

Background: β3-bound c-Src initiates outside-in signaling in platelets.
Results: After platelet stimulation, β3 binds to the c-Src SH3 domain at a site overlapping with its PPII helix binding site.
Conclusion: The interaction of c-Src with β3 requires c-Src activation to vacate the PPII helix binding site.
Significance: The mode of c-Src binding to β3 prevents c-Src-mediated signaling in circulating platelets.
Keywords: integrin, platelet, signal transduction, Src, Src homology 3 domain (SH3 domain)
Abstract
It is currently believed that inactive tyrosine kinase c-Src in platelets binds to the cytoplasmic tail of the β3 integrin subunit via its SH3 domain. Although a recent NMR study supports this contention, it is likely that such binding would be precluded in inactive c-Src because an auto-inhibitory linker physically occludes the β3 tail binding site. Accordingly, we have re-examined c-Src binding to β3 by immunoprecipitation as well as NMR spectroscopy. In unstimulated platelets, we detected little to no interaction between c-Src and β3. Following platelet activation, however, c-Src was co-immunoprecipitated with β3 in a time-dependent manner and underwent progressive activation as well. We then measured chemical shift perturbations in the 15N-labeled SH3 domain induced by the C-terminal β3 tail peptide NITYRGT and found that the peptide interacted with the SH3 domain RT-loop and surrounding residues. A control peptide whose last three residues where replaced with those of the β1 cytoplasmic tail induced only small chemical shift perturbations on the opposite face of the SH3 domain. Next, to mimic inactive c-Src, we found that the canonical polyproline peptide RPLPPLP prevented binding of the β3 peptide to the RT- loop. Under these conditions, the β3 peptide induced chemical shift perturbations similar to the negative control. We conclude that the primary interaction of c-Src with the β3 tail occurs in its activated state and at a site that overlaps with PPII binding site in its SH3 domain. Interactions of inactive c-Src with β3 are weak and insensitive to β3 tail mutations.
Introduction
Platelet spreading on fibrinogen-coated surfaces and platelet-induced fibrin clot retraction are mediated by the integrin αIIbβ3 interposed between immobilized fibrinogen or fibrin and the platelet cytoskeleton and occur when αIIbβ3-initiated “outside-in” signaling causes platelet cytoskeletal contraction (1, 2). αIIbβ3-mediated outside-in signaling is initiated by activation of the tyrosine kinase c-Src that is bound to the cytoplasmic tail (CT) of the β3 subunit of αIIbβ3 (3, 4). c-Src is a member of a widely expressed family of non-receptor tyrosine kinases that participate in a variety of essential biological processes (5–8) and is the best studied among nine Src family kinases (SFK)3 in humans (9). However, because of the redundancy of SFK paralog expression, the phenotypes of single-gene SFK knockouts have often been unremarkable. This is the case for blood platelets (8) where it was necessary to generate platelets deficient in six SFKs (c-Src, Fyn, Fgr, Hck, Lyn, and Yes) to observe a phenotype of decreased platelet spreading on fibrinogen (10). Although SFK-deficient platelets, as well as wild-type platelets treated with SFK inhibitors, do not manifest defects in αIIb β3 activation, αIIbβ3-dependent tyrosine phosphorylation and cytoskeletal reorganization are affected, implying that SFKs participate in αIIbβ3-mediated outside-in signaling. This is supported by observations that SFKs are recruited to the αIIbβ3-associated cytoskeleton after platelet aggregation (11, 12).
Like other SFKs, c-Src has a conserved domain structure consisting of a myristoylated N-terminal SH4 domain followed by a flexible unique domain and flexible linker, an SH3 domain, a flexible linker, an SH2 domain, a third flexible linker, a kinase domain, and a C-terminal tail (13). Inactive c-Src adopts a compact structure that is stabilized by intramolecular interactions that include binding of the phosphorylated C-terminal tail to the SH2 domain, packing of the SH3 and SH2 domains against the N- and C-terminal lobes of the kinase domain, respectively, and binding of the linker connecting the SH2 and kinase domains to the SH3 domain (14) (Fig. 1). With regard to the latter interaction, crystal structures of inactive c-Src and the inactive conformation of other Src family kinases reveal that the linker adopts a polyproline type II helical conformation that packs against the protein recognition surface of the SH3 domain (13, 15).
FIGURE 1.
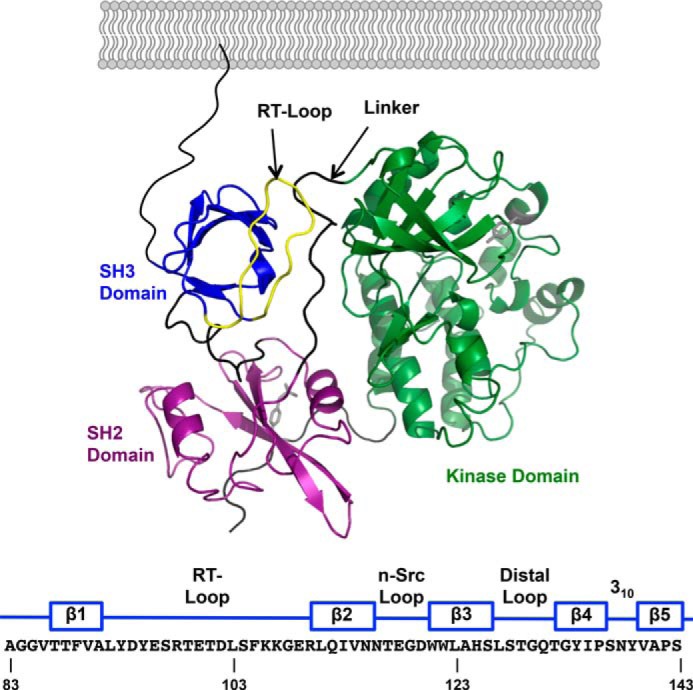
A, domain structure of inactive c-Src (PDB 2SRC). From N- to C terminus: SH3 domain (blue); SH2 domain (purple); kinase domain (green). The linker between the SH2 and kinase domains is colored black, and the RT-loop in the SH3 domain is colored yellow. The amino acid sequence of the human c-Src SH3 domain is numbered according to UniProtKB/Swiss-Prot entry P12931 and indicates the location of its five β strands and the interconnecting loops.
