

Background: The N111G and D74N mutations bias the AT1 receptor for the Gq/11 and β-arrestin pathways, respectively.
Results: Structural rearrangements of theAT1 receptor are induced by the N111G mutation and AngII.
Conclusion: Activation of the Gq/11 and β-arrestin pathways is associated with a decreased and increased stability, respectively, of the ground state of the receptor.
Significance: Distinct conformations of AT1 receptor are associated with distinct pathways.
Keywords: angiotensin II, G protein-coupled receptor (GPCR), molecular dynamics, protein conformation, receptor structure-function, AT1 receptor, AT1R, activation mechanism, biased signaling, functional selectivity
Abstract
Biased signaling represents the ability of G protein-coupled receptors to engage distinct pathways with various efficacies depending on the ligand used or on mutations in the receptor. The angiotensin-II type 1 (AT1) receptor, a prototypical class A G protein-coupled receptor, can activate various effectors upon stimulation with the endogenous ligand angiotensin-II (AngII), including the Gq/11 protein and β-arrestins. It is believed that the activation of those two pathways can be associated with distinct conformations of the AT1 receptor. To verify this hypothesis, microseconds of molecular dynamics simulations were computed to explore the conformational landscape sampled by the WT-AT1 receptor, the N111G-AT1 receptor (constitutively active and biased for the Gq/11 pathway), and the D74N-AT1 receptor (biased for the β-arrestin1 and -2 pathways) in their apo-forms and in complex with AngII. The molecular dynamics simulations of the AngII-WT-AT1, N111G-AT1, and AngII-N111G-AT1 receptors revealed specific structural rearrangements compared with the initial and ground state of the receptor. Simulations of the D74N-AT1 receptor revealed that the mutation stabilizes the receptor in the initial ground state. The presence of AngII further stabilized the ground state of the D74N-AT1 receptor. The biased agonist [Sar1,Ile8]AngII also showed a preference for the ground state of the WT-AT1 receptor compared with AngII. These results suggest that activation of the Gq/11 pathway is associated with a specific conformational transition stabilized by the agonist, whereas the activation of the β-arrestin pathway is linked to the stabilization of the ground state of the receptor.
Introduction
The angiotensin-II type 1 (AT1)2 receptor, a class A GPCR, and its cognate ligand, the octapeptide hormone angiotensin II (AngII), are part of the renin-angiotensin-aldosterone system, responsible for controlling blood pressure and water retention via vascular smooth muscle contraction. The AT1 receptor can also activate steroidogenesis in the adrenal gland, neurosecretion, neuronal activity, cell growth, and proliferation (1). The AT1 receptor is classically known to signal through the Gq/11 pathway, but as is the case with many GPCRs, it also activates other pathways, including β-arrestins, G12/13 proteins, and the epidermal growth factor receptor (2–5). This ability to activate different pathways can be biased by certain ligands (5, 6) or by mutating key amino acids in the receptor (4, 7), a phenomenon known as functional selectivity or biased signaling. Of particular interest is a region within the receptor identified as the major H-bond network (MHN). It is composed of several functionally important and conserved polar residues among class A GPCRs (8, 9). This region has also been identified as a sodium-binding site in some GPCRs (10, 11). Mutations within the MHN can impact the receptor's ability to signal via certain pathways. The N111G mutation, known to increase the constitutive activity of the receptor on the Gq/11 pathway, impedes its phosphorylation by G protein-coupled receptor kinases and diminishes its coupling to β-arrestin1 after stimulation with AngII (7, 12, 13). However, the D74N mutation reduces signaling through the Gq/11 pathway but maintains wild-type level of signaling through the β-arrestin pathway when stimulated with AngII. The D74N-AT1 receptor shows increased β-arrestin signaling compared with the WT receptor when stimulated with the β-arrestin-biased agonist [Sar1,Ile4,Ile8]AngII (4). These data suggest that the MHN is important to both the Gq/11 and the β-arrestin pathways and that mutations in this region of the receptor can tip the scale of functional selectivity in either direction. Although other regions are likely to play key roles in the biased signaling of the AT1 receptor, the characterization of the impact of the N111G and D74N mutations on the MHN and the structure of the receptor in presence and in absence of AngII is of the utmost importance for understanding the underlying mechanisms of biased signaling of the AT1 receptor and GPCRs in general, which is poorly understood at the structural level.
In previous work, we showed that residues N1113.35 and D742.50 are part of a relatively stable H-bond network in the ground state. The N111G mutation destabilized this H-bond network by removing the NH2 moiety involved in the internal stabilization of D742.50 carboxylate, thus favoring a reorientation of the side chain of D742.50 to form a new H-bond with residue N461.50. The importance of the Asp74–Asn46 interaction for Gq/11 signaling was confirmed in vitro (9). This study led us to postulate that the mutation of residue D742.50 to an asparagine could potentially stabilize the MHN and limit the reorganization of the MHN. Here, we further hypothesize that the destabilization and reorganization of the MHN (caused by the N111G mutation) and local structural changes of AT1 receptor are favoring Gq/11 signaling, whereas the stabilization of the MHN (caused by the D74N mutation) is favoring β-arrestin signaling. To verify this hypothesis, we used microsecond time scale MD simulations of the WT-AT1 receptor, N111G-AT1 receptor, and D74N-AT1 receptor to explore their conformational landscape by looking at specific structural determinants. Furthermore, based on a previously developed model of the AT1 receptor in complex with AngII (14), we looked at how the presence of AngII in the binding pocket modified the conformational landscapes of the WT-AT1, D74N-AT1, and N111G-AT1 receptors and also how the β-arrestin-biased agonist [Sar1,Ile8]AngII (SI8) (15, 16) modified the conformational landscapes of the WT-AT1 receptor. The simulations suggest that the N111G mutation destabilizes the ground state of the receptor. This destabilization favors conformational changes consistent with a transition from an inactive to an active state of the receptor engaged with a G protein. Conversely, we observe that the D74N mutation stabilizes the ground state, thus reducing the conformational landscape explored by the receptor. In accordance with our hypothesis, the presence of AngII in the WT-AT1 receptor favors the same conformational transitions as the N111G mutation. However, AngII further stabilizes the ground state of the β-arrestin-biased D74N-AT1 receptor. In the WT-AT1 receptor, the ligand SI8 preferentially stabilized the ground state of the receptor compared with AngII.
Experimental Procedures
Materials
Desktop computers were used for the preparation and equilibration phase of the simulations. Production MD computations were made on the supercomputer Mammouth Parallèle II3 from the Université de Sherbrooke, managed by Calcul Québec and Compute Canada. All reagents were from Sigma unless otherwise indicated. Culture media, trypsin, FBS, penicillin, and streptomycin were from WISENT (St-Bruno, Quebec, Canada). Opti-MEM was from Invitrogen. Polyethyleneimine (PEI) was from Polysciences (Warrington, PA). Coelenterazine 400A was from Gold Biotechnology (St. Louis, MO).
Residue Numbering Scheme
Residues of the AT1 receptor are given two numbering schemes. First, residues are numbered according to their positions in the AT1 receptor sequence. Second, residues are also indexed according to their position relative to the most conserved residue in the TMD where they are located. By definition, the most conserved residue is assigned the position index “50,” e.g. in TMD2, Asp74 is the most conserved residue and is designated D742.50, whereas the upstream residue is designated A732.49 and the downstream residue is designated L752.51. This indexing simplifies the identification of aligned residues in different GPCRs (17).
Homology Modeling
We used the I-TASSER server to generate multiple template homology structures of the AT1 receptor. The resulting five best structures provided in the output had near identical orientations of the side chains of the H-bond network. We selected the only structure that featured both known disulfides bonds, which had a high confidence score of 0.99 (18, 19). The backbone of the model is very similar to the crystal structure of the CXCR4 receptor (PDB code 3ODU), with a root mean square deviation (r.m.s.d.) distance of 0.900 Å between the positions of Cα atoms. Sequence alignment between AT1 and CXCR4 and superposition of the two structures (Fig. 3A) can be found in our previous work using this model (9). The homology model was also analyzed with ProCheck (20), and the Ramachandran plot indicated that over 97% of the residues were in the “most favored” and “additionally allowed” regions. The rest of the stereochemistry was also of high quality. The unstructured N- and C-terminal portions of the model were truncated by removing residues 1–14 and 319–359, respectively, to keep the simulation box as small as possible. This enables better performances for the MD simulations. AngII's initial conformation and position inside the binding pocket were determined in a previous work (14). Models of the N111G-AT1 receptor and D74N-AT1 receptor were generated by replacing residue Asn111 or Asp74 by the corresponding residue using the mutagenesis feature in PyMOL.
FIGURE 3.

Snapshots from the MD simulations showing interactions in the MHN in the Gq-inactive and Gq-active states. A, schematic representation of the backbone of the AT1 receptor homology model produced by the I-TASSER server (colored) closely matches the crystal structure of the CXCR4 receptor (gray). of the residues of the MHN are visible as sticks. B and E, in the WT-AT1 receptor, the side chains of S2526.47 form an H-bond with the side chain of N2947.45. The side chains of N1113.35 and N2957.46 form an H-bond with D742.50. C and F, after the conformational change in the N111G-AT1 receptor, AngII-N111G-AT1 receptor, and AngII-WT-AngII receptor; the side chain of S2526.47 can form H-bonds with backbone amines of Phe293 and Asn294 or the backbone carbonyl of Ala291 (thin line representation) to stabilize the conformational change of TMD7 (red ribbon). The side chain of D742.50 can form H-bonds with the side chains of N2947.45, N2957.46, and N461.50. D and G, in the D74N-AT1 receptor and AngII-D74N-AT1 receptor, interactions are as described for B, but additionally include an H-bond between residue D74N2.50 and N461.50 and between N2957.46 and S1153.39. Transmembrane domains are shown as colored ribbons (TMD1 = dark blue; TMD2 = light blue; TMD3 = aqua; TMD6 = orange, and TMD7 = red). Side chains are show as sticks. Oxygen atoms are red; nitrogen atoms are blue; hydrogen atoms are white, and carbon atoms are colored according to their TMD. H-bonds predicted by PyMOL are shown as yellow dashed lines.
