

Background: RhoA·GTP activates mDia1 resolving an autoinhibited state leading to formation of unbranched actin filaments in cells.
Results: Liprin-α3 uses an α-helical region to bind to mDia1, counteracting mDia1 activation by RhoA.
Conclusion: Liprin-α3 competes with RhoA and mDia1 autoinhibition to modulate its activity.
Significance: Knowing how mDia1 is inactivated is essential to understand its biology and for therapeutic approaches.
Keywords: liprin; Ras homolog gene family, member A (RhoA); actin; formin; mDia1; cytoskeleton
Abstract
Diaphanous-related formins are eukaryotic actin nucleation factors regulated by an autoinhibitory interaction between the N-terminal RhoGTPase-binding domain (mDiaN) and the C-terminal Diaphanous-autoregulatory domain (DAD). Although the activation of formins by Rho proteins is well characterized, its inactivation is only marginally understood. Recently, liprin-α3 was shown to interact with mDia1. Overexpression of liprin-α3 resulted in a reduction of the cellular actin filament content. The molecular mechanisms of how liprin-α3 exerts this effect and counteracts mDia1 activation by RhoA are unknown. Here, we functionally and structurally define a minimal liprin-α3 core region, sufficient to recapitulate the liprin-α3 determined mDia1-respective cellular functions. We show that liprin-α3 alters the interaction kinetics and thermodynamics of mDiaN with RhoA·GTP and DAD. RhoA displaces liprin-α3 allosterically, whereas DAD competes with liprin-α3 for a highly overlapping binding site on mDiaN. Liprin-α3 regulates actin polymerization by lowering the regulatory potency of RhoA and DAD on mDiaN. We present a model of a mechanistically unexplored and new aspect of mDiaN regulation by liprin-α3.
Introduction
Formins are Rho effector proteins that are present from yeast to humans to bundle and nucleate actin filaments, resulting in the formation of unbranched actin filaments (1–3). As leaky capping proteins, formins stay processively bound at the filaments' barbed ends during polymerization, allowing actin dynamics to proceed (4–6). In total, 15 formins can be distinguished in mammals, which can be subdivided into seven groups (7–9).
Members of the subclass of the Diaphanous-related formins are regulated by proteins of the Rho family of small GTP-binding proteins (10). Binding of Rho proteins to the regulatory N terminus, mDiaN, relieves an autoinhibited state maintained by binding of the C-terminal Diaphanous-autoregulatory domain (DAD)2 to mDiaN (11–16).
Structurally, mDiaN can be subdivided into the subdomains GBDN (GTPase-binding domain; aa 69–134), essential for RhoA binding; the Armadillo repeat region/Diaphanous-inhibitory domain (DID; aa 135–369); and the dimerization domain (DD; aa 370–451) (14). Adjacent to the DD, there is a coiled-coil (CC) domain spanning residues 452–570. The DAD contains the DAD core region (DCR; aa 1175–1195) essential for DID binding and a subsequent lysine/arginine-rich region (aa 1196–1199) also contributing to the binding (11, 17). Both the GBDN and DID subdomains are needed for RhoA·GTP interaction, but the DID is sufficient for DAD binding (11, 14).
Most formins contain central formin homology (FH) domains (18). The proline-rich FH1 domain serves as binding site for Src homology 3 and WW domain-containing proteins and furthermore for the G-actin-binding protein profilin (19–21).
Recent single particle electron microscopy data support previous biochemical studies to show that full-length formins are dimeric (22). The FH2 domain forms a dimeric ring, which contacts three actin molecules to form an actin nucleus for actin polymerization (23–25). Both FH1 and FH2 are needed for formin function in vivo. The activation of formins by Rho proteins has been extensively studied (11, 14, 15, 26). Actin filaments assembled in vitro using recombinantly expressed FH1-FH2 can grow up to 50 μm in length (27, 28). However, considering an average length of actin filaments in cells of about 1 μm shows that additional mechanisms next to autoinhibition must exist to inactivate formins in a spatiotemporal manner.
Recently, Sakamoto et al. (29) identified liprin-α3 as a direct mDia1 binding partner. Liprins can be found in vertebrates, flies, and worms (30). They play important roles in regulation of synapse function and assembly but are also involved in regulation of cell motility (31, 32). Depletion of liprin-α leads to decreased cell migration and cell spreading, whereas its overexpression results in the opposite (33, 34).
Liprins can be subdivided into four types based on their primary structure: liprin-α, liprin-β, KazrinE, and liprin-γ. In mammals, there are four liprin-αs (α1–4), two liprin-βs (β1 and β2), and KazrinE (35, 36). Additionally, liprin-γ has recently been identified as another member of the liprin family in Drosophila (32). Liprin-α1, -β1, and -β2 are ubiquitously expressed, whereas the remaining are mainly expressed in the brain (37, 38). Liprins have an N-terminal coiled-coil domain, which is followed by three sterile α-motif or liprin homology domains. It was shown that the second sterile α-motif domain binds to the intracellular domain of the LAR receptor tyrosine phosphatase (39). The sterile α-motif domains are used to form heterodimers between α- and β-liprins, whereas the N-terminal coiled-coil domains mediate homooligomerization (40). Thereby, liprins are able to form multiprotein complexes needed to regulate numerous cellular activities (41, 42).
A region in the liprin-α N terminus overlapping the coiled-coil domain has recently been mapped as directly binding the open form of mDia1 contacting the DID of the regulatory N terminus (mDiaN). The expression of this liprin-α3 domain in HeLa cells displaces mDia1 from the plasma membrane and reduces actin stress fiber formation (29). Binding of active RhoA is not sufficient to fully activate formins, suggesting that additional events, such as post-translational modifications or binding to proteins and phospholipids, are needed (22, 43). To switch off formin function, these activating events must be counterbalanced.
Here, we aim at elucidating how liprin-α3 binds to mDia1 and regulates its function counteracting RhoA activation, resulting in reduction in cellular actin filament formation. Does it use the same binding site as DAD or active RhoA on mDiaN, and how is RhoA and DAD binding affected by the presence of liprin-α3?
Experimental Procedures
Expression and Purification of mDiaN, Liprin-α3, and DAD Fragments
Human RhoA and mouse mDia1 fragments were expressed and purified as described in the final buffer A (50 mm Tris/HCl, pH 7.4, 100 mm NaCl, 5 mm MgCl2, 2 mm β-mercaptoethanol) (26). Liprin-α3 fragments and mutants were purified accordingly. Point mutations in mDia1, liprin-α3, and DAD were created using the QuikChange protocol (Stratagene). All protein concentrations of mDiaN, DAD, and liprin-α3 were determined using the absorption at 280 nm (A280). For DAD(1145–1200)/DAD(1145–1209) and for liprin-α3 fragments with a low extinction coefficient at A280, tryptophan residues were added at the N terminus for concentration determination. The RhoA concentration was determined using the Bradford assay (Expedeon).
Liprin-α3 Peptide Synthesis
Peptides were synthesized on a Rink amide resin using an automated solid phase peptide synthesizer (MultiSynthec) and following standard Fmoc/tert-butanol strategy. The resin load (Rink amide resin) was 15 μmol, and for each coupling step, seven equivalents of Fmoc-amino acid and seven equivalents of activation reagents (ethyl 2-cyano-2-(hydroxyimino)acetate and diisopropylcarbodiimide) were used. N-terminal fluorescein labeling was achieved by using 3 eq of 5,6-carboxyfluorescein (CF) and 3 eq of ethyl 2-cyano-2-(hydroxyimino)acetate and diisopropylcarbodiimide, respectively. All reactions were performed in N,N-dimethylformamide. For final cleavage of the peptides, the resin was treated with TFA/triisopropylsilane/water (95:2.5:2.5 (v/v/v)) for 3 h. Subsequently, peptides were precipitated by the addition of ice-cold diethylether, washed, and lyophilized. All peptides were purified using preparative reverse phase HPLC and analyzed by LC-electrospray ionization-MS (LCQ, FinniganMAT, Thermo) with a gradient of 20–70% acetonitrile (0.1% TFA) in water (0.08% TFA) for 15 min. Lip(567–587) (TPRSARLERMAQALALQAGSP): 2223.6 Da (calc.), 2223.9 Da (exp.); F-Lip(567–587) (CF-GSG-TPRSARLERMAQALALQAGSP): 2784.11 Da (calc.), 2784.32 Da (exp.); F-Lip(567–582) (CF-GSG-TPRSARLERMAQALAL): 2343.65 Da (calc.), 2343.62 Da (exp.).
Stopped-flow Kinetics
The experiments were performed at 20 °C using a SX20 Applied Photophysics spectrometer (Leatherhead, UK). The mant-GppNHp nucleotide was excited at 350 nm, and the emission was recorded using a 420-nm cut-off filter. For the association kinetics, a constant concentration of 250 nm (final) RhoA·mant-GppNHp was titrated with increasing concentrations of mDiaN (final 0.6–40.7 μm) without/with the presence of a 2-fold molar excess of liprin-α3. The obtained data follow a pseudo-first-order binding kinetics. The fluorescence was plotted as a function of time and fitted to a single exponential function to result in the observed rates (kobs) for each concentration. The slope of the linear fit of the plot of kobs versus protein concentration resulted in the association rate constant (ka). The dissociation rate constants (kd) were obtained by mixing a preformed 100 nm (final) mDiaN·RhoA·mant-GppNHp complex with a 100-fold excess of unlabeled RhoA Q63L (final 10 μm). In the experiments with liprin-α3, 10 μm (final) of liprin-α3 was added to RhoA Q63L. GraphPad Prism version 6.0 was used for data analysis.
