

Background: Intracellular parts of GPCRs have yet to be effectively exploited as allosteric modulators.
Results: The structure and mechanism of action of a lipidated pepducin agonist is determined.
Conclusion: The pepducin requires the H8 helix and TM7 tyrosine propeller to stabilize the on-state and trigger receptor-G protein signaling.
Significance: This work provides critical insight into the identification of allosteric modulators to this major drug target class.
Keywords: biotechnology, cell surface receptor, cell-penetrating peptide (CPP), chemical biology, drug delivery system, G protein-coupled receptor (GPCR), nuclear magnetic resonance (NMR), receptor structure-function, signal transduction, thrombin
Abstract
G protein-coupled receptors (GPCRs) are remarkably versatile signaling systems that are activated by a large number of different agonists on the outside of the cell. However, the inside surface of the receptors that couple to G proteins has not yet been effectively modulated for activity or treatment of diseases. Pepducins are cell-penetrating lipopeptides that have enabled chemical and physical access to the intracellular face of GPCRs. The structure of a third intracellular (i3) loop agonist, pepducin, based on protease-activated receptor-1 (PAR1) was solved by NMR and found to closely resemble the i3 loop structure predicted for the intact receptor in the on-state. Mechanistic studies revealed that the pepducin directly interacts with the intracellular H8 helix region of PAR1 and allosterically activates the receptor through the adjacent (D/N)PXXYYY motif through a dimer-like mechanism. The i3 pepducin enhances PAR1/Gα subunit interactions and induces a conformational change in fluorescently labeled PAR1 in a very similar manner to that induced by thrombin. As pepducins can potentially be made to target any GPCR, these data provide insight into the identification of allosteric modulators to this major drug target class.
Introduction
G protein-coupled receptors have evolved to respond to a diverse repertoire of extracellular ligands but share a highly conserved molecular architecture of seven transmembrane domains (1). Upon activation, the receptors undergo a large conformational change that transmits signals from the outside of the cell to the G proteins on the inside (2). Recent structures of dozens of different GPCRs2 have provided an unprecedented understanding of ligand binding in the transmembrane core, also known as the classical orthosteric site (3). How the intracellular loop domains mediate signaling to G proteins is not as well understood, and the intracellular parts of the receptor have yet to be effectively exploited as drug targets. To address this issue, a new technology known as “pepducins” has emerged that targets this inside part of GPCRs (4).
Pepducins are highly stable lipidated intracellular loops derived from GPCRs that specifically act on their cognate receptor (5). The rapid and efficient cell membrane flipping and membrane tethering of pepducins (6–10) makes them well suited for interrogation of the role of intracellular regions of receptors in G protein coupling and cell signaling. Pepducin agonists are allosteric modulators that typically exhibit biphasic agonist and inhibitory activity on receptor-G protein signaling (5) and can favor receptor-G protein engagement of one class of G proteins over others, so-called biased agonism (11, 12). Allosteric agonists and antagonists that bind at alternative sites to the orthosteric site (13) can provide certain advantages such as a greater GPCR subtype specificity and G protein-selective pathway engagement (14). Libraries of pepducins can be readily targeted to a GPCR of interest based on the sequences of the intracellular loops, including orphan GPCRs, and tested for allosteric activity by high throughput screening (15, 16). This approach has proven useful in delineating the functional significance of GPCRs in cancer (17–20) and cardiovascular (6, 21–24) and inflammatory diseases (25–28), and now pepducins have just entered human clinical trials (29); however, their mechanism of activation is largely unknown. Here, we define the structure of a PAR1 agonist pepducin, P1pal-19, that together with biochemical analyses and assays that monitor conformational changes reveal an allosteric activation mechanism for pepducins at the receptor-G protein interface.
Experimental Procedures
Cell Culture and Reagents
N-Palmitoylated pepducins, N-biotinylated PAR1 third intracellular (i3) loop peptides, and PAR1 peptide agonist SFLLRN were synthesized with C-terminal amides by standard Fmoc solid-phase methods as described previously (5) by the Tufts University Peptide Core Facility. Peptides were purified by C4 or C18 reverse-phase high pressure liquid chromatography. The following reagents were used: thrombin (Hematologic Technologies), pertussis toxin (Sigma), myo[3H]inositol (PerkinElmer Life Sciences), GDP, GTPγS, avidin-agarose (Sigma-Aldrich), T7-agarose beads and mouse monoclonal T7-Ab (Novagen), rabbit anti-Gαi1/2 antibody (PerkinElmer Life Sciences), and rabbit Myc antibody (Cell Signaling Technology). The rabbit polyclonal PAR1 antibody (SFLLR-Ab) was generated as described previously (30). PAR1 mutants were made by site-directed mutagenesis in pcDEF3 with QuikChange (Qiagen) or PCR mutagenesis and sequenced to verify the fidelity of the construct as described (31). Cercopithecus aethiops kidney cells (COS7) and human embryonic cells (HEK293) were transfected with T7-PAR1 mutants using Lipofectamine (31) for inositol phosphate (InsP) and fluorescence assays and calcium phosphate for binding studies, and stable PAR1 Rat 1 cells (5) and Rat 1 PAR1-null cells were cultured in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin in 5% CO2 at 37 °C.
NMR Structural Determination of P1pal-19
P1pal-19 (palmitate-RCLSSSAVANRSKKSRALF-NH2) pepducin was synthesized by standard Fmoc solid-phase methods and purified to 99.1% by reverse-phase high performance liquid chromatography. NMR samples were prepared by dissolving lyophilized P1pal-19 in a buffer comprising 2.2 mm P1Pal-19, 180 mm dodecylphosphocholine-d38, 5% glucose-d7, 10 mm acetate-d4, 0.02% NaN3, 1.1 mm deuterated dithiothreitol (DTT), and 0.1 mm 4,4-dimethyl-4-silapentane-1-sulfonic acid at pH 5.12. NMR spectra included total correlation spectroscopy with a mixing time of 40 ms, nuclear Overhauser effect spectroscopy (NOESY) with a mixing time of 100 ms, and natural abundance 13C heteronuclear single quantum coherence spectroscopy. Data were collected at 600 and 800 MHz at 25 °C. Resonances were assigned, and distance restraints were obtained by standard methods. Proton chemical shifts, carbon chemical shifts, and distance restraints were used in CNSSolve version 1.3 to generate 32 structures through simulated annealing, energy minimization, and refinement (32).
PAR1 Modeling
The PAR1 off-state was modeled based on the vorapaxar-PAR1 x-ray structure (Protein Data Bank code 3VW7) (33) using the highly homologous H8 helix and i3 loop structures from the rhodopsin off-state (Protein Data Bank code 3CAP) (34). The PAR1 on-state was modeled from the β2-adrenergic receptor·Gs complex (Protein Data Bank code 3SN6) (35) with the i3 loop and H8 helix from the rhodopsin·Gt peptide complex (Protein Data Bank code 3PQR) (36). The G protein was positioned such that regions of α5 known to interact with i2, TM5, and TM6 in the structure of the β2-adrenergic receptor·Gs complex were replaced with the intact heterotrimeric G protein transducin (Protein Data Bank code 1GOT) (37). The chemical geometries of all models were refined using Coot and Refmac5, and molecular graphics figures were generated using PyMOL.
InsP Production Assay
COS7 cells expressing PAR1 mutants were split into 12-well plates at 250,000 cells/well. Myo[3H]inositol (2 mCi/ml) was added to the cells 24 h prior to the experiment. Phospholipase C-β (PLC-β)-dependent accumulation of [3H]InsP was measured in the presence of 20 mm LiCl. The cells were stimulated with agonist (in duplicate or triplicate) for 30 min and then extracted with cold methanol and chloroform. The extracts were loaded onto columns containing 1 ml of anion exchange resin AG1X8 (formate form, 100–200 mesh size; Bio-Rad). After loading, the columns were washed twice with 10 ml of H2O and twice with 10 ml of 60 mm ammonium formate, 5 mm borax. Column fractions were eluted with 4 ml of 2 m ammonium formate, 0.1 m formic acid into vials containing 7.5 ml of scintillation fluid and then counted. The mean of two to five determinations was expressed as the -fold stimulation above vehicle-treated cells and converted to a percentage using maximal activity for each agonist with WT receptor or as described in the figure legends.
Platelet Aggregation
Venous blood samples were collected from healthy volunteers using an 18-gauge needle and a 30-ml syringe prefilled with 3 ml of 4% sodium citrate following approved protocols from the Tufts Institutional Review Board. Whole blood was transferred into 15-ml polypropylene tubes with 0.25 mm EDTA, and 0.1 unit/ml apyrase was added as an anticoagulant. Platelet-rich plasma was harvested by centrifuging blood at 700 × g for 20 min at 30 °C. Gel-filtered platelets were further prepared from platelet-rich plasma using Sepharose 2B columns in modified PIPES buffer (38). Samples were recalcified with CaCl2 (2.5 mm), and fibrinogen (300 μg/ml) was added before addition of pepducins. All reactions were conducted in final volumes of 250 μl at 37 °C while stirring at 900 rpm. Platelet aggregation was measured with a Chronolog 560VS/490 aggregometer with PIPES buffer serving as a blank as described before (39, 40).
