

Background: NAMPT inhibition leads to attenuation of glycolysis in cancer cells.
Results: NAMPT inhibition also perturbs other carbohydrate metabolism, resulting in elevated fructose 1-phosphate, sedoheptulose 1-phosphate, glyceraldehyde, and erythrose levels.
Conclusion: Condensation of dihydroxyacetone phosphate/glyceraldehyde and dihydroxyacetone phosphate/erythrose by aldolase leads to increased fructose 1-phosphate and sedoheptulose 1-phosphate, respectively.
Significance: NAMPT plays a key role in regulating glycolysis-related carbohydrate metabolism in cancer cells.
Keywords: carbohydrate metabolism, glucose metabolism, glycolysis, nicotinamide adenine dinucleotide (NAD), pharmacology
Abstract
Nicotinamide phosphoribosyltransferase (NAMPT) has been extensively studied due to its essential role in NAD+ biosynthesis in cancer cells and the prospect of developing novel therapeutics. To understand how NAMPT regulates cellular metabolism, we have shown that the treatment with FK866, a specific NAMPT inhibitor, leads to attenuation of glycolysis by blocking the glyceraldehyde 3-phosphate dehydrogenase step (Tan, B., Young, D. A., Lu, Z. H., Wang, T., Meier, T. I., Shepard, R. L., Roth, K., Zhai, Y., Huss, K., Kuo, M. S., Gillig, J., Parthasarathy, S., Burkholder, T. P., Smith, M. C., Geeganage, S., and Zhao, G. (2013) Pharmacological inhibition of nicotinamide phosphoribosyltransferase (NAMPT), an enzyme essential for NAD+ biosynthesis, in human cancer cells: metabolic basis and potential clinical implications. J. Biol. Chem. 288, 3500–3511). Due to technical limitations, we failed to separate isotopomers of phosphorylated sugars. In this study, we developed an enabling LC-MS methodology. Using this, we confirmed the previous findings and also showed that NAMPT inhibition led to accumulation of fructose 1-phosphate and sedoheptulose 1-phosphate but not glucose 6-phosphate, fructose 6-phosphate, and sedoheptulose 7-phosphate as previously thought. To investigate the metabolic basis of the metabolite formation, we carried out biochemical and cellular studies and established the following. First, glucose-labeling studies indicated that fructose 1-phosphate was derived from dihydroxyacetone phosphate and glyceraldehyde, and sedoheptulose 1-phosphate was derived from dihydroxyacetone phosphate and erythrose via an aldolase reaction. Second, biochemical studies showed that aldolase indeed catalyzed these reactions. Third, glyceraldehyde- and erythrose-labeling studies showed increased incorporation of corresponding labels into fructose 1-phosphate and sedoheptulose 1-phosphate in FK866-treated cells. Fourth, NAMPT inhibition led to increased glyceraldehyde and erythrose levels in the cell. Finally, glucose-labeling studies showed accumulated fructose 1,6-bisphosphate in FK866-treated cells mainly derived from dihydroxyacetone phosphate and glyceraldehyde 3-phosphate. Taken together, this study shows that NAMPT inhibition leads to attenuation of glycolysis, resulting in further perturbation of carbohydrate metabolism in cancer cells. The potential clinical implications of these findings are also discussed.
Introduction
The NAD+ cofactor is essential for a number of enzymes and regulatory proteins involved in a variety of cellular processes. In mammals, NAD+ can be synthesized from nicotinamide, nicotinic acid, or tryptophan (2–5). However, the nicotinamide salvage pathway represents the major route to NAD+ biosynthesis in mammals (6–8). NAMPT,3 a rate-limiting enzyme in the conversion of nicotinamide to NAD+ (9–10) in cancer cells, is crucial to several physiological processes, including metabolism, energy generation, survival, apoptosis, DNA repair, and inflammation (2, 11–13).
NAMPT is overexpressed in several types of tumors (14–17), and its expression is associated with tumor progression (18). The down-regulation of NAMPT suppresses tumor cell growth in vitro and in vivo and sensitizes cells to oxidative stress and DNA-damaging agents (8, 14, 17, 19–21). The inhibition of NAMPT also leads to the attenuation of tumor growth and induction of apoptosis due to NAD+ depletion (8, 20–23). Taken together, NAMPT represents a promising therapeutic target for the development of potential novel cancer drugs (24–26).
NAD+ is a substrate for dehydrogenases, poly(ADP-ribose) polymerases, sirtuins (SIRT), mono ADP-ribosyltransferases, and ADP-ribosyl cyclases (2, 4, 11). In most cancer cells, poly(ADP-ribose) polymerase is activated due to DNA damage and genome instability (2, 26–28). The activation of poly(ADP-ribose) polymerases leads to NAD+ depletion in cancer cells (2, 8, 26–28). Similarly, aberrant expression of SIRT, mono(ADP-ribosyl) transferases, and ADP-ribosyl cyclases can lead to consumption of NAD+ in cancer cells (2, 4, 11).
To better understand the metabolic basis of NAMPT inhibition, we have recently shown that inhibition of NAMPT leads to attenuation of glycolysis at the glyceraldehyde-3-phosphate dehydrogenase step (1). The attenuation of glycolysis results in an accumulation of glycolytic intermediates before and at the glyceraldehyde 3-phosphate dehydrogenase step (1). The attenuation of glycolysis also leads to a decrease of glycolytic intermediates after the glyceraldehyde-3-phosphate dehydrogenase step (1). However, due to throughput considerations and technical difficulties, we were unable to separate isomers of glycolytic intermediates, such as glucose 6-phosphate and fructose 6-phosphate, and glyceraldehyde 3-phosphate and dihydroxyacetone phosphate, or their geometrical isomers, such as glucose 1-phosphate and fructose 1-phosphate. To further investigate the effects of NAMPT inhibition on cancer cell metabolism, we developed an LC-MS method enabling us to separate each isomer. This new method also resolves sedoheptulose 7-phosphate from its 1-isomer and glyceraldehyde 3-phosphate from dihydroxyacetone phosphate, which allows us to identify the source of triose. We now report that NAMPT inhibition leads to an accumulation of fructose 1-phosphate and sedoheptulose 1-phosphate, but not fructose 6-phosphate and sedoheptulose 7-phosphate, in different cancer cells and tumors. Glucose-labeling studies indicated that increased levels of fructose 1-phosphate and sedoheptulose 1-phosphate are derived from dihydroxyacetone phosphate and glyceraldehyde and from erythrose, respectively, via an aldolase condensation reaction, a hypothesis later confirmed by biochemical and direct labeling studies. The addition of uniformly labeled glyceraldehyde and erythrose led to the formation of 3-carbon-labeled fructose 1-phosphate and four-carbon-labeled sedoheptulose 1-phosphate, respectively, in the cell. Consistent with this, NAMPT inhibition also led to an accumulation of glyceraldehyde and erythrose in cancer cells and tumors. Taken together, these studies suggest that NAMPT inhibition leads to attenuation of glycolysis, resulting in further alteration of carbohydrate metabolism in the cell. The findings from this study also have potential clinical implications because increased formation of fructose 1-phosphate and sedoheptulose 1-phosphate can be used as PD markers for evaluating NAMPT inhibitors in the clinic.
Experimental Procedures
Materials
The following materials were purchased from Sigma-Aldrich: formic acid (MS/HPLC grade), 1-ethyl-3-(3-imethylaminopropyl)carbodiimide, glucose, O-benzylhydroxylamine, sedoheptulose 7-phosphate lithium salt, glyceraldehyde 3-phosphate solution, fructose 1,6-bisphosphate trisodium salt, glucose 6-phosphate sodium salt, fructose 6-phosphate disodium salt, glucose 1-phosphate, fructose 1-phosphate, hydrochloric acid (12.1 m HCl), d-erythrose, and dihydroxyacetone phosphate (hemimagnesium salt). d-Glyceraldehyde, d-[1,2,3-13C3]glyceraldehyde, d-[U-13C4]erythrose, and d-[U-13C6]glucose were purchased from Omicron Biochemicals (South Bend, IN). Ethyl acetate and methanol (HPLC grade) were purchased from VWR International (Plainview, NY). Water (OptimaTM LC/MS grade) was purchased from Thermo Fisher Scientific. Tissue Lyser (II) and stainless steel beads (5 mm) were purchased from Qiagen (Hilden, Germany). RPMI 1640, Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum (FBS), and dialyzed FBS were purchased from Life Technologies, Inc. The cancer cell line KM-12 was purchased from NCI, and other cell lines were purchased from ATCC. HCT-116 was cultured in McCoy's 5A Medium modified (Hyclone) containing 10% FBS, NCI-H1155, and DMEM (25 mm glucose) in the presence of 10% FBS and 5% CO2 at 37 °C. Other cell lines were cultured in RPMI containing 10% FBS. Athymic nude mice (female) were purchased from Harlan Laboratories (Indianapolis, IN). FK866 was obtained and formulated as described (1).