Molecular Dynamics Simulations
The GROMACS software suite (21–24) was used to prepare and run the simulations. The AT1 receptor, N111G-AT1 receptor, and D74N-AT1 receptor models were inserted in a lipid bilayer consisting of 128 molecules of DOPC using the InflateGRO approach (25). Simulation parameters were based on previous work (26–28). The membrane-receptor system was solvated with the SPC water model (29). Counter-ions were added at random positions, replacing water molecules, to keep the net charge of the system at 0. The ffg53a6 force field, modified to use the Berger lipids parameter (30), was used for the calculations. Parameters for the DOPC molecules and the PDB file of the bilayer, developed by the Tieleman group (31–33), were obtained from Peter Tieleman's website. A first equilibration phase was performed under conditions of constant pressure, temperature, and number of molecules (NPT) for 1 ns while gradually heating the system for the first 500 ps to reach the desired temperature of 310 K. During this first phase, the phosphate head group of the DOPC molecules was restrained. This was followed by a second equilibration in NPT conditions for 15 ns with the pressure set at 1 bar, without the restraints on the DOPC molecules. Such a long equilibration is necessary for proper equilibration of the lipids after embedding a protein in the membrane (34). The positions of all heavy atoms of the receptor and ligand were restrained during equilibration. Unrestrained MD simulations were run in 2-fs steps for 1 μs of total MD simulation time, in the form of 10 simulations of 100 ns in length for each receptor. For the WT-AT1 receptor, an additional 20 simulations of ∼125 ns were performed, for a combined total of around 3.5 μs of MD simulation time. The 100-ns simulation length was deemed long enough for our study, which aims at analyzing side-chain re-orientation in the core of the AT1 receptor and, to a certain extent, backbone rearrangements caused by mutations or ligand binding. These motions occur on the nanosecond time scale (35). Using multiple shorter MD trajectories rather than a single long one prevents known problems that can occur with long trajectories in current force fields (36). Random seed was used for velocity generation during the initial equilibration phase. Thus, all MD trajectories of a given system (e.g. AngII-WT-AT1 receptor) share the same initial conformation and velocities at the start of the production MD runs. Divergence in the multiple trajectories is the result of the inherent imprecision of the calculations, which originate from the single-precision floating point format used as well as differences in the order of addition of force caused by dynamic load balancing ((a + b) + c ≠ a + (b + c) due to rounding-off). The simulations were run in periodic boundary conditions at constant temperature (310 K) and pressure (1 bar) using the Nose-Hoover thermostat (37, 38) with τT = 0.2 ps and the Parrinello-Rahman barostat with τP = 5 ps, respectively. Simulation data were saved every 20 ps, for a total of 50,001 frames/μs. Stability of the systems was assessed by calculating the r.m.s.d. distance between the positions of Cα atoms of the TMDs during the simulations. In all trajectories, the r.m.s.d. converged to values between 2.5 and 3.5 Å independently of the receptor mutants, indicating that equilibrium was reached.
Trajectory Analysis
MD trajectory output from GROMACS were converted to PDB files for visual inspection with PyMOL (39) and to compressed XTC trajectory files for other analyses. Data regarding distances, dihedral angles, and solvent-accessible surface were performed, respectively, with the g_dist, g_angle, and g_sas tools within GROMACS. The r.m.s.d. for the region of TMD7 undergoing a conformational transition was calculated using the g_rms tool after performing a least squares fit on the backbone atoms of residues 288–301. Two-dimensional probability density functions describing the populations of substrates defined by the selected metrics were calculated using g_sham with a grid of 50 × 50 bins and nlevels = 200.
Constructs
The cDNA clone for the human AT1 receptor was kindly provided by Dr. Sylvain Meloche (University of Montréal). The AT1-GFP10 construct was built by inserting the GFP10 sequence at the C terminus of the AT1 construct, joined by the linker GSAGT, using the In-Fusion® PCR cloning system (Clontech) as recommended by the manufacturer. The RLuc-βarrestin1 and RLuc-βarrestin2 constructs were kindly provided by Dr. Michel Bouvier (University of Montréal). The N111G-AT1-GFP10 and D74N-AT1-GFP10 constructs were built using the QuikChange II XL mutagenesis kit (Stratagene, La Jolla, CA) as recommended by the manufacturer. Briefly, forward and reverse oligonucleotides were constructed to introduce either the N111G or D74N mutation in the AT1-GFP10 receptor background. Site-directed mutations were then confirmed by automated DNA sequencing by aligning the AT1 receptor sequence with MultAlin (40).
Cell Culture and Transfection
HEK293 cells were maintained in DMEM supplemented with 10% FBS, 100 IU/ml penicillin, and 100 μg/ml streptomycin at 37 °C in a humidified 5% CO2 atmosphere. The day prior to transfection, cultured cells were washed with PBS at room temperature, trypsinized, and seeded at 150,000 cells/well in a 6-well plate. For transfection, 2 μg of the DNA construct containing the appropriate AT1 receptor construct was added to 100 μl of Opti-MEM medium containing 6 μg of PEI, and the mixture was incubated for 20 min before being added to the cultured cells, as described previously (41). For β-arrestin recruitment assays, HEK293 cells (3 × 106 cells) were transiently transfected with 8700 ng of AT1-GFP10 or mutant receptors and either 300 ng of Rluc-βarrestin1 or 300 ng of Rluc-βarrestin2 using linear PEI (1 mg/ml) (PEI/DNA ratio 4:1).
Inositol Phosphate Production
Inositol monophosphate (IP1) production was determined using the IP-One assay (CisBio Bioassays, Bedford, MA). Necessary dilutions of the agonist AngII were prepared in stimulation buffer (Hepes 10 mm, CaCl2 1 mm, MgCl2 0.5 mm, KCl 4.2 mm, NaCl 146 mm, glucose 5.5 mm, LiCl 50 mm, pH 7.4). 48 h after transfection, the cells were washed with phosphate-buffered saline (PBS) at room temperature. The cells were trypsinized and distributed at 20,000 cells/well (7 μl) in a white 384-well plate in stimulation buffer. Cells were stimulated at 37 °C for 30 min with increasing concentrations of AngII. Cells were then lysed with 3 μl of IP1-d2. After addition of 3 μl of anti-IP1-cryptate antibody and cells were incubated for 1 h at room temperature under agitation. FRET signal was measured with a TECAN M1000 plate reader.
β-Arrestin Recruitment in BRET-based Biosensor Assays
At 48 h post-transfection, cells were washed with PBS and resuspended in stimulation buffer. For the β-arrestin recruitment assays, the proximity of fusion protein RLuc-βarrestin to the reporter AT1-GFP10 is evaluated. Upon stimulation, RLuc-βarrestin is recruited to the AT1-GFP10 fusion protein, whereby the BRET signal is increased. Cells transfected with the appropriate constructs were stimulated with the indicated ligands in 96-well white plates (50,000 cells/well) for 8 min, and then coelenterazine 400A was added at a final concentration of 5 μm. All BRET signals were measured using a TECAN M1000 fluorescence reader (TECAN, Austria). The BRET ratio was calculated as the GFP10 emission over luminescence emission. Net BRET ratio was calculated by subtracting the BRET ratio upon maximal stimulation with the BRET ratio under basal conditions. All data were expressed as a percentage of maximal AngII response toward AT1-GFP10.
Results
Experimental Validation of the Biases Caused by the N111G and D74N Mutations
We verified that the WT-AT1, the N111G-AT1, and the D74N-AT1 receptors can associate with β-arrestins (1 and 2) using the BRET2 assay. The N111G-AT1 receptor displayed a higher basal signal than the other two receptors (0.075 versus 0.045). After stimulation with AngII, the N111G-AT1 receptor showed poor BRET ratio increases of about 0.020 with both β-arrestins (Fig. 1, A and C). The D74N-AT1 receptor showed a BRET ratio increase of 0.060 with β-arrestin1 and a BRET ratio increase of 0.075 with β-arrestin2. The WT-AT1 receptor showed a BRET ratio increase of 0.075 for β-arrestin1 and a BRET ratio increase of 0.100 with β-arrestin2. We used the data to establish the maximum increase in β-arrestin recruitment relative to the WT-AT1 receptor after normalizing for each receptor's level of expression (Bmax). Fig. 1, B and D, shows that stimulation of the D74N-AT1 receptor with AngII produced 74 and 77% of the signal of the WT-AT1 receptor for β-arrestin1 and β-arrestin2 recruitment, respectively. Stimulation of the N111G-AT1 receptor with AngII produced 59 and 55% of the signal of the WT-AT1 receptor for β-arrestin1 and β-arrestin2 recruitment, respectively. Inositol phosphate production assays confirmed the constitutive activity of the N111G-AT1 receptor and the poor activation of the Gq/11 pathway by the D74N-AT1 receptor (Fig. 1E). These results confirmed the bias of the N111G-AT1 receptor toward the Gq/11 pathway and the bias of the D74N-AT1 receptor toward the β-arrestin pathway.
FIGURE 1.

Signaling properties of the WT-AT1 receptor and the biased mutants on the β-arrestins and inositol phosphate pathways. HEK293 cells were transfected with the indicated receptor, and their recruitment of β-arrestin1 (A), β-arrestin2 (C), and IP1 production (E) was assayed as described under “Experimental Procedures.” Each point represents the mean ± S.D. of duplicate determinations of a typical experiment, which is representative of at least three independent experiments. Bar graphs represent the mean ± S.E. for the maximum increase in β-arrestin1 (B) and β-arrestin2 (D) recruitment relative to the WT-AT1 receptor after normalizing for each receptor's level of expression.
MD Simulations of the N111G Mutant Suggest That the Activation of the Gq/11 Pathway Is Linked to a Destabilization of the Helical Structure of TMD7 of the AT1 Receptor
As described previously, we simulated the molecular dynamics of an experimentally validated homology model of the “resting” state of AT1 receptor (and mutants) embedded in a hydrated DOPC bilayer (9). Each system (WT-AT1, N111G-AT1, and D74N-AT1) was simulated for at least 1 μs of MD time carried out as 10 × 100-ns MD simulations. We also simulated each receptor in the presence of AngII. We used ligand poses satisfying experimentally determined receptor contacts previously obtained by photoaffinity labeling as starting structure of the three complexes (14). In our previous MD simulation of the N111G receptor (9), we had observed a conformational change in TMD7, between residues I2887.39 and N2957.46. In fact, we had witnessed a transition from a helical to an extended configuration of the backbone. We thus monitored this region of the receptor in our most recent simulations to see whether it was consistent and reproducible.