Isothermal Titration Calorimetry (ITC) Measurements
The interactions of liprin-α3/mDiaN, mDiaN/DAD, and mDiaN/RhoA; the mutational analysis of the liprin-α3-mDia1 interface; and the competition experiments were performed on an ITC200 instrument at 20 °C (GE Healthcare) (65). For a typical experiment, 2 μl of protein in the syringe (200–500 μm) was stepwise injected into the cell solution of protein in concentrations of 20–50 μm, depending on the interaction analyzed. In the competition experiments, 270 μm protein in the syringe was titrated to a complex of a 2-fold molar excess of liprin-α3 (54 μm) over mDia1 (27 μm) in the cell. The binding isotherms obtained were fitted to a one-site or competitive binding model using Origin version 7.0 and the integrated MicroCal software, in which stoichiometry (N), enthalpy change (ΔH), and the equilibrium association constant (KA) are directly obtained from the experimental data (44). We used the standard EDTA-CaCl2 sample tests as described by MicroCal to assess the statistical significance of individual observations. These gave values within the manufacturer's tolerances of ±20% for KA values and ±10% in ΔH.
Fluorescence Polarization Experiments
All experiments were done using a Paradigm Detection Platform (Beckman Coulter). The CF-FITC-labeled F-Lip(567–587) and F-Lip(567–582) peptides were excited at 485 nm, and the emission was detected at 535 nm. The experiments were performed in buffer A at 37 °C, and the integration time was 500 ms. F-Lip(567–587) and F-Lip(567–582) (100 nm) were incubated with 10 μm mDiaN or 10 μm mDiaN plus 15 μm RhoA Q63L. The solutions were preincubated until the equilibrium was reached, and the polarization signal was stable. Measurements without liprin-α3 peptides served as blank values. Data analysis was done with GraphPad Prism version 6.0.
Crystallization and Structure Determination
Crystals of DID (aa 135–369) and Lip(567–587) grew in mother liquor containing 0.2 m NaCl, 0.1 m Tris/HCl, pH 8.0, 20% PEG 6000 (buffer C) overnight at 20 °C using the sitting drop/vapor diffusion method (150 nl of protein at 10 g/liter plus 150 nl of buffer C). The crystals had a final size of ∼50 × 50 × 50 μm and were shock-frozen in liquid nitrogen in buffer C containing 30% (w/v) of d-glucose as cryoprotectant. A native data set was collected at 100 K on beamline X06DA at a wavelength of 1.0 Å using a Dectris Pilatus 2M detector at the Swiss Light Source (SLS), Paul Scherrer Institute (Villigen, Switzerland). The detector distance was 165 mm, the oscillation range was 0.1°, and 1800 frames were collected. The program suite CCP4 was used for structure determination (45). Data were indexed and integrated with MOSFLM version 7.0.9 and scaled with SCALA version 3.3.20 (46–48). The crystals belonged to the monoclinic space group C2 (C121) and contained two Lip(567–587)·DID heterodimers in the asymmetric unit. Initial phases were determined with the program Phaser using residues 135–369 of chain B of the mDiaN·RhoC structure (PDB code 1Z2C) to search for two molecules (49). The program Coot version 0.7.1 was used to build the model into the 2Fo − Fc and Fo − Fc electron density maps in iterative rounds of refinement with REFMAC5 (50, 51). In the final model, 100% of the residues are in the allowed regions of the Ramachandran plot, as judged by MolProbity (52, 53). All structure figures presented here were made with PyMOL version 1.6.9.0 (54). Data collection and refinement statistics are given in Table 2. As an example, a part of the electron density for the liprin-α3-peptide (chain D) is shown as an omit map in Fig. 2I. Rwork is calculated as follows, Rwork = Σ|Fo − Fc|/ΣFo. Fo and Fc are the observed and calculated structure factor amplitudes. Rfree is calculated as Rwork using the test set reflections only.
TABLE 2.
Data collection and refinement statistics and validation of the geometry of the Lip(567–587)·DID structure (molecular replacement)
Values shown in parenthesis for data collection and refinement statistics are for the highest resolution shell. One single crystal was used for data collection. For the definitions of Rfree and Rwork, see “Experimental Procedures.”
| Lip(567–587)·DID | |
|---|---|
| Data collection | |
| Space group | C121 |
| Cell dimensions | |
| α, β, χ (Å) | 121.09, 49.38, 106.37 |
| a, b, g (degrees) | 90.0, 97.86, 90.0 |
| Resolution (Å) | 32.30–1.65 (1.74–1.65) |
| Rsym or Rmerge | 0.066 (0.730) |
| I/σI | 9.4 (1.6) |
| Completeness (%) | 99.7 (99.9) |
| Redundancy | 3.1 (3.2) |
| Refinement | |
| Resolution (Å) | 32.32 (1.65) |
| No. of reflections | 71,251 |
| Rwork/Rfree | 17.55/ 21.01 |
| No. of atoms | |
| Protein | 3926 |
| Ligand/ion | 16 |
| Water | 394 |
| B factors | |
| Protein | 26.56 |
| Ligand/ion | |
| Ni | 22.98 |
| Tris | 31.94 |
| Water | 35.62 |
| Root mean square deviations | |
| Bond lengths (Å) | 0.022 |
| Bond angles (degrees) | 2.007 |
| Geometry | |
| Poor rotamers | 0 |
| Ramachandran | |
| Outliers (%) | 0 |
| Favored (%) | 99.38 (480/483 aa) |
| Allowed (%) | 100 (483/283 aa) |
| Cβ deviations >0.25Å | 0 |
| Clashscorea | 2.39 |
a Clashscore is the number of serious steric overlaps (>0.4 Å) per 1000 atoms.
FIGURE 2.

Structure of the Lip(567–587)·DID complex. A, ribbon representation of the Lip(567–587)·DID (aa 135–369) complex (left) and a 90° clockwise rotated view (right). Liprin-α3 (green) binds to the ARM3 to -5 region of DID (gray) in an orientation antiparallel to ID α17. B, close-up of the Lip(567–587)·DID structure. Liprin-α3 makes electrostatic interactions with the ID (Arg-575L–Glu-358D and Arg-572L–Glu-362D) as well as hydrophobic interactions using Leu-573L, Met-576L, and other residues. C, schematic presentation of the interactions in the Lip(567–587)·DID structure generated with LigPlot+ version 1.4.5. Liprin-α3 makes hydrophobic as well as specificity-creating salt bridges with the mDia1 DID. D, superposition of the Lip(567–587)·DID (gray/green) structure with the mDiaNΔG·DAD (light orange/dark orange) structure (PDB code 2BAP). The liprin-α3 and DAD-binding sites overlap significantly. E, detailed view of the superposition of Lip(567–587)·DID and mDiaNΔG·DAD. The C-terminal half of liprin-α3 overlaps with the C-terminal half of the DCR. Liprin-α3 follows the path of the DBR along the mDiaN ID. F, composition of the putative ternary complexes of liprin-α3·RhoC·mDiaN (green/blue/gray; PDB code 2BAP) and liprin-α3·DAD·DID (green/orange/gray; PDB code 1Z2C). The subdomain GBDN from the RhoC·mDiaN (PDB code 1Z2C) structure is shown in dark gray. RhoC does not make any obvious steric or electrostatic clashes with liprin-α3. G, structural similarity of DID from the complexes of RhoC·mDiaN (PDB code 1Z2C), DAD·mDiaNΔG (PDB code 2BAP), mDiaN (PDB code 2BNX), and Lip(567–587)·DID. The mDiaN fragments shown were superposed on the DID. The structures are highly similar in the DID with root mean square deviation values ranging from 0.401 to 0.583 Å for the peptide backbone and from 0.367 to 0.541 Å for the Cα atoms. Structural differences are only visible in the ID and the following DD (aa 369–451) not present in the structure presented here. Lip(567–587)·DID resembles most the uncomplexed mDiaN structure (PDB code 2BNX). H, electrostatic surface potential of DID as generated by APBS. RhoC, DAD, and liprin-α3 are shown as they are located on the mDiaN surface. RhoC uses switch I and II to interact with a positively charged groove on the DID surface. As a second binding site, the Rho insert helix is within interaction distance to the ARM4/5 of the DID. Lip(567–587) is located along ID α17 connecting the DID and the DD of mDiaN. Arg-572L and Arg-575L make salt bridges toward negatively charged residues Glu-362D and Glu-358D of the mDiaN ID. The C-terminal part of the ID is highly negatively charged. Blue, positive charge; red, negative charge. The figure is scaled from −10 to +10 kbTeC−1 (kb, Boltzmann's constant; T, temperature in Kelvin; eC−1, elementary charge). I, FO − FC omit difference map of the N-terminal part of the liprin-α3 peptide covering residues Thr-567 to Arg-572 (chain D) countered at 3σ.