Preparation of Cellular Extracts
Cancer cells (50,000/well in 100 μl) were seeded in 96-well plates and cultured overnight as described above. Cells were rinsed with DMEM or McCoy's 5A containing 10% dialyzed FBS (Gibco) three times as described above and treated with FK866 (0–100 nm) in the same medium (150 μl/well) for 24 h. For metabolite analysis, cultural medium was discarded, and metabolites were extracted with 80% methanol as described below. For 13C-labeling studies, the culture medium was replaced with a freshly prepared medium containing the following: 0.0–100 nm of FK866, 12.5 mm glucose, and 10% dialyzed FBS as well as 1.0 mm d-[1,2,3-13C3]glyceraldehyde, 1.0 mm d-[U-13C4]erythrose, or 12.5 mm d-[U-13C6]glucose, as indicated. Cells were incubated for 0.25–6 h. At each time point, 80% methanol (100 μl each) was added to each well to extract the metabolites after the removal of the medium. After incubation at room temperature for 15 min, the methanol extracts were transferred to a 96-deep well plate and rinsed with 100% methanol (100 μl/well). Samples were dried under a stream of nitrogen at 40 °C using a homemade 96-well plate dryer and reconstituted in 100 μl of water. The extracts were ready for further analysis.
Preparation of Tumor Samples
NCI-H1155 cells (ATCC) were grown as described above. The cells (106 cells/animal) were mixed with Matrigel (1:1) and implanted subcutaneously into the rear flank of the mice (female CB17 SCID, Charles River). The implanted tumor cells grew as solid tumors. The animals (6 animals/group) were dosed orally with 5 and 10 mg/kg of FK866 in 20% Captisol and 25 mm phosphate buffer, pH 2, twice a day for 6 days after tumors reached 500 mm3. The tumor volume and body weight were measured twice a week (1). The tumor samples (∼50 mg) were homogenized in 1 ml of ice-cold 80% methanol for 30 s with a TissueLyser II and 5-mm stainless steel beads at 4 °C. The homogenates were centrifuged at 4,000 × g for 10 min at 5 °C (5417C Centrifuge, Eppendorf, Westbury, NY). The pellets were discarded, and the tumor extracts were collected into a 96-deep well plate (2 ml). The tumor extracts (100 μl) were dried under nitrogen at 40 °C as described above and reconstituted in 100 μl of water. The extracts were ready for further analysis.
Determination of Cellular Protein Concentrations
For determination of protein concentrations, an extra set of plates was used. Cells were grown and treated as described before and only used for protein determination. After treatment, growth medium was removed, and cells were washed three times with PBS buffer (200 μl each). Then 75 μl of a lysis buffer (1% Triton X-100, 25 mm Tris, pH 7.5, 150 mm NaCl, 1 mm EDTA, and 1 mm EGTA) containing 1× protease inhibitor mixture (Complete, Mini, EDTA-free, Roche Applied Science) was added to each well. The plate was kept on ice for 10 min, followed by shaking (750 rpm) for 10 s. Protein concentrations were determined using a BCA protein assay kit (Pierce) according to the manufacturer's instructions.
Detection and Quantitation of Erythrose and Glyceraldehyde
Pyridine buffer (pH 5.0) was prepared by combining 12.1 m HCl (5.4 ml), pyridine (8.6 ml), and water (86 ml). The reconstituted cell extracts and tumor extracts (100 μl) in water were mixed with 1 m O-benzylhydroxylamine (50 μl) and 1 m 1-ethyl-3-(3-imethylaminopropyl)carbodiimide (50 μl) in the pyridine buffer. After incubation at room temperature for 1 h, ethyl acetate (300 μl) was added, and the plates were shaken for 10 min (VWR® multitube vortexer). The organic layer was taken into a 96-deep well plate (2 ml). Then the aqueous layer was extracted again with ethyl acetate (300 μl each). The organic layers were combined into a 96-deep well plate (2 ml). The 96-deep well plate was dried as described above and reconstituted in 100 μl of 10% methanol/water. The derivatized samples (10 μl) were injected onto an LC-triple quadrupole mass spectrometer (Qtrap 5500, AB Sciex, Framingham, CA), coupled with a pair of Shimadzu 10ADVP pumps, and a Shimadzu SIL-20A autosampler (Shimadzu, Columbia, MD). Samples were separated by HPLC using a Waters Xbridge 2.1 × 50-mm C18 column (Waters, Milford, MA) with a two-solvent system (A, water with 0.1% formic acid; B, methanol) at a flow rate of 0.6 ml min−1. The gradient was programmed as follows: 0–1.0 min, 0–30% B; 1.0–5.5 min, 30–85% B; 5.5–5.6 min, 85–98% B; 5.6–6.5 min, 98% B; and 6.5–8.0 min, 0% B. The MS acquisition methods used positive electrospray ionization and analysis in a multiple-reaction monitoring (MRM) mode. MRM transitions for derivatized glyceraldehyde and erythrose are 196/91 and 226/91. MRM transitions for isotopomers (M1–M3) of derivatized glyceraldehyde are 197/91, 198/91, and 199/91. MRM transitions for isotopomers (M1–M4) of derivatized erythrose are 227/91, 228/91, 229/91, and 230/91. The collision energy parameters are 15. Ion spray voltage was 4,500 V. Nebulizer gas (GS1), auxiliary gas (GS2), curtain gas (CUR), and collision gas (CAD) were 70, 70, 30, and 5 (arbitrary units), respectively. The ion source temperature was maintained at 600 °C. The curtain and collision gas was nitrogen. Declustering potential, entrance potential, and cell exit potential were 60, 10, and 15, respectively.
Analysis of Sugar Phosphates and NAD(H)
The reconstituted cell and tumor extracts (100 μl) in water were injected into an LC-triple quadrupole mass spectrometer (API 4000, AB Sciex, Framingham, CA), coupled with a pair of Shimadzu 10ADVP pumps, and a Shimadzu SIL-20A autosampler (Shimadzu, Columbia, MD). Samples were separated by HPLC using a Waters T3 2.1 × 50-mm column with a two-solvent system (A, 10 mm tributyl amine and 15 mm acetic acid in 3% methanol/water; B, methanol) at a flow rate of 0.6 ml min−1. The gradient was programmed as follows: 0–0.5 min, 0% B; 0.5–1.8 min, 0–70% B; 1.8–3.0 min, 90% B; and 3.0–6.0 min, 0% B. The injection volume was 10 μl, and the method used negative electrospray ionization mode. The MRM transitions for glucose 6-phosphate/fructose 6-phosphate, fructose 1-phosphate/glucose 1-phosphate, fructose 1,6-bisphosphate, dihydroxyacetone phosphate/glyceraldehyde 3-phosphate, sedoheptulose 1-phosphate/sedoheptulose 7-phosphate, phosphoglycerate, and phosphoenolpyruvate are 259/97, 259/241, 339/79, 169/79, 289/97, 185/97, and 165/97, respectively. Collision energies are −20, −16, −86, −46, −26, −49, and −25, respectively. Structural isomers were separated based on retention time. MRM transitions for isotopomers generated from [13C]glucose labeling experiments were adjusted based on the numbers of 13C- labeled carbons. Ion spray voltage was −4,500 V. Nebulizer gas (GS1), auxiliary gas (GS2), curtain gas (CUR), and collision gas (CAD) were 60, 60, 30, and 6 (arbitrary units), respectively. The ion source temperature was maintained at 600 °C. The curtain and collision gas was nitrogen. Declustering potential, entrance potential, and cell exit potential were −40, −10, and −10, respectively. The analysis of NAD(H) was described previously (1). Commercial standards were used for quantification of endogenous metabolites.
