X25 of the National Synchrotron Light Source (Brookhaven National Laboratory). These data were processed and scaled using HKL2000 (28). Using the full resolution range (to 1.2 Å) with the high resolution structure of FKBP51 (PDB code 3O5E (22)) as a search model, only one clear solution with a correlation coefficient of 67% was found with the PHASER molecular replacement program within the PHENIX suite (29). The Rfactor of the initial model was 48.6%. Structural refinement was carried out using PHENIX (30). Model rebuilding was carried out using Coot (31). Figures of crystallographic structures were generated using Chimera software (32) and PyMol ((33)).
Results
Structural Analysis of the Interface between the β4-β5 Loop and the β2+β3a Strands in the FK1 Domain of FKBP51
For the 22 residues spanning Cys-107 at the end of the β4 strand to Leu-128 at the beginning of the β5 strand (Fig. 1), human FKBP51 and FKBP52 differ only at residues 119 and 124 (L119P and S124P). The five residues of the β3a strand (Lys66-Ser70) are completely conserved not only for FKBP51 and FKBP52 but for the close structural homologs FKBP12 and FKBP12.6 (Lys35-Ser39). Beyond these two segments, the FK1 domains of FKBP51 and FKBP52 exhibit only a 61% sequence identity, highly suggestive of a substantial functional role for this region of the protein.
FIGURE 1.

Backbone structure of the FK1 domain of FKBP51 and sequence alignment of homologous β4-β5 loop segments. The β4-β5 loop segment is indicated in black as is Phe-67 (PDB code 3O5Q (22)). The analogous domain of FKBP52 and the single domain proteins FKBP12 and FKBP12.6 are the evolutionarily closest FKBP domains to the FK1 domain of FKBP51 in the human genome (63). Strands β1 to β5 are numbered in accord with the FKBP12 alignment. Residues 119 and 124 are the only positions in the β4-β5 loop that differ between FKBP51 and FKBP52.
The tip of the β4-β5 loop is positioned by a set of hydrophobic interactions involving the Cβ from residue 121 and the side chains of Ile-122 and Pro-123, which are packed against the phenyl ring of Phe-67 in the β3a strand and the side chain of Leu-61 at the end of the β2 strand (Fig. 2A). This set of interacting residues is conserved in FKBP52, FKBP12, FKBP12.6, and in many of the other 18 FKBP domains within the human genome (34).
FIGURE 2.
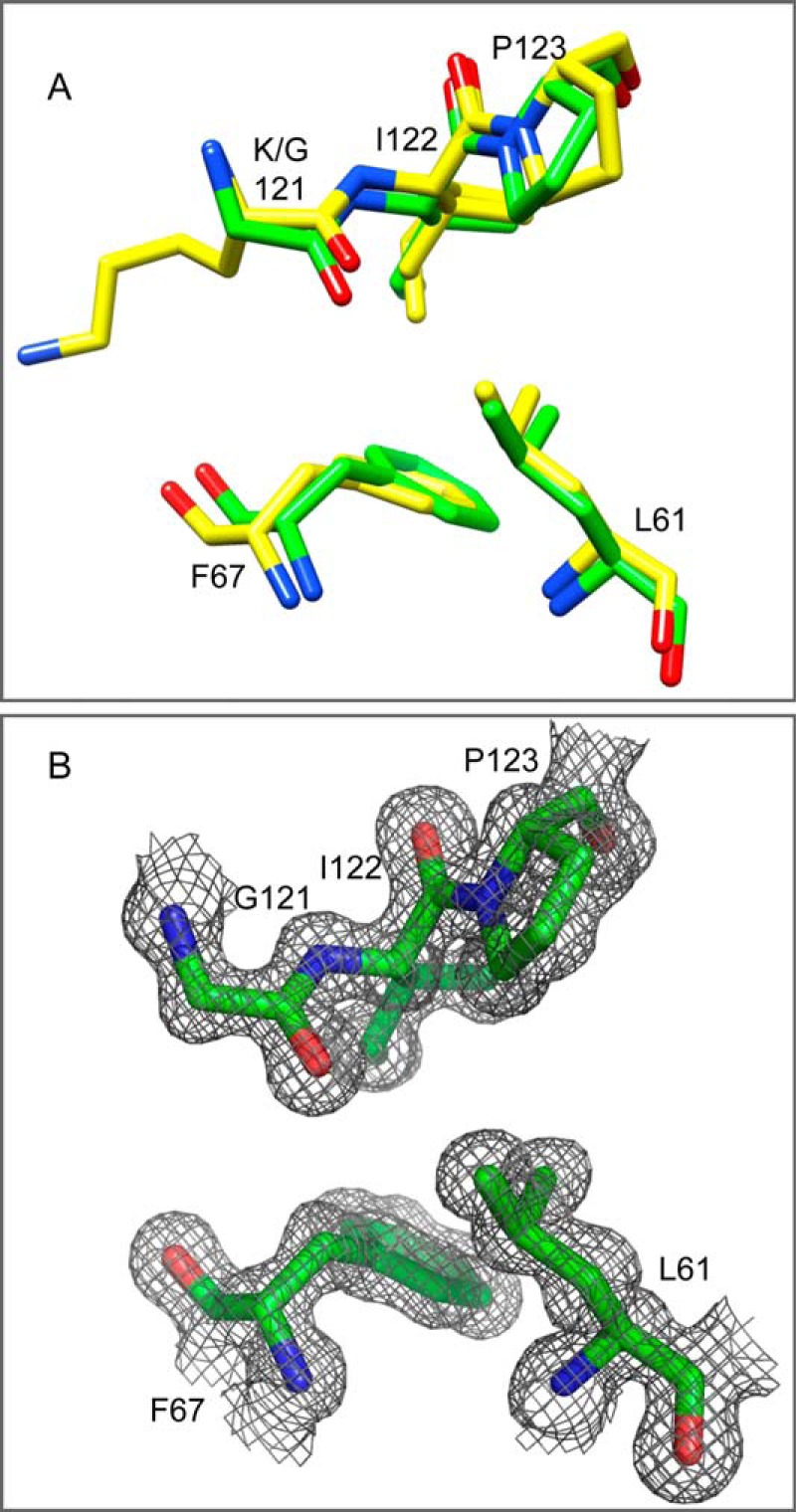
The hydrophobic packing interactions between the β4-β5 loop and the β2+β3a strands in the wild type and K121G variant of the FK1 domain of FKBP51. A, the packing interactions between Ile-122 and Pro-123 of the β4-β5 loop, Leu-61 of the β2 strand, and Phe-67 of the β3a strand are conserved among FKBP51 and FKBP52 as well as among FKBP12 and FKBP12.6 family members as illustrated in the high resolution structures of FKBP51 (yellow, PDB code 3O5Q (22)). These packing interactions are closely maintained in the 1.24-Å resolution structure of the K121G variant (green, PDB code 4R0X). B, the 2Fo − Fc electron density contour map for the K121G variant is displayed at the 1σ level.
In hindsight, an early indication of the functional significance for FKBP51 and FKBP52 of the interface between the β4-β5 loop and the underlying β2 and β3a strands was the widespread use of a F67D,D68V double mutant as a presumed indicator for the participation of the peptidyl prolyl isomerase activity for these proteins in their biological functions, in particular for steroid receptor activity (15, 35–38). More recently, in the context of their analysis of the effects of the L119P and P119L variants of FKBP51 and FKBP52 in the transcriptional activity of the steroid hormone receptor discussed above, Smith and colleagues (16) compared the F67D,D68V double mutant to other mutations that lay within the catalytic cleft to conclude that the steroid receptor activity does not depend upon prolyl isomerization catalysis.
In the effort to better understand how structural and dynamic effects propagate across the hydrophobic interface between the β4-β5 loop and the β2 and β3a strands, we have endeavored to identify mutational variants of the FK1 domain of FKBP51 that selectively modulate the conformational equilibria and kinetics along this interface. Such an approach has proven quite useful in the analysis of various conformational processes in the homologous FKBP12 (39–41).