Immunofluorescence Analysis of F-actin Content
HeLa cells were grown in DMEM and N2a cells were grown in 1:1 DMEM/Opti-MEM (plus 10% FCS, 100 units/ml penicillin, and 100 μg/ml streptomycin). For transfection, the DNA constructs (0.5 μg of pEGFP-N3-RhoA Q63L and mCherry-C1-liprin-α3) were mixed with 1.5 μl of Lipofectamine LTX with 1 μl of Plus reagent (Invitrogen) in 100 μl of Opti-MEM. Staining of filamentous actin was done with CF647-phalloidin (1:60; Biotium). Immunofluorescence images were taken using an UltraView VoX, PerkinElmer Life Sciences spinning disc confocal microscope. Maximum projection of a confocal Z-stack was used for data processing. The quantification of CF647-phalloidin intensity was done using ImageJ software (National Institutes of Health), determining the mean total fluorescence. Normalization was done to non-transfected cells. The mock-transfected cells were used as a reference to analyze the effect of RhoA G14V or the RhoA G14V/liprin-α3 overexpression. At least 20 cells were used for analysis per construct. Each construct was transfected and analyzed at least two times.
Results
Definition of the Liprin-α3 mDia1 Interaction Sites
We expressed different N-terminal mDia1 fragments in Escherichia coli and purified them to high purity in multimilligram yield (Fig. 1A). To thermodynamically characterize the mDia1 liprin-α3 interaction, we started with the liprin-α3 fragment 457–737 (Lip(457–737)) defined by Sakamoto et al. (29). Lip(457–737) associates with the N-terminal fragment of mDia1, mDiaNCC (aa 69–570), with an affinity of 5.9 μm, as shown by ITC (Table 1). The same fragment lacking the subdomain GBDN, mDiaNΔGCC (aa 135–570), binds with a similar affinity (3 μm), reflecting that the GBDN is dispensable (Table 1). The longest liprin-α3 fragment solubly expressible in E. coli, Lip(217–587), showed a 2–3-fold reduced affinity toward mDia1 (Table 1).
FIGURE 1.

Definition of the binding sites in liprin-α3 and mDia1. A, domain organization of liprin-α3 and mDia1 and the constructs used in this study. +, interactions of liprin-α3 fragments with mDia1; −, fragments not interacting. B, thermodynamic characterization of the shortest liprin-α3 and mDia1 fragments needed for full interaction using ITC. Lip(567–587) and DID bind with 6.9 μm, showing that those fragments contain all residues essential for the interaction. C, coiled-coil prediction of mDia1 and mouse liprin-α3 using COILS version 2.1. For mDia1, the prediction shows a high score for aa 460–562 forming a coiled-coil. For liprin-α3, extended coiled-coil regions were predicted in the N terminus covering residues aa 26–493 and a second patch in the C terminus from aa 863 to 889. D, analytical size exclusion chromatography of mDia1 and liprin-α3 fragments as indicated. Shown is the molecular size calculated by calibration (MW calc.) and the molecular weights for the respective monomers (MW mon.). For all proteins, the absorption at 280 nm is shown except for Lip(567–587) (for which A220 nm is shown). Aprotinin (6.5 kDa), ribonuclease A (13.7 kDa), carbonic anhydrase (29.0 kDa), ovalbumin (43.0 kDa), and conalbumin (75.0 kDa) were used for calibration of the S75 10/300, and for S200 10/300, additionally aldolase (158 kDa), ferritin (440 kDa), and thyroglobulin (669 kDa) were used.
TABLE 1.
Thermodynamic characterization of liprin-α3 and mDia1 by ITC
KD is the equilibrium dissociation constant, ΔH is the enthalpy, ΔS is the entropy change, and N is the stoichiometry of the interaction. The reaction partner in the syringe (S) (300–400 μm) was titrated to the protein in the cell (C) (30–40 μm). In the cases of the competition assays, a 270 μm concentration of the protein in the cell was titrated to 27 μm complex in the cell.
To further narrow down the mDia1-binding site in liprin-α3, we constructed several liprin-α3 fragments and analyzed their interaction with mDia1 by ITC (Fig. 1, A and B, and Table 1). The liprin-α3 fragments aa 443–491 and aa 491–560 (Lip(443–491) and Lip(491–560)) lost all binding affinity toward mDia1, whereas the fragments 561–587 (Lip(561–587)) and 567–587 (Lip(567–587)) bound mDiaN and DID with 2.6–8.3 μm affinity (Fig. 1B and Table 1).
The DID contains all essential liprin-α3 interaction residues because Lip(561–587) and Lip(567–587) bind with 8.3 and 6.9 μm affinity, respectively. Both liprin-α3 fragments are not part of the coiled-coil domain (55). Mouse liprin-α3 has an N-terminal coiled-coil domain spanning residues 26–493 (UniProt; Fig. 1, A and C).
Analytical size exclusion chromatography runs suggest that Lip(561–587), Lip(567–587), and Lip(457–587) are trimeric in solution (Fig. 1D). Lip(457–587) overlaps in its N terminus with the coiled-coil domain, supporting an oligomerization. However, a low content of secondary structure could also explain the elution profiles observed (Fig. 1D). Lip(217–587) forms a higher oligomer (Fig. 1D). For liprin-α3 aa 190–275, a trimeric coiled-coil region is predicted (MultiCoil; data not shown), supporting our analytical gel filtration experiments. The mDia1 DID forms an elongated monomer in solution. Both mDiaN and mDiaNCC elute as higher oligomers, but from structural data, it is known to form elongated dimers in solution (Fig. 1D). This is supported by coiled-coil predictions (Fig. 1C) (55). Predictions using MultiCoil showed a low probability for mDia1 to form trimeric coiled-coils (data not shown) (56). We used the DID and a synthesized Lip(567–587) peptide for crystallization trials.
The Liprin-α3-mDia1 Structure
We determined the atomic structure of the complex of Lip(567–587) and the DID (Lip(567–587)·DID) by x-ray crystallography up to a final resolution of 1.65 Å (Fig. 2 and Table 1). The crystals belonged to the space group C121 with two copies of Lip(567–587)·DID per asymmetric unit. This is the first structure of a part of the liprin-α3 N-terminal domain in complex with an interaction partner, namely the mDia1 DID. The data showed well defined electron density for the DID as well as for most residues of the Lip(567–587) peptide (Fig. 2I). However, the C-terminal residues 583–587 of liprin-α3 were not visible in the electron density. Using ITC, we found that these residues did not contribute to mDiaN binding under the assay conditions because Lip(561–582) binds with 4.5 μm affinity (Table 1).
The structure shows that the liprin-α3 aa 567–582 (Lip(567–582)) forms an α-helix upon binding to the DID Armadillo repeat motifs 3–5 (ARM3 to -5) with Lip(567–582) contacting the third α-helices (α3) of the three-α-helix-containing ARMs (Fig. 2A). The third α-helices of the ARMs form a concave surface for protein-protein interactions. As stated earlier, the first ARM of the DID is incomplete and contains only helices α2 and α3 of ARM1 and can therefore be considered as a heat repeat (26, 57). The third helix of ARM5 is the extended interdomain helix (ID; α17, α35) connecting the DID with the DD (Fig. 2A). Liprin-α3 makes extensive contacts toward the ID of mDia1, forming salt bridges between Glu-358D and Glu-362D (where the superior “D” represents mDia1) in mDia1 and Arg-575L and Arg-572L (where the superior “L” represents liprin-α3) in liprin-α3 (Fig. 2, B and C). The superposition of the mDiaNΔG·DAD (Protein Data Bank code 2BAP) structure with Lip(567–587)·DID shows that the binding sites of liprin and DAD for mDia1 closely match (Fig. 2, D–F). As shown in Fig. 2E, liprin-α3 overlaps with the DCR (DAD residues 1175–1195) in its C terminus but also with residues following the DCR, including the DAD basic region (DBR; aa 1196–1209) not visible in the mDiaNΔG·DAD structure but shown biochemically to take the path along the ID (PDB code 2BAP; DADvisible: aa 1196–1200) (11, 26). The presence of the complete DBR in DAD substantially increases the affinity toward mDia1. DAD(1145–1195) binds with a 70-fold reduced affinity compared with DAD(1145–1200), as shown previously (11). Under the conditions used here, the affinity of DAD(1145–1200) toward mDiaN is further increased from 37 to 3 nm for DAD(1145–1209) as judged by a competitive ITC-binding assay (Table 1). Although the major contacts in the DAD-mDia1 interaction were hydrophobic, liprin-α3 forms hydrophobic interactions as well as specificity-creating salt bridges with DID residues (Fig. 2, B and C). The validation of the geometry of the structure was performed by MolProbity and is shown in Table 2 (52).
Mutational Characterization of the Liprin-α3 DID Interface
We mutated several hydrophobic residues in mDia1 and found that residues that were reported to completely abolish binding toward DAD, namely A256DD and I259DD (11, 14), also strongly interfered with liprin-α3 binding. Although no binding toward liprin-α3 could be observed with mDiaN I259DD, A256DD reduced the affinity ∼10-fold from 4 to 42 μm (Table 1). Ile-259D is located directly within the hydrophobic interface with liprin-α3, whereas Ala-256D is located at its edge (Fig. 2B). Interestingly, N165DD, which abolishes RhoA·GTP binding without affecting DAD binding, also does not affect liprin-α3 binding (Table 1). For liprin-α3, the mutations M576EL, L573EL, and L580EL within the hydrophobic core of interaction did completely block binding toward mDia1, as did the mutations R569EL, R572EL, and R575EL, the latter two resolving the salt bridges toward Glu-362D and Glu-358D in mDia1. Opposite charge mutations of E358RD and E362RD also completely abolished binding toward liprin-α3, showing the importance of these electrostatic interactions (Table 1). The integrity of the mutant mDia1 proteins (N165DD, A256DD, I259DD, R317ED, E362D, and E358D) used was shown previously examining RhoA and DAD affinities by stopped flow and ITC (14, 26). We analyzed the DID and liprin-α3 mutants by analytical gel filtration (data not shown). All proteins showed a running behavior and elution profile similar to those of the wild type proteins (Fig. 1D). To analyze how liprin-α3 mechanistically impacts the interaction dynamics of mDiaN and Rho/DAD, we performed kinetic studies using stopped flow.