Enzymatic Synthesis of Sedoheptulose 1-Phosphate and Fructose 1-Phosphate as Standards
Synthesis of sedoheptulose 1-phosphate was carried out as described (30) by coupling aldolase, transketolase, and triose-phosphate isomerase except that rabbit muscle aldolase (Sigma) was used. Sedoheptulose 1-phosphate and fructose 1-phosphate were also synthesized using an aldolase under the following conditions. For synthesis of sedoheptulose 1-phosphate, reaction mixtures (100 μl each) contained aldolase (∼0.01 mg or 1.25 units/ml), 50 mm Tris-HCl, pH 7.4, 250 mm erythrose, 0.0–40 mm dihydroxyacetone phosphate or 100 mm dihydroxyacetone phosphate, and 0.0–250 mm erythrose. The reaction mixtures were incubated at 37 °C for 60 min. For the synthesis of fructose 1-phosphate, reaction mixtures (100 μl each) contained 0.00082 mg or 0.1 units/ml aldolase, 50 mm Tris-HCl, pH 7.4, 50 mm glyceraldehyde, 0.0–40 or 50 mm dihydroxyacetone phosphate, and 0.0–50 mm glyceraldehyde. The reaction mixtures were incubated at 37 °C for 30 min. The products formed were confirmed by LC-MS/MS analysis. The structural identification of sedoheptulose 1-phosphate from the aldolase reactions has been reported previously (29). Fructose 1-phosphate formed from the enzymatic reaction exhibited the same retention time and MS/MS spectra as the commercial standard (fructose 1-phosphate). Sedoheptulose 1-phosphate synthesized using the two different methods also exhibited the same retention times and MS/MS spectra as reported previously (29). Fructose 1-phosphate formed from the enzymatic reaction exhibited the same retention time and MS/MS spectra as the commercial standard (fructose 1-phosphate). Sedoheptulose 1-phosphate synthesized using the two different methods also exhibited the same retention times and MS/MS spectra as reported previously (29). The commercial standard of fructose 1-phosphate was used for quantification of the endogenous metabolic intermediates. The concentrations of synthetic sedoheptulose 1-phosphate standard after enzymatic reaction were calculated using sedoheptulose 7-phosphate due to structural similarity. The synthetic sedoheptulose 1-phosphate standards made from 40 mm dihydroxyacetone phosphate (DHAP) and 250 mm erythrose were used to quantify the endogenous metabolite levels.
Statistical Analysis
Data analysis was performed using MultiQuant version 2.1 (AB Sciex). Calibration curves were calculated by least-squares linear regression with 1/x weighting. The metabolites were quantified using the analyte peak area from the standard curves. Comparisons between groups were made with one-way analysis of variance, followed by Dunnett's t test using JMP version 9.0 software. p < 0.05 was considered as the significant level of difference.
Results
NAMPT Inhibition Led to Accumulation of Fructose 1-Phosphate and Sedoheptulose 1-Phosphate as Well as Fructose 1,6-Bisphosphate and Dihydroxyacetone Phosphate
We have shown previously that NAMPT inhibition results in the blockade of glyceraldehyde 3-phosphate dehydrogenase, a key enzyme in glycolysis dependent on NAD+ for activity (1). As a result, this leads to an accumulation in glycolytic intermediates before and at the glyceraldehyde 3-phosphate dehydrogenase step and a decrease in intermediates after the step (1). Using the methodology developed at the time, we were unable to resolve isomers or their geometrical isomers, such as glucose 6-phosphate/fructose 6-phosphate, dihydroxyacetone phosphate/glyceraldehyde 3-phosphate, and sedoheptulose 7-phosphate/sedoheptulose 1-phosphate. To further confirm this hypothesis, we developed an LC-MS methodology (see “Experimental Procedures”) enabling us to separate and detect these isomers. Further, this new method also allowed us to separate the two geometrical isomers (1- versus -6) for each sugar phosphate. Using this methodology, we were able to detect and quantify fructose 6-phosphate, fructose 1-phosphate, glucose 6-phosphate, glucose 1-phosphate, dihydroxyacetone phosphate, glyceraldehyde 3-phosphate (data not shown), sedoheptulose 7-phosphate, and sedoheptulose 1-phosphate synthesized biochemically or produced in cells and tumor xenografts treated with FK866, a specific NAMPT inhibitor (data not shown). To further validate this methodology, we treated HCT-116 cells with FK866 as described (see “Experimental Procedures”) and analyzed the glycolytic and pentose phosphate pathway intermediates in the cell. We observed a significant increase in fructose 1,6-bisphosphate and dihydroxyacetone phosphate levels and a decrease in 1,3-bisphosphoglycerate, 2- and 3-phosphoglycerate, and phosphoenolpyruvate levels (data not shown) as reported before (1). To our surprise, we also observed a significant increase in fructose 1-phosphate and sedoheptulose 1-phosphate but not glyceraldehyde 3-phosphate, fructose 6-phosphate, glucose 6-phosphate, and sedoheptulose 7-phosphate (data not shown) as previously thought (1).
To confirm that these observed metabolic changes were indeed due to NAMPT inhibition, we treated NCI-H1155, a NAPRT-negative cell line, and HCT-116, a NAPRT-positive cell line (1), with different concentrations of FK866 for 24 h in the presence or absence of nicotinic acid and analyzed the metabolite levels as described above. We showed that FK866 alone caused a dose-dependent increase in fructose 1,6-bisphosphate, dihydroxyacetone phosphate, fructose 1-phosphate, and sedoheptulose 1-phosphate levels (Fig. 1, A and B), and a decrease in phosphoglycerate, phosphoenolpyruvate, NAD+, and NADH levels in both cell lines (Fig. 1, C and D). The addition of nicotinic acid completely abolished these effects observed in HCT-116 (Fig. 1, B and D) but not in NCI-H1155 (Fig. 1, A and C). This is expected because HCT-116, but not NCI-H1155, can use nicotinic acid to produce NAD+ (6, 7), thereby bypassing the NAMPT-mediated pathway. Together, these studies have confirmed the previous finding that NAMPT inhibition attenuates glycolysis at the glyceraldehyde 3-phosphate dehydrogenase step and have also shown that NAMPT inhibition leads to increased fructose 1-phosphate and sedoheptulose 1-phosphate levels in cancer cells.
FIGURE 1.

NAMPT inhibition leads to an increase in fructose 1,6-bisphosphate (FBP), DHAP, fructose 1-phosphate (F1P), and sedoheptulose 1-phosphate (S1P) and a decrease in phosphoglycerate (PG), phosphoenolpyruvate (PEP), NAD+, and NADH. NCI-H1155 (B and D) and HCT-116 cells (A and C) were grown and treated with FK866 (0–100 nm) and with or without nicotinic acid (NA) (10 μm) in duplicates as described (see “Experimental Procedures”). After the treatment, cells were processed for the analysis of metabolites by LC-MS as described (see “Experimental Procedures”). The metabolites measured were expressed as nmol/mg total cellular protein. Error bars, S.D.
Elevated Levels of Fructose 1-Phosphate and Sedoheptulose 1-Phosphate in Cancer Cells in Response to NAMPT Inhibition Are Derived from Dihydroxyacetone Phosphate and Glyceraldehyde and from Erythrose, Respectively, via an Aldolase Condensation Reaction; Evidence from the Glucose-labeling Study
To investigate how NAMPT inhibition leads to increased fructose 1-phosphate and sedoheptulose 1-phosphate levels in the cell, we carried out a glucose-labeling study. In this study, we treated HCT-116 cells with FK866 for 24 h and then incubated the cells in the presence of U-13C6-labeled and unlabeled glucose (1:1). By doing so, we were able to discern the metabolic fates of trioses produced from fructose 1,6-bisphosphate via aldolase by taking advantage of their facile interconversion through the analysis of metabolite isotope labeling patterns. As shown in Fig. 2A, isotopomers of fructose 1-phosphate were mainly present as a mixture of M0 (0 carbon labeled), M3 (3 carbons labeled), and M6 (6 carbons labeled) in the FK866-treated cells. Significantly, the three isotopomers were distributed roughly at a ratio of 1:2:1 (M0/M3/M6), indicating that fructose 1-phosphate was synthesized from two trioses (1:1 for M0 and M3), such as dihydroxyacetone phosphate and glyceraldehyde. The treatment with FK866 led to a dose-dependent increase in M0, M3, and M6 of fructose 1-phosphate (Fig. 2B). Similarly, sedoheptulose 1-phosphate was mainly present as M0, M3, M4, and M7 (Fig. 2A). Interestingly, and also more revealingly, the isotopomers are roughly distributed at a ratio of 2:1:2:2:1:2 for M0, M1, M3, M4, M6, and M7. No M2 or M5 was detected (data not shown). The M0, M3, M4, and M7 pattern is consistent with sedoheptulose 1-phosphate being synthesized from two precursors, one being a triose (1:1 for M0 and M3), such as dihydroxyacetone phosphate, and another being a tetraose (1:1 for M0 and M4), such as erythrose. Furthermore, the data also revealed that there was a minor pool of M1 and M6 isotopomers for sedoheptulose 1-phosphate. The ratio between the major (M0 and M7) and the minor forms (M1 and M6) of sedoheptulose 1-phosphate is ∼2:1. The interpretation of the isotopomer distribution of the sedoheptulose 1-phosphate will be addressed in detail under “Discussion.” The treatment with FK866 also led to a dose-dependent increase in M0, M3, M4, and M7 (Fig. 2B). Interestingly, fructose 1,6- bisphosphate was found to exhibit exactly the same pattern as fructose 1-phosphate (i.e. M0, M3, and M6 present at a ratio of ∼1:2:1) (Fig. 2A), again indicating that the majority of fructose 1,6-bisphosphate was originated from two trioses: dihydroxyacetone phosphate and glyceraldehyde 3-phosphate in the treated cells. The treatment with FK866 led to a dose-dependent increase in M0, M3, and M6 (Fig. 2B). As expected, dihydroxyacetone phosphate was solely present as M0 and M3 (1:1) (Fig. 2A), and the treatment with FK866 led to a dose-dependent increase in M0 and M3 (Fig. 2B).