PAR1-i3 Loop Binding Assays
HEK cells transfected with PAR1 were lysed in lysis buffer (100 mm NaCl, 25 mm Hepes, pH 7.2, 1 mm PMSF, 0.2 mm leupeptin), sonicated, and centrifuged at 40,000 × g for 40 min at 4 °C. Membrane pellets were resuspended in 20 mm KPO4, 100 mm NaCl, 0.1 mm EDTA, 10% glycerol and incubated overnight at 4 °C with avidin-agarose beads that were precoupled to N-biotin-PAR1 i3 peptide (RCLSSSAVANRSKKSRALF; Bio19) or negative control i3 peptide Bio19E (RCESSSAEANRSKKERELF) at room temperature for 4 h in binding buffer (20 mm Tris, pH 7.2, 70 mm NaCl, 1 mm DTT). Agarose beads were collected by centrifugation and washed three times with binding buffer. Immunoprecipitated protein was eluted with 40 μl of SDS loading buffer for 1 h at 42 °C. Samples were centrifuged, and 25 μl of the protein supernatants was analyzed by Western blot.
PAR1-PAR1 Immunoprecipitations
A pcDEF3-PAR1 construct was tagged at the N terminus with a T7 epitope (MASMTGGQQMGT) as described (30) or at the C terminus with a Myc epitope (FFEQKLISEEDL). COS7 cells were transiently transfected with T7-PAR1 alone, PAR1-Myc alone, or both. Cell lysates were prepared 48 h after transfection in lysate buffer (100 mm NaCl, 25 mm Hepes, pH 7.2, 1 mm PMSF plus Halt protease inhibitor mixture (Thermo Scientific)), and protein was quantified using a Bradford assay. Cell lysates were precleared with 25 μl of protein A-agarose (Calbiochem) for 1 h and then incubated overnight at 4 °C with 25 μl of T7-agarose beads in a final volume of 500 μl. Agarose beads were collected by centrifugation (500 × g) and washed three times with 1% lysis buffer. Immunoprecipitated protein was eluted with 50 μl of 10 mm acetic acid, pH 2.2 in SDS loading buffer for 30 min at 37 °C. Samples were centrifuged (10,000 × g) for 10 min, and 10 μl of neutralization buffer (1.5 m Tris, pH 8.3) was added to the protein supernatants before gel electrophoresis and Western blotting.
Fluorescence Resonance Energy Transfer (FRET) between PAR1-CFP and PAR1-YFP
COS7 fibroblasts were transiently transfected using the DEAE-dextran method. Scanning fluorometry of transfected COS7 cells was performed on a Perkin Elmer Life Sciences LS50B fluorescence spectrophotometer to analyze expression levels of PAR1-CFP and PAR1-YFP (22, 26) and to detect FRET interactions between donor and acceptor receptors. Transfected cells expressing both PAR1-CFP and PAR1-YFP were excited at 425 nm, and the emission spectrum was collected from 450 to 650 nm. This spectrum consisted of CFP donor fluorescence, YFP acceptor fluorescence due to cross-excitation at 425 nm, and FRET between donor and acceptor. FRET was calculated by subtracting two components as described previously (22, 26). The first subtracted component was the emission spectrum of cells expressing CFP (donor) normalized to the CFP emission maximum of the cells co-expressing donor and acceptor. The second subtracted component was the emission spectrum of YFP (acceptor) only via cross-excitation by 425 nm at the same expression level as YFP in the cells co-expressing donor and acceptor receptors.
Bimane Labeling of PAR1
HEK 293 cells were transiently transfected with either C378S or T7-PAR1C296S/C378S/Δ396 constructs in pcDEF3. Cell lysates were prepared 48 h after transfection in lysis buffer (100 mm NaCl, 25 mm Hepes, pH 7.2, 1 mm PMSF plus Halt protease inhibitor mixture (Thermo Scientific)) with 1% Nonidet P-40, and total protein concentration was determined using a Bradford assay. Bimane-labeled PAR1 was prepared by incubating the cell lysates (protein concentration, 1.5 mg/ml) with 500 μm monobromobimane (Molecular Probes) for 2 h at room temperature with gentle mixing. For the isolation of the T7-tagged/bimane-labeled PAR1, the lysates were incubated with 25 μl of T7-agarose beads (Abcam) in a final volume of 500 μl overnight at 4 °C. Agarose beads were collected by centrifugation (500 × g) and washed with 1% lysis buffer (no Nonidet P-40). The T7-PAR1 protein attached to the beads was eluted with 0.2 mm glycine, pH 2.2 and neutralized immediately with an equal volume of 75 mm Tris, pH 10 for a final pH of 7.5. The fluorescence intensity changes of the bimane-labeled PAR1 induced by agonists were measured by excitation at 370 nm and recording emission from 430 to 490 nm using a PerkinElmer Life Sciences LS50B spectrofluorometer.
Statistical Analysis
All quantified in vitro assay results are presented as mean ± S.D. Comparisons were made with the Student's t test. Statistical significance was defined as p < 0.05 (*) or p < 0.01 (**).
Results
Structure of a PAR1 Agonist Pepducin and Interrogation of Intracellular Residues of PAR1 Required for Activation by P1pal-19
The structure of the P1pal-19 pepducin derived from the i3 loop of PAR1 was determined using 551 NMR-derived distance restraints, including those to the proximal portion of the N-terminal palmitate lipid (Table 1). The i3 loop pepducin was highly structured with a root mean square deviation of 0.54 ± 0.19 Å for an ensemble of 30 structures (Fig. 1, A–C). The i3 loop pepducin structure resembles that predicted for the intact receptor with a short loop centered on residue Arg305 flanked by interacting helices at the N- and C-terminal ends of the peptide that correspond to the ends of TM5 and TM6 (Fig. 1, A and B). The angle between the helices, however, differs slightly by 20° from that in the intact receptor in the on-state (35).
TABLE 1.
NMR structural parameters of P1pal-19
NMR spectra included total correlation spectroscopy with a mixing time of 40 ms, NOESY with a mixing time of 100 ms, and natural abundance 13C heteronuclear single quantum coherence spectroscopy. Data were collected at 600 and 800 MHz at 25 °C. r.m.s., root mean square.
| P1pal-19 | |
|---|---|
| Experimental restraints | |
| Distance restraints from NOEs | 551 |
| Dihedral angle restraints | 0 |
| Hydrogen bond restraints | 0 |
| Total no. of experimental restraints | 551 |
| r.m.s. deviations from experimental data | |
| Average distance restraint violation (Å) | 0.0183 ± 0.0005 |
| Dihedral restraint violations >5° | 0.0 ± 0.0 |
| r.m.s. deviations from ideal stereochemistry | |
| Bonds (Å) | 0.0033 ± 0.00011 |
| Angles (°) | 0.5863 ± 0.0162 |
| Impropers (°) | 0.4788 ± 0.0133 |
| Ramachandran analysis of the structures | |
| Residues in favored regions (%) | 78.6 |
| Residues in additionally allowed regions (%) | 21.4 |
| Residues in disallowed regions (%) | 0 |
| Lennard-Jones potential energies | |
| Ensemble average (kcal/mol) | 99.74 ± 8.17 |
| Coordinate precision (Å) | |
| Backbone | 0.543 ± 0.1394 |
| All heavy atoms | 1.178 ± 0.2023 |
FIGURE 1.

Structure of an i3 loop pepducin agonist. A, the NMR structure of the P1pal-19 pepducin was determined by simulated annealing methods using 551 NOE-based distance restraints. The α carbons of the pepducin residues were numbered corresponding to the intact receptor. B, the structure of P1pal-19 (red) corresponded closely (backbone root mean square deviation, 2.1 Å) with the analogous i3 loop region of the on-state of the β2-adrenergic receptor·Gs complex (35) (yellow) comprising the cytoplasmic α-helical extensions of TM5 and TM6 and an overall root mean square deviation of 3.6 Å with the entire i3 loop region of the PAR1 model. The PAR1-vorapaxar (VPX) structure of the off-state is shown in white (33). After cleavage by thrombin or MMP-1 (23), the new N-terminal tethered peptide ligand SFLLRN--- or PRSFLLRN---, respectively, binds to the outside surface of PAR1 in an intramolecular mode (42) to activate receptor-G protein signaling. C, a superimposed ensemble of 30 individual energy-minimized structures of P1pal-19.