The c-Src SH3 domain also binds to the C-terminal Arg-Gly-Thr (RGT) residues of the β3 CT in an interaction that can be inhibited by proline-rich peptides selective for the SH3 domain (3). The C terminus of the β3 CT is quite different from the polyproline sequences usually recognized by SH3 domains (16, 17). Nonetheless, in a recent NMR study, Katyal et al. concluded that like polyproline helices, the CT of inactive β3 interacts with the c-Src SH3 domain by binding to a site located between the RT and n-Src loops of the SH3 domain and with its RGT motif in a pocket formed by the n-Src loop (18). However, it is quite likely that such binding would be precluded in inactive c-Src because the polyproline helix binding site in the SH3 domain is already occupied by the linker connecting the SH2 and kinase domains (14).
Here, we have reassessed the interaction of the SH3 domain with the β3 CT using immunoprecipitation to study the interaction of c-Src with β3 in platelets and NMR spectroscopy to directly study the interaction of the β3 CT with the c-Src SH3 domain. Using a peptide corresponding to the C-terminal seven residues of the β3 CT, we identified a weak interaction between β3 and the c-Src SH3 domain. It is often the case that binding interactions are weak when they represent only two components of a ternary or higher order complex. Nevertheless, they can be very specific, as we and others have shown for the interaction of the β3 CT with the talin head domain (19). In this case, to demonstrate that the interaction is both specific and biologically relevant, we also synthesized and studied a mutant peptide previously shown to be unable to interact with and activate c-Src (3, 20, 21).
Experimental Procedures
Protein and Peptide Synthesis
cDNAs corresponding to the human c-Src SH3 domain (residues Ala-83 to Ser-143) and the human c-Src SH4-SH3 domains (residues Met-1 to Ser-143) were cloned between the NdeI and XhoI restriction sites of the pET-28a vector (Novagen) such that they contained an N-terminal 6×His tag followed by a thrombin cleavage site. The cDNAs were then expressed overnight at 20 °C in BL21-CodonPlus(DE3) cells (Agilent Technologies) induced with 1 mm IPTG and isolated from bacterial lysates by affinity chromatography using a HisTrap HP column (GE Healthcare) on an ÄKTApurifier 10 system. Following cleavage by thrombin to remove the 6×His tag, the recombinant proteins were purified by ion exchange chromatography. 7-mer peptides were purchased from Mimotopes.
Immunoprecipitation and Immunoblotting of c-Src Bound to β3
To measure the association of c-Src with β3 in unstimulated and stimulated platelets, human platelets obtained from blood anticoagulated at a ratio of 5:1 with ACD (sodium citrate 65 mm, citric acid 77 mm, glucose 95 mm, pH 4.4) were washed with 10 mm HEPES buffer, pH 6.5, containing 150 mm NaCl, 3 mm EDTA, 1 μm PGE1, and 0.3 units/ml apyrase. The washed platelets were resuspended in modified Tyrode's buffer (20 mm HEPES, pH 7.35, containing 135 mm NaCl, 2.7 mm KCl, 3 mm NaH2PO4, 5 mm glucose, 10 units/ml apyrase, and 0.1% BSA). 500-μl aliquots of the washed platelets were then stimulated with 1 unit/ml thrombin for up to 90 s and lysed with 125 μl of lysis buffer containing 1% Nonidet P-40, 50 mm Tris, 150 mm NaCl, and 1 mm EDTA. β3 and associated proteins were immunoprecipitated from lysates from equal numbers of platelets using integrin β3 (D-11) monoclonal antibody (Santa Cruz Biotechnology) and protein G magnetic beads (Millipore). The immunoprecipitated proteins were separated on 4–12% NuPAGE Bis-Tris gels (Life Technologies), transferred to nitrocellulose membranes (Life Technologies), and immunoblotted for phosphorylated c-Src residue 419 (pTyr419) with a pTyr419-specific antibody (Cell Signaling) followed by an incubation with horseradish peroxidase-conjugated anti-IgG. Protein-bound antibody was visualized using Enhanced Chemiluminescence (Amersham Biosciences). Membranes were then stripped of protein-bound antibody using Restore Western blot Stripping Buffer (Thermo Scientific), blocked with 5% nonfat dry milk (Bio-Rad) and immunoblotted again with either integrin β3 (D-11) or a c-Src-specific antibody (Cell Signaling).
Surface Plasmon Resonance (SPR)
All SPR experiments were performed on a Biacore 3000 (GE Healthcare). The c-Src SH4-SH3 domain protein was coupled to a CM5 SPR sensor chip using EDC/NHS. Injections of the C-terminal β3 CT peptide NITYRGT and the class 1 PPII core peptide RPLPPLP (22, 23) were performed at a flow rate of 10 μl/min for 5 min in 20 mm HEPES buffer, pH 7.4, containing 150 mm NaCl. Each peptide was injected as a dilution series in concentrations ranging from 0 to 5 mm (0, 0.039, 0.078, 0.156, 0.313, 0.625, 1.25, 2.5, and 5 mm). The resulting sensorgrams were evaluated using BIAevaluation software and fitted to a 1:1 steady-state binding model.