FIGURE 2.

Glucose-labeling studies suggest that fructose 1-phosphate (F1P) and sedoheptulose 1-phosphate (S1P) are derived from DHAP and glyceraldehyde and from erythrose, respectively, via aldolase. A, HCT-116 cells were grown and treated with FK866 (100 nm) in triplicates for 24 h. After the treatment, the growth medium was removed and replaced with a freshly prepared medium containing FK866 (100 nm) and 13C-labeled glucose and unlabeled glucose (1:1) as described (see “Experimental Procedures”). The cells were incubated for 6 h. After the treatment, cells were collected and processed for the analysis of fructose 1,6-bisphosphate (FBP), DHAP, F1P, and S1P levels by LC-MS (see “Experimental Procedures”). B, HCT-116 cells were grown in a medium containing 13C-labeled glucose and unlabeled glucose (1:1), treated with FK866 (0.0–100 nm) in triplicates for 24 h and processed for the analysis of metabolite as described above. C, HCT-116 cells were grown and treated with FK866 (100 nm) in triplicates for 24 h. After the treatment, the growth medium was removed and replaced with a freshly prepared medium containing FK866 (100 nm) and 13C-labeled and unlabeled glucose (1:1) as described (see “Experimental Procedures”). The cells were incubated for a different period of time (0.25–6 h) and processed for the analysis of metabolites as described above. M0–M7, metabolites with 0–7 carbons labeled. The metabolites measured were expressed as nmol/mg total cellular protein. Error bars, S.D.
To further investigate the kinetics of the isotopomer formation, we carried out a time course study. In this study, we first treated HCT-116 cells with FK866 for 24 h and then U-13C6-labeled glucose and unlabeled glucose (1:1) for a different period of time. For fructose 1-phosphate, the addition of labeled glucose caused a rapid decrease in M0 (from ∼45 to ∼5 nmol/mg protein) in the first 2 h followed by a rebound (Fig. 2C). At 6 h, fructose 1-phosphate was mainly present as M0, M3, and M6 (∼1:2:1) (Fig. 2C). These results indicated that the formation of fructose 1-phosphate from two trioses was very rapid and reversible. After the first 2 h, the metabolite pools of M0 and M6 reached to the equilibrium (Fig. 2C). Similarly for sedoheptulose 1-phosphate, the addition of labeled glucose caused a time-dependent decrease in M0 (from ∼16 to ∼1 nmol/mg protein) but a slow increase in M3, M4, and M7. At 6 h, M0, M3, M4, and M7 were the most abundant isomers present at similar levels (Fig. 2C). These results indicated that the formation of sedoheptulose 1-phosphate was much slower than that of fructose 1-phosphate. Consistent with this, the metabolite pools of M0, M3, M4, and M7 reached equilibrium after 6 h (Fig. 2C). For fructose 1,6-bisphosphate, the addition of labeled glucose caused a very rapid decrease in M0 in the first 15 min (from ∼170 to ∼10 nmol/mg protein) followed with a slow rebound (Fig. 2C). At 6 h, M3 was about twice amount of M0 and M6 (Fig. 2C), indicating that the majority of fructose 1,6-bisphosphate was derived from glyceraldehyde 3-phosphate and dihydroxyacetone phosphate. Consistent with this, the rate of M3 increase was higher than those of M0 and M6 (Fig. 2C). Finally, for dihydroxyacetone phosphate, the addition of labeled glucose caused a very rapid decrease in M0 (from ∼110 to ∼40 nmol/mg protein) in the first 15 min accompanied with an increase in M3 (Fig. 2C). On the basis of these results and the fact that dihydroxyacetone phosphate is also accumulated, we hypothesized that fructose 1-phosphate is derived from the elevated levels of dihydroxyacetone phosphate and glyceraldehyde and that sedoheptulose 1-phosphate is derived from the elevated levels of dihydroxyacetone phosphate and erythrose via an aldolase condensation reaction.
Increased Levels of Fructose 1-Phosphate and Sedoheptulose 1-Phosphate in Cancer Cells in Response to NAMPT Inhibition Are Derived from Elevated Levels of Dihydroxyacetone Phosphate and Glyceraldehyde and of Erythrose, Respectively, via an Aldolase Condensation Reaction; Evidence from the Biochemical Studies
To test this hypothesis, we carried out biochemical studies using an aldolase (see “Experimental Procedures”). As shown in Fig. 3A, when the enzymatic reactions were carried out in the presence of glyceraldehyde and dihydroxyacetone phosphate, the formation of fructose 1-phosphate, but not sedoheptulose 1-phosphate, was observed and increased with dihydroxyacetone phosphate concentrations to about 9.0 mm and then plateaued. Similar results were obtained when reactions were carried out at a fixed concentration of dihydroxyacetone phosphate and various concentrations of glyceraldehyde (Fig. 3A). When the enzymatic reactions were carried out in the presence of erythrose and dihydroxyacetone phosphate, the formation of sedoheptulose 1-phosphate, but not fructose 1-phosphate, was observed and increased with dihydroxyacetone phosphate concentrations to about 10.0 mm and then plateaued (Fig. 3B). Similar results were obtained when reactions were carried out at a fixed concentration of dihydroxyacetone phosphate and various concentrations of erythrose (Fig. 3B). On the basis of these kinetic results, the rate of formation for fructose 1-phosphate is much higher (7–20-fold) than that for sedoheptulose 1-phosphate. To confirm the identity of these enzymatic products, we showed that the fructose 1-phosphate formed exhibited a retention time and MS/MS spectra identical to those of the standard of fructose 1-phosphate. Similarly, the sedoheptulose 1-phosphate synthesized was also shown to exhibit a retention time and MS/MS spectra identical to those previously reported (29). In addition, these products exhibited retention times and MS/MS spectra identical to those of fructose 1-phosphate and sedoheptulose 1-phosphate produced in the cancer cells and tumor xenografts treated with FK866 (data not shown). Taken together, these biochemical studies confirmed that mammalian aldolase can indeed catalyze the formation of fructose 1-phosphate and sedoheptulose 1-phosphate from dihydroxyacetone phosphate and glyceraldehyde and from dihydroxyacetone phosphate and erythrose, respectively.
FIGURE 3.

Fructose 1-phosphate (F1P) and sedoheptulose 1-phosphate (S1P) are synthesized from DHAP and glyceraldehyde and from erythrose, respectively, via aldolase. The synthesis of F1P from DHAP and glyceraldehyde (A) was carried out in triplicates as described (see “Experimental Procedures”). Reaction mixtures containing aldolase (0.00082 mg), DHAP, and glyceraldehyde were incubated at room temperature for 30 min, and the product formed was analyzed by LC-MS as described (see “Experimental Procedures”). The synthesis of S1P from DHAP and erythrose (B) was carried out in triplicates as described (see “Experimental Procedures”). Reaction mixtures containing aldolase (0.01 mg), DHAP, and erythrose were incubated at room temperature for 60 min, and the product formed was analyzed as described above. The products formed were expressed as nmol/min/mg aldolase. Error bars, S.D.
Increased Levels of Fructose 1-Phosphate and Sedoheptulose 1-Phosphate in Cancer Cells in Response to NAMPT Inhibition Are Derived from Elevated Levels of Dihydroxyacetone Phosphate and Glyceraldehyde and of Erythrose, Respectively, via an Aldolase Condensation Reaction; Evidence from Glyceraldehyde- and Erythrose-labeling Studies
To further test this hypothesis, we treated HCT-116 cells with FK866 in the presence of [U-13C]glyceraldehyde or [U-13C]erythrose for a different period of time and analyzed 13C-labeled and unlabeled metabolites. The addition of labeled glyceraldehyde to growth media led to a significant increase in M3 of fructose 1-phosphate (Fig. 4A) but not M3 of sedoheptulose 1-phosphate and fructose 1,6-bisphosphate in the treated cells. Similarly, the addition of [U-13C]erythrose led to a significant increase in M4 of sedoheptulose 1-phosphate (Fig. 4A) but not M4 of fructose 1-phosphate and fructose 1,6-bisphosphate in the FK866-treated cells. These results indicated that fructose 1-phosphate was derived from dihydroxyacetone phosphate and glyceraldehyde and sedoheptulose 1-phosphate from dihydroxyacetone phosphate and erythrose. The rate of sedoheptulose 1-phosphate (M4) formation appears to be much slower than that of fructose 1-phosphate (Fig. 4A). This finding is consistent with that of the biochemical study showing that the rate of sedoheptulose 1-phosphate formation catalyzed by the aldolase is 7–20-fold slower than that of fructose 1-phosphate (Fig. 3).