Liprin-α3 Alters mDia1-RhoA Interaction Dynamics
Association
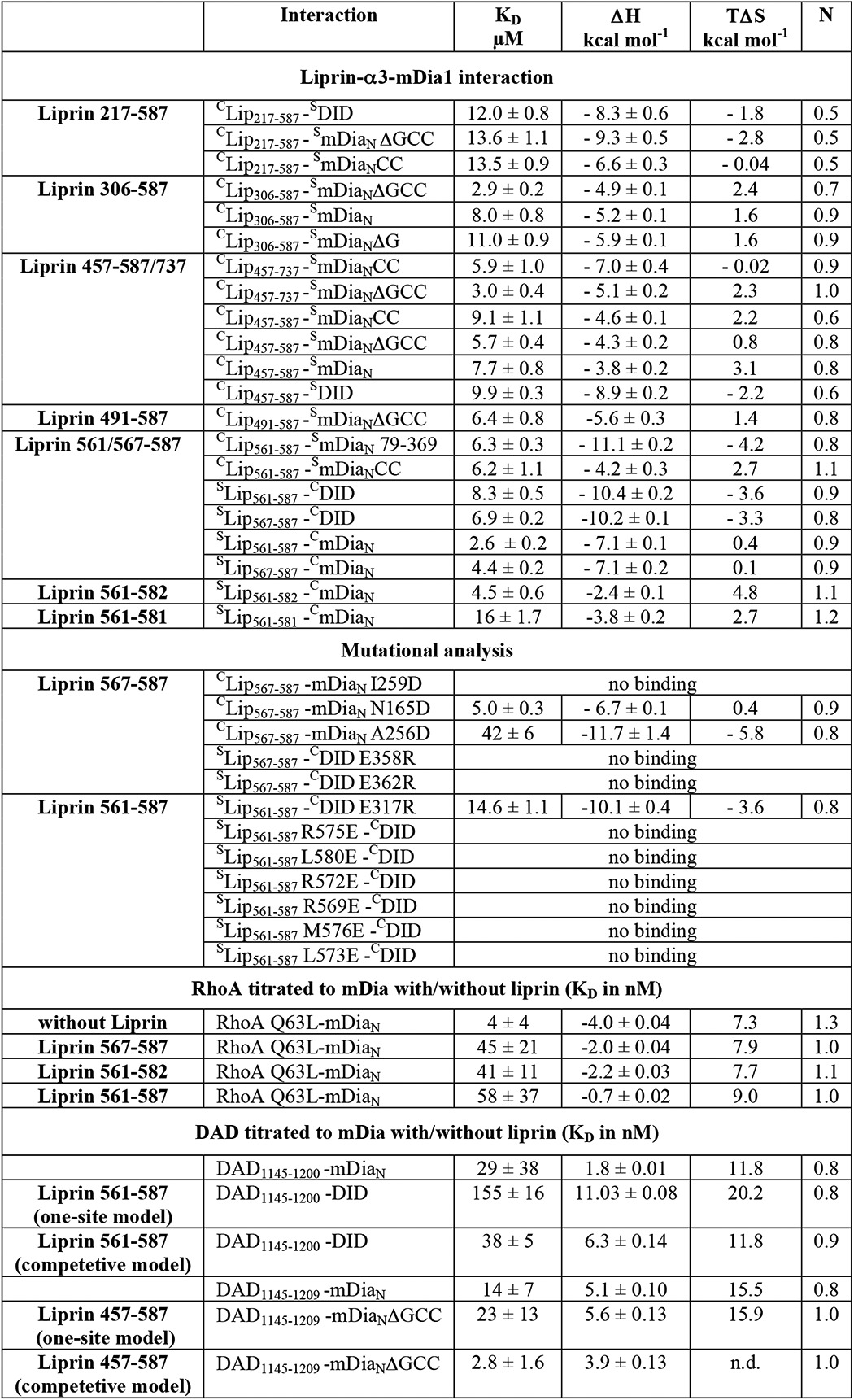
Based on the superimposed structures of RhoC·mDiaN (PDB code 1Z2C) and Lip(567–587)·DID, it is not directly obvious whether and how liprin-α3 interferes with RhoA binding to mDia1 (Fig. 2F). Sakamoto et al. (29) concluded from their cell biological data that on the one hand, liprin-α3 does only bind to the open, non-autoinhibited form of mDia1. On the other hand, RhoA, mDia1, and liprin-α3 were not able to form a ternary complex. We wondered how RhoA displaces liprin-α3 from mDia1, either using an allosteric mechanism or influencing liprin-α3 binding sterically or through long range electrostatic effects. Both might involve the liprin-α3 C-terminal residues not visible in the crystal structure (aa 583–587). Because RhoA shows a more than 1000-fold tighter association with mDia1 compared with liprin-α3 (4 nm versus ∼5 μm; Table 1), we tested whether the presence of liprin-α3 at all influences the mDiaN-RhoA interaction kinetics. To this end, we performed pre-equilibrium stopped-flow experiments (Fig. 3 and Table 3).
FIGURE 3.
Stopped-flow analysis of the influence of liprin-α3 on the mDiaN·RhoA interaction dynamics. A, association rate constant. Mant-GppNHp-loaded RhoA was titrated with increasing concentrations of mDiaN together with/without different liprin-α3 fragments. The observed association rate constants (kobs) were plotted against the mDiaN concentration. The association rate constants (ka) correspond to the slope of the linear fit. B, dissociation rate constant. A preformed mDiaN·RhoA·mant-GppNHp complex was mixed with a 100-fold excess of active RhoA Q63L, and the increase in fluorescence was followed over time. The data followed a single exponential behavior. Error bars, S.D.
TABLE 3.
Effect of different liprin-α3 fragments on the RhoA-mDia1 interaction dynamics as determined by stopped-flow analysis
The ka is the association rate constant, and kd is the dissociation rate constant.
| Liprin fragment | ka | kd | KD |
|---|---|---|---|
| μm−1 s−1 | ms−1 | nm | |
| Without fragment | 1.00 ± 0.01 | 2.35 ± 0.09 | 2.4 |
| 217–587 | 0.51 ± 0.01 | 2.82 ± 0.07 | 5.5 |
| 567–587 | 0.37 ± 0.01 | 3.22 ± 0.04 | 8.7 |
| 561–582 | 0.46 ± 0.01 | 3.22 ± 0.07 | 7.0 |
| 561–587 | 0.27 ± 0.01 | 2.84 ± 0.11 | 10.5 |
RhoA loaded with fluorescently labeled mant-GppNHp (a non-hydrolyzable GTP analogue) was titrated with increasing concentrations of mDiaN (pseudo-first order conditions) in absence/presence of a 2-fold molar excess of different liprin-α3 fragments (Lip(217–587), Lip(561–587), Lip(567–587), and Lip(561–582)). The observed association rates (kobs) were obtained by a single exponential fit of the primary data (not shown). For the reaction in the absence of liprin-α3, linear fitting of kobs against increasing mDiaN concentrations yielded the second-order association rate constant, ka = 1.0 μm−1 s−1 (Fig. 3A). This linear behavior was also observed for the mDiaN-RhoA associations in the presence of all liprin-α3 fragments tested. Interestingly, all liprin-α3 fragments similarly reduced the association rates (Lip(217–587), 0.51 μm−1 s−1; Lip(567–587), 0.37 μm−1 s−1; Lip(561–582), 0.46 μm−1 s−1; Lip(561–587), 0.27 μm−1 s−1; Table 3). This suggests that the functionally important region in liprin-α3 mediating this effect is located within the liprin-α3 residues 567–582, the residues visible in the crystal structure.
Dissociation
We determined how liprin-α3 fragments influence the RhoA·mDiaN dissociation rate constant (kd) by preforming a complex of mant-GppNHp-labeled RhoA and mDiaN and mixing this with a 100-fold excess of unlabeled active RhoA Q63L in the absence/presence of equimolar liprin-α3 concentrations. We found that the primary dissociation kinetics data followed a single exponential behavior as reported earlier under slightly different reaction conditions (here, 100 mm NaCl; earlier, 300 mm NaCl; Fig. 3B). The dissociation rate (2.35 ms−1) obtained here for the RhoA·mDiaN dissociation agrees with the dissociation rate reported earlier (26). We see that the overall dissociation rate, kd, is only slightly increased by the presence of each liprin-α3 fragment tested (2.82–3.22 ms−1; Fig. 3B and Table 3).
In summary, the presence of liprin-α3 influences the dynamics of the mDiaN-RhoA interaction, leading to a much slower RhoA-mDiaN association and an only marginally faster dissociation. This holds to a comparable extent for all of the liprin-α3 fragments tested, again showing that Lip(567–582) does contain all residues mediating the observed effects on the mDiaN-RhoA interaction dynamics. To quantify the inhibitory potency of liprin-α3 on the interactions of mDia1 and RhoA/DAD under equilibrium conditions, we performed competition measurements using ITC.