Nuclear Magnetic Resonance (NMR) Spectroscopy
The c-Src SH3 domain was labeled with 15N by bacterial expression in M9 minimal media supplemented with 15N ammonium chloride (Cambridge Isotopes). For titration of the SH3 domain with the β3 CT peptide NITYRGT, NMR data were collected at 20 °C on a Bruker DMX-600 spectrometer with a 5-mm x,y,z-shielded pulse-field gradient triple-resonance probe. Initially, two NMR samples were prepared in 20 mm NaPO4 buffer, pH 6.0, containing 100 mm NaCl. One contained 1 mm 15N-labeled SH3 domain only and the other contained 15.7 mm NITYRGT. A series of titration experiments were then performed at NITYRGT concentrations ranging between 5.4 mm and 15.7 mm. For titrations using the mutant β3/β1 peptide NITYEGK and RPLPPLP, ligands were prepared at 100 mm in 50 mm NaPO4 buffer, pH 6.3, containing 50 mm NaCl. The stocks were added into 0.33 mm 15N SH3 in 20 mm NaPO4 buffer, pH 6.0, containing 100 mm NaCl at ligand concentrations of 0, 0.10, 0.30, 0.89, 1.09, 1.28, and 1.48 mm for NITYEGK, and 0, 0.02, 0.06, 0.12, 0.22, 0.42, 0.81, 1.40, and 2.17 mm for RPLPPLP, respectively. For titration of NITYRGT in presence of RPLPPLP, 6 eq RPLPPLP (1.96 mm) was added into the 0.33 mm 15N SH3 sample first, then 100 mm NITYRGT stock was added to the sample to reach concentrations of 0, 0.10, 0.29, 0.88, 2.76, 4.58, 6.33, and 8.03 mm. Two-dimensional 15N-HSQC spectra were recorded along t1(15N) and t2(1H), respectively, with 256 or 128 and 4096 complex points, and t1,max(15N) = 72 or 36 ms and t2,max(1H) = 297 ms, 12 or 16 scans, at 25 °C on a Bruker 600 MHz Avance II spectrometer equipped with a cryogenic probe. Spectra were processed and analyzed using the programs NMRPipe (24) and Sparky (T. D. Goddard and D. G. Kneller, SPARKY 3, University of California, San Francisco). The 15N carrier frequency was set to 118.5 ppm. Proton chemical shifts were referenced to residual water signal (4.74 ppm) while 15N chemical shifts were indirectly referenced. Prior to Fourier Transform, time domain data were multiplied by sine square bell window functions shifted by 90° and zero-filled once. Forward-backward linear prediction was applied in the indirect dimension once. Chemical shift assignments for the SH3 domain were referenced to the assignments in almost identical conditions (100 mm phosphate pH 6.0, 100 mm NaCl and 25 °C, BMRB ID 3433) (25). Thresholds for chemical shift changes indicative of specific protein-protein interactions were calculated using the method of Schumann et al. (26).
Results
c-Src Binding to β3 in Platelets Increases Progressively following αIIbβ3 Activation
The putative β3 CT binding site on the SH3 domain of inactive c-Src is occupied by the linker connecting the c-Src SH2 and kinase domains (13, 18). Nonetheless, c-Src activity is stimulated after β3 CT binding (3). Therefore, we sought to understand how β3 CT binding to c-Src regulates its enzymatic activity. To begin to address this question, we measured the time course of c-Src binding to β3 and its subsequent activation following platelet stimulation by thrombin. Human platelets were isolated from platelet-rich plasma in the presence of PGE1 to prevent factitious platelet activation during the isolation procedure and were stimulated by 1 unit/ml thrombin. The stimulated platelets were lysed with Nonidet P-40, β3 was immunoprecipitated from the lysates, and was immunoblotted for c-Src and for phosphorylated c-Src residue Tyr-419, an indicator of c-Src activation. As shown in Fig. 2, there was little c-Src binding to β3 in unstimulated platelets. However, c-Src binding to β3 was detectable within 10 s after thrombin stimulation, reached a maximum at 30 s, and declined thereafter. Further, similar to the rapid and transient c-Src activation that Clark and Brugge observed in detergent extracts of thrombin-stimulated platelets (12), we detected the phosphorylation of Tyr-419 of β3-bound c-Src within 20 s following thrombin stimulation. Tyr-419 phosphorylation then increased progressively, reaching a maximum at 60 s, after which it also declined. Thus, these results indicate that the vast majority of c-Src binding to β3 occurs subsequent to αIIbβ3 activation, after which the β3-bound c-Src becomes enzymatically active.
FIGURE 2.

A, time course of c-Src binding to the integrin β3 subunit and its subsequent activation after platelet stimulation by thrombin. Following the stimulation of washed human platelets by 1 unit/ml thrombin for the indicated times, platelets were lysed with 1% Nonidet P-40, β3 was immunoprecipitated (IP) from the lysates using a β3-specific monoclonal antibody, and the immunoprecipitated β3 was serially immunoblotted (IB) for β3, c-Src, and phosphorylated c-Src residue 419 (pTyr419). B, densitometry using NIH ImageJ software of the immunoblot shown in Fig. 2A demonstrating the time course of c-Src binding to the integrin β3 subunit and its subsequent activation after platelet stimulation by thrombin. The densitometry for β3-bound c-Src and for β3-bound c-Src containing phosphorylated tyrosine residue 419 (pY419) at each time point was normalized using the densitometry for the corresponding β3 band.