FIGURE 4.

Inhibition of NAMPT leads to formation of corresponding labeled fructose 1-phosphate (F1P) and sedoheptulose 1-phosphate (S1P) from labeled glyceraldehyde and erythrose in cancer cells. HCT-116 cells were grown and treated in triplicates with FK866 (100 nm) for 24 h. After the treatment, the growth medium was replaced with a freshly prepared medium containing FK866 (100 nm), [13C]glyceraldehyde (1.0 mm), or [13C]erythrose (1.0 mm). The cells were incubated for a different period of time as described (see “Experimental Procedures”). After the treatment, cells were processed for analysis of the effects of glyceraldehyde and erythrose on the formation of fructose 1-phosphate (M3) and sedoheptulose 1-phosphate (M4) (A) and unlabeled F1P, S1P, fructose 1,6-bisphosphate (FBP), and DHAP (B) as described (see “Experimental Procedures”). The metabolites measured were expressed as nmol/mg total cellular protein. Error bars, S.D.
Next, we further investigated how the addition of glyceraldehyde affected the kinetics of the isotopomer formation. As shown in Fig. 4B, the addition of [U-13C]glyceraldehyde rapidly reduced mainly M0 of dihydroxyacetone phosphate, fructose 1,6-bisphosphate, and fructose 1-phosphate and more slowly reduced M0 of sedoheptulose 1-phosphate. These results again indicated that fructose 1-phosphate was derived from dihydroxyacetone phosphate and glyceraldehyde via a rapid condensation reaction, resulting in the immediate depletion of dihydroxyacetone phosphate (M0). The depletion of dihydroxyacetone phosphate (M0) caused by the addition of glyceraldehyde is also correlated with the rapid formation of fructose 1-phosphate (M3) (Fig. 4A). The rapid disappearance of fructose 1,6-bisphosphate (M0) caused by the addition of glyceraldehyde was due to the depletion of dihydroxyacetone phosphate (M0), again supporting the notion that fructose 1,6-bisphosphate was mainly derived from dihydroxyacetone phosphate and glyceraldehyde 3-phosphate in the treated cells. The rapid disappearance of fructose 1-phosphate (M0) caused by the addition of glyceraldehyde suggests a rather rapid equilibrium between the forward and backward condensation reactions. Finally, the slow disappearance of sedoheptulose 1-phosphate (M0) (Fig. 4B) was also consistent with its slow rate of formation (Figs. 3 and 4A).
Finally, due to the very slow rate of sedoheptulose 1-phosphate synthesis, the addition of [U-13C]erythrose, unlike [U-13C]glyceraldehyde, did not significantly alter levels of dihydroxyacetone phosphate (M0), fructose 1,6-bisphosphate (M0), fructose 1-phosphate (M0), and sedoheptulose 1-phosphate (M0) (Fig. 4B). Taken together, these results support the hypothesis that the increased formation of fructose 1-phosphate and sedoheptulose 1-phosphate in cancer cells in response to NAMPT inhibition is derived from elevated levels of dihydroxyacetone phosphate and glyceraldehyde and of erythrose, respectively.
On the basis of these results, we reasoned that NAMPT inhibition may also lead to increased formation of glyceraldehyde and erythrose in the cell, which also contributes to increased formation of fructose 1-phosphate and sedoheptulose 1-phosphate, respectively. To test this, we treated HCT-116 cells with FK866 in the presence or absence of NA (10 μm) and analyzed glyceraldehyde and erythrose levels. As shown in Fig. 5, the treatment with FK866 alone indeed led to a dose-dependent increase in glyceraldehyde and erythrose levels, whereas the addition of NA abolished the effects, showing that the increased glyceraldehyde and erythrose were due to NAMPT inhibition. The level of glyceraldehyde was ∼4-fold higher than that of erythrose, consistent with a lower level of sedoheptulose 1-phosphate detected in the cell. Together, these results further support the hypothesis that the increased formation of fructose 1-phosphate and sedoheptulose 1-phosphate that is derived from the elevated levels of dihydroxyacetone phosphate and glyceraldehyde and of erythrose, respectively.
FIGURE 5.
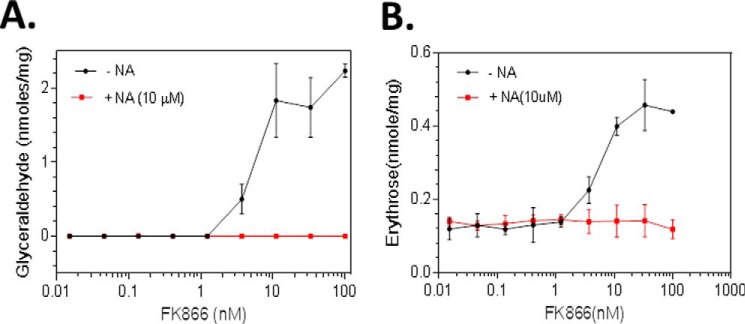
NAMPT Inhibition results in accumulation of glyceraldehyde and erythrose in cancer cells. HCT-116 cells were grown and treated in triplicates with FK866 (0.0–100.0 nm) and with or without nicotinic acid (NA) as described (see “Experimental Procedures”). After the treatment, cells were collected and processed for the analysis of glyceraldehyde (A) and erythrose (B) levels by LC-MS (see “Experimental Procedures”). The metabolites measured were expressed as nmol/mg total cellular protein. Error bars, S.D.
Inhibition of NAMPT Led to Increased Formation of Fructose 1-Phosphate and Sedoheptulose 1-Phosphate in Other Types of Cancer Cell Lines
We have assessed the metabolic consequences of NAMPT inhibition in HCT-116 and NCI-H1155 cells. To further investigate whether NAMPT inhibition leads to similar metabolic changes in other types of cancer cells, we treated the following cancer cell lines with FK866 (0–100 nm) for 24 h and analyzed fructose 1-phosphate, sedoheptulose 1-phosphate, and other glycolytic intermediate levels as described above: A2780 (ovarian cancer), KM-12 (colon cancer), HGC27 and SNU 484 (stomach cancer); PC-3 (prostate cancer); and SK-N-SH (neuroblastoma). NAMPT inhibition also led to accumulation of fructose 1-phosphate, sedoheptulose 1-phosphate, and other glycolytic intermediates in these cells (data not shown). These results indicate that the physiological function of NAMPT is conserved in different types of cancer cells.
Inhibition of NAMPT Led to Accumulation of Fructose 1-Phosphate, Sedoheptulose 1-Phosphate, Glyceraldehyde, and Erythrose in Tumor Xenografts
To confirm that the effects of NAMPT inhibition on cancer cell metabolism observed in vitro also occur in tumors in vivo, we analyzed FK866-treated tumor xenografts derived from NCI-H1155. The treatment of animals bearing tumors with FK866 at 5 and 10 mg/kg led to a dose-dependent increase in fructose 1-phosphate, sedoheptulose 1-phosphate, fructose 1,6-bisphosphate, dihydroxyacetone phosphate, glyceraldehyde, and erythrose (Fig. 6) but not glyceraldehyde 3-phosphate, fructose 6-phosphate, glucose 6-phosphate, and sedoheptulose 7-phosphate (data not shown). When dosed at 5 and 10 mg/kg, FK866 concentrations in the mouse plasma were determined to be ∼39 and 114 nm, respectively. These results showed that the in vivo concentrations of FK866 used were very similar to those of in vitro concentrations (0–100 nm) in these studies. As a result, both studies yielded similar results (Figs. 1 and 6). Furthermore, there was no obvious toxicity observed after 5 days of dosing. This is consistent with the observations that NAD+ turns over a lot faster in cancer cells than normal cells, NAMPT is a rate-limiting enzyme, and cancer cells are highly dependent on NAD+ for their rapid growth (6–9, 11–12). Taken together, these in vivo and in vitro studies have confirmed the previous findings and have also shown that NAMPT inhibition leads to elevated levels of fructose 1-phosphate and sedoheptulose 1-phosphate in cancer cells.
FIGURE 6.