Given that the Cβ atom of Lys-121 is packed tightly against the Cβ atom of Phe-67 at the site of the inter-strand bifurcated hydrogen bonding discussed above, we wished to analyze how alterations in that steric interaction might modulate the conformational dynamics on either side of the interface. Because removal of the Cβ atom at residue 121 would eliminate this steric interaction, we solved the crystal structure of the K121G variant of FKBP51 to a resolution of 1.24 Å (Table 1) to determine whether the glycine substitution might introduce variations into the backbone conformation that could complicate the structural interpretation of the NMR relaxation measurements. The targeted steric interaction was eliminated as expected (Fig. 2), whereas the crystallographic structure throughout the rest of this interface remained closely similar to that observed in the highest resolution structure reported for the wild type protein (22). The absence of discernible structural perturbation arising from the K121G substitution enhances the reliability with which our NMR relaxation results for the residue 121 side chain substitutions could be interpreted.
TABLE 1.
Crystallographic data collection, refinement, and model details (PDB code 4R0X)
| Statistics | |
|---|---|
| Data collection | |
| Resolution range (Å) | 27–1.24 (1.27–1.24) |
| No. of unique reflections | 30,633 |
| Redundancy | 6.4 (3.1) |
| Completeness (%) | 97.6 (74.2) |
| Average I/σ(I) | 47.1 (4.4) |
| Rmerge (%) | 4.4 (30.1) |
| Refinement | |
| Resolution limits (Å) | 27–1.24 |
| No. reflections | 30,633 |
| Minimum Fo/σFo | 1.39 |
| Rwork (%) | 15.8 |
| Rfree (%) | 16.7 |
| Non-H atoms | |
| Protein | 953 |
| Water | 199 |
| Average B (Å2) | 17.7 |
| All atoms | 15.3 |
| Solvent | 29.1 |
| Wilson B (Å2) | 12.4 |
| Geometry | |
| Root mean square deviation bond length (Å) | 0.005 |
| Root mean square deviation bond angle (°) | 1.034 |
| Ramachandran Plot | |
| Favored | 97.5 |
| Allowed | 2.5 |
| Outliers | 0 |
The Exchange Line Broadening of the β4-β5 Loop and the β2+β3a Strands Arise from Distinct Conformational Transitions Which Are Energetically Coupled
Each of the three standard NMR relaxation experiments, R1, R2 (or R1ρ) and heteronuclear NOE, are sensitive to conformational motion that is faster than the molecular rotational correlation time (∼10 ns for smaller globular proteins). Previous studies on the wild type FK1 domain of FKBP51 revealed three segments in which both the 15N R1 relaxation rates and the heteronuclear NOE values were reduced, relative to the rest of protein backbone, indicative of motion in the picosecond-nanosecond time frame. However, for two of these three segments, the β3a-β3b loop and the tip of the long β4-β5 loop, the 15N R2 relaxation rates are increased (Fig. 3A). These increased rates reflect the fact that the R2 transverse relaxation experiment is also sensitive to motion in the conformational exchange line broadening regime (microsecond-millisecond). The residues at the tip of the β4-β5 loop have also been reported to have elevated crystallographic B-factors (22), which is consistent with the common observation that the orientational disorder implied by residues exhibiting reduced relaxation rates and NOE values is often reflected in local crystallographic disorder.
FIGURE 3.
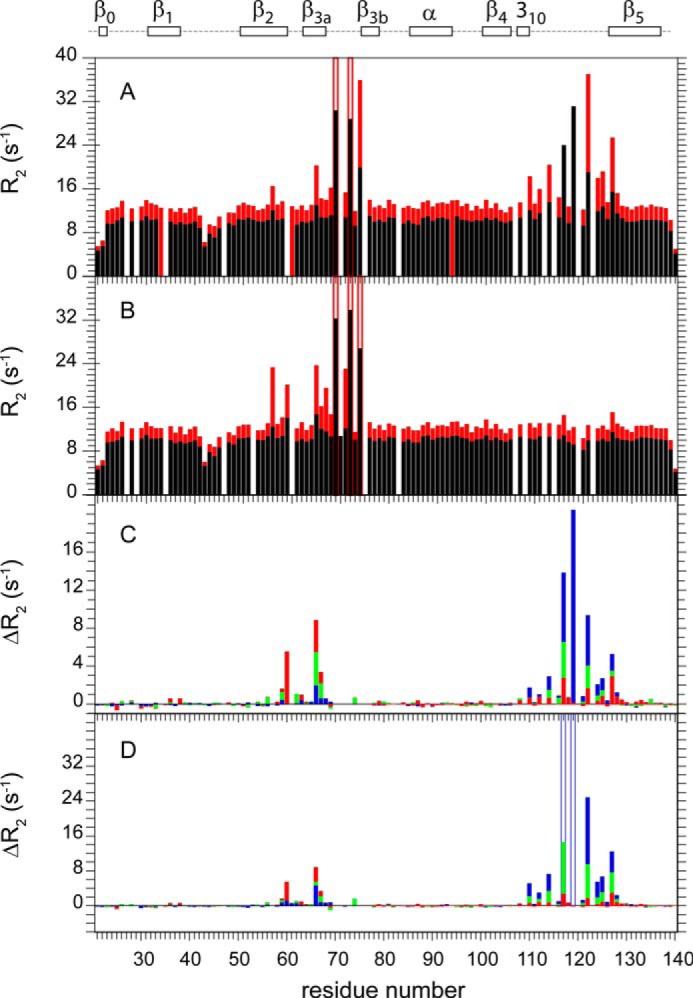
15N transverse relaxation measurements for the backbone amide resonances in the FK1 domain of wild type and β4-β5 loop variants of FKBP51. Transverse (R2) relaxation rates at 600 MHz 1H (black) and 900 MHz 1H (red) are shown for the wild type (A) and K121G variant (B) of the FK1 domain. For the differential R2 comparison at 600 MHz 1H (C) and 900 MHz (D), the median R2 values for the non-exchange broadened residues of the wild type (blue), K121A (green), and K121G (red) variant data sets were scaled to that of the L119P variant to correct for small variations in the global molecular correlation time due to minor differences in sample temperature and concentration. Given the increased experimental uncertainty of the fast relaxing resonances in the β3a-β3b loop (Ser-70, Arg-73, and Glu-75), these residues were not included in the differential analysis. For the residues outside of segments 57–77 and 108–128, the median average differences for ΔR2 (s−1) were 0.083 and 0.091 for K121G, 0.088 and 0.113 for K121A, and 0.093 and 0.090 for wild type at 600 MHz and 900 MHz, respectively.
Upon introducing the K121G mutation, the conformational exchange line broadening for the β4-β5 loop of FKBP51 is almost completely eliminated (Fig. 3B). Interestingly, the small degree of exchange line broadening in the β2 and β3a strands previously reported for the wild type protein (20) is significantly enhanced in the K121G variant.
These effects are more clearly illustrated when the transverse relaxation rates for the samples are directly analyzed in differential mode. In the present case (Fig. 3, C and D), the L119P data set was used as reference due to the strong attenuation of exchange line broadening in the β4-β5 loop of this variant (20). In comparing protein samples for which it is warranted to assume that the differences in internal motion affecting the R2 values occur exclusively within the conformational exchange line broadening time regime, the resultant cancellation of errors provided approximately a 3-fold increase in precision for the data of this study, relative to that obtained by applying the standard model-free analysis approach of Lipari and Szabo (42) to the individual protein data sets. In illustration, for the 72 backbone amide resonances outside of segments 57–77 and 108–128, which contain the residues exhibiting systematic differences in line broadening, the median average difference for the ΔR2 values of the K121G and L119P variants at 600 (Fig. 3C) and 900 MHz (Fig. 3D) were ∼0.8% of the median R2 values. The line broadening effects in the β2+β3a strands arising from the K121G substitution are up to 80-fold larger than this median average difference for the ΔR2 values outside of the two affected segments.
For the conformational exchange broadening both in the β4-β5 loop and within the β2 and β3a strands, the effects of the K121A substitution were intermediate between those of the K121G variant and the wild type protein. In contrast to the suppression of exchange line broadening in both the β4-β5 loop and β2 and β3a strands caused by the L119P variant, the K121G and K121A substitutions enhance the exchange line broadening in the β2 and β3a strands, whereas at the same time suppressing line broadening in the β4-β5 loop. This opposite pattern of response to the substitutions at residue 121 indicates that the differential exchange line broadening of the β2 and β3a strands does not reflect the propagation of dynamics from the β4-β5 loop, but rather a distinct conformational transition underlies the exchange line broadening of β2 and β3a strands.