Displacement of Liprin-α3 from mDia1 by Active RhoA and DAD(1145–1200)/DAD(1145–1209)
For quantification of the inhibitory potency of liprin-α3 on the mDiaN-RhoA and mDiaN-DAD interactions, we performed ITC experiments. We preformed a complex between mDiaN and liprin-α3 using a molar excess of liprin-α3 over mDiaN and titrated this with active RhoA Q63L or DAD.
DAD
Structurally, it is obvious that mDiaN cannot simultaneously bind to liprin-α3 and DAD because they use a highly overlapping binding site on mDiaN (Fig. 2, D–F). We performed competition ITC experiments by titrating DID with Lip(561–587) until saturation and injected DAD(1145–1200) into this complex. The presence of liprin-α3 decreases the DID-DAD(1145–1200) binding affinity from 37 to 155 nm, as judged from fitting the data with a competitive and one-site fit model (Fig. 4B). The affinity obtained by the competitive fit model agrees with a direct measurement of DAD(1145–1200) mDiaN interaction under the same reaction conditions (29 nm; Table 1). Notably, this competitive fit model is only an approximation because, although the liprin-α3 DAD(1145–1200) interaction sites highly overlap, they are not identical. The reaction is mainly entropically driven (TΔS = +20.2 kcal mol−1) with an unfavorable enthalpic contribution. The enthalpy of the competition reaction (ΔH) corresponds to the enthalpy of direct Lip(561–587) binding to DID but with the opposite sign (from +11.0 to −10.4 kcal mol−1; Fig. 4B and Table 1). To show that the DAD(1145–1200) displaces liprin-α3 from mDiaN, we analyzed the ITC cell content by analytical size exclusion chromatography (S75 10/300; Fig. 4C). Indeed, DAD(1145–1200) eluted with DID in the same peak, whereas liprin-α3 eluted alone in the second peak. We furthermore showed that the longer Lip(457–587) can be displaced from mDiaNΔGCC using the DAD(1145–1209) fragment, including the DBR (Table 1). These experiments explain why liprin-α3 is only capable of binding to the open, active form of mDia1, because the affinity of mDiaN toward DAD(1145–1200) and DAD(1145–1209) is more than 2 and 3 orders of magnitude higher compared with liprin-α3, respectively. As a conclusion, liprin-α3 is dissociated by DAD using a highly overlapping binding site on mDiaN due to its significantly higher binding affinity toward mDiaN.
FIGURE 4.

Competition of RhoA and DAD(1145–1200) with liprin-α3 for binding toward mDiaN. A, the competition of RhoA Q63L with Lip(567–587) for mDiaN binding was studied by ITC. RhoA Q63L was titrated on a preformed complex of Lip(567–587) and mDiaN. The interaction showed a negative reaction enthalpy, ΔH, mainly driven by the favorable entropy, TΔS. The presence of liprin-α3 reduced the RhoA affinity toward mDiaN from 4 to 45 nm. B, competition of DAD(1145–1200) and Lip(561–587) for DID binding. The presence of liprin-α3 reduced the binding of DAD(1145–1200) to DID 4-fold (37 versus 153 nm). C, the ITC cell content from B analyzed by analytical gel filtration (S75 10/300). DAD(1145–1200) quantitatively displaces Lip(561–587) from DID. The SDS-PAGE (inset) shows that the high molecular weight peak at 11.06 ml contained only DID·DAD(1145–1200), whereas liprin-α3 eluted in the second peak. D, fluorescence polarization assay. The addition of 10 μm mDiaN to 100 nm F-Lip(567–587) and F-Lip(567–582) led to an increase in the polarization signal, reflecting complex formation. The addition of 15 μm active RhoA Q63L dissociates the Lip·mDiaN complexes shown by the decrease of the polarization signal to the starting level. No ternary Lip·mDiaN·RhoA complexes are formed. Values are shown as mean ± S.D. (error bars) of three replicates.
RhoA
Because both RhoA and liprin-α3 use distinct binding sites on mDiaN, we did not apply a competitive model as described for DAD but instead used a one-site model to fit the data. This shows that the presence of liprin-α3 does reduce the binding affinity (KD) to active RhoA Q63L from 4 nm to an apparent affinity of 40–60 nm (Fig. 4A and Table 1). The stoichiometry for the RhoA-mDiaN-Lip(567–587) interaction was 1:1, and the reaction shows an exothermic heat profile at 20 °C with a total enthalpy change of −2.0 kcal mol−1. The reduction in favorable enthalpy compared with the mDiaN-RhoA interaction in the absence of liprin-α3 (from −4.0 to −2.0 kcal mol−1) agrees with the release of liprin-α3 from mDiaN by RhoA binding. The reaction is mainly driven by the favorable, positive entropic contribution (TΔS = 7.9 kcal mol−1). As a control, we performed these experiments with Lip(561–587) and Lip(561–582) to show that the C and N termini of liprin-α3 have no additional contribution. C-terminally truncated Lip(561–582) reduces the RhoA-mDiaN affinity similarly to 41 nm, and Lip(561–587) reduces it to 58 nm (Table 1). Again, these reactions show exothermic heat profiles, and the reactions are entropically favored. These data show that the presence of liprin-α3 interferes with the association of RhoA and mDiaN, although both use a distinct binding site on mDiaN (Fig. 2F). As a control, we excluded a direct binding of liprin-α3 toward RhoA by ITC (data not shown). Our results support an allosteric displacement mechanism of liprin-α3 by RhoA rather than competing for the same site on mDiaN. However, because the ITC data for RhoA do not exclude a ternary complex formation between mDiaN·liprin-α3 and RhoA, we next performed fluorescence polarization experiments to test whether RhoA displaces liprin-α3 from mDiaN.
Does Liprin-α3 Form a Ternary Complex with mDiaN and RhoA?
In order to examine whether liprin-α3, mDiaN, and RhoA can form a ternary complex, we synthesized N-terminally CF-labeled peptides of Lip(567–587) (F-Lip(567–587)) as well as Lip(567–582) (F-Lip(567–582)), which lacks the C-terminal residues 583–587 not visible in the structure but oriented toward RhoA in a putative ternary complex (Fig. 4D). In fluorescence polarization experiments, we used a 100 nm concentration of the fluorescently labeled liprin-α3 peptide and added 10 μm mDiaN. We observed an increase in the fluorescence polarization signal for both liprin-α3 peptides tested, reflecting a complex formation between both liprin-α3 peptides and mDiaN. To test whether RhoA dissociates the complexes of the liprin-α3 peptides and mDiaN, we added 15 μm active RhoA Q63L. This led to a drop of the fluorescence polarization signal to the baseline for Lip(567–587) and Lip(567–582), indicating the dissociation of the liprin-α3·mDiaN complexes. These results show that the liprin-α3 residues 583–587 are dispensable for the displacement of liprin-α3 from mDiaN by active RhoA. As a support, we also performed analytical size exclusion chromatography with mDiaN, RhoA Q63L, and Lip(457–587) to exclude the formation of a ternary complex (data not shown).
Liprin-α3(567–587) Is a Negative Regulator of Actin Filament Formation
To show that our liprin-α3 fragments are also functional in cells, we analyzed their impact on actin filament formation. We transiently transfected HeLa cells with various N-terminal mCherry-C1-liprin-α3 fragments together with constitutively active EGFP-N3-RhoA G14V constructs. Expression of constitutively active RhoA significantly induced actin stress fiber formation, as judged by quantification of phalloidin staining (Fig. 5, A and B). This effect was reduced by co-expression of mCherry-C1-Lip(1–817), -Lip(561–587), and -Lip(561–582). However, neither full-length liprin-α3, Lip(561–587) L580E, nor the shortest Lip(567–587) fragment led to a reduction in actin filament formation in HeLa cells (Fig. 5, A and B).
FIGURE 5.

The minimal liprin-α3 fragment Lip(567–582) reduces the actin filament content in HeLa cells overexpressing constitutively active RhoA Q63L. A, HeLa cells were transiently transfected with mCherry-C1-liprin-α3 and pEGFP-N3-RhoA G14V expression constructs as indicated. Filamentous actin was stained with CF647-phalloidin. The presence of liprin-α3 fragments reduced the cellular actin filament content in a RhoA G14V overexpression background. Images for selected liprin-α3 fragments are shown; all fragments are depicted in the quantification diagram. The images are shown as a single 0.2-μm-thick optical section of the bottom. For full-length liprin-α3, a close-up representing the merged image of DAPI, EGFP-N3-RhoA G14V, and mCherry-liprin-α3 fluorescence is shown. Scale bars, 50 μm. B, quantification of filamentous actin in HeLa cells. The expression of full-length liprin-α3 did not influence the actin filament content. Of the N-terminal fragments compared, Lip1–817 had the mildest effect on filamentous actin content. C, quantification of F-actin in N2a cells. Full-length liprin-α3 and Lip(561–582) and Lip(3GS)567–587 significantly reduced the F-actin content in RhoA G14V-expressing cells. For B and C, the counted cell numbers are shown in parentheses. The experiment was repeated independently at least two times, and one representative experiment is shown. The data are represented as mean ± S.D. (error bars). **, p < 0.01; ***, p < 0.001 for the indicated comparison; ###, p < 0.001 compared with mock (one-way analysis of variance). a.u., arbitrary units. D, full-length liprin-α3 is localized to the cell periphery in N2a cells (inset of merged image as in A). N2a cells were transfected with mCherry-C1-liprin-α3 full-length and EGFP-N3-RhoA G14V. F-actin was stained with CF647-phalloidin. E, working model for the inhibitory effect of liprin-α3 on mDia1 function. The LCR (aa 567–582) binds to the DID only if the formin is in the open conformation. Binding to the DID lowers the DAD affinity moderately, whereas the affinity toward RhoA is more strongly affected. In this way, the DAD is able to compete with RhoA to re-establish the autoinhibited conformation. Moreover, Rho regulators, such as RhoGAPs, could compete with mDiaN for RhoA binding, leading to subsequent RhoA and thereby mDia1 inactivation. The mDia1 membrane translocation could be achieved by liprin-α3 competing for DID/formin interaction partners, such as IQGAP, and by RhoA dissociation and inactivation. The LCR might also interfere with the interplay of the DBR, PIP2, and ID, resulting in formin inhibition and reduction in cellular filamentous actin. For simplicity, the model does not include the subcellular localization of mDia1/liprin-α3 and the dimeric state of mDia1 and possible oligomeric (homo- and/or heterooligomers) states of liprin-α3. a.u., arbitrary units.