By measuring the r.m.s.d. of the Cα atoms of residues I2887.39 to N2957.46 following a superposition of TMD7 in all frames of the MD simulations, it is possible to monitor the local structural change in the helix while ignoring rigid body movements. This analysis of the trajectories of the WT-AT1 receptor without ligand showed that residues I2887.39 through N2957.46 had a stable helical conformation. Indeed, a single population (with maximum probability (pmax) of 0.160 at r.m.s.d. = 0.040 nm) can be seen on the one-dimensional probability distribution function (Fig. 2). The D74N mutation showed higher probability (0.230) and narrower distribution around the same r.m.s.d., suggesting an increased stability of that region of TMD7 (Fig. 2A). The N111G mutation, as expected, showed a much broader r.m.s.d. distribution (reaching beyond 0.30 nm) with the appearance of new populations (pmax = 0.058), indicating a change in conformation of this region of TMD7 (Fig. 2B). An r.m.s.d. beyond 0.16 nm indicates a transition from an α-helical conformation toward the extended configuration (Fig. 3C). Below this threshold, intrahelical H-bonds are still present to stabilize a helical conformation. In the presence of AngII, the stability of the α-helical conformation of TMD7 was decreased for the WT-AT1 receptor as the population of low r.m.s.d. conformations was diminished (p = 0.023 at r.m.s.d. = 0.040 nm, pmax = 0.094 at r.m.s.d. = 0.096) to the favor of higher r.m.s.d. populations (r.m.s.d. > 0.30 nm) (Fig. 2). The presence of SI8 in the WT-AT1 receptor caused a slight reduction, narrowing and rightward shift of the low r.m.s.d. population compared with the WT receptor (p = 0.053 at r.m.s.d. = 0.04 nm and pmax = 0.157 at r.m.s.d. = 0.056 nm) along with the appearance of higher r.m.s.d. populations (Fig. 2A). Similarly to its nonliganded state, the N111G-AT1 receptor in complex with AngII showed a wide distribution of populations (Fig. 2B). The AngII-D74N-AT1 receptor showed a higher and narrower population (pmax = 0.276) at r.m.s.d. = 0.04 nm compared with its nonliganded state, which suggests that the α-helical conformation was further stabilized by the presence of AngII (Fig. 2A). It thus appears that activation of the Gq/11 pathway is associated with a loss of helical structure between residues 2887.39 and 2957.46 in TMD7, whereas maintaining the helical structure is associated with β-arrestin signaling.
FIGURE 2.

Probability distribution of the r.m.s.d. of the Cα atoms of residues I2887.39 through N2957.46 on TMD7 measured from the MD simulations of each identified receptor. r.m.s.d. was calculated following a superposition of TMD7 in all frames of the MD simulations to ignore rigid body movements of the helix.
New Interhelical H-bonds Stabilize the Conformational Change in TMD7
In the resting state of the WT-AT1 receptor, an inter-helical H-bond between the side chains of S2526.47 and N2947.45 as well as H-bonds involving the side chains of N1113.35 and N2957.46 with the carboxylate of D742.50 were shown to stabilize the MHN (Fig. 3, B and E). However, analysis of the trajectories revealed that these inter-helical H-bonds were reorganized in the N111G-AT1 receptor. For example, the side chain of N2947.45 was re-oriented toward the side chain of D742.50 to form new H-bonds (Fig. 3, C and F). Concomitantly, the side chain of S2526.47 formed new stabilizing H-bonds with backbone atoms of residues 2917.42–2947.45, which underwent a conformational transition. Note that these new inter-TMD H-bonds replaced the intra-helical H-bonds in their α-helical state (Fig. 3, B and D). The re-orientation of residue D742.50 toward N461.50, previously observed in the N111G-AT1 receptor and identified as important for Gq/11 signaling (9), was also observed in the trajectories (Fig. 3, C and F).
To verify the potential correlation between these switches in inter-TMD H-bonds and the conformational transition in TMD7, we generated two-dimensional probability distribution functions. In the first one (Fig. 4), we monitored the correlation between the conformational transition (r.m.s.d. of the backbone atoms of residues 2887.39–2957.46) and the formation of H-bonds between S2526.47 and TMD7 over all frames of the MD simulation. The formation of the H-bonds was monitored by measuring the distance between the Oγ atom of S2526.47 and the center-of-mass of the backbone amide hydrogens of N2947.45 and F2937.44 and carbonyl oxygen of A2917.42. In the second one (Fig. 5), we monitored the potential coupling between the conformational transition and the re-orientation of the side chain of N2947.45 as measured by the distance between the Nδ atom of N2947.45 and the Cγ atom of D742.50 over all frames of the MD simulations. The resulting two-dimensional probability distribution functions indicate that as the r.m.s.d. of the backbone atoms of residues 2887.39–2957.46 increases from around 0.05 nm in the resting state to beyond 0.30 nm (conformational transition in TMD7), the distance between the side chain of S2526.47 and the backbone atoms of residues 2917.42–2947.45 diminishes from about 0.54 to 0.36 nm (Fig. 4). As this occurs, the distance between the side chains of residues N2947.45 and D742.50 is also decreased from about 1.00 to 0.50 nm (Fig. 5). The conformational landscapes of the three systems activating the Gq/11 pathway (AngII-WT-AT1, N111G-AT1 and AngII-N111G-AT1 receptors) all show a similar transition away from the initial resting conformation observed in the WT-AT1 receptor. However, the D74N-AT1 receptor, with and without AngII, is more stable than the WT-AT1 receptor without ligand in the initial resting state. Conformational landscapes generated for the SI8-WT-AT1 receptor depict the same transition observed with the AngII-WT-AT1 receptor but also suggest that the initial resting state is favored by SI8 compared with AngII as it has a similar relative population than in the WT-AT1 receptor without AngII (Figs. 4 and 5). These results further suggest a correlation between the conformational transition in TMD7 and the activation of the Gq/11 pathway and provide explanations as to how this transition is stabilized by new inter-helical interactions.
FIGURE 4.

Probability landscape generated by sorting frames of the MD simulations according to two measurements as follows: x axis, distance between the Oγ atom of residue S2526.47 and the center-of-mass of backbone atoms of A2917.42 (carbonyl O), F2937.44 (amine hydrogen) and N2947.45 (amine hydrogen); y axis, r.m.s.d. of the Cα atoms of residues 288–295 on TMD7.
FIGURE 5.

Probability landscape generated by sorting frames of the MD simulations according to two measurements as follows: x axis, distance between the Oγ atom of residue Ser252 and the center-of-mass of backbone atoms of A2917.42 (carbonyl O), F2937.44 (amine hydrogen), and N2947.45 (amine hydrogen); y axis, distance between the Cγ atom of N2947.45 and the Cγ atom of D742.50 (or the mutated D74N).
Conformational Transition in TMD7 Is Associated with a Higher Probability of Opening the G Protein-binding Site
Crystal structures of GPCRs in complex with a Gα-subunit (42–45) have revealed that the coupling involves an opening between TMD3 and TMD6 of the receptors that allows for the insertion of the C-terminal helix of the Gα-subunit. Consequently, the coupling causes an increase in the interhelical distance between TMD3 and TMD6 at the cytosolic interface. This has led to the hypothesis that when a GPCR couples to and activates G proteins, this is accompanied, among other potential conformational changes, by a relative displacement between TMD3 and TMD6. To unveil a potential link between the conformational transition in TMD7 and an increase in the interhelical distance between TMD3 and TMD6, we monitored the distance between the cytosolic ends of TMD3 (center-of-mass of the backbone atoms of residues S1233.47 to Y1273.51) and TMD6 (center-of-mass of the backbone atoms of residues I2386.33 to I2426.37) during the MD simulations. We then calculated the probability distribution functions between this distance and the r.m.s.d. of the backbone atoms of residues 2887.39–2957.46. Fig. 6 shows that all simulated systems sampled an open conformation as indicated by the increase of the distance between TMD3 and TMD6 from about 1.0 to 1.1 nm in the initial resting state to more than 1.5 nm. There is, however, an interesting trend emerging from our analysis; it appears there is an increased probability of existence of the open state concurrent with the conformational transition in TMD7. Indeed, once the r.m.s.d. in TMD7 increases above a value of about 0.16 nm, the difference in probability between the open and closed states becomes less pronounced and favors the open state. This trend is particularly striking in the N111G-AT1 and AngII-N111G-AT1 receptors. Interestingly, the presence of the biased agonist SI8 in the WT-AT1 receptor increases the relative probability of finding the receptor with a stable helical TMD7 while decreasing the probability of the relative displacement between TM3 and TMD6 when compared with the AngII-WT-AT1 receptor. Therefore, the conformational transition in TMD7 seems to relieve structural constraints facilitating the opening between TMD3 and TMD6, but beyond this point, a higher r.m.s.d. between the backbone atoms of residues 2887.39–2957.46 is not associated with a larger probability of opening. In other words, although the conformational transition in TMD7 and the opening between TMD3 and TMD6 can occur independently, there appears to be a relationship between the loss of helical structure in TMD7 and the probability of opening the G protein-binding site between TMD3 and TMD6.
FIGURE 6.

Probability landscape generated by sorting frames of the MD simulations according to two measurements as follows: x axis, distance between the center-of-mass of backbone atoms of residues S1233.47 to Y1273.51 (intracellular extremity of TMD3) and residues I2386.33 to I2426.37 (intracellular extremity of TMD6); y axis, r.m.s.d. of the Cα atoms of residues I2887.39 through N2957.46 on TMD7.
Loss of α-Helical Conformation in TMD7 Is Associated with a Higher Probability of Opening the Hydrophobic Core Adjacent to the Binding Pocket
We had previously identified a hydrophobic core between TMD3 and TMD7 (see Fig. 7 in Ref. 9). The stability of that hydrophobic core (probability of being formed during MD simulations) was correlated with the inability of an AT1 receptor mutant (N111W) to engage the Gq/11 pathway. Moreover, an MD simulation of the N111G-AT1 receptor showed that this hydrophobic core was destabilized and opened, effectively expanding the binding pocket (9). To substantiate this movement, we monitored the distance between TMD3 (backbone atoms of residues V1083.32 to L1123.36) and TMD7 (backbone atoms of residues 2887.39 to Y2927.43) in the region harboring the hydrophobic core in the different variations of the AT1 receptor. The two-dimensional probability distribution functions between the conformational transition from the r.m.s.d. of residues I2887.39 to N2957.46 of TMD7 and the distance between TMD3 and TMD7 in the hydrophobic core indicated that the distance between TMD3 and TMD7 can increase from about 1.1 nm in the ground state to reach 1.6 nm when the conformational transition in TDM7 is complete in the AngII-WT-AT1, N111G-AT1, AngII-N111G-AT1, and SI8-WT-AT1 receptors (Fig. 7). However, the calculations showed that the hydrophobic core has an increasing probability of staying closed in WT-AT1, D74N-AT1, and AngII-D74N-AT1 receptors (Fig. 7). Despite the conformational transition in TMD7 being observed in the SI8-WT-AT1 receptor, the initial resting conformation is more stable than with the AngII ligand and appears nearly as stable as the WT-AngII receptor without ligand. These results suggest that the hydrophobic core is destabilized in Gq-active receptors and that this destabilization is correlated with the conformational transition in TMD7.
FIGURE 7.

Probability landscape generated by sorting frames of the MD simulations according to two measurements as follow: x axis, distance between the center-of-mass of backbone atoms of residues V1083.32–L1123.36 (region of the hydrophobic core on TMD3) and residues I2887.39–Y2927.43 (region of the hydrophobic core on TMD7); y axis, r.m.s.d. of the Cα atoms of residues I2887.39 through N2957.46 on TMD7.