Measuring the Affinity of Peptide Ligands for the c-Src SH3 Domain
At a minimum, the c-Src binding site on αIIbβ3 consists of the three C-terminal β3 CT residues RGT (3). Accordingly, we used SPR to compare the affinities of a β3 C-terminal heptapeptide NITYRGT and the canonical polyproline helix heptapeptide RPLPPLP for a recombinant human c-Src SH3 domain immobilized on a CM5 SPR chip. As shown by the equilibrium binding sensorgrams in Fig. 3, NITYRGT bound weakly to the SH3 domain. Although NITYRGT binding did not reach saturation, an estimated Kd calculated using BIAevaluation software was 320 ± 120 mm. By contrast, RPLPPLP binding to the SH3 domain was saturable and substantially stronger with an estimated Kd of 259 ± 52 μm.
FIGURE 3.

Representative SPR measurements of steady state NITYRGT (A) and RPLPPLP (B) binding to the c-Src SH3 domain. Average dissociation constants (Kd), calculated using BIAevaluation software, from 9–12 steady state measurements, were 259 ± 52 μm and 320 ± 120 mm for RPLPPLP and NITYRGT, respectively.
Detection of NITYRGT Binding to the c-Src SH3 Domain by Two-dimensional NMR
To determine how the β3 CT interacts with c-Src, we turned to NMR, measuring NITYRGT binding to the c-Src SH3 domain labeled with 15N. Because our SPR measurements indicated that NITYRGT binding to the SH3 domain was weak, we measured chemical shift perturbations (CSP) induced by NITYRGT in the two-dimensional HSQC spectrum of the SH3 domain, an NMR technique able to detect and localize weak protein-protein interactions (27). CSP for each SH3 domain residue, calculated using the relationship Δppm = (Δ1H2 + (Δ15N/5)2)1/2, were determined from recorded HSQC spectra when we serially titrated NITYRGT at concentrations up to 15.7 mm against a single 1 mm concentration of the 15N-labeled c-Src SH3 domain. The interactions appeared to be in fast exchange, as only shifts in peak location were observed and peaks remained sharp and intense throughout the course of the titration (Fig. 4, A and B).
FIGURE 4.

The C-terminal β3 peptide NITYRGT binds to the RT-Loop of the c-Src SH3 domain. A, superimposition of HSQC spectra of 1 mm 15N-labeled c-Src SH3 domain in the presence of 0, 5.4, 6.7, 7.5, 8.3, 9.2, 10.3, 12.6, and 15.7 mm NITYRGT. B, subspectra for c-Src SH3 domain residues Leu-92 and Arg-98 at NITYRGT concentrations of 0, 5.4, 6.7, 8.3, 9.2, 10.3, 12.6, and 15.7 mm. C, plot of CSP in ppm for residue Arg-98 as a function of NITYRGT concentration. D, CSP induced in each residue of the c-Src SH3 domain by 15.7 mm NITYRGT. The dotted line represents 2 standard deviations for CSP induced in the c-Src SH3 HSQC, determined by iteratively fitting the CSP to normal distributions as described by Schumann et al. (26). The calculated standard deviation of NITYRGT-induced CSP was 0.027 ppm. E, CSP induced by NITYRGT in the c-Src SH3 domain are color-coded by magnitude and mapped onto an NMR structure of the SH3 domain (PDB 1NLP).
The specificity of the CSP induced by NITYRGT is illustrated in Fig. 4B, which compares the CSP of Leu-92 and Arg-98 as a function of NITYRGT concentration. Although there were essentially no CSP for Leu-92, there was a direct linear relationship between the concentration of NITYRGT and the CSP induced for Arg-98. Nonetheless, the titration of Arg-98 did not reach saturation, and the majority of the NITYRGT was not bound to the c-Src SH3 domain, even at 15.7 mm (Fig. 4C). Under these conditions, [SH3]total approximates [SH3]free and [β3]total approximates [β3]free. Thus, the relationship Δppm = Δppmmax × ([SH3]free × [β3]free)/(Kd × [SH3]total) simplifies to Δppm = [β3]total × (Δppmmax/Kd). The term Δppmmax represents the change in chemical shift when the SH3 domain is fully bound by ligand. As saturation was not reached at the highest concentration, it is likely that the Kd is greater than 10 mm.
Further analysis of the NITYRGT titration results revealed fourteen SH3 domain residues [Glu-96, Ser-97, Arg-98, Thr-99, Glu-100, Thr-101, Asp-102, Leu-103, Asn-115, Asn-116, Glu-118, Ser-126, Gly-130, and Thr-132] with CSP exceeding the threshold of >2 standard deviations, indicative of specific protein-protein interaction (Fig. 4D) (27). The majority of these residues are located in the n-Src- and RT-loops, the specificity pocket, and in the region occupied by the linker connecting the SH2 and kinase domains in the compact structure of inactive c-Src (Fig. 4E). The largest CSP occurred in residues in or adjacent to the RT-loop, especially residues Arg-98 (0.25 ppm), Glu-100 (0.15 ppm), and Asp-102 (0.13 ppm). It is noteworthy that Asp-102, which forms a salt-bridge with the Arg of PPII helices (17, 28) has a significant chemical shift change, suggesting that Asp-102 also interacts with residue Arg-760 in the β3 CT. On the other hand, there were minimal CSP for the aromatic residues Tyr-93, Tyr-95, Trp-122, and Tyr-139.