For Lip(567–587), we placed a longer polypeptide linker between the Lip(567–587) coding region and the N-terminal mCherry (Lip(3GS)(567–587)), preventing the possibility that the mCherry protein sterically occludes the binding of Lip(567–587) to mDia1. Indeed, placing the linker between the mCherry and the Lip(567–587) restored the actin-reducing effect, showing the importance of the N terminus of Lip(567–587) for its function (Fig. 5, A and B). This reflects the mechanistic relevance of the N-terminal region contacting the mDiaN ID/DD.
Notably, Sakamoto et al. (29) showed a reduction in cellular F-actin for endogenous full-length liprin-α3 by siRNA knockdown in HeLa cells. However, we could not observe this overexpressing full-length liprin-α3 in HeLa cells. To show whether this is cell type-dependent, we overexpressed full-length liprin-α3 in murine neuroblastoma N2a cells. Indeed, we observed that full-length liprin-α3 significantly reduces the actin filament content similarly to Lip(561–587) and Lip(3GS)(567–587) (Fig. 5C). As a control, we analyzed the overexpression levels of full-length liprin-α3 in HeLa and N2a cells by immunoblotting. Both cell types expressed liprin-α3 substantially and to a comparable extent, with the N2a cells showing an only marginally stronger expression (data not shown). This excludes the possibility that the differences observed in the F-actin content in the two cell types are due to prominent divergent expression levels. Additionally, full-length liprin-α3 accumulates at the cell periphery in N2a cells in contrast to HeLa cells (Fig. 5, A and D).
Discussion
The Minimal and Functional Liprin-α3
Lip(567–582) (liprin-α3 core region; LCR) affects the mDiaN-RhoA interaction dynamics, competes with RhoA and DAD for binding to mDiaN, is not able to form a ternary complex with RhoA/DAD and mDiaN, and leads to an actin-reducing effect similar to that of longer liprin-α3 fragments. We showed that DAD and active RhoA can displace liprin-α3 from mDia1.
Under physiological conditions, the oligomeric states of mDia1 and liprin might strongly affect its capacity to modulate the interactions of mDia1 and RhoA/DAD. We determined in ITC experiments that the N-terminally longest fragment tested, Lip(217–587), binds mDia1 with a 2–3-fold reduced affinity compared with the shorter fragments with a stoichiometry of 0.5 (shorter fragments 1:1). This indicates that one mDia1 molecule binds two liprins, independently of the mDia1 oligomeric state of the fragments tested (Table 1). Additionally, the stopped-flow data showed the lowest effect on the mDiaN-RhoA kinetics for Lip(217–587). The formation of a higher oligomeric state in Lip(217–587) might render one mDia-binding site inaccessible. For full-length liprin-α3, we only found a reduction of the cellular F-actin content in neuronal N2a cells. Furthermore, liprin-α3 was mostly localized to the cell periphery, in contrast to HeLa cells (Fig. 5D). Therefore, cell type-dependent unidentified mechanisms must exist in cells to bring liprin-α3 to an mDia1 binding-competent and/or -inactivating state. These may involve liprin oligomerization or a functionality residing in the liprin C terminus, such as recruitment to membrane receptors.
Interplay of Liprin-α3 and DAD
Deletion and/or mutation of residues (1196RRKR1199) within the DBR (aa 1196–1209) influences actin filament formation (17). Opposite charge mutations E358RD, E362RD, and D366RD in the mDiaN ID decreased binding toward DAD(1145–1200) (26). The Lip(567–587)·DID structure presented here shows that Lip(567–587) overlaps significantly with the C-terminal part of the DCR (aa 1175–1195) on mDiaN and that it takes a path along the negatively charged ID also used by the DBR (Fig. 2, B and E). For the DBR, it was postulated that it clusters plasma membrane PIP2 and inserts into the lipid bilayer partially responsible for an inhibition of mDia1-mediated actin filament elongation (58). The presence of liprin-α3 might support this by competing with the DBR for binding to the negatively charged ID, reducing the competitive potency of the ID for PIP2 to bind to the DBR (Fig. 2, B and E). Whether liprin-α3 has the same inhibitory potency in vivo is not clear, taking into account the intermolecular interaction of mDiaN and DAD as studied here, whereas it is intramolecular in vivo and therefore most likely even more potent.
In this context, a comparison of the consensus sequence defined for the DCR, (G/A)(V/A)MDXLLEXL(K/R/Q)X(G/A)(S/G/A)(A/P), and Lip(567–587) shows that the residues 583–587 not visible in the structure are a perfect match with the C-terminal DCR consensus (Lip(567–587), N-TPRSARLERMAQALALQAGSP-C) (16). However, DAD and liprin-α3 are oriented in the opposite direction on mDia1 (Fig. 2E). Determination of whether this has any physiological regulatory role will require further investigation.
Notably, Sakamoto et al. (29) used a DAD peptide covering residues 1075–1196, which was displaced by Lip(457–737) from mDia1. However, this truncated DAD fragment only binds with 15 μm to mDia1 (11). We show here that, in fact, DAD(1145–1209) binds nearly 3 orders of magnitude tighter (Fig. 4 and Table 1). In ITC competition experiments, preforming a DAD(1145–1209)·mDia1 complex, we observed neither any direct liprin-α3 binding to the complex nor a dissociation of DAD from mDia1 by liprin-α3 (data not shown).
Displacement of Liprin-α3 by RhoA
From the structure presented here, it is not directly obvious how RhoA displaces Lip(567–582) from DID because both use completely independent binding sites (Fig. 2F). Although RhoA binds 3 orders of magnitude tighter to mDiaN than liprin-α3, its presence can weaken the RhoA-mDiaN interaction about 10–15-fold. These structural and equilibrium ITC data support an allosteric displacement mechanism of liprin-α3 from mDiaN by RhoA.
The displacement of liprin-α3 from mDiaN by RhoA is mainly entropically driven. Lip(567–587) forms an α-helix in complex with mDiaN, but it might be unstructured in the unbound form, which could explain the thermodynamics observed. In initial BioSAXS experiments, we found that unbound liprin-α3 shows a high degree of flexibility in solution indicative of a low amount of secondary structure (data not shown). The mDia1 DID from the structure presented here superposes well with DID domains from other structures (Fig. 2G). However, there is conformational flexibility within the C-terminal half of the mDiaN ID and the DD (Fig. 2G). Liprin-α3 forms important salt bridges with residues from the ID (Fig. 2, B, C, and E). Small alterations in the conformation of the ID/DD induced by RhoA-binding to mDiaN might interfere with liprin-α3 binding and might support liprin-α3 dissociation.
Our kinetic data show that the presence of liprin-α3 reduces the association rates of mDiaN and RhoA, whereas the dissociation is nearly unaffected with the concentrations used. It was reported earlier (i.e. for the interaction of Raf-RBD and Ras) that electrostatics play an important role for the association rates in protein complex formation (59, 60). The opposite charge mutation K133E in the Rho insert helix region was shown to strongly reduce the association rate of RhoA toward mDiaN (26). Binding of liprin-α3 to DID occurs at a highly negatively charged patch along the ID, which is in spatial proximity to the positively charged Rho insert helix in the complexes of mDiaN·RhoC (PDB code 2Z2C) and mDiaN (TSH)·Cdc42 (PDB code 3EG5) (Fig. 2H) (14, 26). Liprin-α3 might interfere with the electrostatic attraction of RhoA by mDiaN in a first association step.
The association rates obtained for mDia1 and RhoA are 2–3 orders of magnitude lower than expected for a diffusion-controlled interaction. This suggests that there might be at least a two-step binding with a rate-limiting step possibly involving conformational changes, which we could not observe under the conditions used.
Mechanisms of How Liprin-α3 Inhibits Formin Function
The shortest still functional liprin-α3 fragment, Lip(567–582), is presumably not capable of sterically interfering with the catalytic FH1 and FH2 domains. It is unlikely that it simultaneously recruits further factors such as RhoGAPs to down-regulate RhoA activity or factors mediating mDia1 membrane translocation. We postulate that liprin-α3 down-regulates mDia1 function by several possible mechanisms (Fig. 5E).
First, liprin-α3 directly interferes with the regulatory machinery to inactivate mDia1 by modulating the RhoA interaction and DAD autoinhibition. The RhoA binding to mDia (KD = 4 nm) is slightly higher compared with the DAD(1145–1209), including the DBR (KD = 3–14 nm). However, the presence of liprin-α3 reduces the mDia1-RhoA interaction (10–15-fold to 40–60 nm) more strongly than the mDia-DAD interaction (3–4-fold to 23 nm). Thereby, the capacity of DAD to compete with RhoA for mDiaN binding and to re-establish the autoinhibited, closed conformation of the formin is increased. Liprin-α3 might also directly affect the DBR function and its interplay with membrane-bound PIP2 and would thereby result in subsequent formin inactivation.