Loss of α-Helical Conformation in TMD7 Increases the Accessibility of the Side Chain of Residue Y3027.53 in the NPXXY Motif
Because the conserved NPXXY motif lies at the end of TMD7, we monitored how the conformational transition observed just above impacted the movement of the bottom portion of TMD7. We measured the distance between the NPXXY motif (taken as the center-of-mass of the backbone atoms of residues N2987.49 to Y3027.53) and a fixed region in the middle of TMD3 (taken as the center-of-mass of the backbone atoms of residues N1113.35 to S1153.39) to evaluate the movement of the NPXXY motif relative to the TM bundle of the receptor. The two-dimensional probability distribution functions between this distance and the r.m.s.d. of the Cα of residues I2887.39 to N2957.46 (Fig. 8) indicate that the basal distance in the WT-AT1 receptor is about 1.5 nm, with a secondary population at about 1.75 nm. A population centered at d = 1.5 nm was observed with the SI8-WT-AT1 receptor but with a slightly narrower distribution and higher probability. However, conformations centered at d = 1.75 nm display a much broader distribution in r.m.s.d. indicating the occurrence of the conformational transition in TMD7. Both the N111G mutation and the presence of AngII allow the distance between TMD3 and the NPXXY motif to increase beyond 2.0 nm, and the increase in distance appears facilitated by the increase in the r.m.s.d. of residues I2887.39 to N2957.46. The D74N-AT1 receptor with and without AngII shows an increased probability of sampling the initial resting state, and no significant secondary population at 1.75 nm is observed. The MD simulations were further analyzed to verify whether this movement could be associated with changes in the interactions formed by residues N2987.49 or Y3027.53 of the NPXXY motif. Although the analysis did not identify new specific interactions, it did reveal a change in solvent accessibility for the side chain of residue Y3027.53. Indeed, Y3027.53 has a small solvent accessibility surface (SAS) in the initial resting state as it is buried between hydrophobic residues V491.53, A632.39, F662.42, L672.43, L702.46, L1223.46, I2386.33, I2416.36, and I2426.37 (Fig. 9A). Fig. 10 shows that the most populated states have an SAS of 0.4 nm2 in the WT-AT1 receptor and the SI8-WT-AT1 receptor. The SAS is reduced to 0.3 nm2 for the D74N-AT1 receptor and 0.2 nm2 for the AngII-D74N-AT1 receptor. In opposition, the SAS in the AngII-WT-AT1 receptor has higher probabilities of reaching higher values. The most populated states are at SAS = 0.9 nm2 for the N111G-AT1 receptor with and without AngII. These results indicate that the conformational transition in TMD7 favors a movement of the NPXXY motif away from the TM bundle, which increases the solvent accessibility of Y3027.53 (Fig. 9B).
FIGURE 8.

Probability landscape generated by sorting frames of the MD simulations according to two measurements as follows: x axis, distance between the center-of-mass of the backbone atoms of residues N1113.35 to S1153.39 (middle of TMD3) and the center-of-mass of the backbone atoms of residues N2987.49 to Y3027.53 (NPXXY motif), y axis, r.m.s.d. of the Cα atoms of residues I2887.39 through N2957.46 on TMD7.
FIGURE 9.
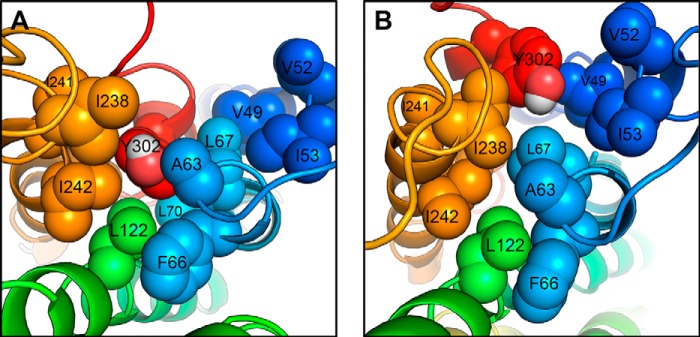
Snapshots from MD simulations showing different SAS of the Y3027.53 side chain from the NPXXY motif as it is surrounded by hydrophobic side chains. A, buried configuration, with SAS of 0.21 nm2 for Y3027.53. B, a more exposed configuration, with SAS of 0.82 nm2 for Y3027.53.
FIGURE 10.

Probability landscape generated by sorting frames of the MD simulations according to two measurements as follows: x axis, distance between the center-of-mass of the backbone atoms of residues N1113.35 to S1153.39 (middle of TMD3) and the center-of-mass of the backbone atoms of residues N2987.49 to Y3027.53 (NPXXY motif); y axis, solvent-accessible surface of the side chain of residue Y3027.53.
β-Arrestin Bias Induced by the D74N Mutation and the SI8 Ligand Is Associated with a Restriction of the Conformational Landscape Explored by AT1 Receptor
So far, our results focused on structural changes associated with Gq/11 signaling caused by either the presence of the endogenous agonist AngII or the N111G mutation. The MD simulations of the β-arrestin-biased D74N-AT1 receptor with and without AngII revealed that it was conformationally more stable. Indeed both systems did not display significant conformational transitions as observed with the other systems. In fact, all the probability distribution functions calculated indicate that the D74N-AT1 receptor explores a much smaller conformational landscape than the others. They also unveiled the existence of a highly populated state, which corresponds to the most probable and initial resting state of the nonliganded WT-AT1 receptor. To understand the molecular mechanism for such stabilization, we monitored how the D74N mutation affects the MHN. We observed that the presence of the asparagine at position 742.50 allowed simultaneous interactions with the asparagines at position 1113.35 and position 461.50 (Fig. 3, D and G). It also caused residue N2957.46 to be oriented slightly more toward TMD3 than TMD2 compared with the WT-AT1 receptor, allowing it to interact with the side chain of residue S1153.39 (Fig. 3, D and G). This interaction tightened the helical conformation in this region of TMD7. By measuring the distance between the Nδ atom of Asn295 and the Oγ atom of S1153.39, we observed that the tendency of N2957.46 to be close to S1153.39 was increased by the D74N mutation (pmax = 0.210 at 0.28 nm) when compared with the WT-AT1 receptor (p = 0.082 at 0.28 nm and pmax = 0.198 at 0.39 nm). This tendency was further increased in the AngII-liganded D74N-AT1 receptor (pmax = 0.327 at 0.28 nm) (Fig. 11A). However, this was decreased by the presence of AngII in the WT-AT1 receptor (p = 0.033 at 0.28 nm and pmax = 0.066 at 0.68 nm) or by the N111G mutation (p = 0.047 at 0.28 nm and pmax = 0.066 at 0.62 nm without AngII and p = 0.063 at 0.28 nm and pmax = 0.084 at 0.60 nm with AngII) (Fig. 11B). The distance between S115 and N295 was also decreased, to a lesser extent, in the SI8-WT-AT1 receptor (pmax = 0.147 at 0.41 nm) (Fig. 11A). The results observed here with the SI8-WT-AT1 receptor are representative of what has been observed so far with the SI8 ligand in that it does not restrict the conformational landscape of the receptor as the D74N mutation does, but it shows a clear preference for the initial resting state compared with the AngII ligand. These results suggest that the D74N-AT1 receptor is more stable in the initial resting state than the WT-AT1 receptor and that the presence of AngII might further stabilize this conformation. Furthermore, the SI8 ligand is less destabilizing for the initial resting state of the WT-AT1 receptor than the AngII ligand.
FIGURE 11.

Probability distribution of the distance between the Nδ atom of N2957.46 and the Oγ atom of S1153.39 measured from the MD simulations of each identified receptor.
Residue Phe-8 of AngII Is Inserted in the Hydrophobic Core of Gq-active Receptors
The MD simulations of the WT-AT1, N111G-AT1, and D74N-AT1 receptors in complex with AngII provided a lot of structural insights on how AngII can stabilize different states of the WT-AT1 receptor and the two biased mutants. In all cases and in accordance with previous studies (14, 46–49), we have found that all AT1-AngII complexes are rather dynamic. However, we noted distinct sectors of the AT1 receptor suited to accommodate specific interactions with the side chains of the AngII ligand. For example, we noted that Arg2 of AngII generally formed H-bonds with residues D2636.58 and D2817.32 (Fig. 12A) and that Val3 faced hydrophobic residues on ECL2 (Fig. 12B). Residue His6 in AngII was usually positioned between TMD1, -2, and -7 where it formed H-bonds with Y351.39 and sometimes Y2927.43. Residue His6 also interacted with W842.60 (Fig. 12C). Y351.39, Y2927.43, and W842.60 were also observed to interact with each other independently of the presence or absence of AngII. Residue R1674.64 of the AT1 receptor could occasionally reach this binding area and form an H-bond with His6 in its neutral state. Residue Ile5 of AngII was above the usual position of His6 and surrounded by the side chains of residues I271.31, F281.32, I311.35, T882.64, P2857.36, and I2887.39 (Fig. 12D). The C-terminal moiety of AngII was stabilized by an extensive network of polar residues and formed H-bonds with Y1133.37, K1995.42, N2005.43, H2566.51, Q2576.52, and T2606.55 (Fig. 12E). The side chains of residues Asp1 and Tyr4 were much more mobile and interacted with various polar groups in the extracellular domains of the receptor as well as with water molecules (data not shown). The main difference in ligand binding that could be observed between Gq-active and Gq-inactive receptors was the propensity for the phenyl moiety of the Phe8 side chain of AngII to be inserted within the hydrophobic core when it was in the “open” configuration after the conformational transition in TMD7 had occurred (Fig. 12F). In such a case, the phenyl moiety of Phe8 contacted all residues of the hydrophobic core (F772.53, V1083.32, L1123.36, I2887.39, A2917.41, and Y2927.42), as well as W2536.48 and H2566.51. When the hydrophobic core was closed, the phenyl moiety of Phe8 was adjacent to the hydrophobic core and made contacts mostly with residues among V1083.32, L1123.36, I2887.39, W2536.48, and H2566.51, although not simultaneously (Fig. 12G). The insertion in the hydrophobic core was not observed with the smaller Ile8 residue of the SI8 ligand. To monitor the insertion of Phe8 in the hydrophobic core, we measured the distance between the center-of-mass of the Phe8 side chain and the center-of-mass of the side chain of residues forming the hydrophobic core (F772.53, V1083.32, L1123.36, I2887.39, A2917.41, and Y2927.42). The two-dimensional probability distribution functions showed that the distance between Phe8 and the hydrophobic core tends to decrease from about 0.74 to 0.40 nm as the hydrophobic core opens (Fig. 13, left) or the r.m.s.d. of TMD7 increases (Fig. 13, right). The landscape for the AngII-N111G-AT1 receptor showed the two populations more distinctly, one with the hydrophobic core closed (d = 1.15 nm) and the Phe8 further from it (d = 0.74 nm), and one with the hydrophobic core open (d = 1.4 nm) and Phe8 inserted within it (d = 0.40 nm). That second population is not observed in simulations with the SI8 ligand despite the opening of the hydrophobic core and the increase in the r.m.s.d. of TMD7. These results suggest that the side chain of residue Phe8 of AngII can stabilize the open conformation of the hydrophobic core but that the side chain of residue Ile8 of SI8 cannot.