Verifying the Specificity of NITYRGT Binding using the Mutant Peptide NITYEGK
The β1 integrin CT does not interact with the c-Src SH3 domain (3, 20, 21). Accordingly, to address the specificity of the interaction of NITYRGT with the SH3 domain, we repeated the NMR titration using the peptide NITYEGK in which we replaced the C-terminal three residues of NITYRGT with the corresponding residues of β1. When the SH3 domain was titrated with NITYEGK, chemical shifts were perturbed for a completely different set of residues than when the titration was performed using NITYRGT (Fig. 5, A–C). There were no large CSP in or around residues 91–107, as was the case for NITYGRT; instead small, but statistically significant, CSP were seen in residues Gln-112, His-125, Ser-126, Gly-130, and Thr-132. These residues are located on the 2nd, 3rd, and 4th β strands of the SH3 domain, distant from the face of the SH3 domain that binds PPII ligands and NITYRGT (Fig. 5D). These results indicate that the negative control peptide NITYEGK fails to interact with the specific polypeptide binding site of the SH3 domain.
FIGURE 5.

Interaction of the mutant β3/β1 peptide NITYEGK with the c-Src SH3 domain. A, CSP induced in each residue of the c-Src SH3 domain by NITYEGK. The dotted line represents 2 standard deviations for NITYEGK-induced CSP. The calculated standard deviation of NITYEGK-induced CSP was 0.004 ppm. B, plot of the magnitude of CSP in ppm for residue Gly-130 as a function of NITYEGK concentration. C, superimposition of normalized CSP induced by NITYEGK (blue) and NITYRGT (red, taken from Fig. 4). NITYEGK CSP were normalized by dividing the maximal CSP for each SH3 domain residue in NITYEGK spectra by the maximal CSP for Gly-130. NITYRGT CSP were normalized by dividing the maximal CSP for each residue in the NITYRGT spectra by the maximal CSP for Arg-98. D, CSP induced in the c-Src SH3 domain by NITYEGK are color-coded by magnitude and mapped onto an NMR structure of the SH3 domain (PDB 1NLP).
Comparing NITYRGT and PPII Peptide RPLPPLP Binding to the c-Src SH3 Domain
Because polyproline peptides selective for the c-Src SH3 domain blocked the interaction of the SH3 domain with the β3 CT in solid phase assays (3), we compared CSP induced in the c-Src SH3 domain by the class 1 PPII core peptide RPLPPLP with those induced by NITYRGT. Serial titration of RPLPPLP into a solution containing 15N-labeled SH3 caused significant CSP of up to 1 ppm in the SH3 HSQC spectra (Fig. 6A). Global fitting of CSP for Val-90, Leu-92, Val-114, Asn-115, and Ile-135, residues whose peaks were in the fast exchange mode as a function of RPLPPLP concentration and did not overlap with others, yielded a Kd of 83 ± 27 μm for RPLPPLP binding to the SH3 domain binding, similar to the Kd for RPLPPLP binding determined by SPR. Major CSP occurred in three regions: residues 98–103 (RT-loop), 117–122 (n-Src loop and specificity pocket including Trp-121 and Trp-122), and residues 137–138. It is noteworthy that the binding surface for RPLPPLP defined by CSP overlaps with that expected from the SH3 domain crystal structure, although negligible CSP patterns were observed for the aromatic cluster Tyr-93, Tyr-95, and Tyr-139 (Fig. 6B) (13, 17).
FIGURE 6.

NITYRGT and the PPII core peptide RPLPPLP bind to the same region of the c-Src SH3 domain. A, CSP caused by the peptide RPLPPLP for each residue in the HSQC spectra of the 15N-labeled c-Src SH3 domain. The dotted line represents 2 standard deviations for RPLPPLP-induced CSP. The calculated standard deviation of RPLPPLP-induced CSP was 0.100 ppm. B, magnitude of CSP are color-coded and mapped onto an NMR structure of the c-Src SH3 domain (PDB 1NLP). C, superimposition of normalized CSP for RPLPPLP (blue) and NITYRGT (red, taken from Fig. 4). CSP were normalized by dividing the maximal CSP for each residue by the maximal CSP for Arg-98, a residue whose chemical shift was most perturbed by each peptide. D, correlation between the normalized CSP induced in c-Src SH3 domain residues 83–113 by NITYRGT and RPLPPLP (R2 = 0.87).
To directly compare the binding sites for NITYRGT and RPLPPLP on the c-Src SH3 domain, we normalized the CSP induced by each peptide by dividing the CSP for each residue by the CSP for Arg-98 whose chemical shift was perturbed by both peptides. There was extensive overlap in the CSP induced by NITYRGT and RPLPPLP (Fig. 6C). Further, the CSP for SH3 residues 83–113 were highly correlated (R2 = 0.87) (Fig. 6D). Thus, both NITYRGT and RPLPPLP interact with residues located in the RT-loop of the SH3 domain, although RPLPPLP binds with substantially greater affinity.
Because of its relatively higher affinity, it is likely that RPLPPLP binding to the PPII binding site on the SH3 domain will block binding of the NITYRGT peptide. To demonstrate that this is the case, we mixed 0.33 mm 15N-labeled c-Src SH3 domain with 1.96 mm RPLPPLP (6 eq.) and serially titrated the mixture with NITYRGT at concentrations up to 8.03 mm. HSQC spectra were recorded, and CSP for each residue were calculated as described above. Under these conditions, CSP were induced by NITYRGT, but the chemical shifts for residues located in the RPLPPLP binding site were unperturbed (Fig. 7A). Instead, CSP was observed for residues that non-specifically interacted with NITYEGK (Fig. 7, B and C) with an essentially perfect correlation between the CSP induced by both peptides (R2 = 0.96) (Fig. 7D). Thus, these results indicate that when the PPII binding site in the SH3 domain is occupied by a PPII helix, the β3 CT is unable to specifically interact with c-Src.