Second, the 10–15-fold reduction of RhoA-mDia1 affinity might be sufficient in vivo for GAP proteins or other regulators to compete with mDia1 for RhoA binding and its following inactivation. Affinities of many RhoGAPs toward Rho proteins loaded with GTP or its transition state mimic GDP·AlFx are reported to be in the low nanomolar range (61).
Third, the presence of liprin-α3 was shown to translocate an N-terminal mDia1 fragment from the plasma membrane (29). This effect could be due to interference with RhoA binding. Alternatively, it was shown that IQGAP recruits mDia1 to the plasma membrane in phagocytic cups by direct binding to the DID (62, 63) (Fig. 5E). The LCR directly targeting the same binding site as IQGAP on mDiaN could thereby interfere with mDia1 membrane translocation. Moreover, neurochondrin, anilin, and the recently identified F-Bar protein CIP-4/Toca-1 have been shown to be involved in formin membrane translocation (64). Determination of whether this is affected by liprins will require further investigation.
Conclusion
Here, we present a detailed mechanistic view down to atomic resolution of how the liprin-α3 regulates the interplay of mDia1, RhoA, and the mDia1-autoinhibitory switch. However, it is still unclear how liprin-α3 can bind to mDiaN in the first place, considering the much higher affinities of DAD and RhoA for mDiaN. Therefore, events that stabilize the formin's open conformation are a prerequisite. Binding to phospholipids and adaptor proteins or post-translational modifications could cooperate in this process. Future investigations will show how liprin-α3 regulates mDia1 function under physiological conditions. This includes the determination of the effective cellular concentrations of liprin-α3, mDia1, and RhoA, dependent on the cell and tissue type. Additionally, the oligomeric states of liprins and the formins and its subcellular localization (receptor/membrane-bound versus cytosolic) might also affect its interplay. The results presented here might be of therapeutic interest to inactivate formin function and thereby actin filament formation in processes such as tumor invasion and metastasis.
Acknowledgments
We thank the beamline groups at the SLS Villingen/Switzerland, whose outstanding efforts have made these experiments possible. Support by the European BIOSTRUCTX_5870 is gratefully acknowledged. We thank the ESRF/Grenoble for being able to perform and support the first BioSAXS measurements at beamline BM29 (BAG: MX-1479). We thank Anncharlott Berglar for the design of the graphical abstract and Astrid Schauss and Nikolay Kladt (CECAD Imaging Facility) for critical discussions of the cell biological experiments. We also thank Prof. Dr. A. Wittinghofer for providing the RhoA and mDia constructs.
This work was supported by the Emmy Noether Program of the German Research Foundation (Deutsche Forschungsgemeinschaft (DFG) Grant LA 2984/1-1).
The atomic coordinates and structure factors (code 4UWX) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- DAD
- Diaphanous-autoinhibitory/autoregulatory domain
- Fmoc
- N-(9-fluorenyl)methoxycarbonyl
- GBDN
- GTPase-binding domain
- DID
- Diaphanous-inhibitory domain
- DD
- dimerization domain
- FH
- formin homology domain
- CC
- coiled-coil domain
- SAM
- sterile α-motif domain
- DCR
- DAD core region
- CF
- 5,6-carboxyfluorescein
- ITC
- isothermal titration calorimetry
- ARM
- Armadillo repeat motif
- ID
- interdomain helix
- DBR
- DAD basic region
- LCR
- liprin-α3 core region
- aa
- amino acids
- PIP2
- phosphatidylinositol 4,5-bisphosphate
- PDB
- Protein Data Bank
- mant-GppNHp
- 2′/3′-O-(N-methyl-anthraniloyl)-guanosine-5′-[(β,γ)-imido]triphosphate.
References
- 1. Li F., Higgs H. N. (2005) Dissecting requirements for auto-inhibition of actin nucleation by the formin, mDia1. J. Biol. Chem. 280, 6986–6992 [DOI] [PubMed] [Google Scholar]
- 2. Machaidze G., Sokoll A., Shimada A., Lustig A., Mazur A., Wittinghofer A., Aebi U., Mannherz H. G. (2010) Actin filament bundling and different nucleating effects of mouse Diaphanous-related formin FH2 domains on actin/ADF and actin/cofilin complexes. J. Mol. Biol. 403, 529–545 [DOI] [PubMed] [Google Scholar]
- 3. Zigmond S. H. (2004) Formin-induced nucleation of actin filaments. Curr. Opin. Cell Biol. 16, 99–105 [DOI] [PubMed] [Google Scholar]
- 4. Pring M., Evangelista M., Boone C., Yang C., Zigmond S. H. (2003) Mechanism of formin-induced nucleation of actin filaments. Biochemistry 42, 486–496 [DOI] [PubMed] [Google Scholar]
- 5. Kovar D. R., Pollard T. D. (2004) Progressing actin: formin as a processive elongation machine. Nat. Cell Biol. 6, 1158–1159 [DOI] [PubMed] [Google Scholar]
- 6. Zigmond S. H., Evangelista M., Boone C., Yang C., Dar A. C., Sicheri F., Forkey J., Pring M. (2003) Formin leaky cap allows elongation in the presence of tight capping proteins. Curr. Biol. 13, 1820–1823 [DOI] [PubMed] [Google Scholar]
- 7. Higgs H. N. (2005) Formin proteins: a domain-based approach. Trends Biochem. Sci. 30, 342–353 [DOI] [PubMed] [Google Scholar]
- 8. Higgs H. N., Peterson K. J. (2005) Phylogenetic analysis of the formin homology 2 domain. Mol. Biol. Cell 16, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schönichen A., Geyer M. (2010) Fifteen formins for an actin filament: a molecular view on the regulation of human formins. Biochim. Biophys. Acta 1803, 152–163 [DOI] [PubMed] [Google Scholar]
- 10. Kühn S., Geyer M. (2014) Formins as effector proteins of Rho GTPases. Small GTPases 5, e29513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lammers M., Rose R., Scrima A., Wittinghofer A. (2005) The regulation of mDia1 by autoinhibition and its release by Rho*GTP. EMBO J. 24, 4176–4187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nezami A., Poy F., Toms A., Zheng W., Eck M. J. (2010) Crystal structure of a complex between amino and carboxy terminal fragments of mDia1: insights into autoinhibition of diaphanous-related formins. PLoS One 10.1371/journal.pone.0012992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Seth A., Otomo C., Rosen M. K. (2006) Autoinhibition regulates cellular localization and actin assembly activity of the diaphanous-related formins FRLα and mDia1. J. Cell Biol. 174, 701–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rose R., Weyand M., Lammers M., Ishizaki T., Ahmadian M. R., Wittinghofer A. (2005) Structural and mechanistic insights into the interaction between Rho and mammalian Dia. Nature 435, 513–518 [DOI] [PubMed] [Google Scholar]
- 15. Watanabe N., Madaule P., Reid T., Ishizaki T., Watanabe G., Kakizuka A., Saito Y., Nakao K., Jockusch B. M., Narumiya S. (1997) p140mDia, a mammalian homolog of Drosophila diaphanous, is a target protein for Rho small GTPase and is a ligand for profilin. EMBO J. 16, 3044–3056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Alberts A. S. (2001) Identification of a carboxyl-terminal diaphanous-related formin homology protein autoregulatory domain. J. Biol. Chem. 276, 2824–2830 [DOI] [PubMed] [Google Scholar]
- 17. Wallar B. J., Stropich B. N., Schoenherr J. A., Holman H. A., Kitchen S. M., Alberts A. S. (2006) The basic region of the diaphanous-autoregulatory domain (DAD) is required for autoregulatory interactions with the diaphanous-related formin inhibitory domain. J. Biol. Chem. 281, 4300–4307 [DOI] [PubMed] [Google Scholar]
- 18. Westendorf J. J., Mernaugh R., Hiebert S. W. (1999) Identification and characterization of a protein containing formin homology (FH1/FH2) domains. Gene 232, 173–182 [DOI] [PubMed] [Google Scholar]
- 19. Courtemanche N., Pollard T. D. (2012) Determinants of Formin Homology 1 (FH1) domain function in actin filament elongation by formins. J. Biol. Chem. 287, 7812–7820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Paul A. S., Pollard T. D. (2008) The role of the FH1 domain and profilin in formin-mediated actin-filament elongation and nucleation. Curr. Biol. 18, 9–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Neidt E. M., Scott B. J., Kovar D. R. (2009) Formin differentially utilizes profilin isoforms to rapidly assemble actin filaments. J. Biol. Chem. 284, 673–684 [DOI] [PubMed] [Google Scholar]
- 22. Maiti S., Michelot A., Gould C., Blanchoin L., Sokolova O., Goode B. L. (2012) Structure and activity of full-length formin mDia1. Cytoskeleton 69, 393–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Otomo T., Tomchick D. R., Otomo C., Panchal S. C., Machius M., Rosen M. K. (2005) Structural basis of actin filament nucleation and processive capping by a formin homology 2 domain. Nature 433, 488–494 [DOI] [PubMed] [Google Scholar]