FIGURE 12.
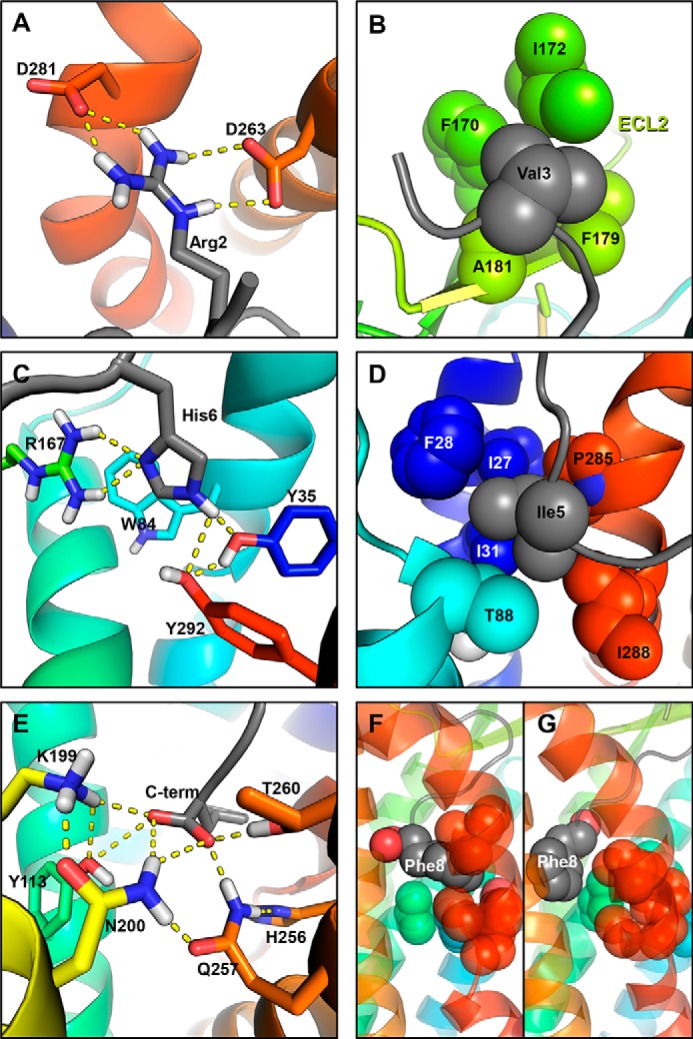
Snapshot from MD simulations showing common interactions between side chains or the C-terminal carboxyl of AngII and specific sectors of the AT1 receptor. A, side chain of Arg2 forms H-bonds with D2636.58 and D2817.32 at the top of TMD6 and TMD7. B, side chain of Val3 interacts with the hydrophobic side chains of F170ECL2, I172ECL2, F179ECL2, and A181ECL2. C, side chain of His6 is positioned between TMD1, TMD2, and TMD7 and can form H-bonds with Y351.39, Y2927.43, and R1674.64 and also π-stacking interactions with W842.60. D, Ile5 is positioned between TMD1, TMD2, and TMD7 (above His6) and contacts residues I271.31, F281.32, I311.35, T882.64, P2857.36, and I2887.39. E, C-terminal moiety is positioned between TMD3, TMD5, and TMD6 and can form H-bonds with the side chains of residues Y1133.37, K1995.42, N2005.43, H2566.51, Q2576.52, and T2606.55. F, side chain of residue Phe8 is inserted in the open hydrophobic core (observed in certain trajectories from the MD simulations of the N111G-AT1, AngII-N111G-AT1, and AngII-WT-AT1 receptors). G, side chain of Phe8 is adjacent to the closed hydrophobic core (observed in all trajectories). Transmembrane domains are showed as colored ribbons (TMD1 = dark blue; TMD2 = light blue; TMD3 = aqua, TMD4/ECL2 = green; TMD5 = yellow; TMD6 = orange, and TMD7 = red). Angiotensin-II is colored gray. Side chains are shown as sticks for polar interactions and spheres for hydrophobic interactions. Oxygen atoms are red, nitrogen atoms are blue, hydrogen atoms are white and carbon atoms are colored according to their TMD. H-bonds predicted by PyMOL (ranging between 1.7 and 2.6 Å between hydrogen and acceptor) are shown as yellow dashed lines.
FIGURE 13.

Probability landscape generated by sorting frames of the MD simulations according to two measurements as follows: x axis, distance between the center-of-mass of the side chain of residue Phe8 and the center-of-mass of the side chains of residues V1083.32, L1123.36, F772.53, I2887.39, A2917.41, and Y2927.42 of the hydrophobic core; y axis, graphs on the left, distance between the center-of-mass of backbone atoms of residues V10832–L1123.36 (region of the hydrophobic core on TMD3) and residues I2887.39–Y2927.43 (region of the hydrophobic core on TMD7); y-axis, graphs on the right, r.m.s.d. of the Cα atoms of residues I2887.39 through N2957.46 on TMD7.
Discussion
In this study, we used molecular dynamic simulations to unveil structural features that could explain the biased signaling properties of AT1 receptor mutants (N111G-AT1 receptor and D74N-AT1 receptor) and of an AT1 receptor ligand (SI8) observed experimentally. More precisely, we further confirmed and expanded our understanding of the molecular basis of N111G-AT1's biased signaling. Upon stimulation with its agonist AngII, this mutant efficiently activates the Gq pathway but βarrestin1 and -2 are recruited to a lesser extent than the WT-AT1 receptor. Similarly, we established that the D74N-AT1 receptor mutant efficiently recruits the βarrestin1 and -2 but does not activate the Gq/11 pathway (Fig. 1). These observations support the previous suggestions that the N111G-AT1 receptor had limited coupling to βarrestin1 (12) and that the D74N-AT1 receptor was biased for the βarrestin2 pathway and could not activate the Gq/11 pathway (4). However, we cannot overlook the increased basal BRET ratio of the N111G receptor, which could be indicative of a basal coupling between the N111G-AT1 receptor and β-arrestins. It was shown that an increase in phosphorylation of the C-terminal tail of the AT1 receptor is required for high affinity binding of β-arrestins and internalization (50, 51) and that the C-terminal tail of the N111G-AT1 receptor, even after stimulation with AngII, is not phosphorylated beyond the basal level of the WT-AT1 receptor (7). These observations support the notion that this basal coupling could be a transient, low affinity interaction caused by the increased opening of the G protein-binding site between TMD3 and TMD6, which is now known to be a common interface for the binding of β-arrestins and G proteins to the receptor (52, 53).
MD simulations (exploring the micro-second time scale) of the WT-AT1, N111G-AT1, and D74N-AT1 receptors showed distinct structural and dynamic features of the AT1 receptor associated with signaling through the Gq/11 pathway and the β-arrestin pathway. These structural differences span the whole receptor from top to bottom, including the binding pocket and hydrophobic core in the more extracellular region, the arrangement of the major H-bond network, and the structure of TMD7 in the middle of the receptor and the G protein binding domain in the extracellular extremity of the receptor.
MD simulations of the constitutively active N111G-AT1 receptor have further validated our previous results obtained with shorter MD simulations. Briefly, a re-arrangement in the MHN and a conformational transition from a helical to an extended state change in a section of TMD7 occurring concurrently with the opening of an hydrophobic core (above the MHN) were proposed to lead to the activation of the Gq/11 pathway (9). Such a transition is validated by experimental data showing that the WT-AT1 receptor can be photolabeled on residues 2937.44 to 2977.48 (46, 54), and such a pattern can only be rationalized if this segment is in an extended conformation rather than an α-helix. We now show that the loss of α-helical conformation in a portion of TMD7 (Fig. 2), which is also linked to an opening of the hydrophobic core (Fig. 7), is promoted by new H-bonds involving S2526.47 and N2947.45. Although the side chain of N2947.45 forms an H-bond with the side chain of S2526.47 in the initial resting state (observed in the MD simulations of the WT-AT1, D74N-AT1, and AngII-D74N-AT1 receptor), the conformational change in TMD7 allows the side chain of N2947.45 to re-orient toward and form an H-bond with residue D742.50 and also allows the side chain of S2526.47 to form H-bonds with the backbone atoms of residues A2917.42, F2937.44, and N2947.45 that are no longer forming an α-helix (Figs. 3–5). The possible interactions of N2947.45 with S2526.47 or D742.50 have been tested experimentally through reciprocal mutations of the amino acids (55). Based on the lack of activity of the receptor with reciprocal mutations (N294S/S252N and N294D/D74N), the authors suggested that theses residues do not interact in the WT-AT1 receptor. Our MD simulations, however, clearly suggest (Fig. 3) that the side chains of the polar residues within the MHN do not form mutually exclusive interactions in pairs. This can explain why reciprocal mutations of side chains that do interact with each other (occasionally) might not rescue the impairment to Gq/11 signaling caused by the single mutations. The conformational change of TMD7 also appears associated with an increased probability of opening the G protein-binding site at the intracellular side of the receptor, between TMD3 and TMD6 (Fig. 6), which is coherent with the constitutively active nature of the N111G-AT1 receptor on the Gq/11 pathway (Fig. 1). Residues from the conserved (E/D)RY motif at the intracellular extremity of TMD3 have been suggested to stabilize the inactive state of GPCRs either through a salt bridge, referred to as the “ionic lock,” between R3.50 and a conserved E6.39 at the intracellular end TMD6, or between R3.50 and (E/D)3.49 (56). The AT1 receptor, however, does not feature the conserved glutamate at position 6.39, which is instead an asparagine that did not interact significantly with R1263.50 in our MD simulations. We also monitored H-bonds formed between D1253.39 and R1263.50 and between residues of the DRY motif and other neighboring residues, but no interaction involving the DRY motif of AT1 was linked to the opening of the G protein-binding site. This is in line with the alternative role proposed to be played by these residues for the AT1 receptor and other GPCRs (56–59). Indeed, they have been proposed to be rather involved in the direct interaction with the G protein rather than modulating the conformational stability of the receptor. For the highly conserved NPXXY motif, we observed that the conformational transition in TMD7 allowed increased movement of the bottom portion of the helix containing this motif. This was also associated with an increased solvent accessibility of the side chain of residue Y3027.53 (Figs. 8–10). This increased accessibility could be important for G protein signaling by the AT1 receptor. Indeed, as reported elsewhere, Tyr302 is important to activate the Gq/11 pathway but not for the internalization of the AT1 receptor (60–62).