Comparison of the ΔR2 values at 600 and 900 MHz indicates an approximate 2.25-fold increase for residues in both the β2 and β3a strands and the β4-β5 loop (Fig. 3, C and D). Variation of conformational exchange line broadening with the square of the magnetic field strength is indicative of these conformational transitions occurring in the submillisecond time frame approaching the fast exchange limit (21), as had previously been reported for the β4-β5 loop in FKBP12 (43–45).
For a two-state transition near the fast exchange time regime, the magnitude of the exchange line broadening is proportional to pApBΩ2/kex, where pA and pB are the state populations, Ω is the difference in 15N resonance frequency for the two states, and kex = kAB + kBA is the conformational exchange rate. The fact that the exchange line broadening effects for the various residues within the β4-β5 loop scale proportionally in going from K121G to K121A to the wild type protein strongly indicates that the differential 15N chemical shifts and therefore presumably the conformation of the interchanging states are largely unaffected by these substitutions. As a result, the marked decrease in exchange line broadening within the β4-β5 loop in going from wild type to K121A to K121G variants arises from a large decrease in the population of the minor conformational state and/or from a large increase in the rate of exchange. Given the increase in flexibility that could be expected from decreasing the steric bulk of the side chain at residue 121, it is anticipated that an increased rate of conformational exchange likely dominates these line broadening effects.
The packing of the β4-β5 loop against the β2 and β3a strands plays a significant role in determining the dynamics of the exchange line broadening transition for residues in those strands. The line broadening effects for residues within the β2 and β3a strands also scale nearly proportionally going from the wild type to the K121A and K121G variants, consistent with the conformations of interchanging states being largely unaffected by these substitutions. In contrast to the β4-β5 loop, the magnitude of the exchange line broadening in the β2 and β3a strands increases in going from wild type to the K121A and K121G variants. This increase predominately reflects either a decreased rate for the conformational transition of the β2 and β3a strands or an increase in the population of the minor conformational state for that transition. As the K121G and K121A variants reduce the packing interaction between the β4-β5 loop and the β3a strand, an increase in the minor state population offers a more plausible explanation for the increased exchange line broadening.
Selective Suppression of the β2+β3a Strand Exchange Line Broadening Transition in FKBP51
As noted above, the introduction of the FKBP51-like P119L mutation into the FK1 domain of FKBP52 caused the β4-β5 loop to exhibit a FKBP51-like pattern of exchange line broadening effects, albeit with a 5-fold lower intensity at the various residues along that loop. Introducing a second FKBP51-like mutation P124S into the P119L variant of FKBP52 enhances this pattern of exchange line broadening to a level of 60% that of the wild type FKBP51 (20). On the other hand, introduction of the P119L and P124S mutations into FKBP52 had no effect on the line broadening behavior of the β2 and β3a strands, indicating that dynamical coupling to the β4-β5 loop differs significantly from that observed for wild type FKBP51. It may be noted that the bifurcated main chain hydrogen bond interactions between the carbonyl oxygen of Gly-59 in the β2 strand and the amides of Phe-67 and Asp-68 in the β3a strand (Fig. 4) is present in the unliganded crystal structures of both FKBP51 and FKBP52.
FIGURE 4.
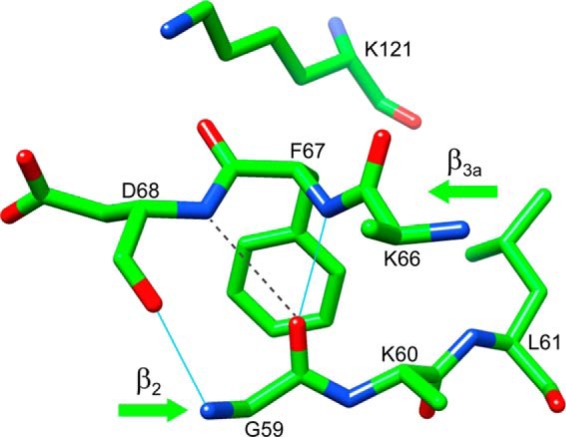
Bifurcated interaction disrupting the canonical antiparallel hydrogen bonding pattern between the β2 and β3a strands in the FK1 domain of FKBP51. As illustrated from the 1.30-Å resolution crystal structure (PDB code 3O5L (22)), the carbonyl oxygen of Gly-59 is hydrogen bonded to the amide of Phe-67, in addition to forming a weaker interaction with the amide of Asp-68 (N to O distance of 3.3 Å, beyond the default cutoff of 3.0 Å).
If a rearrangement of this bifurcated main chain hydrogen bonding interaction underlies the conformational line broadening transition, which is being selectively modulated by the mutations at residues 119 and 121 in FKBP51, attention is drawn to residues that mediate the interactions between the β2 and β3a strands, which differ between FKBP51 and FKBP52. In each of the reported high resolution structures of the unliganded FKBP52 FK1 domain (PDB codes 1N1A (46), 1Q1C (47), 4LAV, and 4LAW (48)), a hydrogen bond between the Oγ of Ser-69 and the carbonyl oxygen of Tyr-57 is further stabilized by a second hydrogen bond between the side chain Oγ atoms of Ser-69 and Thr-58 (Fig. 5). The presence of Lys-58 in FKBP51 eliminates the possibility of an interaction analogous to the hydrogen bond between the Oγ atoms of Ser-69 and Thr-58 seen in the FKBP52 structures. Furthermore, the hydrogen bond between the side chain Oγ of Ser-69 and the carbonyl oxygen of Tyr-57 is disrupted (N to O distance of 3.8 Å) in the highest resolution crystal structures for the unliganded FK1 domain of FKBP51 (22).
FIGURE 5.
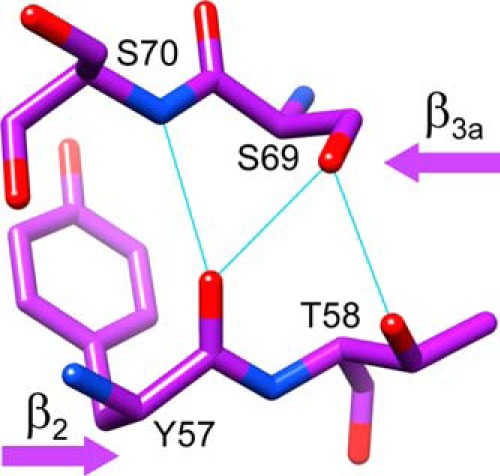
Side chain hydrogen bonding interactions between the β2 and β3a strands in the FK1 domain of FKBP52 surrounding Thr-58. Thr-58 is the only residue displayed that differs from the sequence of FKBP51. As illustrated in the 1.90-Å resolution crystal structure (PDB code 1Q1C (47)), the hydrogen bonding between the side chain hydroxyl of Thr-58 and the Oγ of Ser-69 indirectly stabilizes the hydrogen bond between the Ser-69 Oγ and the carbonyl oxygen of Tyr-57, which is not observed in the highest resolution structures of FKBP51.
The L119P variant only partially suppresses the exchange line broadening in the β2 and β3a strands (20). To test the premise that introducing the K58T substitution into FKBP51 might stabilize the β2-β3a strand interactions seen in the crystal structures of the unliganded protein, 15N relaxation measurements were carried out on this variant. Qualitatively, the 15N R2 data for the K58T-substituted FKBP51 (Fig. 6A) offers no apparent evidence for conformational exchange line broadening in the β2 and β3a strands. More rigorously, the model-free analysis (42, 49) of the 15N T1, T1ρ and heteronuclear NOE data for K58T variant predicted exchange broadening at both magnetic fields only for Lys-60, and these predicted values are only slightly above the estimated experimental uncertainty. Interestingly, the K58T substitution also results in a 3-fold reduction in the much larger exchange line broadening effects within the β3a-β3b loop (Fig. 6A), suggesting that the conformational transition that gives rise to those line broadening effects also involves the disruption of the hydrogen bonding interaction between the side chains of Thr-58 and Ser-69. At the same time, the exchange line broadening in the β4-β5 loop of the K58T variant (Fig. 6A) is only modestly reduced from that observed for the wild type protein (Fig. 3A).
FIGURE 6.