FIGURE 7.

Occupation of the PPII helix binding site in the SH3 domain by RPLPPLP prevents NITYRGT binding. A, CSP caused by NITYRGT in the HSQC spectra of the 15N-labeled c-Src SH3 domain when RPLPPLP is pre-bound to the SH3 domain. B, sub-spectra for residues Glu-100, Lys-107, Gln-112, Thr-117, Ser-126, and Gly-130 illustrating the absence of CSP in the RPLPPLP binding site and the presence of CSP in the NITYEGK interaction site. C, superimposition of normalized CSP in the c-Src SH3 domain induced by NITYEGK (blue) and by NITYRGT (red) when RPLPPLP is pre-bound to the c-Src SH3 domain. CSP were normalized by dividing the maximum CSP for each residue by the maximum CSP for Gly-130. D, correlation between the normalized CSP induced in c-Src SH3 domain by NITYEGK and by NITYRGT in the presence of RPLPPLP (R2 = 0.96).
Discussion
αIIbβ3-mediated outside-in signaling in platelets appears to be initiated by c-Src bound to the β3 CT via its SH3 domain. However, when and how c-Src binds to β3 remains unclear, in particular since the ligand binding surface on the SH3 domain is already occupied in inactive c-Src (3) and the affinity of PPII helices for the SH3 domain is several orders of magnitude greater than that of β3. Here, we have addressed these questions, finding first that c-Src binding to β3, as well as c-Src activation, increase with time following platelet stimulation and second that the binding site for the β3 CT on the c-Src SH3 domain overlaps with the binding site for PPII helices.
Recently, Xiao et al. reported a crystal structure in which the tri-peptide RGT was bound to residues 119–122 in the n-Src loop of the c-Src SH3 domain (numbering according to UniProtKB/Swiss-Prot entry P12931) and speculated that by binding at this location, RGT might disrupt the intramolecular interaction between the SH3 domain and the linker between the SH2 and kinase domains (29). We found that both RPLPPLP and NITYRGT caused CSP in these residues, but the CSP were substantially smaller than those induced in residues 98–103 and those caused by NITYRGT were no longer present when the SH3 domain was occupied by RPLPPLP, implying that NITYRGT binding is not sufficiently strong to displace a PPII helix from the SH3 domain. Subsequently, Katyal et al. reported NMR and paramagnetic relaxation enhancement (PRE) studies that localized the β3 CT binding site in the SH3 domain to an extended interface consisting of residues in both the RT and n-Src loops, as well as an isolated interaction with Tyr-134 in the 4th β strand (18). Similar to our results, the largest CSP they detected involved residues 98–103 (numbering according to UniProtKB/Swiss-Prot entry P12931). Nevertheless, they focused their attention on residues 138–139, as well as residues 118–121 in the n-Src loop, because the PRE experiments suggested that the β3 C-terminal Thr binds in proximity to these residues. Further, in contrast to the data shown in Fig. 2, they concluded that the SH3 domain interacts constitutively with inactive αIIbβ3 because they did not detect CSP when the β3 CT was phosphorylated at tyrosine residues 747 and 759, phosphorylations that occur subsequent to αIIbβ3 activation (30).
Our SPR and NMR measurements of β3 CT binding to the c-Src SH3 domain indicated that the interaction is substantially weaker than had been reported previously when c-Src SH3 binding was measured to the β3 CT immobilized on a solid support. One possible explanation for this discrepancy is that we used the seven C-terminal β3 residues for our measurements, whereas the entire β3 CT was immobilized for the solid-state measurements (3). Nonetheless, it appears that the C-terminal β3 residues are sufficient to mediate binding to the SH3 domain (3, 18). A second possibility is that the increased affinity was a reflection of an increased avidity of dimeric GST-SH3 fusion proteins (3, 31). But the third and most likely possibility is that additional interactions contribute to the interaction of β3 with c-Src under physiologic conditions and were absent in our measurements. For example, sequences in the β3 CT adjacent to those essential for c-Src binding also recognize members of the kindlin and filamin protein families, and these proteins could cooperate to enhance c-Src binding. Further, we recently found that in solution, the β3 CT has weak affinity for the talin-1 FERM domain, whereas appending it to acidic phospholipids increased its affinity for the FERM domain by three orders of magnitude (19). Since all Src family kinases are myristoylated (32, 33) and c-Src contains a conserved patch of positively-charged Arg and Lys residues that are necessary for its binding to bilayers containing negatively-charged phospholipids (34), it is possible that the interaction of c-Src with β3 is a ternary interaction in which protein-lipid interactions play an important role. Lastly, in the presence of membranes, c-Src binding to β3 would be restricted to a 2-dimensional surface where binding partners have much higher probability of collision than in 3-dimensional space.
We found that the β3 CT peptide NITYRGT binds to a site on the RT-loop of c-Src SH3 domain that includes residues 98–103 and that can also be occupied by the canonical polyproline II helical peptide RPLPPLP, although RPLPPLP binds to the site with significantly greater affinity (22, 23). CSP for acidic residues located in the RT-loop were particularly sensitive to the presence of NITYRGT and were not present when we replaced the C-terminal residues of NITYRGT with EGK. Thus, it is likely that the conserved β3 residue Arg-760 makes an important contribution to NITYRGT binding, perhaps by forming a salt-bridge with SH3 domain residue Asp-102. We also detected overlapping CSP for NITYRGT and RPLPPLP for more C-terminal SH3 domain residues, but the magnitude of the CSP induced by NITYRGT were quite small, implying that the predominant binding site for β3 CT encompasses SH3 domain residues 98–103 (27). This contrasts with PPII helices like RPLPPLP whose binding interface on the SH3 domain is much more extensive (17) and likely responsible for their substantially greater affinity for SH3 domain binding.