To further delineate the mechanism of activation by the pepducin, an array of 29 single, double, and triple mutations was constructed across the intracellular surface of PAR1 comprising the i1–i3 loops and the proximal H8 helix region of the C-terminal i4 domain and in the extracellular ligand. These mutant PAR1 receptors were interrogated for the ability to be activated by the P1pal-19 intracellular pepducin and compared with the classical extracellular agonist of PAR1, namely thrombin, which cleaves the receptor at the Arg41-Ser42 peptide bond to generate the tethered ligand SFLLRN (Fig. 1B). In addition, the array of intracellular loop mutations were tested for the ability to be activated by a soluble SFLLRN peptide agonist, and activity for each mutant was scored using a heat map from 0 to 120% relative to WT PAR1 for each agonist. As shown in Fig. 2 and Table 2, mutation of i1, i2, and i3 residues had a highly similar pattern of effects on activation of PAR1 by thrombin, SFLLRN, and P1pal-19, suggesting that all three agonists utilize the i1–i3 loops to activate G protein in a similar manner. In particular, i2 loop residues that come into direct contact with critical Gα protein Cα and Nα helices and Switch I (35, 41) are essential for activation of G protein for all three agonists (Fig. 2). Consistent with previous studies (5), the wild-type P1pal-19 did not compensate for deleterious mutations in the receptor i3 loop and therefore is not likely to activate G protein signaling by acting as a direct replacement for the intact receptor third intracellular loop.
FIGURE 2.
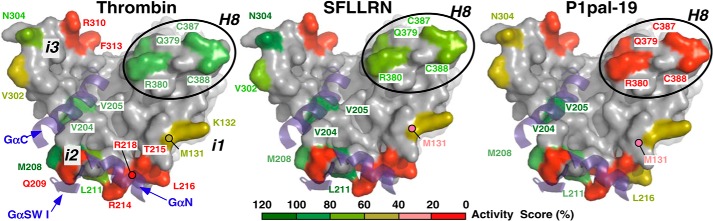
Interrogation of intracellular receptor residues required for activation of PAR1-G protein signaling by pepducin versus extracellular agonists. An array of 29 single, double, and triple mutants covering the i1, i2, i3, and H8 intracellular loop regions of PAR1 were transfected into COS-7 cells. Cells were then challenged with various concentrations of the extracellular agonists thrombin, SFLLRN peptide, and P1pal-19 pepducin. Maximal PLC-β activity was determined by measuring total [3H]InsP formation for 30 min (5) as detailed in Table 2. The effects of each mutation for the respective agonists are depicted according to the color activity scale at the bottom. SW I, Switch I.
TABLE 2.
Effect of intracellular i1–i4 loop mutations on PAR1 activation of Gq-PLC-β signaling by extracellular versus intracellular ligands
COS7 fibroblasts were transiently transfected with PAR1 mutants, and Gq activation of PLC-β was measured by accumulation of InsP over 30 min following addition of 10−12–10−8 m thrombin, 10−9–10−4 m SFLLRN, and 10−9–10−5 m P1pal-19 in triplicate. Experiments were repeated two to five times.
| Maximal signala |
Receptor expressionb | |||
|---|---|---|---|---|
| Thrombin | SFLLRN | P1pal-19 | ||
| % | ||||
| WT PAR1 | 100 | 100 | 100 | 1.0 |
| i1 loop | ||||
| M131A | 48 ± 5 | 38 ± 3 | 37 ± 2 | 1.2 |
| K132A | 55 ± 6 | 50 ± 1 | 48 ± 1 | 1.0 |
| i2 loop | ||||
| V204A | 93 ± 2 | 125 ± 5 | 107 ± 4 | 1.0 |
| V205A | 89 ± 2 | 116 ± 10 | 129 ± 8 | 0.9 |
| M208A | 108 ± 8 | 86 ± 15 | 84 ± 10 | 0.8 |
| Q209A | 16 ± 1 | 5 ± 1 | 0 ± 2 | 1.0 |
| L211A | 73 ± 1 | 140 ± 13 | 90 ± 2 | 0.7 |
| R214A | 0 ± 2 | 0 ± 2 | 0 ± 1 | 1.0 |
| T215A | 5 ± 1 | 0 ± 1 | 16 ± 2 | 0.7 |
| L216A | 10 ± 1 | 5 ± 1 | 43 ± 1 | 1.2 |
| R218A | 13 ± 2 | 0 ± 1 | 5 ± 1 | 1.3 |
| i3 loop | ||||
| V302A | 60 ± 1 | 68 ± 6 | 60 ± 1 | 1.2 |
| N304A | 76 ± 1 | 114 ± 6 | 59 ± 6 | 1.3 |
| R305S | 120 ± 2 | 102 ± 5 | 75 ± 6 | 1.0 |
| R310S | 18 ± 1 | 0 ± 1 | 16 ± 2 | 1.1 |
| F313A | 0 ± 1 | 0 ± 2 | 0 ± 2 | 0.9 |
| TM7 | ||||
| L369A | 69 ± 5 | 57 ± 6 | 118 ± 11 | 1.1 |
| Y371A | 0 ± 1 | 3 ± 1 | 0 ± 2 | 0.9 |
| Y372A | 62 ± 6 | 52 ± 11 | 84 ± 20 | 0.9 |
| Y373A | 8 ± 2 | 3 ± 1 | 7 ± 1 | 1.1 |
| H8 helix | ||||
| ΔH8 | 41 ± 3 | 38 ± 5 | 0 ± 5 | 1.0 |
| S377A/E378A | 67 ± 3 | 45 ± 2 | 64 ± 4 | 0.8 |
| C387S/C388S | 100 ± 2 | 70 ± 3 | 10 ± 2 | 0.9 |
| Q379A | 69 ± 1 | 67 ± 3 | 30 ± 4 | 1.3 |
| R380A | 81 ± 2 | 63 ± 5 | 60 ± 3 | 0.8 |
| Q379A/R380A | 81 ± 6 | 66 ± 7 | 0 ± 1 | 0.9 |
| Y383A | 22 ± 4 | 16 ± 2 | 6 ± 1 | 0.7 |
| S384A/I385A/L386A | 81 ± 5 | 90 ± 8 | 20 ± 5 | 1.0 |
| e1 domain | ||||
| F43A | 11 ± 11 | 102 ± 24 | 42 ± 16 | 1.0 |
a Maximum signal is the 30-min-accumulated InsP signal of the mutant receptor relative to WT and was not corrected for receptor expression relative to WT.
b Mean surface PAR1 mutant expression level (S.D. was ±5–15%) on transiently transfected COS7 cells relative to WT PAR1 was determined by FACS analysis using the SFLLR-Ab.
Alternatively, the pepducin agonist could be proposed to function by stabilizing active conformations of PAR1 that normally are driven inefficiently by the uncleaved tethered ligand of the receptor. In other words, the pepducin may enhance what is otherwise a very weak partial agonist activity of the uncleaved tethered ligand. To test this possibility, we determined whether P1pal-19 was active toward PAR1 bearing a defective tethered ligand with a mutation in the critical Phe43 of SFLLRN in the e1 domain (22). The Phe43 phenyl side chain interacts with ligand-binding site-1 located in the e1 domain of PAR1 (42). As expected, the F43A PAR1 lost 90% of activity to thrombin but retained full activity to the SFLLRN peptide agonist (Table 2). However, the P1pal-19 pepducin was still able to act as a partial agonist (42%) to the F43A mutant, suggesting that extracellular ligand/ligand-binding site interactions may partially contribute to but are not essential for the allosteric activity of the i3 loop pepducin. These data are consistent with previous data (43) that showed that the affinity of PAR1 for its extracellular ligand was weakened in the absence of coupled G protein that allosterically stabilizes the receptor in the high affinity state.
Direct Interaction of the i3 Loop Pepducin Agonist with the H8 Helix of PAR1
In striking contrast, mutation of Gln379/Arg380 and palmitoylated Cys387/Cys388 (44) residues in the H8 helix region (31, 45) of the intracellular i4 domain gave 95–100% loss of activation by P1pal-19 with little or no effect on activation by extracellular agonists thrombin and SFLLRN (Fig. 2). Deletion of the PAR1 H8 helix (ΔH8 mutant) caused a complete loss of ability to be activated by P1pal-19 (Fig. 3A) but retained 40% activity for thrombin and SFLLRN extracellular agonists (Table 2). These data indicate that unlike the extracellular agonists the P1pal-19 pepducin requires an intact PAR1 H8 helix to activate PAR1-G protein coupling.
FIGURE 3.

The i3 loop peptide directly interacts with the PAR1 H8 helix region. A, deletion of the PAR1 H8 helix causes complete loss of the receptor-PLC-β (InsP) response to P1pal-19 in COS7 cells. B, binding of WT PAR1 or ΔH8 PAR1 and Gαi to avidin-N-biotin-i3 loop peptides. HEK293 cells were transfected with WT PAR1 or ΔH8, or PAR1-null Rat 1 cells were used. Membrane lysates were incubated with avidin beads that were precoated with the biotinylated WT i3 loop peptide (Bio19) or negative control i3 loop (Bio19E) in the presence of 10 μm GDP or GTPγS. Eluted proteins were resolved by 10% SDS-PAGE, and Western blotting was performed to detect PAR1 (SFLLR-Ab; 1:500) or Gαi (Gαi1/2-Ab; 1:1000). C and D, Gαi inhibitor pertussis toxin (PTX) suppresses PAR1-PLC-β activity of PAR1 Rat 1 cells stimulated by either thrombin or P1pal-19. E, top, P1pal-19 and thrombin enhance binding of Gαi to PAR1. PAR1 Rat 1 cells were stimulated for 10 min with P1pal-19 or thrombin (Thr). Cell lysates were then incubated with PAR1-Ab-coated protein A beads, and bound Gαi protein was detected by Western blotting. Bottom, schematic representation of the PAR1·G protein complex bound to P1pal-19. Error bars represent S.D. IP, immunoprecipitation.