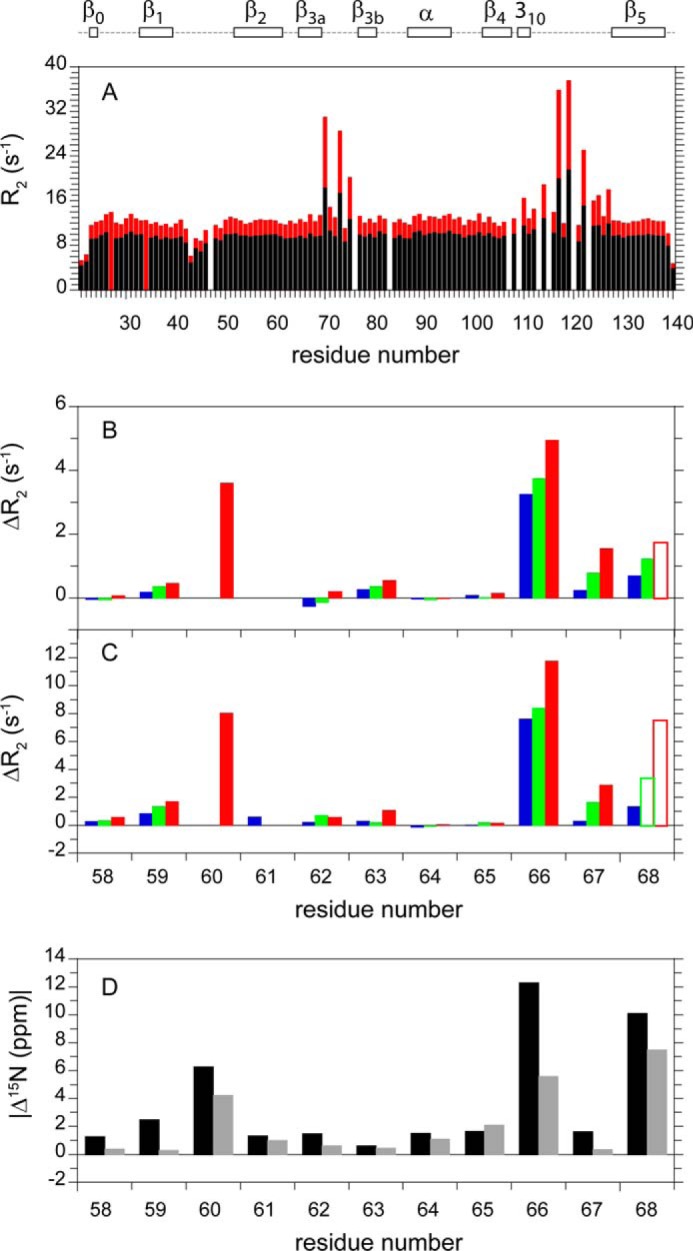
15N transverse relaxation in the K58T variant of FKBP51 and structural interpretation of the ΔR2 values for wild type and the K121A and K121G variants. R2 relaxation rates at 600 MHz 1H (black) and 900 MHz 1H (red) are shown for the K58T variant of the FK1 domain (A). For the differential R2 comparison within the β2 and β3a strands at 600 (B) and 900 (C) MHz, the median R2 values for the wild type (blue), K121A (green), and K121G (red) data sets were scaled to that of the K58T variant to correct for small variations in the global molecular correlation time. A subset of the Asp68 relaxation data are denoted with open bars indicating root mean square deviation fits to the optimal decay constants larger than the 1.5% cutoff used for the other ΔR2 values. Overlap with the resonance of Val-94 precluded the use of the Lys-60 data for wild type and the K121A variant. SPARTA+-based differential 15N chemical shift values were predicted for the iFit1-bound (black) and iFit4-bound (gray) domains, as compared with the reference FK506-bound FK1 domain of FKBP51 (D).
15N Chemical Shift Analysis of the Exchange Line Broadening Transition in the β2 and β3a Strands
Given the suppression of exchange line broadening observed for the K58T variant of FKBP51 in the β2 and β3a strands, this protein provides an appropriate reference state for comparing the differences in exchange broadening for this segment in the wild type and K121A and K121G variants. The largest exchange line broadening effects at 600 (Fig. 6B) and 900 MHz (Fig. 6C) were observed for the amides of Lys-60, Lys-66, Phe-67, and Asp-68, strongly suggestive of a transition involving the bifurcated main chain hydrogen bond (Fig. 4). In such a transition, the substantial exchange line broadening of the Lys-60 amide might reflect not only changes in the local backbone geometry of the β2 strand, it could also reflect alterations in the hydrogen bonding of the Gly-59 carbonyl oxygen, which serves as the acceptor for the bifurcated hydrogen bonds to the β3a strand. Disruption of hydrogen bonding at a peptide C = O group is known to give rise to changes in the chemical shift for the peptide nitrogen resonance of several ppm (50).
Early studies of FKBP12 replaced Phe-36 with valine so as to introduce a hydrophobic pocket along the side of the packing interface between the β4-β5 loop and the underlying β3a strand (51, 52). This F36V variant was then used to develop a series of selective inhibitors, including Shield1 (53), which have been applied to numerous mammalian cellular expression studies (e.g. Refs. 54 and 55). Introducing the analogous F67V mutation to the FK1 domains of FKBP51 and FKBP52, Hausch and colleagues (23) recently reported the development of FKBP51-selective inhibitors using Shield1 as their initial scaffold. In contrast to earlier studies on the F36V variant of FKBP12, which exhibited minimal structural perturbation upon inhibitor binding (51), Hausch and colleagues (23) found that their iFit inhibitors bound to a conformation of wild type FKBP51 and FKBP52 in which the Phe-67 side chain is reoriented away from its typical position packed underneath the tip of the β4-β5 loop (Fig. 2).
As illustrated for the complex with the iFit1 inhibitor (PDB code 4TW6 (23)) (Fig. 7), the resultant backbone conformation of the β2 and β3a strands entails a flipping of the Phe-67 peptide linkage. In contrast to Phe-67 and Asp-68 forming a bifurcated hydrogen bond interaction with the carbonyl of Gly-59, as seen in the reference FK506-inhibited structure (PDB code 3O5R (22)), the amide of Asp-68 in the iFit1-inhibited structure now forms a linear hydrogen bond with the carbonyl oxygen of Gly-59. The resultant rearrangement in the β3a strand causes a major repositioning of the Cα-Cβ bond vector of Phe-67 so that the phenyl ring is now directed out toward the solvent phase (Fig. 7B). The binding of the iFit1 inhibitor is also accompanied by an appreciable shift in position for the tip of the β4-β5 loop, which is moved away from the β3a strand.
FIGURE 7.
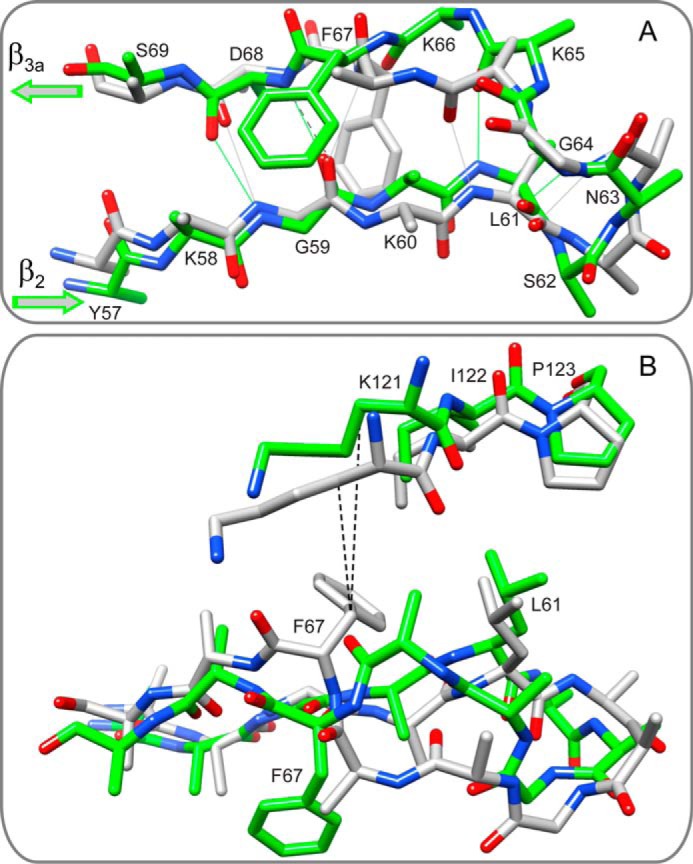
Superposition of the backbone conformation of the β2 strand, the β3a strand, and the connecting loop from the iFit1-inhibited and the FK506-inhibited FK1 domain of FKBP51. A, in contrast to the FK506-inhibited structure (gray, PDB code 3O5R (22)), in the iFit1-inhibited structure (green, PDB code 4TW6 (23)) the side chain of Phe-67 is reoriented out toward the solvent phase and the backbone of the β3a strand is shifted so as to enable a direct hydrogen bonding interaction between the amide of Asp-68 and the carbonyl oxygen of Gly-59. Other side chains were removed to more clearly illustrate the backbone conformation. B, a 90° rotation of the β2 and β3a strands illustrates the major repositioning of the Cα-Cβ bond vector for Phe-67 that accompanies the binding of the iFit1 inhibitor that reflects the rearrangement of the β3a strand backbone surrounding the site of bifurcated hydrogen bonding interaction in the FK506-inhibited structure. The binding of the iFit1 inhibitor is also accompanied by a substantial upward shift at the tip of the β4-β5 loop. With respect to the Cβ of Phe-67 in the FK506-inhibited structure in this global superpositioning, the distance to the Cβ of Lys-121 is 2.5 Å larger in the iFit1-inhibited structure.