The model shown in Fig. 8, based on the results described above, illustrates the behavior of c-Src in unstimulated and stimulated platelets. In unstimulated platelets, inactive c-Src does not interact with inactive αIIbβ3 when the PPII helix binding on the SH3 domain is occupied. However, the very high concentrations of both proteins in platelets, as well as interactions with other partner proteins and with membranes would tend to co-localize c-Src to the vicinity of the β3 CT (10, 34). Following agonist-stimulated platelet activation, c-Src is “unlatched” by dephosphorylation of pTyr530 (31), relieving the inhibitory packing of the SH2 and SH3 against the lobes of the kinase domain (14). The β3 CT is now able to bind with substantially greater affinity to SH3 domain residues 98–103 vacated by the linker between the c-Src SH2 and kinase domains. Subsequent αIIbβ3 clustering dramatically increases the concentration of c-Src, enabling the auto-transphosphorylation of Tyr-419 in the activation loop of the c-Src kinase domain, the completion of c-Src activation, and the initiation of outside-in platelet signaling.
FIGURE 8.

A proposed mechanism for αIIbβ3-mediated c-Src activation. In circulating platelets, where both αIIbβ3 and c-Src are present in their inactive conformations, there is no specific interaction between the β3 CT and c-Src. When platelets are stimulated, αIIbβ3 is activated, c-Src is extended, and the β3 CT is able to interact with the PPII helix binding surface of the c-Src SH3 domain that is no longer occupied by the linker between the SH2 and kinase domains. Activated αIIbβ3 then oligomerizes, enabling the trans-autophosphorylation of Tyr-419 responsible for c-Src activation.
The work was supported by National Institutes of Health Grants P01 HL40387 (to J. S. B. and W. F. D.), R01 GM056250 (to H. R.), P01 CA06927 to the Fox Chase Cancer Center, and GM054616. The Spectroscopy Support Facility at the Fox Chase Cancer Center provided access to NMR instrumentation. The authors declare that they have no conflicts of interest with the contents of this article.
- SFK
- Src family kinase
- SPR
- surface plasmon resonance
- CSP
- chemical shift perturbation.
References
- 1. Shattil S. J., Kashiwagi H., Pampori N. (1998) Integrin signaling: the platelet paradigm. Blood 91, 2645–2657 [PubMed] [Google Scholar]
- 2. Haling J. R., Monkley S. J., Critchley D. R., Petrich B. G. (2011) Talin-dependent integrin activation is required for fibrin clot retraction by platelets. Blood 117, 1719–1722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Arias-Salgado E. G., Lizano S., Sarkar S., Brugge J. S., Ginsberg M. H., Shattil S. J. (2003) Src kinase activation by direct interaction with the integrin beta cytoplasmic domain. Proc. Natl. Acad. Sci. U.S.A. 100, 13298–13302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Senis Y. A., Mazharian A., Mori J. (2014) Src family kinases: at the forefront of platelet activation. Blood 124, 2013–2024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yeatman T. J. (2004) A renaissance for SRC. Nat. Rev. Cancer 4, 470–480 [DOI] [PubMed] [Google Scholar]
- 6. Ingley E. (2008) Src family kinases: regulation of their activities, levels and identification of new pathways. Biochim. Biophys. Acta 1784, 56–65 [DOI] [PubMed] [Google Scholar]
- 7. Kim L. C., Song L., Haura E. B. (2009) Src kinases as therapeutic targets for cancer. Nature Reviews. Clinical Oncology 6, 587–595 [DOI] [PubMed] [Google Scholar]
- 8. Lowell C. A., Soriano P. (1996) Knockouts of Src-family kinases: stiff bones, wimpy T cells, and bad memories. Genes Dev. 10, 1845–1857 [DOI] [PubMed] [Google Scholar]
- 9. Stehelin D., Varmus H. E., Bishop J. M., Vogt P. K. (1976) DNA related to the transforming gene (s) of avian sarcoma viruses is present in normal avian DNA. Nature 260, 170–173 [DOI] [PubMed] [Google Scholar]
- 10. Obergfell A., Eto K., Mocsai A., Buensuceso C., Moores S. L., Brugge J. S., Lowell C. A., Shattil S. J. (2002) Coordinate interactions of Csk, Src, and Syk kinases with [alpha]IIb[beta]3 initiate integrin signaling to the cytoskeleton. J. Cell Biol. 157, 265–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pumiglia K., Feinstein M. (1993) Thrombin and thrombin receptor agonist peptide induce tyrosine phosphorylation and tyrosine kinases in the platelet cytoskeleton. Translocation of pp60c-src and integrin αIIb β3 (glycoprotein IIb/IIIa) is not required for aggregation, but is dependent on formation of large aggregate structures. Biochem. J. 253, 253–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Clark E. A., Brugge J. S. (1993) Redistribution of activated pp60c-src to integrin-dependent cytoskeletal complexes in thrombin-stimulated platelets. Mol. Cell. Biol. 13, 1863–1871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Xu W., Harrison S. C., Eck M. J. (1997) Three-dimensional structure of the tyrosine kinase c-Src. Nature 385, 595–602 [DOI] [PubMed] [Google Scholar]