NAMPT inhibition results in accumulation of fructose 1-phosphate (F1P), sedoheptulose 1-phosphate (S1P), fructose 1,6-bisphosphate (FBP), DHAP, glyceraldehyde, and erythrose in tumor xenografts. NCI-H1155 cells were grown and implanted as described (see “Experimental Procedures”). Animals bearing the established tumor xenografts were then treated with FK866 (0.0, 5.0, and 10 mg/kg) twice daily for 6 days as described (see “Experimental Procedures”). After the treatment, tumors were collected and processed for the analysis of fructose 1,6-bisphosphate, DHAP, fructose 1-phosphate, sedoheptulose 1-phosphate, glyceraldehyde, and erythrose by LC-MS (see “Experimental Procedures”). The metabolites measured were expressed as nmol/mg tumor tissue. Error bars, S.D.
Discussion
In this study, we have further investigated the metabolic basis of NAMPT inhibition. We have confirmed the blockade of glycolysis at the glyceraldehyde 3-phosphate dehydrogenase step as the central metabolic basis of NAMPT inhibition (1). We also show that NAMPT inhibition leads to the accumulation of fructose 1-phosphate, sedoheptulose 1-phosphate, glyceraldehyde, and erythrose but not glucose 6-phosphate, fructose 6-phosphate, and sedoheptulose 1-phosphate as previously thought (1). Furthermore, our combined biochemical and cellular studies indicate that the increased formation of fructose 1-phosphate and sedoheptulose 1-phosphate is derived from elevated levels of dihydroxyacetone phosphate and glyceraldehyde and of erythrose, respectively, via an aldolase activity. Together, this study shows that inhibition of NAMPT leads to significant changes in carbohydrate metabolism, and these metabolic changes, such as elevated levels of fructose 1-phosphate and sedoheptulose 1-phosphate, may be used as unique pharmacodynamics markers for evaluating on-target effects of NAMPT inhibitors in tumors in the clinic.
There are several lines of evidence that the increased formation of fructose 1-phosphate and sedoheptulose 1-phosphate is derived from elevated levels of dihydroxyacetone phosphate and glyceraldehyde and of erythrose, respectively, via an aldolase activity. First, the glucose-labeling study showed that fructose 1-phosphate mainly existed as M0, M3, and M6 isomers (1:2:1) at 6 h, which were derived from M0 and M3 of dihydroxyacetone phosphate and glyceraldehyde (Fig. 7). The labeling pattern of sedoheptulose 1-phosphate is such that M0, M3, M4, and M7 are the major and M1 and M6 are the minor isotopomers (Fig. 2A). The absence of M2 and M5 isotopomers strongly supports the notion that sedoheptulose 1-phosphate is not derived from sedoheptulose 7-phosphate, which would predict the presence of M2 and M5. A rationalization of the biogenesis and incorporation of labeled species into various isomers of sugar phosphates is depicted in Fig. 7. NAMPT inhibition is known to cause depletion of NADP+ (1), thereby limiting the carbon flow into the oxidative branch of the pentose phosphate pathway. At the same time, the accumulation of fructose 1-phosphate and possibly glyceraldehyde 3-phosphate could generate xylulose 5-phosphate and erythrose 4-phosphate via transketolase. Subsequently, dephosphorylation of erythrose 4-phosphate leads to erythrose formation. The erythrose generated is expected to have an isotopomer distribution pattern of M0, M1, M3, and M4 (M1 and M3 derived from M3 of fructose 6-phosphate and M0 and M4 derived from M6 of fructose 6-phosphate) and then coupled with DHAP (M0 and M3) by the action of transaldolase to form sedoheptulose 1-phosphate with isotopomers of M0, M1, M3 M4, M6, and M7 (Fig. 7). As is the case for the origin of erythrose labeling, M0, M3, M4, and M7 are derived from M0 and M6 of fructose 6-phosphate, whereas M1 and M6 are derived from M3 of fructose 6-phosphate. Of interest, the observation that the amounts of M1 and M6 are about half of those of M0 and M7 suggests that M0 and M6 of fructose 6-phosphate are present at about twice the amount of M3. Second, the addition of labeled glyceraldehyde, but not erythrose, led to increased fructose 1-phosphate with a corresponding decrease in dihydroxyacetone phosphate levels because the rate of sedoheptulose 1-phosphate synthesis is much slower than that of fructose 1-phosphate. Third, all three human aldolase isozymes are known to convert dihydroxyacetone phosphate and glyceraldehyde to fructose 1-phosphate (30–32). In addition, a rabbit aldolase was shown to convert dihydroxyacetone phosphate and erythrose to sedoheptulose 1-phosphate (30), and this finding has been confirmed in this study. Furthermore, the labeling and biochemical studies showed that the rate of sedoheptulose 1-phosphate synthesis was much slower than that of fructose 1-phosphate, consistent with their cellular levels (Figs. 3, 4A, and 5). Fourth, the absence of detectable levels of fructose and sedoheptulose in the FK866-treated cells (data not shown) ruled out a direct phosphorylation event involving enzymes like ketohexokinase and sedoheptulokinase (33, 34). In addition, the expression of ketohexokinase is diminished in the human clear cell type of renal cell carcinoma (35). Taken together, these results indicate that NAMPT inhibition leads to accumulation of fructose 1-phosphate and sedoheptulose 1-phosphate synthesized via an aldolase condensation reaction (Fig. 7).
FIGURE 7.

Proposed pathways lead to the formation of fructose 1-phosphate and sedoheptulose 1-phosphate isotopomers in cancer cells treated with NAMPT inhibitor. For all metabolites, each 13C-labeled carbon is marked as a solid circle, and each unlabeled carbon is marked as an open circle. G3P, glyceraldehyde 3-phosphate; F6P, fructose 6-phosphate; FBP, fructose 1,6-bisphosphate; F1P, fructose 1-phosphate; X5P, xylulose 5-phosphate; E4P, erythrose 4-phosphate; S1P, sedoheptulose 1-phosphate; TA, transaldolase; TK, transketolase, Ptase, phosphatase; M0–M7, metabolites with 0–7 carbons labeled.
The increased glyceraldehyde and erythrose levels in cancer cells have been reported and are metabolically intriguing. The interconversion of dihydroxyacetone phosphate and glyceraldehyde 3-phosphate is catalyzed by triose-phosphate isomerase, one of the most efficient enzymes (36, 37). In the forward direction, the kcat for the enzyme is near 500 s−1, and in the reverse direction, it is about 5,000 s−1 (37). Based on this, the overall equilibrium constant calculated from the total dihydroxyacetone phosphate/glyceraldehyde 3-phosphate ratio is 22 (37). Thus, the physiological concentration of glyceraldehyde 3-phosphate is only a fraction of dihydroxyacetone phosphate. This might have explained why glyceraldehyde 3-phosphate is barely detectable in either FK866-treated or untreated cells. However, due to the significant accumulation of dihydroxyacetone phosphate in the treated cells, this may push the equilibrium toward glyceraldehyde 3-phosphate formation, resulting in dephosphorylation to glyceraldehyde. Similarly, the accumulation of erythrose 4-phosphate (1) may also lead to its dephosphorylation to erythrose because erythrose 4-phosphate is an intermediate of the pentose phosphate pathway and has no other metabolic function. Finally, the significant absence of M1, M2, M4, and M5 of fructose 1,6-bisphosphate (data not shown) indicates that the contribution from the oxidative branch of the pentose phosphate pathway to the formation of fructose 1,6-bisphosphate is very limited. This also indicates that the contribution from the non-oxidative branch of the pathway is the main source of the formation of fructose 1,6-bisphosphate, especially M3. Therefore, the significant conversion of fructose 6-phosphate to glyceraldehyde 3-phosphate and erythrose 4-phosphate (Fig. 7) may have explained why fructose 1,6-bisphosphate was accumulated but fructose 6-phosphate was not.
Fructose 1-phosphate has been reported in renal cell carcinoma (38), but sedoheptulose 1-phosphate, to our knowledge, has not been reported in any cancer cells. The physiological relevance of these metabolites to cancer cells is therefore not clear. Fructose 1-phosphate is well known for its important role in regulating glucokinase activity in the liver by binding to the regulatory protein of glucokinase (39). The binding of fructose 1-phosphate to the regulatory protein prevents the regulatory protein from inhibiting glucokinase activity, thereby promoting glucose phosphorylation (39). It is possible that fructose 1-phosphate may play a similar role in cancer cells to increase glucose phosphorylation in order to compensate for the reduced glycolytic activity as the blockade at the glyceraldehyde 3-phosphate dehydrogenase step imposed by NAMPT inhibition significantly reduces glycolysis.