MD simulations of the WT-AT1 receptor in presence of its ligand AngII indicated that AngII induces the same conformational changes as those caused by the N111G mutation (Figs. 4–8 and 10). Moreover, our data suggest that the phenyl moiety of residue Phe8 of AngII could insert in the hydrophobic core and stabilize its open configuration, although Ile8 of SI8 could not (Figs. 12F and 13). The ability to insert in the hydrophobic core and elicit the conformational change could be unique to the phenyl moiety, as mutation of this residue to a tyrosine or diphenylalanine are the only tolerated changes that do not lead to an AngII analog with an antagonist profile on the Gq/11 pathway. Indeed, substitution of Phe8 for other hydrophobic residues, such as alanine, leucine, or isoleucine, turns the ligand into an antagonist on the Gq/11 pathway (15, 63–65). However, a larger side chain such as (pentabromo)Phe-8 or a change of orientation from l-Phe-8 to d-Phe-8 also results in conferring an antagonistic nature on the Gq/11 pathway (66, 67). Whether the side chain of Phe8 is inserted in the hydrophobic core or not, it can interact with the side chain of H2566.51, which is consistent with previously proposed contact points between the ligand and receptor (5, 68). Furthermore, analysis of the trajectories has unveiled distinct sectors well suited to accommodate certain side chains and the C-terminal moiety of the ligand (Fig. 12). The side chain of residue Arg2 forms H-bonds with the side chains of residues D2636.58 and D2817.32, which is in agreement with previous reports (5, 69–71). However, in our receptor model, access to the other side chain suggested to interact with Arg2, D2787.29, is hampered by the N-terminal domain that is constrained in that area by the conserved disulfide bond between cysteines 18 and 274. The side chains of residues Val3 and Ile5 are both stabilized by hydrophobic clusters, formed by F170ECL2, I172ECL2, F179ECL2, and A181ECL2 for Val3 and I271.31, F281.32, I311.35, T882.64, P2857.36, and I2887.39 for Ile5. The side chain of residue His6 forms H-bonds with the side chains of residues Y351.39, Y2927.43, and R1674.64 and also interacts with W842.60. The C-terminal moiety had a wide variety of partners with which to form H-bonds, within a polar sector formed of Y1133.37, K1995.42, N2005.43, H2566.51, Q2576.52, and T2606.55 (Fig 12). It was previously suggested that residue K1995.42 could form H-bonds with the C-terminal moiety of AngII, and this was also supported by docking experiments (68, 71–73). In a study in which residue R1674.64 was shown to be important for the binding of AngII, the authors suggested that the side chain of R167 interacts with the hydroxyl group of Tyr4 (70). In our MD simulations, such an interaction only rarely occurred. Actually, we observe that the side chain Tyr4 is highly mobile and found more often to interact with water molecules and extracellular loops. This conformation is similar to some of the poses obtained recently through molecular docking of AngII and ab initio reconstruction of the peptide within the binding pocket of AT1 (71). Other reports have suggested that the hydroxyl moiety of Tyr4 could form an H-bond with the side chain of residue N1113.35, but we have not witnessed such an event in our simulations (74, 75).
Overall, the broader area explored in the probability landscapes by the Gq/11 active systems (AngII-WT-AT1 receptor, N111G-AT1 receptor, and AngII-N111G-AT1 receptor) support the notion of a destabilization of the initial resting state of the receptor model. In addition, no simulation of these systems has converged to one single stable state or conformation. It is important to keep in mind that the MD simulations are most likely and inherently biased from a conformational standpoint as they all start from the same initial resting state. Because of this limitation and the short time scale covered in the MD simulations, it is important to emphasize that we have sampled only a portion of the conformational landscape of the AT1 receptor. We are looking forward to using the different conformations reached in the current MD simulations as starting points for a multitude of other trajectories to explore more extensively the conformational landscape of the receptor and approach ergodicity to obtain a more accurate thermodynamic characterization of its different states. For example, it will be interesting to see whether trajectories where the conformational transition in TDM7 occurred will explore other accessible “active” states or revert back to the resting state. Furthermore, the addition of an intracellular effector in the simulations, such as a trimeric G protein, should allow for the stabilization of specific conformations sampled in the current MD simulations and therefore help in the discrimination of conformations of the intracellular portion compatible with the engagement of the effector. Nonetheless, the results presented here are in agreement with the activation model of the β2-adrenergic receptor by agonists proposed by Nygaard et al. (76) from a combination of solution state NMR (cHSQC of [ϵ-13C]methionines) and MD simulations. More specifically, they have used line shape analysis of assigned [ϵ-13C]methionines to estimate the dynamic nature of the receptors under different conditions. From these data, they suggest that agonist binding to this GPCR (in absence of an intracellular effector) is associated with a conformational heterogeneity and flexibility following the destabilization of the inactive state.
Results obtained with the β-arrestin-biased D74N-AT1 receptor indicate that there are no major conformational changes from the initial resting state unlike those observed for the N111G-AT1 receptor. There is a difference in the configuration of the side chains at the MHN due to the D74N mutation allowing it to interact with both N461.50 and N1113.35 simultaneously, which appears to diminish movement within the MHN (Fig. 3D). This interaction also causes a reorientation of the side chain of residue N2957.46 toward S1153.39 of TMD3 (Fig 11), potentially tightening the α-helical conformation of TMD7 and further reducing movement in that region of the receptor. Interestingly, an equivalent interaction is present between residues S1423.39 and S3737.46 in the crystal structure of the serotonin 5-HT2B receptor in presence of ergotamine (PDB code 4ib4), a β-arrestin biased ligand-receptor complex (77), but absent in the crystal structure of the unbiased 5-HT1B-ergotamine complex (PDB code 4iar). This suggests that this interaction might indeed be associated with the stabilization of a β-arrestin-biased conformation of the receptor.
The aforementioned reduction of movement caused by the increased interactions within the MHN of the AT1 receptor was evident in the probability landscapes as the D74N-AT1 receptor always displayed an increased probability of being in the initial resting state compared with the WT-AT1 receptor, which indicates a conformation of lower energy (Figs. 2, 4–8, and 10). This stability was often enhanced by the presence of AngII in the binding pocket of the D74N-receptor (Figs. 2, 6–8, and 10). There was one of the 10 trajectories of the AngII-D74N-AT1 receptor that displayed a slight kink in TMD7, causing an increase in r.m.s.d. that was visible as a small secondary population on the probability landscapes. We ran 10 more 100-ns MD simulations from the final frame of that trajectory, and because the kink disappeared in all of them, it was interpreted as a random event not linked to the activation of the β-arrestin pathway (data not shown). MD simulations with the β-arrestin-biased SI8 ligand in the WT-AT1 receptor did not show such a drastic stabilization of the initial resting state as the D74N mutation, but the probability landscapes did display a preference for the initial resting state of the receptor when compared with MD simulations of the WT-AT1 receptor in presence of AngII.
Therefore, the results suggest that the β-arrestin recruitment requires the receptor to be stabilized in the initial resting conformation. This resting state, however, does not preclude the opening of the intracellular G protein/β-arrestin binding site between TMD3 and TMD6 (Fig. 6). This is similar to what was observed in simulations of the β2-adrenergic receptor showing that the ligand-binding site, connector region (in the middle of the receptor), and G protein-binding site were only weakly coupled and could fluctuate between active and inactive conformations independently (78). Because of the limited duration of the MD simulations, the possibility that a rare conformational change not sampled yet could be linked to the activation of the β-arrestins pathway cannot be dismissed. Also, we focused our observations on specific conformational properties that we first identified with the N111G-AT1 receptor and associated with the activation of the Gq/11 pathway.
Interestingly, it was noted in the first ever released crystal structure of bovine rhodopsin that “H-VII is considerably elongated in the region from Ala295 to Tyr301” (79), which corresponds to residues A2917.41 to L2977.48 of the AT1 receptor. This is similar to the conformational transition we observed with the AT1 receptor. Although this is not observed in other crystal structures of class A GPCRs, rhodopsin is the only known crystal structure that features a second proline residue in TMD7, other than in the NPXXY motif (79). So although it is believed that class A GPCRs should share common activation mechanisms due to the presence of some highly conserved residues (80–84), the conformational transition in TMD7 might require the presence of a second, nonconserved proline in the upper portion of TMD7.
In conclusion, MD simulations of the AT1 receptor have revealed that the D74N mutation, biasing the receptor toward the β-arrestins pathways, stabilizes the initial and resting conformation through additional H-bonds formed within the MHN. Adding the AngII agonist to the D74N-AT1 receptor further stabilizes the initial resting conformation. At the opposite, the N111G mutation, biasing the receptor toward the Gq/11 pathway, destabilizes the initial conformation and favors various conformational changes, including the re-orientation of side chains and H-bonds formed within the MHN, the loss of the regular α-helical structure in a part of TMD7, the opening of a hydrophobic core toward the ligand-binding pocket, and the opening of the G protein-binding site at the cytosolic side of the receptor. Adding the unbiased agonist AngII to the WT-AT1 receptor causes the same conformational changes and possibly stabilizes them through the insertion of the side chain of residue Phe8 within the hydrophobic core. Moreover, the biased agonist SI8 displays a preference for the stabilization of the initial resting state of the WT-AT1 receptor.
This work was supported by the Canadian Institutes of Health Research.
The operation of the supercomputer Mammouth Parallèle II is funded by the Canada Foundation for Innovation, NanoQuébec, RMGA, and the Fonds de Recherche du Québec-Nature et Technologies.
- AT1
- angiotensin II type 1
- GPCR
- G protein-coupled receptor
- TMD
- transmembrane domain
- AngII
- angiotensin II
- SI8
- [Sar1,Ile8]AngII
- CXCR4
- CXC chemokine receptor type 4
- DOPC
- dioleoylphosphatidylcholine
- MD
- molecular dynamics
- r.m.s.d.
- root mean square deviation
- MHN
- major H-bond network
- SAS
- solvent-accessible surface
- PDB
- Protein Data Bank
- IP1
- inositol monophosphate
- BRET
- bioluminescence resonance energy transfer.