We compared the ability of the WT and ΔH8 mutant receptors to directly bind the i3 loop peptide of PAR1. The WT PAR1 i3 loop peptide was biotinylated at the N terminus (Bio19), bound to avidin beads, and incubated with membranes from cells expressing WT or ΔH8 mutant PAR1. G proteins were stabilized in either the off-state with GDP or in the on-state with GTPγS. As shown in Fig. 3B, PAR1 preferentially bound to the WT i3 loop peptide Bio19 when G proteins were in the GTPγS-induced on-state with relatively low levels bound in the GDP-stabilized off-state or to the negative control PAR1 i3 loop Bio19E. Likewise, the corresponding palmitoylated peptide P1pal-19E has no agonist activity to PAR1 (5). The ΔH8 mutant PAR1 exhibited low level nonspecific background binding to Bio19 beads or beads alone.
i3 Loop Agonist Pepducin Stabilizes the GTP-on State of Gα Subunit in Complex with PAR1
PAR1·i3 loop complexes were also probed for the presence or absence of bound Gα subunit. Eluates were probed for Gαi as P1pal-19 gave robust biphasic stimulation of Gαi-dependent InsP production (suppressed by 75% by pertussis toxin), similar to that observed with stimulation by the classical agonist thrombin (Fig. 3, C and D). Addition of GTPγS induced a marked increase in Gαi bound to the PAR1 receptor·i3 loop complex with little or no detectable binding of Gαi in the GDP off-state (Fig. 3B). Low levels of Gαi-GTPγS were bound to the negative control Bio19E i3 loop. No Gαi subunit was detected bound to the Bio19 or Bio19E beads in membranes expressing the ΔH8 PAR1 mutant or in membranes from PAR1-null cells (Fig. 3B), demonstrating that Gαi does not form a stable complex with the i3 peptide in the absence of the PAR1 receptor H8 helix.
To determine whether Gαi could also stably associate with PAR1 stimulated by i3 loop pepducin agonist, we immunoprecipitated PAR1 from intact cells incubated for 10 min with increasing concentrations of P1pal-19. As shown in Fig. 3E, P1pal-19 induced stable formation between PAR1 receptor and Gαi, peaking at 1–10 μm pepducin and decreasing at 20 μm in a biphasic manner similar to that observed for InsP signaling. Likewise, activation of PAR1 with 20 nm thrombin induced enhanced association of PAR1 with Gαi subunit (Fig. 3E). Together, these data provide direct evidence that the i3 loop pepducin associates with the PAR1 H8 helix, which forms a stable complex with the Gαi subunit in the GTP on-state as depicted schematically in the model in Fig. 3E. These data are highly consistent with bioluminescence resonance energy transfer (BRET) experiments demonstrating large increases in the BRET signal between PAR1-YFP and Gαi-Rluc following activation of PAR1 with thrombin (46).
P1pal-19 Activates PAR1 through the (D/N)PXXYYY Motif in TM7 and Adjacent H8 Helix
To facilitate coupling with the on-state of the G protein, the bottom half of TM7 of the rhodopsin-like GPCRs undergoes a dramatic conformational change and ∼90° rotation along the axes of the helix (35, 47) and is also critical for allosteric activation of Class C GPCRs (48). TM7 contains an essential conserved (D/N)PXXY motif whose tyrosine (Y) residue rotates inside the transmembrane helical bundle to stabilize TM6 in an open on-state (1, 49). This lower region of TM7 also interacts with the eighth helix in a tightly coordinated manner during coupling of receptor to G protein (31, 49, 50). To further delineate the mechanism of activation of PAR1 by the i3 loop pepducin, we tested the activity of a series of point mutants across the entire H8 helix region and adjacent TM7 helix of PAR1 for the ability to be activated by pepducin versus extracellular agonists (Fig. 4, A and B). Single, double, and triple mutations of residues in the H8 helix of PAR1 generally resulted in loss of 80–100% of activity to P1pal-19 pepducin but retained 60–100% of activity to SFLLRN and thrombin extracellular agonists. Single point mutation of H8 helix residue Gln379 resulted in a 70% loss in activity, and double mutation of Gln379/Arg380 resulted in complete loss of activity to P1pal-19 but retained 66–81% of activity to SFLLRN and thrombin (Fig. 4, A and B, and Table 2). This pattern of effects is consistent with an H8 helix-specific requirement for P1pal-19 to activate PAR1-G protein coupling.
FIGURE 4.

The i3 loop pepducin activates the receptor via the TM7 (D/N)PXXY motif and tyrosine propeller. A, identification of essential residues in (D/N)PXXYYY and H8 helix. PAR1 mutants were generated as listed in Table 1, and maximal PLC-β activities ±S.D. for each agonist (P1pal-19, SFLLRN, and thrombin) are depicted by activity score. Bottom, side view of the (D/N)PXXY motif, YYY propeller, and H8 helix. Residues are colored according to the activity score for P1pal-19 agonist. B, PLC-β activity for each PAR1 mutant was converted to a percentage of the full response relative to 1 μm P1pal-19 for WT PAR1 (100%) and plotted as a function of P1pal-19 concentration using a two-site equation that fit the biphasic activation and inhibition profile (5). C, proposed model of activation PAR1 by P1pal-19 by a TM7 tyrosine propeller mechanism. The on-state (silver) and off-state (blue) PAR1 structural models from Fig. 1A were superimposed and docked to the NMR-derived structure of P1pal-19 using the mutagenesis data from A and B. Binding of P1pal-19 to the H8 helix first triggers an inward motion of the N terminus of the H8 helix and C-terminal end of TM7. This inward motion of H8/TM7 leads to a ∼90° rotation of TM7, causing the three tyrosine residues Tyr371/Tyr372/Tyr373 to also rotate in a propeller motion. Residue Tyr373 of the (D/P)XXY motif swings into the back of TM6, thereby facilitating a large outward movement of the TM6 and stabilizing the on-state of the receptor. Error bars represent S.D.
Unlike the H8 helix mutations, nearby TM7 mutations gave a nearly identical profile of activation by P1pal-19, SFLLRN, and thrombin (Fig. 4A). In particular, TM7 residues Tyr371 and Tyr373 ((D/N)PXXYYY) were essential for G protein coupling activity induced by all three agonists. The profile of H8 helix and TM7 mutational effects on P1pal-19 activation of PAR1-G protein coupling is consistent with a mechanism whereby P1pal-19 interacts with the H8 helix, inducing a conformational change in the attached TM7 helix that triggers rotation of the three tyrosine residues in a propeller motion to push out the TM6 into the open on-state of the receptor (Fig. 4, A and C). The outward displacement of TM6 then allows enhanced binding and coupling of PAR1 with the Gα C-terminal helix (e.g. Fig. 3E).
Critical Pharmacophores of the P1pal-19 Agonist
The i3 loop of P1pal-19 is rich in positively charged side chains that may come in direct contact with the PAR1 H8 helix region located at the cytoplasmic interface of the anionic phospholipid bilayer. To further test the pepducin-induced activation mechanism of Fig. 4, side chain substitutions were introduced at the positively charged pepducin pharmacophores predicted to interact with the receptor H8 helix and nearby TM7 residues. The i3 pepducin lacking residue Arg310 (P1pal19R310S) lost all agonist activity to PAR1 as assessed by both InsP assay and platelet aggregation as compared with WT P1pal-19 pepducin (Fig. 5, A and B). Loss of pepducin Arg305 and Lys307 residues or double substitution of Lys307/Lys308 resulted in partial loss (∼50–75%) of agonist activity for PAR1. Therefore, P1pal-19 residues Arg305, Lys307, and especially Arg310 side chains are critical for pepducin activity (Fig. 5C). Consistent with this model, corresponding point mutations in Tyr373, Tyr383, Gln379, and Arg380 of the H8 helix and TM7 in the intact PAR1 receptor resulted in a 40–100% loss of activity for P1pal-19 activation of PAR1 (Fig. 4).
FIGURE 5.

Critical pharmacophores in the P1pal-19 agonist. A, cationic side chain residues of P1pal-19 predicted to potentially interact with the PAR1 H8 helix region were sequentially replaced with serine and tested for PLC-β activity for WT PAR1 expressed in COS7 cells (percentage of InsP relative to full response of 0.1 nm thrombin). B, P1pal-19 pepducins (0.6 μm) were tested for the ability to activate PAR1-dependent human platelet aggregation using gel-purified human platelets and light transmission platelet aggregometry. *, p < 0.05; **, p < 0.01; ***, p < 0.001. C, interactions between P1pal-19 i3 loop residues and the TM7/H8 helix region of PAR1. Error bars represent S.D. Pal, palmitate.