Because the amide of Lys-66 is also oriented away from the β2 strand, the flipping of the Phe-67 peptide linkage in the iFit1-inhibited structure results in the kinked β3a strand conformation being shifted by one residue toward the β2-β3a loop (Fig. 7). Otherwise, the conformation of the β2-β3a loop and the β2 strand in the iFit1-inhibited structure roughly follows that of the reference FK506-inhibited protein. The iFit4-inhibited structure (PDB code 4TW7 (23)) differs more strongly from the reference FK506-inhibited protein. In this case, a more extended canonical antiparallel hydrogen bonding pattern is formed, and the “kink” has been further shifted so as to contribute to an enlarged β2-β3a loop.
Conformational exchange line broadening data provide a measure of the relative magnitude of the differential 15N chemical shift values for the residues affected by this transition. Although these differential 15N chemical shift values alone will generally be insufficient to provide de novo structure determination, given plausible structural models for the transition, experimental ΔR2 values can provide a valuable test for the reliability of those structural models.
If the exchange line broadening effects in the β2 and β3a strands arise from a two-state transition between conformations similar to the crystal structures of the FK506- and iFit-inhibited proteins, as displayed in Fig. 7, prediction of the 15N chemical shifts for those two conformational states would provide an estimate for the relative magnitude of the exchange line broadening for the residues of these β strands. A number of empirical algorithms have been developed for predicting NMR chemical shifts from protein structures (56–59). Although the capability of these algorithms to predict individual chemical shift values for residues within a given protein is still fairly modest, the robustness of this approach can be significantly improved when applied in a differential mode that provides for the cancellation of various modeling uncertainties, as recently illustrated in the analysis of the β4-β5 loop transition in FKBP12 (40).
Using the SPARTA+ program of Shen and Bax (56), the 15N chemical shifts were predicted for the iFit1- and iFit4-inhibited crystal structures of the FKBP51 FK1 domain as well as for the reference FK506-inhibited structure. In addition to the dependence of chemical shift on the local peptide backbone conformation, the SPARTA+ algorithm (56) uses the Haigh-Mallion model (60, 61) for estimating aromatic ring current effects and also incorporates electric field effects for the 1H resonances.
Changes in the backbone conformation thoroughly dominate the predicted differences in 15N chemical shift for these liganded FK1 domains. Despite the marked reorientation of the phenyl ring of Phe-67 upon the binding of the iFit inhibitors, the predicted differential contributions from the aromatic ring current effects are quite small. In particular, for the residues exhibiting the largest differential exchange line broadening (Lys-60, Lys-66, Phe-67, and Asp-68 in Fig. 6, B and C) the maximal ring current contribution to the differential 15N chemical shift is only 0.1 ppm (Fig. 6D).
With respect to the FK506-inhibited reference structure, the iFit1- and iFit4-inhibited structures yield similar patterns of predicted differential 15N chemical shifts for the β2 and β3a strands (Fig. 6D), consistent with their qualitatively similar conformations. The correlation coefficient for differential 15N chemical shifts over these 11 residues is 0.93 between the iFit1- and iFit4-inhibited structures.
Particularly for the iFit1-inhibited structure, the predicted differential 15N shifts correlate rather well with the observed ΔR2 values in the β2 and β3a strands as well as in the intervening loop. Averaging over the ΔR2 values for the 900 MHz datasets (Fig. 6C) of the wild type, K121A, and K121G proteins (data for Lys-60 and Leu-61 normalized by the average for each protein variant), the correlation coefficient with respect to the differential 15N shifts predicted from the iFit1-inhibited structure was also 0.93. Thus within the structural sensitivity of these predicted differential 15N chemical shifts, the transient conformation that gives rise to the exchange line broadening in the β2 and β3a strands would appear to be as similar to the iFit1-inhibited crystal structure as that structure is to the iFit4-inhibited structure.
Discussion
The correspondence between the crystal structure-based prediction of differential 15N chemical shifts and the observed exchange line broadening within the β2 and β3a strands of the unliganded FK1 domain of FKBP51 strongly supports the expectation that this line broadening arises from a transition to a significantly populated conformation that is similar to what is observed in the crystal structures of the iFit-inhibited domain. As such, conformational selection is surely the more appropriate mechanism to assume for the binding interaction of FKBP51 with the iFit family of inhibitors until more detailed thermodynamic and kinetics studies are obtained. More generally, this exchange line broadening analysis for the β2 and β3a strands in the FK1 domain of FKBP51 provides further demonstration of the utility of NMR relaxation analysis in identifying sites of transient conformational transitions that are potentially amenable to systematic drug targeting.
Hausch and colleagues (23) have proposed that the altered conformation that they observed in their crystal structures of the iFit-inhibited FK1 domains is biochemically relevant. One line of potential support for that hypothesis comes from their demonstration that the introduction of either one of two FKBP51-like substitutions, T58K or W60K, into FKBP52 yields a variant that binds the fluorescent iFit-FL ligand more tightly than does the wild type FKBP52 domain by over 100-fold, thus binding nearly as tightly as does FKBP51 (23). Conversely, introducing the K58T or the K60W mutation into FKBP51 markedly reduces the binding affinity for this ligand, consistent with a corresponding alteration in the stability of the conformation in the crystal structures of the iFit-inhibited protein.
In a set of experiments designed to test the regulation of the glucocorticoid receptor, Hausch and colleagues (23) used the brain barrier-permeable analog SAFit2 in mouse studies to inhibit the endogenous FKBP51. They found a reduced corticosterone level at the peak of the circadian cycle and an enhanced suppression of corticosterone levels following the combined treatment with dexamethasone and corticotrophin-releasing factor, results that closely mimic the behavior of FKBP51 null mice. Although indicating that the signaling functions of FKBP51 can be selectively inhibited in vivo, these studies do not address the issue of whether the conformational transition utilized in binding the inhibitor also participates in the physiological functioning of this signaling pathway.
If a conformational transition involving the rearrangement of the Phe-67 side chain and the bifurcated main chain hydrogen bonding interactions of the β2 and β3a strands is critical for mediating steroid receptor interactions, and furthermore, single point mutations can efficiently interchange the conformational sampling of the β3a strand in both FKBP51 and FKBP52, it is interesting that only mutations in the β4-β5 loop were repeatedly isolated in the hormone-induced transcription selection studies of Smith and colleagues (16). In that earlier study, a total of 25 gain-of-function mutants were isolated from eight independent error-prone PCR libraries of FKBP51 variants. In addition to five independent occurrences of the L119P mutation discussed above, at least three independent isolates were found for both the FKBP52-like S124P variant and the nonhomologous A116V variant, which in one isolate was found combined with the L119P mutation. This A116V,L119P double mutant yielded transcription activity levels fully equivalent to those of wild type FKBP52.
Although the results of Smith and colleagues (16) do not exclude the possibility that such a conformational transition in the β3a strand participates in mediating steroid receptor signaling, these results do strongly indicate that such a conformational plasticity-based mechanism should incorporate the additional transition of the β4-β5 loop. The present study demonstrates the energetic coupling between the transition centered in the β4-β5 loop and a second transition centered in the β3a strand. The most physically plausible interpretation of the pattern of mutationally induced variations in the exchange line broadening behavior indicates that an increase in conformational flexibility near the tip of the β4-β5 loop gives rise to an increased population of the reoriented conformation of the β3a strand, fully consistent with the requirement of enabling the side chain of Phe-67 to escape from its burial within the tightly packed interface (Fig. 7).