References
- 1. de Gasparo M., Catt K. J., Inagami T., Wright J. W., Unger T. (2000) International union of pharmacology. XXIII. The angiotensin II receptors. Pharmacol. Rev. 52, 415–472 [PubMed] [Google Scholar]
- 2. Thomas W. G., Qian H. (2003) Arresting angiotensin type 1 receptors. Trends Endocrinol. Metab. 14, 130–136 [DOI] [PubMed] [Google Scholar]
- 3. Benigni A., Cassis P., Remuzzi G. (2010) Angiotensin II revisited: new roles in inflammation, immunology and aging. EMBO Mol. Med. 2, 247–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bonde M. M., Hansen J. T., Sanni S. J., Haunsø S., Gammeltoft S., Lyngsø C., Hansen J. L. (2010) Biased signaling of the angiotensin II type 1 receptor can be mediated through distinct mechanisms. PLoS ONE 5, e14135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Aplin M., Bonde M. M., Hansen J. L. (2009) Molecular determinants of angiotensin II type 1 receptor functional selectivity. J. Mol. Cell. Cardiol. 46, 15–24 [DOI] [PubMed] [Google Scholar]
- 6. Wilson P. C., Lee M. H., Appleton K. M., El-Shewy H. M., Morinelli T. A., Peterson Y. K., Luttrell L. M., Jaffa A. A. (2013) The arrestin-selective angiotensin AT1 receptor agonist [Sar1,Ile4,Ile8]AngII negatively regulates bradykinin B2 receptor signaling via AT1-B2 receptor heterodimers. J. Biol. Chem. 288, 18872–18884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Thomas W. G., Qian H., Chang C. S., Karnik S. (2000) Agonist-induced phosphorylation of the angiotensin II (AT1A) receptor requires generation of a conformation that is distinct from the inositol phosphate-signaling state. J. Biol. Chem. 275, 2893–2900 [DOI] [PubMed] [Google Scholar]
- 8. Zhang X. C., Sun K., Zhang L., Li X., Cao C. (2013) GPCR activation: protonation and membrane potential. Protein Cell 4, 747–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cabana J., Holleran B., Beaulieu M. È., Leduc R., Escher E., Guillemette G., Lavigne P. (2013) Critical hydrogen bond formation for activation of the angiotensin II type 1 receptor. J. Biol. Chem. 288, 2593–2604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Katritch V., Fenalti G., Abola E. E., Roth B. L., Cherezov V., Stevens R. C. (2014) Allosteric sodium in class A GPCR signaling. Trends Biochem. Sci. 39, 233–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liu W., Chun E., Thompson A. A., Chubukov P., Xu F., Katritch V., Han G. W., Roth C. B., Heitman L. H., IJzerman A. P., Cherezov V., Stevens R. C. (2012) Structural basis for allosteric regulation of GPCRs by sodium ions. Science 337, 232–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lee C., Hwang S. A., Jang S. H., Chung H. S., Bhat M. B., Karnik S. S. (2007) Manifold active-state conformations in GPCRs: agonist-activated constitutively active mutant AT1 receptor preferentially couples to Gq compared to the wild-type AT1 receptor. FEBS Lett. 581, 2517–2522 [DOI] [PubMed] [Google Scholar]
- 13. Lee C., Bhatt S., Shukla A., Desnoyer R. W., Yadav S. P., Kim M., Jang S. H., Karnik S. S. (2008) Site-specific cleavage of G protein-coupled receptor-engaged β-arrestin: influence of the AT1 receptor conformation on scissile site selection. J. Biol. Chem. 283, 21612–21620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fillion D., Cabana J., Guillemette G., Leduc R., Lavigne P., Escher E. (2013) Structure of the human AT1 receptor bound to angiotensin II from multiple chemoselective photoprobe contacts reveals a unique peptide binding mode. J. Biol. Chem. 288, 8187–8197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Domazet I., Holleran B. J., Richard A., Vandenberghe C., Lavigne P., Escher E., Leduc R., Guillemette G. (2015) Characterization of angiotensin II molecular determinants involved in AT1 receptor functional selectivity. Mol. Pharmacol. 87, 982–995 [DOI] [PubMed] [Google Scholar]
- 16. Hunyady L. (1999) Molecular mechanisms of angiotensin II receptor internalization. J. Am. Soc. Nephrol. 10, S47–S56 [PubMed] [Google Scholar]
- 17. Ballesteros J. A., Weinstein H. (1995) Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci. 25, 366–428 [Google Scholar]
- 18. Zhang Y. (2007) Template-based modeling and free modeling by I-TASSER in CASP7. Proteins 69, Suppl. 8, 108–117 [DOI] [PubMed] [Google Scholar]
- 19. Roy A., Kucukural A., Zhang Y. (2010) I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc. 5, 725–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Laskowski R. A., Rullmannn J. A., MacArthur M. W., Kaptein R., Thornton J. M. (1996) AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 8, 477–486 [DOI] [PubMed] [Google Scholar]
- 21. Berendsen H. J., van der Spoel D., van Drunen R. (1995) GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 91, 43–56 [Google Scholar]
- 22. van der Spoel D., Lindahl E., Hess B., Groenhof G., Mark A. E., Berendsen H. J. (2005) GROMACS: Fast, flexible, and free. J. Comput. Chem. 26, 1701–1718 [DOI] [PubMed] [Google Scholar]
- 23. Hess B., Kutzner C., van d. S., Lindahl E. (2008) GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 4, 435–447 [DOI] [PubMed] [Google Scholar]
- 24. van der Spoel D., Hess B. (2011) GROMACS: the road ahead. WIREs Comput. Mol. Sci. 1, 710–715 [Google Scholar]
- 25. Kandt C., Ash W. L., Tieleman D. P. (2007) Setting up and running molecular dynamics simulations of membrane proteins. Methods 41, 475–488 [DOI] [PubMed] [Google Scholar]
- 26. Lemkul J. A., Bevan D. R. (2009) Perturbation of membranes by the amyloid β-peptide: a molecular dynamics study. FEBS J. 276, 3060–3075 [DOI] [PubMed] [Google Scholar]
- 27. de Vries A. H., Mark A. E., Marrink S. J. (2004) The binary mixing behavior of phospholipids in a bilayer: a molecular dynamics study. J. Phys. Chem. B. 108, 2454–2463 [Google Scholar]
- 28. Lemkul J. A., Bevan D. R. (2008) A comparative molecular dynamics analysis of the amyloid β-peptide in a lipid bilayer. Arch. Biochem. Biophys. 470, 54–63 [DOI] [PubMed] [Google Scholar]
- 29. Berweger C. D., van Gunsteren W. F., Müller-Plathe F. (1995) Force field parametrization by weak coupling. Re-engineering SPC water. Chem. Phys. Lett. 232, 429–436 [Google Scholar]
- 30. Berger O., Edholm O., Jähnig F. (1997) Molecular dynamics simulations of a fluid bilayer of dipalmitoylphosphatidylcholine at full hydration, constant pressure, and constant temperature. Biophys J. 72, 2002–2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tieleman D. P. (2004) The molecular basis of electroporation. BMC Biochem. 5, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tieleman D. P., Leontiadou H., Mark A. E., Marrink S. (2003) Simulation of pore formation in lipid bilayers by mechanical stress and electric fields. J. Am. Chem. Soc. 125, 6382–6383 [DOI] [PubMed] [Google Scholar]
- 33. MacCallum J. L., Tieleman D. P. (2006) Computer simulation of the distribution of hexane in a lipid bilayer: spatially resolved free energy, entropy, and enthalpy profiles. J. Am. Chem. Soc. 128, 125–130 [DOI] [PubMed] [Google Scholar]
- 34. Anézo C., de Vries A. H., Höltje H., Tieleman D. P., Marrink S. (2003) Methodological issues in lipid bilayer simulations. J. Phys. Chem. B 107, 9424–9433 [Google Scholar]
- 35. Werner T., Morris M. B., Dastmalchi S., Church W. B. (2012) Structural modelling and dynamics of proteins for insights into drug interactions. Adv. Drug Deliv. Rev. 64, 323–343 [DOI] [PubMed] [Google Scholar]
- 36. Lange O. F., van der Spoel D., de Groot B. L. (2010) Scrutinizing molecular mechanics force fields on the submicrosecond timescale with NMR data. Biophys. J. 99, 647–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nose S. (1984) A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 81, 511–519 [Google Scholar]
- 38. Hoover W. G. (1985) Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 31, 1695–1697 [DOI] [PubMed] [Google Scholar]
- 39. DeLano W. L. (2010) The PyMOL Molecular Graphics System, Version 1.3r1, Schrödinger, LLC, New York [Google Scholar]
- 40. Corpet F. (1988) Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res. 16, 10881–10890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ehrhardt C., Schmolke M., Matzke A., Knoblauch A., Will C., Wixler V., Ludwig S. (2006) Polyethyleneimine, a cost-effective transfection reagent. Signal Trans. 6, 179–184 [Google Scholar]
- 42. Scheerer P., Park J. H., Hildebrand P. W., Kim Y. J., Krauss N., Choe H. W., Hofmann K. P., Ernst O. P. (2008) Crystal structure of opsin in its G protein-interacting conformation. Nature 455, 497–502 [DOI] [PubMed] [Google Scholar]
- 43. Deupi X., Edwards P., Singhal A., Nickle B., Oprian D., Schertler G., Standfuss J. (2012) Stabilized G protein binding site in the structure of constitutively active metarhodopsin-II. Proc. Natl. Acad. Sci. U.S.A. 109, 119–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Park J. H., Morizumi T., Li Y., Hong J. E., Pai E. F., Hofmann K. P., Choe H. W., Ernst O. P. (2013) Opsin, a structural model for olfactory receptors?. Angew. Chem. Int. Ed. Engl. 52, 11021–11024 [DOI] [PubMed] [Google Scholar]
- 45. Rasmussen S. G., DeVree B. T., Zou Y., Kruse A. C., Chung K. Y., Kobilka T. S., Thian F. S., Chae P. S., Pardon E., Calinski D., Mathiesen J. M., Shah S. T., Lyons J. A., Caffrey M., Gellman S. H., et al. (2011) Crystal structure of the β2-adrenergic receptor-Gs protein complex. Nature 477, 549–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Clément M., Martin S. S., Beaulieu M. E., Chamberland C., Lavigne P., Leduc R., Guillemette G., Escher E. (2005) Determining the environment of the ligand binding pocket of the human angiotensin II type I (hAT1) receptor using the methionine proximity assay. J. Biol. Chem. 280, 27121–27129 [DOI] [PubMed] [Google Scholar]