The i3 Loop Pepducin and Thrombin Induce Conformational Changes in Cys296 Bimane-labeled PAR1
We evaluated whether the canonical agonist thrombin and the i3 loop pepducin P1pal-19 could induce conformational changes at the intracellular end of TM5 of the receptor that engages Gα protein following activation. To probe the dynamics of PAR1 we specifically labeled Cys296 located at the junction of TM5 and the i3 loop with the fluorophore bimane in a mutant PAR1 that lacked all other exposed cysteine residues (C387S/Δ396 PAR1). Bimane labeling at appropriate sites will allow detection of local conformational changes in the protein as the bimane probe moves to a more hydrophobic or hydrophilic environment upon receptor activation. Such conformational changes typically occur at the intracellular ends of TM5 and TM6 as they interact with G protein during coupling (51–54).
T7-tagged/bimane-labeled PAR1 was affinity-purified from HEK cell lysates with T7-Ab beads. The fluorescence of the Cys296 bimane-labeled PAR1 was 40% more intense than that of fluorescently labeled lysates from untransfected HEK cells (Fig. 6A). Upon addition of the canonical agonist 20 nm thrombin there was a 20% enhancement in the fluorescence signal and a decrease in λmax, indicating a conformational change of the cytoplasmic end of TM5 of PAR1 to a more hydrophobic environment (Fig. 6A). Notably, 1–3 μm i3 loop pepducin P1pal-19 also increased the fluorescence intensity by 20–30% and decreased the λmax in a dose-dependent manner, indicating that the pepducin also induced a local conformational change in the cytoplasmic end of TM5 to a more non-polar environment.
FIGURE 6.

The i3 loop pepducin and thrombin induce conformational changes in the cytoplasmic end of TM5 of PAR1. Monobromobimane is a thiol-reactive fluorescent probe and was used to specifically label Cys296 at the TM5/i3 loop junction of PAR1. T7-Ab-agarose beads were used to affinity-purify Cys296 bimane-labeled T7-PAR1C378S/Δ396 (C296Bim) from transfected versus untransfected (HEK) and PAR1 lacking exposed cysteines T7-PAR1C296S/C378S/Δ396 (C296S) cell lysates. A and B, PAR1 agonist 20 nm thrombin (Thr) or 1 or 3 μm P1pal-19 (P1) or buffer was added to 300-μl volumes of the affinity-purified bimane-labeled PAR1 constructs. 30 s after addition of agonists, fluorescence emission intensity was measured at 430–490 nm with excitation at 370 nm. Maximum fluorescence intensity was normalized to the fluorescence of eluates from the T7-Ab beads of the untransfected HEK cells. C, bottom view of PAR1 in the on-state conformation in complex with the agonist pepducin P1pal-19 and the Gα subunit C-terminal α-helix (GαC). D, bottom view of PAR1 in the off-state conformation.
As an additional negative control, we transfected the HEK293 cells with the triple mutant T7-PAR1C296S/C378S/Δ396 construct such that any intracellular loop cysteine residues in PAR1 were mutated to serine to prevent spurious bimane labeling. There was no observable change in the fluorescence intensity of T7-PAR1C296S/C378S/Δ396 in response to either thrombin or the i3 loop pepducin P1pal-19 as shown in Fig. 6B. These data are consistent with a model (Fig. 6C) in which the i3 loop agonist pepducin activates the receptor to adopt an “on-state” conformation to facilitate G protein binding; however, in the absence of either the agonist pepducin or thrombin, PAR1 adopts the “off-state” (Fig. 6D), which has a less hydrophobic environment at the Cys296 side chain (green; C296Bim).
Pseudodimer Mechanism of Activation of PAR1 by the i3 Loop Pepducin
As the P1pal-19 pepducin is derived from the i3 loop and interacts with the PAR1 H8 helix, PAR1 itself may self-associate to form a homodimer or homo-oligomer with each i3 loop interacting with the H8 helix across a dimer interface. To determine whether a dimer interaction is chemically reasonable, the electrostatic potential was calculated for PAR1, and two copies of the molecule were manually docked together such that the i3 and H8 are adjacent. The i3 loop and H8 helix are on opposite ends of the PAR1 monomer where they lie on the same large face, which is slightly concave (Fig. 7A). This face is less positively charged than the opposite side of the molecule, which is convex. Thus, unlike the i1-i2 loops that are highly positively charged, leading to repulsive and geometrically unlikely interactions, the i3 loop-H8 helix face is less positively charged (and less repulsive), leading to the dimer model of Fig. 7A. The “pseudodimer” model in which the i3 of PAR1 was aligned with the i3 loop pepducin is shown in Fig. 7B. These dimer models suggest that a symmetric PAR1 dimer wherein the i3 and H8 on different molecules interact is chemically plausible.
FIGURE 7.

PAR1 forms homodimers/oligomers. A, a dimer model of PAR1 was constructed by first calculating the electrostatic potential of the receptor using Advanced Poisson-Boltzmann Solver assuming 150 mm salt (62). The most intense blue and red coloring represents a potential in excess of +1 kT/e and −1 kT/e, respectively. The dimer interface was selected by minimizing electrostatic repulsions and maximizing favorable interactions. B, bottom view of proposed dimer models of PAR1 depicting the interaction of the i3 loop region of one PAR1 monomer with the H8 helix region of an adjacent PAR1 monomer using the P1pal-19/H8 helix interaction as a pseudodimer template for monomer/monomer interactions. C, co-immunoprecipitations of T7-PAR1 and PAR1-Myc were conducted in protein lysates from transfected HEK293 cells. T7-Ab-agarose beads were used to immunoprecipitate (IP) T7-PAR1 from cell lysates, and bound PAR1-Myc was detected by anti-Myc immunoblot (top). Immunoblotting (IB) with the T7-Ab (bottom) confirmed the presence of T7-PAR1. Pretreatment of HEK cells with 20 nm thrombin for 10 min prior to the collection of cell lysates resulted in complete cleavage and loss of the N-terminal T7 epitope from T7-PAR1 (and binding to beads), confirming that PAR1-Myc did not nonspecifically bind to the T7-Ab-agarose beads. D, PAR1 forms dimers in live COS7 cells. FRET between PAR1-CFP (donor) and PAR1-YFP (acceptor) was quantified in COS7 cells. Fluorescence measurements used 0.5 × 106 cells/ml with excitation at 425 nm and 10-nm slit widths. Top, yellow dashes and blue dots, respectively, represent the signal for PAR1-YFP and PAR1-CFP expressed singly. The FRET signal (gray) was determined by subtracting the background PAR1-CFP (blue) and PAR1-YFP (yellow) signals from the net uncorrected signal from co-expressed receptors (green) as described previously for PAR1-PAR4 heterodimers (22). Bottom, FRET titration between PAR1-CFP and PAR1-YFP co-expressed at different ratios in COS7 cells. The plasmid concentration of donor was kept constant, whereas the acceptor plasmid was varied. Green squares indicate the increase of the FRET amplitude as a function of fluorescence intensity of the acceptor.
Using a mixture of T7- and Myc-tagged PAR1, we found that T7-PAR1 immunoprecipitated with Myc-PAR1, indicating that PAR1 did form a stable homodimer/oligomer (Fig. 7C). To confirm this result in live cells, PAR1 was tagged with CFP and YFP, and FRET experiments were conducted. As shown in Fig. 7D, a FRET signal was observed when PAR1-CFP and PAR1-YFP were co-expressed in COS7 cells. A titration of increasing PAR1-YFP acceptor expression with the same level of PAR1-CFP donor expression was performed. The FRET signal was saturable, indicating that the FRET occurring between PAR1-YFP and PAR-CFP was a consequence of specific binding of homodimers or oligomers rather than the result of random collisions. These data are highly consistent with previous BRET experiments that showed that PAR1 associates to form a stable homodimer (55) in HEK cells. Similarly, PAR1 has been documented to heterodimerize with the closely related PAR2 and PAR4 receptors on endothelial cells and/or platelets (22, 26).
Discussion
The data presented here provide a mechanistic basis to understand the allosteric activation mechanism by third intracellular loop-derived pepducins on the inside surface of a GPCR. The NMR structure of P1pal-19 revealed a highly structured two-helix i3 loop that closely resembled the analogous i3 loop structure of the intact receptor based on the on-state structure of rhodopsin and the β2-adrenergic receptor bound to G protein (35, 49). The i3 loop-derived pepducin requires the H8 helix from PAR1 and adjacent (D/N)PXXYYY motif on TM7 to stabilize the on-state and trigger receptor-G protein signaling from the inside.