The findings from this study may have significant clinical implications. To effectively assess a molecule in the clinic, it is essential to have a robust clinical diagnostic assay for identifying appropriate patient populations and also a reliable pharmacodynamics biomarker assay for evaluating effects of the molecule on its intended target in tumors. As proposed previously (1), the altered metabolite levels, such as increased fructose 1,6-bisphosphate and dihydroxyacetone phosphate levels, and decreased 2(3)-phosphoglycerate and pyruvate level in tumors can be used to assess on-target effects of NAMPT inhibitors in the clinic. This study suggests that the altered metabolite levels, especially those of fructose 1-phosphate and sedoheptulose 1-phosphate, can be used as ideal pharmacodynamics biomarkers as compared with other altered metabolites because these metabolites are highly elevated upon treatment and yet almost absent in untreated cells. Thus, implementing the LC-MS methodology for detecting and quantifying these metabolites in the clinic may enhance our understanding of the mechanism of action of NAMPT inhibitors in the clinic and subsequently accelerate their clinical development.
In summary (see Fig. 8), the previous study (1) shows that NAMPT inhibition leads to attenuation of glycolysis at the glyceraldehyde 3-phosphate dehydrogenase step due to the reduced availability of NAD+ for the enzyme. The attenuation of glycolysis results in the accumulation of glycolytic intermediates before and at the glyceraldehyde 3-phosphate dehydrogenase step, as evidenced by the increased levels of intermediates, such as fructose 1,6-bisphosphate and dihydroxyacetone phosphate. The attenuation of glycolysis also causes decreased glycolytic intermediates, such as 2(3)-phosphoglycerate and pyruvate, after the glyceraldehyde 3-phosphate dehydrogenase step, thereby reducing carbon flow into serine biosynthesis and the TCA cycle. This study shows that inhibition of NAMPT also leads to the accumulation of fructose 1-phosphate, sedoheptulose 1-phosphate, glyceraldehyde, and erythrose in multiple cancer cell lines and tumor xenografts but not glucose 6-phosphate, fructose 6-phosphate, and glyceraldehyde 3-phosphate as previously thought (1). The increased levels of fructose 1-phosphate and sedoheptulose 1-phosphate are probably derived from elevated levels of dihydroxyacetone phosphate and glyceraldehyde and of erythrose, respectively, via an aldolase reaction.
FIGURE 8.

NAMPT inhibition leads to alterations of different metabolic pathways in cancer cells. NAMPT inhibition affects the glycolytic, pentose phosphate pathway, serine biosynthetic pathways, and the TCA cycle. The affected metabolites are highlighted in either red (increased) or green (decreased). The blockade of the glyceraldehyde-3-phosphate dehydrogenase (G3PDH) step is indicated (×). HK, hexokinase; PGI, phosphoglucose isomerase; PFK, phosphofructose kinase; TPI, triose-phosphate isomerase; PGK, phosphoglycerate kinase; PGM, phosphoglycerate mutase; PKM2, pyruvate kinase 2; LDH, lactate dehydrogenase; G6PDH, glucose-6-phosphate dehydrogenase; G6P, glucose 6-phosphate; 6PGLA, 6-phosphogluconolactonase; Gn6P, 6-phosphogluconate; Gn6PDH, 6-phosphogluconate dehydrogenase; NMN, nicotinamide mononucleotide adenine; NMNAT, nicotinamide mononucleotide adenine transferase; 3PG, 3-phosphoglycerate; 3PGDH, 3-phosphoglycerate dehydrogenase; p-ser, phosphoserine; PSAT, phosphoserine aminotransferase; PSPH, phosphoserine phosphatase; SHMT, serine hydroxymethyltransferase; CS, citrate synthase; IDH, isocitrate dehydrogenase; α-KG, α-ketoglutarate; α-KGDH, α-ketoglutarate dehydrogenase; SDH, succinate dehydrogenase; FH, fumarate hydratase; MDH, malate dehydrogenase; PDH, pyruvate dehydrogenase; F6P, fructose 6-phosphate; FBP, fructose 1,6-bisphosphate; BPG, 1,3-bisphosphoglycerate; 2PG, 2-phosphoglycerate; PEP, phosphoenolpyruvate; 6PGL, 6-phosphogluconolactone; E4P, erythrose 4-phosphate; xylulose5P, xylulose 5-phosphate; ribose5P, ribose 5-phosphate; ribulose5P, ribulose 5-phosphate; THF, tetrahydrofolate; MTHF, 5,10-methylenetetrahydrofolate; 3PHP, 3-phosphohydroxypyruvate; NAM, nicotinamide.
Acknowledgments
We thank Jake Starling, Alfonso De Dios, Tim Burkholder, and Gregory D. Plowman for support, guidance, and critical review of the manuscript.
This work was supported by Eli Lilly and Co. The authors declare that they have no conflicts of interest with the contents of this article.
- NAMPT
- nicotinamide phosphoribosyltransferase
- MRM
- multiple-reaction monitoring
- DHAP
- dihydroxyacetone phosphate.
References
- 1. Tan B., Young D. A., Lu Z. H., Wang T., Meier T. I., Shepard R. L., Roth K., Zhai Y., Huss K., Kuo M. S., Gillig J., Parthasarathy S., Burkholder T. P., Smith M. C., Geeganage S., Zhao G. (2013) Pharmacological inhibition of nicotinamide phosphoribosyltransferase (NAMPT), an enzyme essential for NAD+ biosynthesis, in human cancer cells: metabolic basis and potential clinical implications. J. Biol. Chem. 288, 3500–3511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Garten A., Petzold S., Körner A., Imai S., Kiess W. (2009) Nampt: linking NAD+ biology, metabolism and cancer. Trends Endocrinol. Metab. 20, 130–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kirkland J. B. (2009) Niacin status, NAD distribution and ADP-ribose metabolism. Curr. Pharm. Des. 15, 3–11 [DOI] [PubMed] [Google Scholar]
- 4. Magni G., Amici A., Emanuelli M., Orsomando G., Raffaelli N., Ruggieri S. (2004) Enzymology of NAD+ homeostasis in man. Cell Mol. Life Sci. 61, 19–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sauve A. A. (2008) NAD+ and vitamin B3: from metabolism to therapies. J. Pharmacol. Exp. Ther. 324, 883–893 [DOI] [PubMed] [Google Scholar]
- 6. Olesen U. H., Thougaard A. V., Jensen P. B., Sehested M. (2010) A preclinical study on the rescue of normal tissue by nicotinic acid in high-dose treatment with APO866, a specific nicotinamide phosphoribosyltransferase inhibitor. Mol. Cancer Ther. 9, 1609–1617 [DOI] [PubMed] [Google Scholar]
- 7. Olesen U. H., Christensen M. K., Björkling F., Jäättelä M., Jensen P. B., Sehested M., Nielsen S. J. (2008) Anticancer agent CHS-828 inhibits cellular synthesis of NAD. Biochem. Biophys. Res. Commun. 367, 799–804 [DOI] [PubMed] [Google Scholar]
- 8. Watson M., Roulston A., Bélec L., Billot X., Marcellus R., Bédard D., Bernier C., Branchaud S., Chan H., Dairi K., Gilbert K., Goulet D., Gratton M. O., Isakau H., Jang A., Khadir A., Koch E., Lavoie M., Lawless M., Nguyen M., Paquette D., Turcotte E., Berger A., Mitchell M., Shore G. C., Beauparlant P. (2009) The small molecule GMX1778 is potent inhibitor of NAD+ biosynthesis: strategy for enhanced therapy in nicotinic acid phosphoribosyltransferase 1-deficient tumors. Mol. Cell. Biol. 29, 5872–5888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Revollo J. R., Grimm A. A., Imai S. (2004) The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J. Biol. Chem. 279, 50754–50763 [DOI] [PubMed] [Google Scholar]
- 10. Rongvaux A., Galli M., Denanglaire S., Van Gool F., Drèze P. L., Szpirer C., Bureau F., Andris F., Leo O. (2008) Nicotinamide phosphoribosyl transferase/pre-B cell colony-enhancing factor/visfatin is required for lymphocyte development and cellular resistance to genotoxic stress. J. Immunol. 181, 4685–4695 [DOI] [PubMed] [Google Scholar]
- 11. Imai S. (2009) Nicotinamide phosphoribosyltransferase (NAMPT): A link between NAD biology, metabolism, and diseases. Curr. Pharm. Des. 15, 20–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Khan J. A., Forouhar F., Tao X., Tong L. (2007) Nicotinamide adenine dinucleotide metabolism as an attractive target for drug discovery. Expert Opin. Ther. Targets 11, 695–705 [DOI] [PubMed] [Google Scholar]