- 24. Shimada A., Nyitrai M., Vetter I. R., Kühlmann D., Bugyi B., Narumiya S., Geeves M. A., Wittinghofer A. (2004) The core FH2 domain of diaphanous-related formins is an elongated actin binding protein that inhibits polymerization. Mol. Cell 13, 511–522 [DOI] [PubMed] [Google Scholar]
- 25. Xu Y., Moseley J. B., Sagot I., Poy F., Pellman D., Goode B. L., Eck M. J. (2004) Crystal structures of a Formin Homology-2 domain reveal a tethered dimer architecture. Cell 116, 711–723 [DOI] [PubMed] [Google Scholar]
- 26. Lammers M., Meyer S., Kühlmann D., Wittinghofer A. (2008) Specificity of interactions between mDia isoforms and Rho proteins. J. Biol. Chem. 283, 35236–35246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kovar D. R., Harris E. S., Mahaffy R., Higgs H. N., Pollard T. D. (2006) Control of the assembly of ATP- and ADP-actin by formins and profilin. Cell 124, 423–435 [DOI] [PubMed] [Google Scholar]
- 28. Breitsprecher D., Jaiswal R., Bombardier J. P., Gould C. J., Gelles J., Goode B. L. (2012) Rocket launcher mechanism of collaborative actin assembly defined by single-molecule imaging. Science 336, 1164–1168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sakamoto S., Ishizaki T., Okawa K., Watanabe S., Arakawa T., Watanabe N., Narumiya S. (2012) Liprin-α controls stress fiber formation by binding to mDia and regulating its membrane localization. J. Cell Sci. 125, 108–120 [DOI] [PubMed] [Google Scholar]
- 30. Zhen M., Jin Y. (1999) The liprin protein SYD-2 regulates the differentiation of presynaptic termini in C. elegans. Nature 401, 371–375 [DOI] [PubMed] [Google Scholar]
- 31. Spangler S. A., Hoogenraad C. C. (2007) Liprin-α proteins: scaffold molecules for synapse maturation. Biochem. Soc. Trans. 35, 1278–1282 [DOI] [PubMed] [Google Scholar]
- 32. Astigarraga S., Hofmeyer K., Farajian R., Treisman J. E. (2010) Three Drosophila liprins interact to control synapse formation. J. Neurosci. 30, 15358–15368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nomura H., Tadokoro S., Hirashima N. (2011) Liprin-α is involved in exocytosis and cell spreading in mast cells. Immunol. Lett. 139, 110–116 [DOI] [PubMed] [Google Scholar]
- 34. Astro V., Asperti C., Cangi G., Doglioni C., de Curtis I. (2011) Liprin-α1 regulates breast cancer cell invasion by affecting cell motility, invadopodia and extracellular matrix degradation. Oncogene 30, 1841–1849 [DOI] [PubMed] [Google Scholar]
- 35. Nachat R., Cipolat S., Sevilla L. M., Chhatriwala M., Groot K. R., Watt F. M. (2009) KazrinE is a desmosome-associated liprin that colocalises with acetylated microtubules. J. Cell Sci. 122, 4035–4041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sakamoto S., Narumiya S., Ishizaki T. (2012) A new role of multi scaffold protein Liprin-α: Liprin-α suppresses Rho-mDia mediated stress fiber formation. Bioarchitecture 2, 43–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Spangler S. A., Jaarsma D., De Graaff E., Wulf P. S., Akhmanova A., Hoogenraad C. C. (2011) Differential expression of liprin-α family proteins in the brain suggests functional diversification. J. Comp. Neurol. 519, 3040–3060 [DOI] [PubMed] [Google Scholar]
- 38. Zürner M., Mittelstaedt T., tom Dieck S., Becker A., Schoch S. (2011) Analyses of the spatiotemporal expression and subcellular localization of liprin-α proteins. J. Comp. Neurol. 519, 3019–3039 [DOI] [PubMed] [Google Scholar]
- 39. Pulido R., Serra-Pagès C., Tang M., Streuli M. (1995) The LAR/PTP δ/PTP sigma subfamily of transmembrane protein-tyrosine-phosphatases: multiple human LAR, PTP δ, and PTP σ isoforms are expressed in a tissue-specific manner and associate with the LAR-interacting protein LIP.1. Proc. Natl. Acad. Sci. U.S.A. 92, 11686–11690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Serra-Pagès C., Medley Q. G., Tang M., Hart A., Streuli M. (1998) Liprins, a family of LAR transmembrane protein-tyrosine phosphatase-interacting proteins. J. Biol. Chem. 273, 15611–15620 [DOI] [PubMed] [Google Scholar]
- 41. Wei Z., Zheng S., Spangler S. A., Yu C., Hoogenraad C. C., Zhang M. (2011) Liprin-mediated large signaling complex organization revealed by the liprin-α/CASK and liprin-α/liprin-β complex structures. Mol. Cell 43, 586–598 [DOI] [PubMed] [Google Scholar]
- 42. Taru H., Jin Y. (2011) The Liprin homology domain is essential for the homomeric interaction of SYD-2/Liprin-α protein in presynaptic assembly. J. Neurosci. 31, 16261–16268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Li F., Higgs H. N. (2003) The mouse Formin mDia1 is a potent actin nucleation factor regulated by autoinhibition. Curr. Biol. 13, 1335–1340 [DOI] [PubMed] [Google Scholar]
- 44. Sigurskjold B. W. (2000) Exact analysis of competition ligand binding by displacement isothermal titration calorimetry. Anal. Biochem. 277, 260–266 [DOI] [PubMed] [Google Scholar]
- 45. Collaborative Computational Project, Number 4 (1994) The Ccp4 suite: programs for protein crystallography. Acta Crystallogr. D 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 46. Leslie A. G. W., Powell H. R. (2007) Processing diffraction data with MOSFLM. Evolving Methods Macromol. Crystallogr. 10.1007/978-1-4020-6316-9_4 [DOI] [Google Scholar]
- 47. Evans P. (2006) Scaling and assessment of data quality. Acta Crystallogr. D 62, 72–82 [DOI] [PubMed] [Google Scholar]
- 48. Steller I., Bolotovsky R., Rossmann M. G. (1997) An algorithm for automatic indexing of oscillation images using Fourier analysis. J. Appl. Crystallogr. 10.1107/S0021889897008777 [DOI] [Google Scholar]
- 49. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D 67, 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chen V. B., Arendall W. B., 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Davis I. W., Leaver-Fay A., Chen V. B., Block J. N., Kapral G. J., Wang X., Murray L. W., Arendall W. B., 3rd, Snoeyink J., Richardson J. S., Richardson D. C. (2007) MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35, W375–W383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. DeLano W. L. (2002) The PyMOL Molecular Graphics System, version 1.6.9.0, Schroedinger, LLC, New York [Google Scholar]
- 55. Lupas A., Van Dyke M., Stock J. (1991) Predicting coiled coils from protein sequences. Science 252, 1162–1164 [DOI] [PubMed] [Google Scholar]
- 56. Wolf E., Kim P. S., Berger B. (1997) MultiCoil: a program for predicting two- and three-stranded coiled coils. Protein Sci. 6, 1179–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Schulte A., Stolp B., Schönichen A., Pylypenko O., Rak A., Fackler O. T., Geyer M. (2008) The human formin FHOD1 contains a bipartite structure of FH3 and GTPase-binding domains required for activation. Structure 16, 1313–1323 [DOI] [PubMed] [Google Scholar]
- 58. Ramalingam N., Zhao H., Breitsprecher D., Lappalainen P., Faix J., Schleicher M. (2010) Phospholipids regulate localization and activity of mDia1 formin. Eur. J. Cell Biol. 89, 723–732 [DOI] [PubMed] [Google Scholar]
- 59. Sydor J. R., Engelhard M., Wittinghofer A., Goody R. S., Herrmann C. (1998) Transient kinetic studies on the interaction of Ras and the Ras-binding domain of c-Raf-1 reveal rapid equilibration of the complex. Biochemistry 37, 14292–14299 [DOI] [PubMed] [Google Scholar]
- 60. Vijayakumar M., Wong K. Y., Schreiber G., Fersht A. R., Szabo A., Zhou H. X. (1998) Electrostatic enhancement of diffusion-controlled protein-protein association: comparison of theory and experiment on barnase and barstar. J. Mol. Biol. 278, 1015–1024 [DOI] [PubMed] [Google Scholar]
- 61. Graham D. L., Eccleston J. F., Lowe P. N. (1999) The conserved arginine in rho-GTPase-activating protein is essential for efficient catalysis but not for complex formation with Rho·GDP and aluminum fluoride. Biochemistry 38, 985–991 [DOI] [PubMed] [Google Scholar]
- 62. Shannon K. B. (2012) IQGAP family members in yeast, Dictyostelium, and mammalian cells. Int. J. Cell Biol. 2012, 894817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Brandt D. T., Marion S., Griffiths G., Watanabe T., Kaibuchi K., Grosse R. (2007) Dia1 and IQGAP1 interact in cell migration and phagocytic cup formation. J. Cell Biol. 178, 193–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Bogdan S., Schultz J., Grosshans J. (2013) Formin' cellular structures: physiological roles of Diaphanous (Dia) in actin dynamics. Commun. Integr. Biol. 6, e27634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wiseman T., Williston S., Brandts J. F., Lin L. N. (1989) Rapid measurement of binding constants and heats of binding using a new titration calorimeter. Anal. Biochem. 179, 131–137 [DOI] [PubMed] [Google Scholar]