This same TM7/H8 intracellular site has also been identified as a binding site for small molecule intracellular antagonists of PAR1 (56) and the CCR2 (57) receptors and could represent a hot spot for intracellular allosteric modulators. In the case of CCR2, the most important residues for binding its intracellular antagonist were found to be the highly conserved tyrosine, Tyr(7.53) (numbering in parentheses represents the Ballesteros residue number (50)), and phenylalanine, Phe(8.50), of the NPXXYX5,6F motif as well as Val(6.36) at the bottom of TM6 and Lys(8.49) in the H8 helix. The π-stacking interaction between Phe312(8.50) and Tyr305(7.53) directly links the TM7 and H8 helices and is involved in locking the receptor in an inactive state as observed in several x-ray structures, including that of CCR5 (57). Upon activation of CCR2, the aromatic stacking interaction is disrupted, allowing Tyr305(7.53) to rotate into the helical TM core to permit receptor signaling by stabilizing the outward displacement of TM6.
We found that mutation of the Gln379/Arg380 dyad in the H8 helix caused complete loss of PAR1 activation by its i3 loop pepducin. The hydroxamate moiety of Gln379 from the Gln379/Arg380 dyad forms an H-bond with the backbone carbonyl oxygen of Tyr372 immediately adjacent to the (D/N)PXXYY372 motif in TM7 (31). Elimination of this H-bond network and loss of the connection between the QR dyad and the Tyr372 peptide carbonyl group disrupts coordinated movements between TM7 and the H8 helix. This hydrophilic TM7-H8 interaction, although achieved differently, may perform a function in stabilizing the on-state similar to that found for the Tyr306–Phe313 connection in rhodopsin. Indeed, the QR dyad is a highly conserved motif on the H8 helix and is present in 85% of class A GPCRs (31). In the rhodopsin-1 crystal structures, the H8 helix Arg314 side chain hydrogen bonds to the carbonyl backbone oxygens of Ile307 and Met308, which are adjacent to the (D/N)PXXY306 motif on TM7. Previous studies showed that the H8 helix of rhodopsin is conformationally mobile (58, 59) and that its N-terminal end moves away from the C-terminal portion of the i1 loop/TM2 by 2–4 Å upon receptor activation (60, 61).
Key to our model of Fig. 5, the positively charged pharmacophores Arg305/Lys307/Arg310 of P1pal-19 interact with the H8 helix, inducing a conformational change in the attached TM7 helix that triggers rotation of the three tyrosine residues in a propeller motion to push out the TM6 into the open on-state of the receptor. The outward translation of TM6 then allows enhanced binding and coupling of PAR1 with the Gα subunit via its C-terminal helix. Consistent with this mechanism, corresponding point mutations in Tyr373, Tyr383, Gln379, and Arg380 of the H8 helix and TM7 in PAR1 receptor resulted in a 40–100% loss of activity for P1pal-19 activation of PAR1. Specific bimane labeling of Cys296 demonstrated that the C-terminal end of TM5 of PAR1 undergoes highly similar conformational movements during receptor activation by both the i3 loop pepducin and thrombin. Upon engagement and coupling with G protein, the hydrophobic side chains of flanking TM5 residues Ile293 and Leu297 are located in close proximity to the side chains of Leu344 and Leu349 from the C-terminal α-helix of the Gα subunit. The insertion of the C-terminal α-helix of Gα into the pepducin-activated PAR1 receptor may be responsible for the enhanced non-polar environment experienced by the Cys296 bimane probe.
A recent study of an i3 loop pepducin (ICL3-9) derived from the β2-adrenergic receptor also found that ICL3-9 promoted receptor interactions with Gs and increased Gs signaling (54) requiring the presence of its cognate receptor. ICL3-9 induced an increase in the BRET signal between β2-adrenergic receptor and Gs in contrast to the decreased BRET signal observed with the orthosteric agonist isoproterenol. Contrary to the similar conformational effects induced by thrombin and P1pal-19 on PAR1, they demonstrated that the β2-adrenergic receptor·Gs active conformation induced by their i3 loop pepducin is different from that stabilized by isoproterenol at the orthosteric site. The reduction in the BRET signal upon isoproterenol stimulation may reflect a conformational rearrangement of the precoupled β2-adrenergic receptor·Gs complex that resulted in an increase in the distance between the C terminus of the β2-adrenergic receptor and the N terminus of Gαs. The ICL3-9-stimulated binding of Gs promoted an increase in BRET and thus reflects a reduction in the distance between the N terminus of Gαs and the C terminus of the β2-adrenergic receptor. However, when an alternative construct was monitored using BRET between β2-adrenergic receptor-GFP10 and RLucII inserted in the linker 1 region of Gαs between the helical and GTPase domains, similar BRET changes in direction and efficiency between isoproterenol and ICL3-9 were observed (54). This suggests that this region of Gαs may be oriented similarly in both the i3 pepducin- and orthosteric agonist-stimulated states as we observed for PAR1. Although the exact mechanism of this β2-adrenergic receptor·Gs activator pepducin remains unclear, the N-terminal region of the i3 loop of the β2-adrenergic receptor is a critical interaction site with the α5-β4 loop on Gs and optimally positioned to catalyze G nucleotide exchange on the Gα subunit (35).
The co-immunoprecipitation and FRET data demonstrate that PAR1 self-associates to form a homodimer or homo-oligomer, consistent with previous BRET experiments in HEK cells (55). Although the exact structure and relative orientation of the homodimer are difficult to determine with certainty, the direct binding data presented here indicate that a stable homodimer may form with each i3 loop interacting with an H8 helix across the dimer interface. Several crystal structures indicate that the H8 helix plays a prominent role for homomeric interactions in lipid membrane environments, including β2-adrenergic receptor (63), (rhod)opsin in its G protein-interacting state (49), β1-adrenergic receptor (64), and human κ-opioid receptor (65).
In the case of the CXCR4 homodimer, binding of a CVX15 peptide antagonist induces a tilt in the extracellular part of TM5, bringing the intracellular parts of the CXCR4 homomer into close contact (66). This type of ligand-induced conformational change in TM5 could explain the cooperative binding observed with certain CXCR4 ligands. Specifically, binding of a ligand to one receptor could induce a structural change in TM5 of the second receptor that could modify the ligand binding affinity to the second receptor, resulting in either negative or positive allosterism as seen with some CXCR4 ligands, including i1 and i3 loop-based pepducins (11, 20, 25). These structural and biophysical data are consistent with our observations that a conformation change was induced by the i3 loop pepducin at the cytoplasmic end of TM5. As many GPCRs are known to form transient or obligate dimers in various orientations (1, 48), a characteristic that influences their function in receptor pharmacology, signaling, and disease regulation, the studies here provide a mechanistic blueprint to design allosteric modulators based on the intracellular loop structures that mimic receptor dimer interactions at the G protein interface.
This work was supported, in whole or in part, by National Institutes of Health Grant P50 HL110789 from the NHLBI (to A. K.).
- GPCR
- G protein-coupled receptor
- PAR
- protease-activated receptor
- i3
- third intracellular loop
- H8
- intracellular helix 8
- InsP
- inositol phosphate
- Fmoc
- N-(9-fluorenyl)methoxycarbonyl
- GTPγS
- guanosine 5′-3-O-(thio)triphosphate
- Ab
- antibody
- TM
- transmembrane domain
- PLC-β
- phospholipase C-β
- CFP
- cyan fluorescent protein
- BRET
- bioluminescence resonance energy transfer.