- 13. Luk T., Malam Z., Marshall J. C. (2008) Pre-B cell colony-enhancing factor (PBEF)/visfatin: a novel mediator of innate immunity. J. Leukoc. Biol. 83, 804–816 [DOI] [PubMed] [Google Scholar]
- 14. Bi T. Q., Che X. M., Liao X. H., Zhang D. J., Long H. L., Li H. J., Zhao W. (2011) Overexpression of Nampt in gastric cancer and chemopotentiating effects of the Nampt inhibitor FK866 in combination with fluorouracil. Oncol. Rep. 26, 1251–1257 [DOI] [PubMed] [Google Scholar]
- 15. Hufton S. E., Moerkerk P. T., Brandwijk R., de Bruïne A. P., Arends J. W., Hoogenboom H. R. (1999) A profile of differentially expressed genes in primary colorectal cancer using suppression subtractive hybridization. FEBS Lett. 463, 77–82 [DOI] [PubMed] [Google Scholar]
- 16. Van Beijnum J. R., Moerkerk P. T., Gerbers A. J., De Bruïne A. P., Arends J. W., Hoogenboom H. R., Hufton S. E. (2002) Target validation for genomics using peptide-specific phage antibodies: a study of five gene products overexpressed in colorectal cancer. Int. J. Cancer 101, 118–127 [DOI] [PubMed] [Google Scholar]
- 17. Wang B., Hasan M. K., Alvarado E., Yuan H., Wu H., Chen W. Y. (2011) NAMPT overexpression in prostate cancer and its contribution to tumor cell survival and stress response. Oncogene 30, 907–921 [DOI] [PubMed] [Google Scholar]
- 18. Nakajima T. E., Yamada Y., Hamano T., Furuta K., Gotoda T., Katai H., Kato K., Hamaguchi T., Shimada Y. (2009) Adipocytokine levels in gastric cancer patients: resistin and visfatin as biomarkers of gastric cancer. J. Gastroenterol. 44, 685–690 [DOI] [PubMed] [Google Scholar]
- 19. Busso N., Karababa M., Nobile M., Rolaz A., Van Gool F., Galli M., Leo O., So A., De Smedt T. (2008) Pharmacological inhibition of nicotinamide phosphoribosyltransferase/visfatin enzymatic activity identifies a new inflammatory pathway linked to NAD. PLoS One 3, e2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hasmann M., Schemainda I. (2003) FK866, a highly specific noncompetitive inhibitor of nicotinamide phosphoribosyltransferase, represents a novel mechanism for induction of tumor cell apoptosis. Cancer Res. 63, 7436–7442 [PubMed] [Google Scholar]
- 21. Yang H., Yang T., Baur J. A., Perez E., Matsui T., Carmona J. J., Lamming D. W., Souza-Pinto N. C., Bohr V. A., Rosenzweig A., de Cabo R., Sauve A. A., Sinclair D. A. (2007) Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell 130, 1095–1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Drevs J., Löser R., Rattel B., Esser N. (2003) Antiangiogenic potency of FK866/K22.175, a new inhibitor of intracellular NAD biosynthesis, in murine renal cell carcinoma. Anticancer Res. 23, 4853–4858 [PubMed] [Google Scholar]
- 23. Muruganandham M., Alfieri A. A., Matei C., Chen Y., Sukenick G., Schemainda I., Hasmann M., Saltz L. B., Koutcher J. A. (2005) Metabolic signatures associated with a NAD synthesis inhibitor-induced tumor apoptosis identified by 1H-decoupled-31P magnetic resonance spectroscopy. Clin. Cancer Res. 11, 3503–3513 [DOI] [PubMed] [Google Scholar]
- 24. Giannetti A. M., Zheng X., Skelton N. J., Wang W., Bravo B. J., Bair K. W., Baumeister T., Cheng E., Crocker L., Feng Y., Gunzner-Toste J., Ho Y. C., Hua R., Liederer B. M., Liu Y., Ma X., O'Brien T., Oeh J., Sampath D., Shen Y., Wang C., Wang L., Wu H., Xiao Y., Yuen P. W., Zak M., Zhao G., Zhao Q., Dragovich P. S. (2014) Fragment-based identification of amides derived from trans-2-(pyridin-3-yl)cyclopropanecarboxylic acid as potent inhibitors of human nicotinamide phosphoribosyltransferase (NAMPT). J. Med. Chem. 57, 770–792 [DOI] [PubMed] [Google Scholar]
- 25. Zheng X., Bauer P., Baumeister T., Buckmelter A. J., Caligiuri M., Clodfelter K. H., Han B., Ho Y. C., Kley N., Lin J., Reynolds D. J., Sharma G., Smith C. C., Wang Z., Dragovich P. S., Gunzner-Toste J., Liederer B. M., Ly J., O'Brien T., Oh A., Wang L., Wang W., Xiao Y., Zak M., Zhao G., Yuen P. W., Bair K. W. (2013) Structure-based discovery of novel amide-containing nicotinamide phosphoribosyltransferase (nampt) inhibitors. J. Med. Chem. 56, 6413–6433 [DOI] [PubMed] [Google Scholar]
- 26. Du L., Zhang X., Han Y. Y., Burke N. A., Kochanek P. M., Watkins S. C., Graham S. H., Carcillo J. A., Szabó C., Clark R. S. (2003) Intra-mitochondrial poly(ADP-ribosylation) contributes to NAD+ depletion and cell death induced by oxidative stress. J. Biol. Chem. 278, 18426–18433 [DOI] [PubMed] [Google Scholar]
- 27. Pillai J. B., Isbatan A., Imai S., Gupta M. P. (2005) Poly(ADP-ribose) polymerase-1-dependent cardiac myocyte cell death during heart failure is mediated by NAD+ depletion and reduced Sir2α deacetylase activity. J. Biol. Chem. 280, 43121–43130 [DOI] [PubMed] [Google Scholar]
- 28. Yu S. W., Wang H., Poitras M. F., Coombs C., Bowers W. J., Federoff H. J., Poirier G. G., Dawson T. M., Dawson V. L. (2002) Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science 297, 259–263 [DOI] [PubMed] [Google Scholar]
- 29. Clasquin M. F., Melamud E., Singer A., Gooding J. R., Xu X., Dong A., Cui H., Campagna S. R., Savchenko A., Yakunin A. F., Rabinowitz J. D., Caudy A. A. (2011) Riboneogenesis in yeast. Cell 145, 969–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rellos P., Sygusch J., Cox T M. (2000) Expression, purification, and characterization of natural mutants of human aldolase B: role of quaternary structure in catalysis. J. Biol. Chem. 275, 1145–1151 [DOI] [PubMed] [Google Scholar]
- 31. Kitajima Y., Takasaki Y., Takahashi I., Hori K. (1990) Construction and properties of active chimeric enzymes between human aldolases A and B: analysis of molecular regions which determine isozyme-specific functions. J. Biol. Chem. 265, 17493–17498 [PubMed] [Google Scholar]
- 32. Kusakabe T., Motoki K., Hori K. (1994) Human aldolase C: characterization of the recombinant enzyme expressed in E. coli. J. Biochem. 115, 1172–1177 [DOI] [PubMed] [Google Scholar]
- 33. Bais R., James H. M., Rofe A. M., Conyers R. A. (1985) The purification and properties of human liver ketohexokinase: a role for ketohexokinase and fructose-bisphosphate aldolase in the metabolic production of oxalate from xylitol. Biochem. J. 230, 53–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kardon T., Stroobant V., Veiga-da-Cunha M., Schaftingen E. V. (2008) Characterization of mammalian sedoheptulokinase and mechanism of formation of erythritol in sedoheptulokinase deficiency. FEBS Lett. 582, 3330–3334 [DOI] [PubMed] [Google Scholar]
- 35. Hwa J. S., Kim H. J., Goo B. M., Park H. J., Kim C. W., Chung K. H., Park H. C., Chang S. H., Kim Y. W., Kim D. R., Cho G. J., Choi W. S., Kang K. R. (2006) The expression of ketohexokinase is diminished in human clear cell type of renal cell carcinoma. Proteomics 6, 1077–1084 [DOI] [PubMed] [Google Scholar]
- 36. Albery W. J., Knowles J. R. (1976) Evolution of enzyme function and the development of catalytic efficiency. Biochemistry 15, 5631–5640 [DOI] [PubMed] [Google Scholar]
- 37. Wierenga R. K., Kapetaniou E. G., Venkatesan R. (2010) Triosephosphate isomerase: a highly evolved biocatalyst. Cell Mol. Life Sci. 67, 3961–3982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Catchpole G., Platzer A., Weikert C., Kempkensteffen C., Johannsen M., Krause H., Jung K., Miller K., Willmitzer L., Selbig J., Weikert S. (2011) Metabolic profiling reveals key metabolic features of renal cell carcinoma. J. Cell Mol. Med. 15, 109–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Beck T., Miller B. G. (2013) Structural basis for regulation of human glucokinase by glucokinase regulatory protein. Biochemistry 52, 6232–6239 [DOI] [PMC free article] [PubMed] [Google Scholar]