A model for the differential interaction with the steroid receptor can be posited in which the transition to an alternate conformation in the β4-β5 loop shifts the equilibrium between the Phe-67-in and the Phe-67-out conformations of the β3a strand so as to favor the latter conformation (Fig. 8). The suggestion that the tip of the β4-β5 loop might serve as a latch for preferentially stabilizing the Phe-67-in conformation is in accord with the observation that the binding of the iFit1 inhibitor is accompanied by the β4-β5 loop moving away from the β3a strand (Fig. 7B).
FIGURE 8.
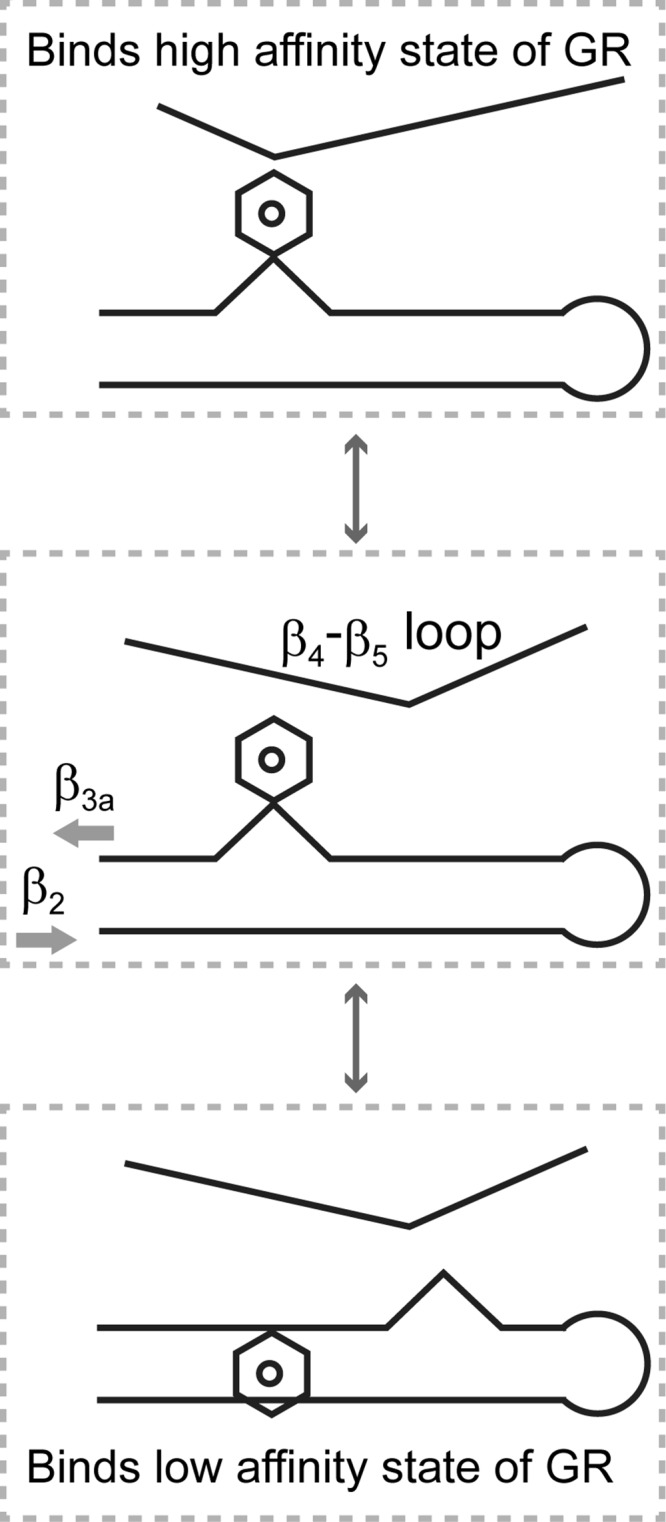
A model for the conformation-dependent modulation of glucocorticoid receptor interactions for the FK1 domains of FKBP51 and FKBP52. The β4-β5 loop and the β3a strand undergo distinct local conformational transitions in which the equilibrium population distributions appear to be energetically coupled. Transition to the alternate conformational state of the β4-β5 loop preferentially stabilizes the Phe-67-out conformation of the β3a strand in which the kink in the antiparallel hydrogen bonding pattern between the β2 and β3a strands is shifted toward the start of the β3a strand. As the transitions at both the β4-β5 loop and the β3a strand are energetically more favorable in FKBP51, the resultant conformational state is assumed to interact more strongly with the glucocorticoid receptor in its low affinity state, whereas FKBP52 primarily exists in a conformation that preferentially binds to the high affinity state of the receptor.
Because the alternate β4-β5 loop conformation and the Phe-67-out conformation are both more readily populated in the FKBP51 domain, that protein more favorably adopts this combination of local conformations, which is assumed to more strongly interact with the steroid receptor complex in a low affinity state for hormone binding. For FKBP52, Pro-119 inhibits the transition of the β4-β5 loop to an alternate conformation, whereas Thr-58 (and perhaps Trp-60) inhibits the transition to the Phe-67-out conformation. Note that even when the FKBP51 sequence for the β4-β5 loop is incorporated into FKBP52 (i.e. P119L,P124S), no exchange line broadening of the β3a strand is observed (20). Hence interactions in both the β4-β5 loop and the β2-β3a strands tend to lock FKBP52 into a conformation which preferentially binds to a high affinity state of the steroid receptor.
Currently, there is no detailed structural model for the conformational transition that underlies the exchange line broadening observed within the β4-β5 loop in the FK1 domain of FKBP51. However, previous studies on both FKBP12 and FKBP12.6 can provide useful insights into this process. The pattern of conformational exchange line broadening for the residues along the β4-β5 loop in the FK1 domain of FKBP51 is similar to that observed for FKBP12 (39). Using a combination of structure-based 15N chemical shift predictions and mutations that selectively modulate the β4-β5 loop transition in FKBP12, we were able to deduce that a central structural element of that transition involves a shift in the backbone φ torsion angle for Gly-89 from the positive value, as observed in all reported crystal structures, to a negative φ angle (40). Given that the homologous residue of FKBP51 is Pro-120, which is covalently constrained to a negative φ torsion angle, the detailed structural mechanism of the exchange line broadening transition must necessarily differ in this case.
Our recent report of the first crystal structures of FKBP12.6 in the unliganded state (62) may provide a more useful initial model for interpreting structural aspects of this exchange line broadening transition in FKBP51. Structures of unliganded FKBP12.6 were obtained from two distinct crystal forms in which the β4-β5 loop conformations primarily differ from each other due to a flipping of the (ψG89,φV90) main chain torsion angles. In the P3121 (PDB code 4IQC) crystal form a side chain methyl group of Val-90 at the tip of the β4-β5 loop is oriented toward the catalytic cleft as is the case for the homologous Ile-90 side chain in all reported crystal structures of FKBP12. In contrast, The P21 (PDB code 4IQ2) crystal form has a Val-90 methyl group directed away from the catalytic cleft. If a similar transition were to occur in FKBP51, the side chain of the homologous Lys-121 (with its Cβ packed snugly against the Cβ of Phe-67 as illustrated in Fig. 2) would undergo a substantial reorientation that could contribute to the differing interactions of FKBP51 with the low and high affinity states of the steroid receptor. It should be noted that the cis peptide linkage of Pro-120 in the FK1 domain of FKBP51 results in crystal structures in which the (ψP120,φK121) torsion angles differ appreciably from homologous (ψG89,φV90) angles of either unliganded FKBP12.6 structure so that an analogous transition must differ in detail. Nevertheless, the transition observed from the crystal structures of unliganded FKBP12.6 may provide a useful initial model for future experimental studies.
Acknowledgments
We acknowledge the use of the NMR facility, x-ray crystallography, and Applied Genomic Technologies cores at the Wadsworth Center as well as the NMR facility at the New York Structural Biology Center and beamline X25 at the National Synchrotron Light Source. The National Synchrotron Light Source Beam line X25 is supported by the United States Dept. of Energy Office of Biological and Environmental Research, the National Institute of General Medical Sciences, and the National Institutes of Health National Center for Research Resources.