References
- 1. Audet M., Bouvier M. (2012) Restructuring G-protein-coupled receptor activation. Cell 151, 14–23 [DOI] [PubMed] [Google Scholar]
- 2. Kruse A. C., Ring A. M., Manglik A., Hu J., Hu K., Eitel K., Hübner H., Pardon E., Valant C., Sexton P. M., Christopoulos A., Felder C. C., Gmeiner P., Steyaert J., Weis W. I., Garcia K. C., Wess J., Kobilka B. K. (2013) Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature 504, 101–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Katritch V., Cherezov V., Stevens R. C. (2012) Diversity and modularity of G protein-coupled receptor structures. Trends Pharmacol. Sci. 33, 17–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. O'Callaghan K., Kuliopulos A., Covic L. (2012) Turning receptors on and off with intracellular pepducins: new insights into G-protein-coupled receptor drug development. J. Biol. Chem. 287, 12787–12796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Covic L., Gresser A. L., Talavera J., Swift S., Kuliopulos A. (2002) Activation and inhibition of G protein-coupled receptors by cell-penetrating membrane-tethered peptides. Proc. Natl. Acad. Sci. U.S.A. 99, 643–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Covic L., Misra M., Badar J., Singh C., Kuliopulos A. (2002) Pepducin-based intervention of thrombin-receptor signaling and systemic platelet activation. Nat. Med. 8, 1161–1165 [DOI] [PubMed] [Google Scholar]
- 7. Wielders S. J., Bennaghmouch A., Reutelingsperger C. P., Bevers E. M., Lindhout T. (2007) Anticoagulant and antithrombotic properties of intracellular protease-activated receptor antagonists. J. Thromb. Haemost. 5, 571–576 [DOI] [PubMed] [Google Scholar]
- 8. Fontanini K. B., Janz J., Looby R., Hamilton J. A. (2010) Rapid binding and transmembrane diffusion of pepducins in phospholipid bilayers. Biophys. J. 98, 278a [Google Scholar]
- 9. Janz J. M., Ren Y., Looby R., Kazmi M. A., Sachdev P., Grunbeck A., Haggis L., Chinnapen D., Lin A. Y., Seibert C., McMurry T., Carlson K. E., Muir T. W., Hunt S., 3rd, Sakmar T. P. (2011) Direct interaction between an allosteric agonist pepducin and the chemokine receptor CXCR4. J. Am. Chem. Soc. 133, 15878–15881 [DOI] [PubMed] [Google Scholar]
- 10. Tsuji M., Ueda S., Hirayama T., Okuda K., Sakaguchi Y., Isono A., Nagasawa H. (2013) FRET-based imaging of transbilayer movement of pepducin in living cells by novel intracellular bioreductively activatable fluorescent probes. Org. Biomol. Chem. 11, 3030–3037 [DOI] [PubMed] [Google Scholar]
- 11. Quoyer J., Janz J. M., Luo J., Ren Y., Armando S., Lukashova V., Benovic J. L., Carlson K. E., Hunt S. W., 3rd, Bouvier M. (2013) Pepducin targeting the C-X-C chemokine receptor type 4 acts as a biased agonist favoring activation of the inhibitory G protein. Proc. Natl. Acad. Sci. U.S.A. 110, E5088–E5097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wisler J. W., Xiao K., Thomsen A. R., Lefkowitz R. J. (2014) Recent developments in biased agonism. Curr. Opin. Cell Biol. 27, 18–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bock A., Merten N., Schrage R., Dallanoce C., Bätz J., Klöckner J., Schmitz J., Matera C., Simon K., Kebig A., Peters L., Müller A., Schrobang-Ley J., Tränkle C., Hoffmann C., De Amici M., Holzgrabe U., Kostenis E., Mohr K. (2012) The allosteric vestibule of a seven transmembrane helical receptor controls G-protein coupling. Nat. Commun. 3, 1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wootten D., Christopoulos A., Sexton P. M. (2013) Emerging paradigms in GPCR allostery: implications for drug discovery. Nat. Rev. Drug Discov. 12, 630–644 [DOI] [PubMed] [Google Scholar]
- 15. Edwards R. J., Moran N., Devocelle M., Kiernan A., Meade G., Signac W., Foy M., Park S. D., Dunne E., Kenny D., Shields D. C. (2007) Bioinformatic discovery of novel bioactive peptides. Nat. Chem. Biol. 3, 108–112 [DOI] [PubMed] [Google Scholar]
- 16. Tchernychev B., Ren Y., Sachdev P., Janz J. M., Haggis L., O'Shea A., McBride E., Looby R., Deng Q., McMurry T., Kazmi M. A., Sakmar T. P., Hunt S., 3rd, Carlson K. E. (2010) Discovery of a CXCR4 agonist pepducin that mobilizes bone marrow hematopoietic cells. Proc. Natl. Acad. Sci. U.S.A. 107, 22255–22259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Boire A., Covic L., Agarwal A., Jacques S., Sherifi S., Kuliopulos A. (2005) PAR1 is a matrix metalloprotease-1 receptor that promotes invasion and tumorigenesis of breast cancer cells. Cell 120, 303–313 [DOI] [PubMed] [Google Scholar]
- 18. Yang E., Boire A., Agarwal A., Nguyen N., O'Callaghan K., Tu P., Kuliopulos A., Covic L. (2009) Blockade of PAR1 signaling with cell-penetrating pepducins inhibits Akt survival pathways in breast cancer cells and suppresses tumor survival and metastasis. Cancer Res. 69, 6223–6231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cisowski J., O'Callaghan K., Kuliopulos A., Yang J., Nguyen N., Deng Q., Yang E., Fogel M., Tressel S., Foley C., Agarwal A., Hunt S. W., 3rd, McMurry T., Brinckerhoff L., Covic L. (2011) Targeting protease-activated receptor-1 with cell-penetrating pepducins in lung cancer. Am. J. Pathol. 179, 513–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. O'Callaghan K., Lee L., Nguyen N., Hsieh M. Y., Kaneider N. C., Klein A. K., Sprague K., Van Etten R. A., Kuliopulos A., Covic L. (2012) Targeting CXCR4 with cell-penetrating pepducins in lymphoma and lymphocytic leukemia. Blood 119, 1717–1725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kuliopulos A., Covic L. (2003) Blocking receptors on the inside: pepducin-based intervention of PAR signaling and thrombosis. Life Sci. 74, 255–262 [DOI] [PubMed] [Google Scholar]
- 22. Leger A. J., Jacques S. L., Badar J., Kaneider N. C., Derian C. K., Andrade-Gordon P., Covic L., Kuliopulos A. (2006) Blocking the protease-activated receptor 1–4 heterodimer in platelet-mediated thrombosis. Circulation 113, 1244–1254 [DOI] [PubMed] [Google Scholar]
- 23. Trivedi V., Boire A., Tchernychev B., Kaneider N. C., Leger A. J., O'Callaghan K., Covic L., Kuliopulos A. (2009) Platelet matrix metalloprotease-1 mediates thrombogenesis by activating PAR1 at a cryptic ligand site. Cell 137, 332–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tressel S. L., Koukos G., Tchernychev B., Jacques S. L., Covic L., Kuliopulos A. (2011) Pharmacology, biodistribution, and efficacy of GPCR-based pepducins in disease models. Methods Mol. Biol. 683, 259–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kaneider N. C., Agarwal A., Leger A. J., Kuliopulos A. (2005) Reversing systemic inflammatory response syndrome with chemokine receptor pepducins. Nat. Med. 11, 661–665 [DOI] [PubMed] [Google Scholar]
- 26. Kaneider N. C., Leger A. J., Agarwal A., Nguyen N., Perides G., Derian C., Covic L., Kuliopulos A. (2007) 'Role reversal' for the receptor PAR1 in sepsis-induced vascular damage. Nat. Immunol. 8, 1303–1312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sevigny L. M., Zhang P., Bohm A., Lazarides K., Perides G., Covic L., Kuliopulos A. (2011) Interdicting protease-activated receptor-2-driven inflammation with cell-penetrating pepducins. Proc. Natl. Acad. Sci. U.S.A. 108, 8491–8496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tressel S. L., Kaneider N. C., Kasuda S., Foley C., Koukos G., Austin K., Agarwal A., Covic L., Opal S. M., Kuliopulos A. (2011) A matrix metalloprotease-PAR1 system regulates vascular integrity, systemic inflammation and death in sepsis. EMBO Mol. Med. 3, 370–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang P., Gruber A., Kasuda S., Kimmelstiel C., O'Callaghan K., Cox D. H., Bohm A., Baleja J. D., Covic L., Kuliopulos A. (2012) Suppression of arterial thrombosis without affecting hemostatic parameters with a cell-penetrating PAR1 pepducin. Circulation 126, 83–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kuliopulos A., Covic L., Seeley S. K., Sheridan P. J., Helin J., Costello C. E. (1999) Plasmin desensitization of the PAR1 thrombin receptor: kinetics, sites of truncation, and implications for thrombolytic therapy. Biochemistry 38, 4572–4585 [DOI] [PubMed] [Google Scholar]
- 31. Swift S., Leger A. J., Talavera J., Zhang L., Bohm A., Kuliopulos A. (2006) Role of the PAR1 receptor 8th helix in signaling: the 7-8-1 receptor activation mechanism. J. Biol. Chem. 281, 4109–4116 [DOI] [PubMed] [Google Scholar]
- 32. Liu Y., Liu Z., Androphy E., Chen J., Baleja J. D. (2004) Design and characterization of helical peptides that inhibit the E6 protein of papillomavirus. Biochemistry 43, 7421–7431 [DOI] [PubMed] [Google Scholar]
- 33. Zhang C., Srinivasan Y., Arlow D. H., Fung J. J., Palmer D., Zheng Y., Green H. F., Pandey A., Dror R. O., Shaw D. E., Weis W. I., Coughlin S. R., Kobilka B. K. (2012) High-resolution crystal structure of human protease-activated receptor 1. Nature 492, 387–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Park J. H., Scheerer P., Hofmann K. P., Choe H. W., Ernst O. P. (2008) Crystal structure of the ligand-free G-protein-coupled receptor opsin. Nature 454, 183–187 [DOI] [PubMed] [Google Scholar]