- 47. Clément M., Cabana J., Holleran B. J., Leduc R., Guillemette G., Lavigne P., Escher E. (2009) Activation induces structural changes in the liganded angiotensin II type 1 receptor. J. Biol. Chem. 284, 26603–26612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Arsenault J., Cabana J., Fillion D., Leduc R., Guillemette G., Lavigne P., Escher E. (2010) Temperature-dependent photolabeling of the human angiotensin II type 1 receptor reveals insights into its conformational landscape and its activation mechanism. Biochem. Pharmacol. 80, 990–999 [DOI] [PubMed] [Google Scholar]
- 49. Clément M., Chamberland C., Pérodin J., Leduc R., Guillemette G., Escher E. (2006) The active and the inactive form of the hAT1 receptor have an identical ligand-binding environment: an MPA study on a constitutively active angiotensin II receptor mutant. J. Recept. Signal Transduct. Res. 26, 417–433 [DOI] [PubMed] [Google Scholar]
- 50. Wei H., Ahn S., Barnes W. G., Lefkowitz R. J. (2004) Stable interaction between β-arrestin2 and angiotensin type 1A receptor is required for β-arrestin 2-mediated activation of extracellular signal-regulated kinases 1 and 2. J. Biol. Chem. 279, 48255–48261 [DOI] [PubMed] [Google Scholar]
- 51. Qian H., Pipolo L., Thomas W. G. (2001) Association of β-arrestin 1 with the type 1A angiotensin II receptor involves phosphorylation of the receptor carboxyl terminus and correlates with receptor internalization. Mol. Endocrinol. 15, 1706–1719 [DOI] [PubMed] [Google Scholar]
- 52. Szczepek M., Beyrière F., Hofmann K. P., Elgeti M., Kazmin R., Rose A., Bartl F. J., von Stetten D., Heck M., Sommer M. E., Hildebrand P. W., Scheerer P. (2014) Crystal structure of a common GPCR-binding interface for G protein and arrestin. Nat. Commun. 5, 4801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Shukla A. K., Westfield G. H., Xiao K., Reis R. I., Huang L. Y., Tripathi-Shukla P., Qian J., Li S., Blanc A., Oleskie A. N., Dosey A. M., Su M., Liang C. R., Gu L. L., Shan J. M., et al. (2014) Visualization of arrestin recruitment by a G protein-coupled receptor. Nature 512, 218–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Pérodin J., Deraët M., Auger-Messier M., Boucard A. A., Rihakova L., Beaulieu M. E., Lavigne P., Parent J. L., Guillemette G., Leduc R., Escher E. (2002) Residues 293 and 294 are ligand contact points of the human angiotensin type 1 receptor. Biochemistry 41, 14348–14356 [DOI] [PubMed] [Google Scholar]
- 55. Nikiforovich G. V., Zhang M., Yang Q., Jagadeesh G., Chen H. C., Hunyady L., Marshall G. R., Catt K. J. (2006) Interactions between conserved residues in transmembrane helices 2 and 7 during angiotensin AT1 receptor activation. Chem. Biol. Drug Des. 68, 239–249 [DOI] [PubMed] [Google Scholar]
- 56. Rovati G. E., Capra V., Neubig R. R. (2007) The highly conserved DRY motif of class A G protein-coupled receptors: beyond the ground state. Mol. Pharmacol. 71, 959–964 [DOI] [PubMed] [Google Scholar]
- 57. Wei H., Ahn S., Shenoy S. K., Karnik S. S., Hunyady L., Luttrell L. M., Lefkowitz R. J. (2003) Independent β-arrestin 2 and G protein-mediated pathways for angiotensin II activation of extracellular signal-regulated kinases 1 and 2. Proc. Natl. Acad. Sci. U.S.A. 100, 10782–10787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Gáborik Z., Jagadeesh G., Zhang M., Spät A., Catt K. J., Hunyady L. (2003) The role of a conserved region of the second intracellular loop in AT1 angiotensin receptor activation and signaling. Endocrinology 144, 2220–2228 [DOI] [PubMed] [Google Scholar]
- 59. Ohyama K., Yamano Y., Sano T., Nakagomi Y., Wada M., Inagami T. (2002) Role of the conserved DRY motif on G protein activation of rat angiotensin II receptor type 1A. Biochem. Biophys. Res. Commun. 292, 362–367 [DOI] [PubMed] [Google Scholar]
- 60. Hunyady L., Catt K. J., Clark A. J., Gáborik Z. (2000) Mechanisms and functions of AT1 angiotensin receptor internalization. Regul. Pept. 91, 29–44 [DOI] [PubMed] [Google Scholar]
- 61. Hunyady L., Bor M., Baukal A. J., Balla T., Catt K. J. (1995) A conserved NPLFY sequence contributes to agonist binding and signal transduction but is not an internalization signal for the type 1 angiotensin II receptor. J. Biol. Chem. 270, 16602–16609 [DOI] [PubMed] [Google Scholar]
- 62. Laporte S. A., Servant G., Richard D. E., Escher E., Guillemette G., Leduc R. (1996) The tyrosine within the NPXnY motif of the human angiotensin II type 1 receptor is involved in mediating signal transduction but is not essential for internalization. Mol. Pharmacol. 49, 89–95 [PubMed] [Google Scholar]
- 63. Khosla M. C., Leese R. A., Maloy W. L., Ferreira A. T., Smeby R. R., Bumpus F. M. (1972) Synthesis of some analogs of angiotensin II as specific antagonists of the parent hormone. J. Med. Chem. 15, 792–795 [DOI] [PubMed] [Google Scholar]
- 64. Aumelas A., Sakarellos C., Lintner K., Fermandjian S., Khosla M. C., Smeby R. R., Bumpus F. M. (1985) Studies on angiotensin II and analogs: impact of substitution in position 8 on conformation and activity. Proc. Natl. Acad. Sci. U.S.A. 82, 1881–1885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Khosla M. C., Hall M. M., Smeby R. R., Bumpus F. M. (1973) Factors that influence the antagonistic properties of angiotensin II antagonists. J. Med. Chem. 16, 829–832 [DOI] [PubMed] [Google Scholar]
- 66. Holck M., Bossé R., Fischli W., Gerold H., Escher E. (1989) An angiotensin II antagonist with strongly prolonged action. Biochem. Biophys. Res. Commun. 160, 1350–1356 [DOI] [PubMed] [Google Scholar]
- 67. Bosse R., Gerold M., Fischli W., Holck M., Escher E. (1990) An angiotensin with prolonged action and blood pressure–lowering properties. J. Cardiovasc. Pharmacol. 16, S50–S55 [DOI] [PubMed] [Google Scholar]
- 68. Noda K., Saad Y., Karnik S. S. (1995) Interaction of Phe8 of angiotensin II with Lys199 and His256 of AT1 receptor in agonist activation. J. Biol. Chem. 270, 28511–28514 [DOI] [PubMed] [Google Scholar]
- 69. Feng Y. H., Noda K., Saad Y., Liu X. P., Husain A., Karnik S. S. (1995) The docking of Arg2 of angiotensin II with Asp281 of AT1 receptor is essential for full agonism. J. Biol. Chem. 270, 12846–12850 [DOI] [PubMed] [Google Scholar]
- 70. Yamano Y., Ohyama K., Kikyo M., Sano T., Nakagomi Y., Inoue Y., Nakamura N., Morishima I., Guo D. F., Hamakubo T. (1995) Mutagenesis and the molecular modeling of the rat angiotensin II receptor (AT). J. Biol. Chem. 270, 14024–14030 [DOI] [PubMed] [Google Scholar]
- 71. Matsoukas M. T., Potamitis C., Plotas P., Androutsou M. E., Agelis G., Matsoukas J., Zoumpoulakis P. (2013) Insights into AT1 receptor activation through AngII binding studies. J. Chem. Inf. Model. 53, 2798–2811 [DOI] [PubMed] [Google Scholar]
- 72. Inoue Y., Nakamura N., Inagami T. (1997) A review of mutagenesis studies of angiotensin II type 1 receptor, the three-dimensional receptor model in search of the agonist and antagonist binding site and the hypothesis of a receptor activation mechanism. J. Hypertens. 15, 703–714 [DOI] [PubMed] [Google Scholar]
- 73. Vauquelin G., Fierens F. L. P., Gàborik Z., Le Minh T., De Backer J., Hunyady L., Vanderheyden P. M. L. (2001) Role of basic amino acids of the human angiotensin type 1 receptor in the binding of the non-peptide antagonist candesartan. JRAAS 10.1177/14703203010020010501 [DOI] [PubMed] [Google Scholar]
- 74. Miura S., Feng Y. H., Husain A., Karnik S. S. (1999) Role of aromaticity of agonist switches of angiotensin II in the activation of the AT1 receptor. J. Biol. Chem. 274, 7103–7110 [DOI] [PubMed] [Google Scholar]
- 75. Noda K., Feng Y. H., Liu X. P., Saad Y., Husain A., Karnik S. S. (1996) The active state of the AT1 angiotensin receptor is generated by angiotensin II induction. Biochemistry 35, 16435–16442 [DOI] [PubMed] [Google Scholar]
- 76. Nygaard R., Zou Y., Dror R. O., Mildorf T. J., Arlow D. H., Manglik A., Pan A. C., Liu C. W., Fung J. J., Bokoch M. P., Thian F. S., Kobilka T. S., Shaw D. E., Mueller L., Prosser R. S., Kobilka B. K. (2013) The dynamic process of beta(2)-adrenergic receptor activation. Cell 152, 532–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Wacker D., Wang C., Katritch V., Han G. W., Huang X. P., Vardy E., McCorvy J. D., Jiang Y., Chu M., Siu F. Y., Liu W., Xu H. E., Cherezov V., Roth B. L., Stevens R. C. (2013) Structural features for functional selectivity at serotonin receptors. Science 340, 615–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Dror R. O., Arlow D. H., Maragakis P., Mildorf T. J., Pan A. C., Xu H., Borhani D. W., Shaw D. E. (2011) Activation mechanism of the β2-adrenergic receptor. Proc. Natl. Acad. Sci. U.S.A. 108, 18684–18689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Palczewski K., Kumasaka T., Hori T., Behnke C. A., Motoshima H., Fox B. A., Le Trong I., Teller D. C., Okada T., Stenkamp R. E., Yamamoto M., Miyano M. (2000) Crystal structure of rhodopsin: A G protein-coupled receptor. Science 289, 739–745 [DOI] [PubMed] [Google Scholar]
- 80. Schwartz T. W., Frimurer T. M., Holst B., Rosenkilde M. M., Elling C. E. (2006) Molecular mechanism of 7TM receptor activation: a global toggle switch model. Annu. Rev. Pharmacol. Toxicol. 46, 481–519 [DOI] [PubMed] [Google Scholar]
- 81. Nygaard R., Frimurer T. M., Holst B., Rosenkilde M. M., Schwartz T. W. (2009) Ligand binding and micro-switches in 7TM receptor structures. Trends Pharmacol. Sci. 30, 249–259 [DOI] [PubMed] [Google Scholar]
- 82. Rosenkilde M. M., Benned-Jensen T., Frimurer T. M., Schwartz T. W. (2010) The minor binding pocket: a major player in 7TM receptor activation. Trends Pharmacol. Sci. 31, 567–574 [DOI] [PubMed] [Google Scholar]
- 83. Ahuja S., Smith S. O. (2009) Multiple switches in G protein-coupled receptor activation. Trends Pharmacol. Sci. 30, 494–502 [DOI] [PubMed] [Google Scholar]
- 84. Worth C. L., Kleinau G., Krause G. (2009) Comparative sequence and structural analyses of G protein-coupled receptor crystal structures and implications for molecular models. PLoS ONE 4, e7011. [DOI] [PMC free article] [PubMed] [Google Scholar]