This work was supported, in whole or in part, by National Institutes of Health Grant GM 088214 (to G. H.). The authors declare that they have no conflicts of interest with the contents of this article.
The atomic coordinates and structure factors (code 4R0X) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- FKBP51
- FK506-binding protein of 51-kDa
- FKBP52
- FK506-binding protein of 52-kDa
- FK506
- immunosuppressive drug primarily used in organ transplant therapies
- FK1
- first FKBP-binding domain
- PDB
- Protein Data Bank.
References
- 1. Tai P. K., Maeda Y., Nakao K., Wakim N. G., Duhring J. L., Faber L. E. (1986) A 59-kilodalton protein associated with progestin, estrogen, androgen and glucocorticoid receptors. Biochemistry 25, 5269–5275 [DOI] [PubMed] [Google Scholar]
- 2. Silverstein A. M., Galigniana M. D., Kanelakis K. C., Radanyi C., Renoir J. M., Pratt W. B. (1999) Different regions of the immunophilin FKBP52 determine its association with the glucocorticoid receptor, hsp90, and cytoplasmic dynein. J. Biol. Chem. 274, 36980–36986 [DOI] [PubMed] [Google Scholar]
- 3. Sanchez E. R. (1990) HSP56: a novel heat shock protein associated with untransformed steroid receptor complexes. J. Biol. Chem. 265, 22067–22070 [PubMed] [Google Scholar]
- 4. Smith D. F., Faber L. E., Toft D. O. (1990) Purification of unactivated progesterone receptor and identification of novel receptor-associated proteins. J. Biol. Chem. 265, 3996–4003 [PubMed] [Google Scholar]
- 5. Denny W. B., Valentine D. L., Reynolds P. D., Smith D. F., Scammell J. G. (2000) Squirrel monkey immunophilin FKBP51 is a potent inhibitor of glucocorticoid receptor binding. Endocrinology 141, 4107–4113 [DOI] [PubMed] [Google Scholar]
- 6. Wochnik G. M., Rüegg J., Abel G. A., Schmidt U., Holsboer F., Rein T. (2005) FK506-binding proteins 51 and 52 differentially regulate dynein interaction and nuclear translocation of the glucocorticoid receptor in mammalian cells. J. Biol. Chem. 280, 4609–4616 [DOI] [PubMed] [Google Scholar]
- 7. Davies T. H., Ning Y. M., Sánchez E. R. (2002) A new first step in activation of steroid receptors: hormone-induced switching of FKBP51 and FKBP52 immunophilins. J. Biol. Chem. 277, 4597–4600 [DOI] [PubMed] [Google Scholar]
- 8. Baughman G., Wiederrecht G. J., Chang F., Martin M. M., Bourgeois S. (1997) Tissue distribution and abundance of human FKBP51, an FK506-binding protein that can mediated calcineurin inhibition. Biochem. Biophys. Res. Commun. 232, 437–443 [DOI] [PubMed] [Google Scholar]
- 9. Binder E. B., Salyakina D., Lichtner P., Wochnik G. M., Ising M., Pütz B., Papiol S., Seaman S., Lucae S., Kohli M. A., Nickel T., Künzel H. E., Fuchs B., Majer M., Pfennig A., Kern N., Brunner J., Modell S., Baghai T., Deiml T., Zill P., Bondy B., Rupprecht R., Messer T., Köhnlein O., Dabitz H., Brückl T., Müller N., Pfister H., Lieb R., Mueller J. C., Lõhmussaar E., Strom T. M., Bettecken T., Meitinger T., Uhr M., Rein T., Holsboer F., Muller-Myhsok B. (2004) Polymorphisms in FKBP5 are associated with recurrence of depressive episodes and rapid response to antidepressant treatment. Nat. Genet. 36, 1319–1325 [DOI] [PubMed] [Google Scholar]
- 10. Galigniana N. M., Ballmer L. T., Toneatto J., Erlejman A. G., Lagadari M., Galigniana M. D. (2012) Regulation of the glucocorticoid response to stress-related disorders by the Hsp90-binding immunophilin FKBP51. J. Neurochem. 122, 4–18 [DOI] [PubMed] [Google Scholar]
- 11. Pei H., Li L., Fridley B. L., Jenkins G. D., Kalari K. R., Lingle W., Petersen G., Lou Z., Wang L. (2009) FKBP51 affects cancer cell response to chemotherapy by negatively regulating Akt. Cancer Cell 16, 259–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fabian A. K., März A., Neimanis S., Biondi R. M., Kozany C., Hausch F. (2013) InterAKTions with FKBPs: mutational and pharmacological exploration. PLoS ONE 8, e57508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stechschulte L. A., Hinds T. D., Jr., Khuder S. S., Shou W., Najjar S. M., Sanchez E. R. (2014) FKBP51 controls cellular adipogenesis through P38 kinase-mediated phosphorylation of GRα and PPARγ. Mol. Endocrinol. 28, 1265–1275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stechschulte L. A., Hinds T. D., Jr., Ghanem S. S., Shou W., Najjar S. M., Sanchez E. R. (2014) FKBP51 reciprocally regulates GRα and PPARγ activation via the AKT-P38 pathway. Mol. Endocrinol. 28, 1254–1264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Riggs D. L., Roberts P. J., Chirillo S. C., Cheung-Flynn J., Prapapanich V., Ratajczak T., Gaber R., Picard D., Smith D. F. (2003) The Hsp90-binding peptidylprolyl isomerase FKBP52 potentiates glucocorticoid signaling in vivo. EMBO J. 22, 1158–1167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Riggs D. L., Cox M. B., Tardif H. L., Hessling M., Buchner J., Smith D. F. (2007) Noncatalytic role of the FKBP52 peptidyl-prolyl isomerase domain in the regulation of steroid hormone signaling. Mol. Cell. Biol. 27, 8658–8669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pfaff S. J., Fletterick R. J. (2010) Hormone binding and co-regulator binding to the glucocorticoid receptor are allosterically coupled. J. Biol. Chem. 285, 15256–15267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Trebble P. J., Woolven J. M., Saunders K. A., Simpson K. D., Farrow S. N., Matthews L. C., Ray D. W. (2013) A ligand-specific kinetic switch regulates glucocorticoid receptor trafficking and function. J. Cell Sci. 126, 3159–3169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kirschke E., Goswami D., Southworth D., Griffin P. R., Agard D. A. (2014) Glucocorticoid receptor function regulated by coordinated action of the Hsp90 and Hsp70 chaperone cycles. Cell 157, 1685–1697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mustafi S. M., LeMaster D. M., Hernández G. (2014) Differential conformational dynamics in the closely homologous FK506-binding domains of FKBP51 and FKBP52. Biochem. J. 461, 115–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Millet O., Loria J. P., Kroenke C. D., Pons M., Palmer A. G., III (2000) The static magnetic field dependence of chemical exchange line broadening defines the NMR chemical shift time scale. J. Am. Chem. Soc. 122, 2867–2877 [Google Scholar]
- 22. Bracher A., Kozany C., Thost A. K., Hausch F. (2011) Structural characterization of the PPIase domain of FKBP51, a cochaperone of human Hsp90. Acta Crystallogr. D Biol. Crystallogr. 67, 549–559 [DOI] [PubMed] [Google Scholar]
- 23. Gaali S., Kirschner A., Cuboni S., Hartmann J., Kozany C., Balsevich G., Namendorf C., Fernandez-Vizarra P., Sippel C., Zannas A. S., Draenert R., Binder E. B., Almeida O. F., Rühter G., Uhr M., Schmidt M. V., Touma C., Bracher A., Hausch F. (2015) Selective inhibitors of the FK506-binding protein 51 by induced fit. Nat. Chem. Biol. 11, 33–37 [DOI] [PubMed] [Google Scholar]
- 24. Boehr D. D., Nussinov R., Wright P. E. (2009) The role of dynamic conformational ensembles in biomolecular recognition. Nat. Chem. Biol. 5, 789–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Palmer A. G., 3rd (2014) Chemical exchange in biomacromolecules: past, present and future. J. Magn. Reson. 241, 3–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sekhar A., Kay L. E. (2013) NMR paves the way for atomic level descriptions of sparsely populated, transiently formed biomolecular conformers. Proc. Natl. Acad. Sci. U.S.A. 110, 12867–12874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lakomek N. A., Ying J., Bax A. (2012) Measurement of 15N relaxation rates in perdeuterated proteins by TROSY-based methods. J. Biomol. NMR 53, 209–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Otwinowski Z., Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 29. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sheldrick G. M., Schneider T. R. (1997) SHELXL: high resolution refinement. Methods Enzymol. 277, 319–343 [PubMed] [Google Scholar]