- 14. Xu W., Doshi A., Lei M., Eck M. J., Harrison S. C. (1999) Crystal structures of c-Src reveal features of its autoinhibitory mechanism. Mol. Cell 3, 629–638 [DOI] [PubMed] [Google Scholar]
- 15. Sicheri F., Moarefi I., Kuriyan J. (1997) Crystal structure of the Src family tyrosine kinase Hck. Nature 385, 602–609 [DOI] [PubMed] [Google Scholar]
- 16. Metcalf D. G., Moore D. T., Wu Y., Kielec J. M., Molnar K., Valentine K. G., Wand A. J., Bennett J. S., DeGrado W. F. (2010) NMR analysis of the αIIb β3 cytoplasmic interaction suggests a mechanism for integrin regulation. Proc. Natl. Acad. Sci. U.S.A. 107, 22481–22486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li S. S. (2005) Specificity and versatility of SH3 and other proline-recognition domains: structural basis and implications for cellular signal transduction. Biochem. J. 390, 641–653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Katyal P., Puthenveetil R., Vinogradova O. (2013) Structural insights into the recognition of beta3 integrin cytoplasmic tail by the SH3 domain of Src kinase. Protein Sci 22, 1358–1365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Moore D. T., Nygren P., Jo H., Boesze-Battaglia K., Bennett J. S., DeGrado W. F. (2012) Affinity of talin-1 for the β3-integrin cytosolic domain is modulated by its phospholipid bilayer environment. Proc. Natl. Acad. Sci. U.S.A. 109, 793–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ablooglu A. J., Kang J., Petrich B. G., Ginsberg M. H., Shattil S. J. (2009) Antithrombotic effects of targeting αIIbβ3 signaling in platelets. Blood 113, 3585–3592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liao Z., Kato H., Pandey M., Cantor J. M., Ablooglu A. J., Ginsberg M. H., Shattil S. J. (2015) Interaction of kindlin-2 with integrin β3 promotes outside-in signaling responses by the αVβ3 vitronectin receptor. Blood 125, 1995–2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sparks A. B., Quilliam L. A., Thorn J. M., Der C. J., Kay B. K. (1994) Identification and characterization of Src SH3 ligands from phage-displayed random peptide libraries. J. Biol. Chem. 269, 23853–23856 [PubMed] [Google Scholar]
- 23. Rickles R. J., Botfield M. C., Zhou X. M., Henry P. A., Brugge J. S., Zoller M. J. (1995) Phage display selection of ligand residues important for Src homology 3 domain binding specificity. Proc. Natl. Acad. Sci. U.S.A. 92, 10909–10913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 25. Yu H., Rosen M. K., Schreiber S. L. (1993) 1H and 15N assignments and secondary structure of the Src SH3 domain. FEBS Lett. 324, 87–92 [DOI] [PubMed] [Google Scholar]
- 26. Schumann F. H., Riepl H., Maurer T., Gronwald W., Neidig K. P., Kalbitzer H. R. (2007) Combined chemical shift changes and amino acid specific chemical shift mapping of protein-protein interactions. J. Biomol NMR 39, 275–289 [DOI] [PubMed] [Google Scholar]
- 27. Williamson M. P. (2013) Using chemical shift perturbation to characterise ligand binding. Progress in Nuclear Magnetic Resonance Spectroscopy 73, 1–16 [DOI] [PubMed] [Google Scholar]
- 28. Yu H., Chen J. K., Feng S., Dalgarno D. C., Brauer A. W., Schreiber S. L. (1994) Structural basis for the binding of proline-rich peptides to SH3 domains. Cell 76, 933–945 [DOI] [PubMed] [Google Scholar]
- 29. Xiao R., Xi X. D., Chen Z., Chen S. J., Meng G. (2013) Structural framework of c-Src activation by integrin β3. Blood 121, 700–706 [DOI] [PubMed] [Google Scholar]
- 30. Law D. A., DeGuzman F. R., Heiser P., Ministri-Madrid K., Killeen N., Phillips D. R. (1999) Integrin cytoplasmic tyrosine motif is required for outside-in αIIbβ3 signaling and platelet function. Nature 401, 808–911 [DOI] [PubMed] [Google Scholar]
- 31. Arias-Salgado E. G., Haj F., Dubois C., Moran B., Kasirer-Friede A., Furie B. C., Furie B., Neel B. G., Shattil S. J. (2005) PTP-1B is an essential positive regulator of platelet integrin signaling. J. Cell Biol. 170, 837–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Resh M. D. (1999) Fatty acylation of proteins: new insights into membrane targeting of myristoylated and palmitoylated proteins. Biochim. Biophys. Acta 1451, 1–16 [DOI] [PubMed] [Google Scholar]
- 33. Satoh K., Matsuki-Fukushima M., Qi B., Guo M. Y., Narita T., Fujita-Yoshigaki J., Sugiya H. (2009) Phosphorylation of myristoylated alanine-rich C kinase substrate is involved in the cAMP-dependent amylase release in parotid acinar cells. American Journal of Physiology. Gastrointestinal and Liver Physiology 296, G1382–1390 [DOI] [PubMed] [Google Scholar]
- 34. Pérez Y., Maffei M., Igea A., Amata I., Gairí M., Nebreda A. R., Bernadó P., Pons M. (2013) Lipid binding by the Unique and SH3 domains of c-Src suggests a new regulatory mechanism. Scientific Reports 3, 1295. [DOI] [PMC free article] [PubMed] [Google Scholar]