

Background: The Wilms tumor suppressor WTX localizes to both nucleus and cytoplasm, but its nuclear function is not understood.
Results: Nuclear WTX co-immunoprecipitates with the transcriptional corepressor TRIM28, modulating its chromatin binding and coregulating the expression of DNA repeat elements.
Conclusion: WTX contributes to silencing of heterochromatic repeats.
Significance: WTX may modulate tumorigenesis and cell differentiation in part through its effect on chromatin regulation.
Keywords: epigenetics, heterochromatin, protein-protein interaction, transcription repressor, tumor suppressor gene, TRIM28, WTX, Wilms tumor
Abstract
WTX encodes a tumor suppressor implicated in the pediatric kidney cancer Wilms tumor and in mesenchymal differentiation with potentially distinct functions in the cytoplasm, at the plasma membrane, and in the nucleus. Although modulating components of the WNT signaling pathway is a proposed function for cytoplasmic and membrane-bound WTX, its nuclear properties are not well understood. Here we report that the transcriptional corepressor TRIM28 is the major binding partner for nuclear WTX. WTX interacted with the coiled coil domain of TRIM28 required for its binding to Krüppel-associated box domains of transcription factors and for its chromatin recruitment through its own coiled coil and proline-rich domains. Knockdown of endogenous WTX reduced the recruitment of TRIM28 to a chromatinized reporter sequence and its ability to repress a target transcript. In mouse embryonic stem cells where TRIM28 plays a major role in repressing endogenous retroviruses and long interspersed elements, knockdown of either TRIM28 or WTX combined with single molecule RNA sequencing revealed a highly significant shared set of differentially regulated transcripts, including derepression of non-coding repetitive sequences and their neighboring protein encoding genes (p < 1e−20). In mesenchymal precursor cells, depletion of WTX and TRIM28 resulted in analogous β-catenin-independent defects in adipogenic and osteogenic differentiation, and knockdown of WTX reduced TRIM28 binding to Pparγ promoter. Together, the physical and functional interaction between WTX and TRIM28 suggests that the nuclear fraction of WTX plays a role in epigenetic silencing, an effect that may contribute to its function as a regulator of cellular differentiation and tumorigenesis.
Introduction
WTX (also known as AMER1/FAM123B) is a tumor suppressor gene inactivated frequently in somatic Wilms tumors (1, 2) and at lower frequency in a subset of other pediatric and adult cancers (3, 4). Germ line inactivation of WTX causes osteopathia striata with cranial sclerosis, an X-linked developmental disorder with macrocephaly and osteosclerosis in human (5), and neonatal lethality with abnormal mesenchymal differentiation in the mouse (6). The role of WTX in tumor suppression may therefore be linked to its contribution to normal differentiation of mesenchymal organs.
Protein interaction studies have suggested that WTX may function as a molecular scaffold in various cellular compartments. WTX interacts with distinct components of WNT signaling in the cytoplasm and at the plasma membrane, resulting in suppression and activation of the pathway, respectively (7, 8). WTX also appears to have nuclear functions: full-length WTX shuttles between the cytoplasm and nucleus, whereas a truncated minor splicing variant is primarily nuclear (9). Proposed nuclear functions of WTX include modulating the transcriptional activity of WT1, another Wilms tumor suppressor and a master transcriptional regulator of kidney and genitourinary development (9). In addition, WTX stabilizes the acetyltransferases CBP2/p300 and enhances the interaction of CBP with p53, thereby positively modulating p53 activity (10). Together, these nuclear protein interactions suggest functional properties for the WTX tumor suppressor that are distinct from modulation of WNT signaling, leading us to undertake an unbiased screen for its binding partners within nuclear fractions.
We demonstrate here, using immunoprecipitation-mass spectrometry analysis, that TRIM28 is a major interaction partner for nuclear WTX. WTX stabilized chromatin binding by TRIM28 and contributed to transcriptional repression of repetitive sequences by TRIM28 in mouse ES cells. Both WTX and TRIM28 had shared effects on differentiation of mesenchymal precursors. Together, these results implicate WTX in transcriptional regulation and chromatin silencing, potentially contributing to its effects on both cellular differentiation and tumorigenesis.
Experimental Procedures
Cell Culture and in Vitro Differentiation
Mouse embryonic stem cell line ES-D3 and mouse preadipocyte 3T3-L1 cells were obtained from ATCC. U2OS and human embryonic kidney (HEK) 293 cells were maintained in Dulbecco modified Eagle's medium (DMEM; Gibco) supplemented with 10% fetal bovine serum (FBS), 2 mm l-glutamine, and 50 units/ml penicillin/streptomycin. ES-D3 cells were grown on a γ-irradiated feeder layer in DMEM supplemented with 15% FBS, 10 ng/ml leukemia inhibitory factor (Millipore), 1× minimum Eagle's medium non-essential amino acids (Gibco), 0.1 mm β-mercaptoethanol, 2 mm l-glutamine, and 50 units/ml penicillin/streptomycin. 3T3-L1 cells were maintained in DMEM supplemented with 10% calf serum (Colorado Serum Co.), 2 mm l-glutamine, and 50 units/ml penicillin/streptomycin. For adipogenic differentiation, 3T3-L1 cells were grown to confluence, stimulated in adipogenic induction medium (DMEM, 10% FBS containing 1 μm dexamethasone (Sigma), 500 μm isobutylmethylxanthine (Sigma), and 1 mg/ml insulin (Sigma)) followed by adipogenic maintenance medium (DMEM and 10% FBS with 1 mg/ml insulin). PNU-74654 (Santa Cruz Biotechnology) was added to the culture at a final concentration of 100 μm, and the medium was changed every 2–3 days.