- 31. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and Development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E. (2004) UCSF Chimera: a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 33. Delano W. (2002) The PyMOL Molecular Graphics System, DeLano Scientific LLC, San Carlos, CA [Google Scholar]
- 34. Galat A. (2008) Functional drift of sequence attributes in the FK506-binding proteins (FKBPs). J. Chem. Inf. Model. 48, 1118–1130 [DOI] [PubMed] [Google Scholar]
- 35. Barent R. L., Nair S. C., Carr D. C., Ruan Y., Rimerman R. A., Fulton J., Zhang Y., Smith D. F. (1998) Analysis of FKBP51/FKBP52 chimeras and mutants for Hsp90 binding and association with progesterone receptor complexes. Mol. Endocrinol. 12, 342–354 [DOI] [PubMed] [Google Scholar]
- 36. Gkika D., Topala C. N., Hoenderop J. G., Bindels R. J. (2006) The immunophilin FKBP52 inhibits the activity of the epithelial Ca2+ channel TRPV5. Am. J. Physiol. Renal Physiol. 290, F1253-F1259 [DOI] [PubMed] [Google Scholar]
- 37. Jiang W., Cazacu S., Xiang C., Zenklusen J. C., Fine H. A., Berens M., Armstrong B., Brodie C., Mikkelsen T. (2008) FK506 binding protein mediates glioma cell growth and sensitivity to rapamycin treatment by regulating NF-κB signaling pathway. Neoplasia 10, 235–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shim S., Yuan J. P., Kim J. Y., Zeng W., Huang G., Milshteyn A., Kern D., Muallem S., Ming G. L., Worley P. F. (2009) Peptidyl-prolyl isomerase FKBP52 controls chemotropic guidance of neuronal growth cones via regulation of TRPC1 channel opening. Neuron 64, 471–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mustafi S. M., Chen H., Li H., Lemaster D. M., Hernández G. (2013) Analyzing the visible conformational substates of the FK506-binding protein FKBP12. Biochem. J. 453, 371–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mustafi S. M., Brecher M., Zhang J., Li H., Lemaster D. M., Hernández G. (2014) Structural basis of conformational transitions in the active site and 80's loop in the FK506-binding protein FKBP12. Biochem. J. 458, 525–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Anderson J. S., Mustafi S. M., Hernández G., LeMaster D. M. (2014) Statistical allosteric coupling to the active site indole ring flip equilibria in the FK506-binding domain. Biophys. Chem. 192, 41–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lipari G., Szabo A. (1982) Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules: 1. theory and range of validity. J. Am. Chem. Soc. 104, 4546–4559 [Google Scholar]
- 43. Brath U., Akke M., Yang D., Kay L. E., Mulder F. A. (2006) Functional dynamics of human FKBP12 revealed by methyl 13C rotating frame relaxation dispersion NMR spectroscopy. J. Am. Chem. Soc. 128, 5718–5727 [DOI] [PubMed] [Google Scholar]
- 44. Brath U., Akke M. (2009) Differential responses of the backbone and side-chain conformational dynamics in FKBP12 upon binding the transition-state analog FK506: implications for transition-state stabilization and target protein recognition. J. Mol. Biol. 387, 233–244 [DOI] [PubMed] [Google Scholar]
- 45. Sapienza P. J., Mauldin R. V., Lee A. L. (2011) Multi-timescale dynamics study of FKBP12 along the rapamycin-mTOR binding coordinate. J. Mol. Biol. 405, 378–394 [DOI] [PubMed] [Google Scholar]
- 46. Li P., Ding Y., Wu B., Shu C., Shen B., Rao Z. (2003) Structure of the N-terminal domain of human FKBP52. Acta Crystallogr. D Biol. Crystallogr. 59, 16–22 [DOI] [PubMed] [Google Scholar]
- 47. Wu B., Li P., Liu Y., Lou Z., Ding Y., Shu C., Ye S., Bartlam M., Shen B., Rao Z. (2004) 3D structure of human FK506-binding protein 52: implications for the assembly of the glucocorticoid receptor/Hsp90/immunophilin heterocomplex. Proc. Natl. Acad. Sci. U.S.A. 101, 8348–8353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bracher A., Kozany C., Hähle A., Wild P., Zacharias M., Hausch F. (2013) Crystal structures of the free and ligand-bound FK1-FK2 domain segment of FKBP52 reveal a flexible inter-domain hinge. J. Mol. Biol. 425, 4134–4144 [DOI] [PubMed] [Google Scholar]
- 49. Mandel A. M., Akke M., Palmer A. G., 3rd (1996) Dynamics of ribonuclease H: temperature dependence of motions on multiple time scales. Biochemistry 35, 16009–16023 [DOI] [PubMed] [Google Scholar]
- 50. Tang S., Case D. A. (2011) Calculation of chemical shift anisotropy in proteins. J. Biomol. NMR 51, 303–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Clackson T., Yang W., Rozamus L. W., Hatada M., Amara J. F., Rollins C. T., Stevenson L. F., Magari S. R., Wood S. A., Courage N. L., Lu X., Cerasoli F., Jr., Gilman M., Holt D. A. (1998) Redesigning an FKBP-ligand interface to generate chemical dimerizers with novel specificity. Proc. Natl. Acad. Sci. U.S.A. 95, 10437–10442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yang W., Rozamus L. W., Narula S., Rollins C. T., Yuan R., Andrade L. J., Ram M. K., Phillips T. B., van Schravendijk M. R., Dalgarno D., Clackson T., Holt D. A. (2000) Investigating protein-ligand interactions with a mutant FKBP possessing a designed specificity pocket. J. Med. Chem. 43, 1135–1142 [DOI] [PubMed] [Google Scholar]
- 53. Banaszynski L. A., Chen L. C., Maynard-Smith L. A., Ooi A. G., Wandless T. J. (2006) A rapid, reversible, and tunable method to regulate protein function in living cells using synthetic small molecules. Cell 126, 995–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Egeler E. L., Urner L. M., Rakhit R., Liu C. W., Wandless T. J. (2011) Ligand-switchable substrates for a ubiquitin-proteasome system. J. Biol. Chem. 286, 31328–31336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Feng H., Zeng Y., Whitesell L., Katsanis E. (2001) Stressed apoptotic tumor cells express heat shock proteins and elicit tumor-specific immunity. Blood 97, 3505–3512 [DOI] [PubMed] [Google Scholar]
- 56. Shen Y., Bax A. (2010) SPARTA+: a modest improvement in empircal NMR chemical shift prediction by means of an artificial neural network. J. Biomol. NMR 48, 13–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Han B., Liu Y., Ginzinger S. W., Wishart D. S. (2011) SHIFTX2: significantly improved protein chemical shift prediction. J. Biomol. NMR 50, 43–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kohlhoff K. J., Robustelli P., Cavalli A., Salvatella X., Vendruscolo M. (2009) Fast and accurate predictions of protein NMR chemical shifts from interatomic distances. J. Am. Chem. Soc. 131, 13894–13895 [DOI] [PubMed] [Google Scholar]
- 59. Moon S., Case D. A. (2007) A new model for chemical shifts of amide hydrogens in proteins. J. Biomol. NMR 38, 139–150 [DOI] [PubMed] [Google Scholar]
- 60. Haigh C. W., Mallion R. B. (1979) Ring current theories in nuclear magnetic resonance. Prog. Nucl. Magn. Reson. Spectrosc. 13, 303–344 [Google Scholar]
- 61. Case D. A. (1995) Calibration of ring-current effects in proteins and nucleic acids. J. Biomol. NMR 6, 341–346 [DOI] [PubMed] [Google Scholar]
- 62. Chen H., Mustafi S. M., LeMaster D. M., Li Z., Héroux A., Li H., Hernández G. (2014) Crystal structure and conformational flexibility of the unligated FK506-binding protein FKBP12.6. Acta Crystallogr. D Biol. Crystallogr. 70, 636–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Blackburn E. A., Walkinshaw M. D. (2011) Targeting FKBP isoforms with small-molecule ligands. Curr. Opin. Pharmacol. 11, 365–371 [DOI] [PubMed] [Google Scholar]