- 35. Rasmussen S. G., DeVree B. T., Zou Y., Kruse A. C., Chung K. Y., Kobilka T. S., Thian F. S., Chae P. S., Pardon E., Calinski D., Mathiesen J. M., Shah S. T., Lyons J. A., Caffrey M., Gellman S. H., Steyaert J., Skiniotis G., Weis W. I., Sunahara R. K., Kobilka B. K. (2011) Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature 477, 549–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Choe H. W., Kim Y. J., Park J. H., Morizumi T., Pai E. F., Krauss N., Hofmann K. P., Scheerer P., Ernst O. P. (2011) Crystal structure of metarhodopsin II. Nature 471, 651–655 [DOI] [PubMed] [Google Scholar]
- 37. Lambright D. G., Sondek J., Bohm A., Skiba N. P., Hamm H. E., Sigler P. B. (1996) The 2.0 Å crystal structure of a heterotrimeric G protein. Nature 379, 311–319 [DOI] [PubMed] [Google Scholar]
- 38. Covic L., Gresser A. L., Kuliopulos A. (2000) Biphasic kinetics of activation and signaling for PAR1 and PAR4 thrombin receptors in platelets. Biochemistry 39, 5458–5467 [DOI] [PubMed] [Google Scholar]
- 39. Kuliopulos A., Mohanlal R., Covic L. (2004) Effect of selective inhibition of the p38 MAP kinase pathway on platelet aggregation. Thromb. Haemost 92, 1387–1393 [DOI] [PubMed] [Google Scholar]
- 40. Kimmelstiel C., Zhang P., Kapur N. K., Weintraub A., Krishnamurthy B., Castaneda V., Covic L., Kuliopulos A. (2011) Bivalirudin is a dual inhibitor of thrombin and collagen-dependent platelet activation in patients undergoing percutaneous coronary intervention. Circ. Cardiovasc. Interv. 4, 171–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hu J., Wang Y., Zhang X., Lloyd J. R., Li J. H., Karpiak J., Costanzi S., Wess J. (2010) Structural basis of G protein-coupled receptor-G protein interactions. Nat. Chem. Biol. 6, 541–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Seeley S., Covic L., Jacques S. L., Sudmeier J., Baleja J. D., Kuliopulos A. (2003) Structural basis for thrombin activation of a protease-activated receptor: inhibition of intramolecular liganding. Chem. Biol. 10, 1033–1041 [DOI] [PubMed] [Google Scholar]
- 43. Swift S., Sheridan P. J., Covic L., Kuliopulos A. (2000) PAR1 thrombin receptor-G protein interactions. Separation of binding and coupling determinants in the gα subunit. J. Biol. Chem. 275, 2627–2635 [DOI] [PubMed] [Google Scholar]
- 44. Canto I., Trejo J. (2013) Palmitoylation of protease-activated receptor-1 regulates adaptor protein complex-2 and -3 interaction with tyrosine-based motifs and endocytic sorting. J. Biol. Chem. 288, 15900–15912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Palczewski K., Kumasaka T., Hori T., Behnke C. A., Motoshima H., Fox B. A., Le Trong I., Teller D. C., Okada T., Stenkamp R. E., Yamamoto M., Miyano M. (2000) Crystal structure of rhodopsin: a G protein-coupled receptor. Science 289, 739–745 [DOI] [PubMed] [Google Scholar]
- 46. Ayoub M. A., Maurel D., Binet V., Fink M., Prézeau L., Ansanay H., Pin J. P. (2007) Real-time analysis of agonist-induced activation of protease-activated receptor 1/Gαi1 protein complex measured by bioluminescence resonance energy transfer in living cells. Mol. Pharmacol. 71, 1329–1340 [DOI] [PubMed] [Google Scholar]
- 47. Liu J. J., Horst R., Katritch V., Stevens R. C., Wüthrich K. (2012) Biased signaling pathways in β2-adrenergic receptor characterized by 19F-NMR. Science 335, 1106–1110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wu H., Wang C., Gregory K. J., Han G. W., Cho H. P., Xia Y., Niswender C. M., Katritch V., Meiler J., Cherezov V., Conn P. J., Stevens R. C. (2014) Structure of a class C GPCR metabotropic glutamate receptor 1 bound to an allosteric modulator. Science 344, 58–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Scheerer P., Park J. H., Hildebrand P. W., Kim Y. J., Krauss N., Choe H. W., Hofmann K. P., Ernst O. P. (2008) Crystal structure of opsin in its G-protein-interacting conformation. Nature 455, 497–502 [DOI] [PubMed] [Google Scholar]
- 50. Ballesteros J. A., Shi L., Javitch J. A. (2001) Structural mimicry in G protein-coupled receptors: implications of the high-resolution structure of rhodopsin for structure-function analysis of rhodopsin-like receptors. Mol. Pharmacol. 60, 1–19 [PubMed] [Google Scholar]
- 51. Yao X., Parnot C., Deupi X., Ratnala V. R., Swaminath G., Farrens D., Kobilka B. (2006) Coupling ligand structure to specific conformational switches in the β2-adrenoceptor. Nat. Chem. Biol. 2, 417–422 [DOI] [PubMed] [Google Scholar]
- 52. Yao X. J., Vélez Ruiz G., Whorton M. R., Rasmussen S. G., DeVree B. T., Deupi X., Sunahara R. K., Kobilka B. (2009) The effect of ligand efficacy on the formation and stability of a GPCR-G protein complex. Proc. Natl. Acad. Sci. U.S.A. 106, 9501–9506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Rasmussen S. G., Choi H. J., Fung J. J., Pardon E., Casarosa P., Chae P. S., Devree B. T., Rosenbaum D. M., Thian F. S., Kobilka T. S., Schnapp A., Konetzki I., Sunahara R. K., Gellman S. H., Pautsch A., Steyaert J., Weis W. I., Kobilka B. K. (2011) Structure of a nanobody-stabilized active state of the β2 adrenoceptor. Nature 469, 175–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Carr R., 3rd, Du Y., Quoyer J., Panettieri R. A., Jr., Janz J. M., Bouvier M., Kobilka B. K., Benovic J. L. (2014) Development and characterization of pepducins as Gs-biased allosteric agonists. J. Biol. Chem. 289, 35668–35684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. McLaughlin J. N., Patterson M. M., Malik A. B. (2007) Protease-activated receptor-3 (PAR3) regulates PAR1 signaling by receptor dimerization. Proc. Natl. Acad. Sci. U.S.A. 104, 5662–5667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Dowal L., Sim D. S., Dilks J. R., Blair P., Beaudry S., Denker B. M., Koukos G., Kuliopulos A., Flaumenhaft R. (2011) Identification of an antithrombotic allosteric modulator that acts through helix 8 of PAR1. Proc. Natl. Acad. Sci. U.S.A. 108, 2951–2956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zweemer A. J., Bunnik J., Veenhuizen M., Miraglia F., Lenselink E. B., Vilums M., de Vries H., Gibert A., Thiele S., Rosenkilde M. M., IJzerman A. P., Heitman L. H. (2014) Discovery and mapping of an intracellular antagonist binding site at the chemokine receptor CCR2. Mol. Pharmacol. 86, 358–368 [DOI] [PubMed] [Google Scholar]
- 58. Imamoto Y., Kataoka M., Tokunaga F., Palczewski K. (2000) Light-induced conformational changes of rhodopsin probed by fluorescent Alexa594 immobilized on the cytoplasmic surface. Biochemistry 39, 15225–15233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Krishna A. G., Menon S. T., Terry T. J., Sakmar T. P. (2002) Evidence that helix 8 of rhodopsin acts as a membrane-dependent conformational switch. Biochemistry 41, 8298–8309 [DOI] [PubMed] [Google Scholar]
- 60. Altenbach C., Cai K., Klein-Seetharaman J., Khorana H. G., Hubbell W. L. (2001) Structure and function in rhodopsin: mapping light-dependent changes in distance between residue 65 in helix TM1 and residues in the sequence 306–319 at the cytoplasmic end of helix TM7 and in helix H8. Biochemistry 40, 15483–15492 [DOI] [PubMed] [Google Scholar]
- 61. Altenbach C., Klein-Seetharaman J., Cai K., Khorana H. G., Hubbell W. L. (2001) Structure and function in rhodopsin: mapping light-dependent changes in distance between residue 316 in helix 8 and residues in the sequence 60–75, covering the cytoplasmic end of helices TM1 and TM2 and their connection loop CL1. Biochemistry 40, 15493–15500 [DOI] [PubMed] [Google Scholar]
- 62. Baker N. A., Sept D., Joseph S., Holst M. J., McCammon J. A. (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Cherezov V., Rosenbaum D. M., Hanson M. A., Rasmussen S. G., Thian F. S., Kobilka T. S., Choi H. J., Kuhn P., Weis W. I., Kobilka B. K., Stevens R. C. (2007) High-resolution crystal structure of an engineered human β2-adrenergic G protein-coupled receptor. Science 318, 1258–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Huang J., Chen S., Zhang J. J., Huang X. Y. (2013) Crystal structure of oligomeric β1-adrenergic G protein-coupled receptors in ligand-free basal state. Nat. Struct. Mol. Biol. 20, 419–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wu H., Wacker D., Mileni M., Katritch V., Han G. W., Vardy E., Liu W., Thompson A. A., Huang X. P., Carroll F. I., Mascarella S. W., Westkaemper R. B., Mosier P. D., Roth B. L., Cherezov V., Stevens R. C. (2012) Structure of the human κ-opioid receptor in complex with JDTic. Nature 485, 327–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wu B., Chien E. Y., Mol C. D., Fenalti G., Liu W., Katritch V., Abagyan R., Brooun A., Wells P., Bi F. C., Hamel D. J., Kuhn P., Handel T. M., Cherezov V., Stevens R. C. (2010) Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science 330, 1066–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]