Plasmids and Antibodies
GAL4 DNA binding sequence-thymidine kinase (GAL4DBS-TK)-luciferase and GAL4 DNA binding domain (GAL4DBD)-KRAB expression constructs were obtained from Dr. David Schultz of the Wistar Institute (11). A doxycycline-inducible lentiviral construct for GAL4DBD-KRAB was generated by cloning the GAL4DBD-KRAB cassette into pINDUCER20 vector (12). Lentiviral hairpin constructs were obtained from The RNAi Consortium, Broad Institute or generated according to their instruction. Antibodies used in this study are: FLAG, WTX (Sigma-Aldrich), HA (Covance), WTX, SUMO-1, SUMO-2/3, IgG (Cell Signaling Technology), β-actin, lamin A/C, GRB2, TRIM28 (Santa Cruz Biotechnology), histone H3 (Active Motif), TRIM28, and SETDB1 (Abcam).
Immunoprecipitation and Mass Spectrometry
Doxycycline-inducible FLAG-WTX was expressed at low level in the HEK293 cell line. Harvested cells were incubated with buffer A (10 mm Tris-HCl, pH 8.0, 1.5 mm MgCl2, 10 mm KCl, 1 mm DTT, and 1× protease inhibitor mixture (Roche Applied Science) for 20 min on ice and then homogenized with a pestle. Nuclei were washed with buffer A and extracted with buffer B (20 mm Tris, 8.0, 20% glycerol, 420 mm NaCl, 1.5 mm MgCl2, 0.2 mm EDTA, 12% sucrose, 1 mm DTT, and 1× protease inhibitor mixture) for 40 min with rotation at 4 °C. After removal of cell debris by centrifugation, nuclear lysates were dialyzed against buffer C (20 mm Tris, 8.0, 20% glycerol, 100 mm KCl, 1.5 mm MgCl2, 0.1 mm EDTA, and 1 mm DTT) at 4 °C. Nuclear lysates were then incubated with FLAG antibody-conjugated agarose beads (Sigma) overnight with rotation at 4 °C. The agarose beads were washed with 10 volumes of wash buffer A (50 mm Tris, pH 8.0, 150 mm NaCl, 0.1% Nonidet P-40, 10% glycerol, and 0.1 mm EDTA). For experiment II, beads were washed with 10 volumes of wash buffer B (50 mm Tris, pH 8.0, 350 mm NaCl, 0.1% Nonidet P-40, 10% glycerol, and 0.1 mm EDTA) and then washed with 5 volumes of wash buffer A. Bound proteins were eluted with FLAG elution buffer (50 μg/ml FLAG peptide (Sigma) in wash buffer A) and concentrated using a SpeedVac (Savant Inc.). The eluates were then subjected to whole-lane liquid chromatography-tandem mass spectrometry (LC/MS/MS) in the Taplin Biological Mass Spectrometry Facility at the Harvard Medical School.
RNA Sequencing
Total RNA was isolated from feeder-depleted mouse ESCs using RNeasy kits (Qiagen) according to the manufacturer's instructions. Purified RNA was processed and analyzed on a HeliScope single molecule sequencer (Helicos BioSciences) as described previously (13). The full RNA sequencing data set has been deposited in Gene Expression Omnibus (GEO) under the accession number GSE60418.
Quantitative Real Time PCR
RNA was extracted using an RNeasy kit, and cDNA was synthesized using SuperScript III (Invitrogen) according to the manufacturer's instructions. Quantitative real time PCR was performed using Power SYBR Green and an ABI 7500 real time PCR system (Applied Biosystems). The ΔΔCt method was used to calculate relative quantity of expression using GAPDH as an internal control.
Chromatin Immunoprecipitation
Briefly, cells were cross-linked with 5 mm dimethyl 3,3′-dithiobispropionimidate (Thermo Scientific) for 30 min followed by 1% formaldehyde fixation for 10 min. Isolated nuclei from cross-linked cells were sonicated with a Covaris S2 sonicator (Covaris). After removing insoluble debris by centrifugation, equal amounts of chromatin were incubated with antibodies and Protein A Dynabeads (Life Technologies). Bound chromatin was eluted from the magnetic beads, reverse cross-linked, purified, and subjected to quantification with real time PCR.
Micrococcal Nuclease Digestion Assay
After counting the cell number, equal amounts of nuclei were incubated with digestion buffer (50 mm Tris-HCl and 5 mm CaCl2, pH 7.9) containing 1 gel unit/μl micrococcal nuclease (New England Biolabs) for 20 min at room temperature. The digested chromatins were extracted with phenol:chloroform, precipitated with ethanol, and separated on a 1.2% agarose gel.
Bioinformatics
To compute digital gene expression (DGE) from the Helicos sequence data for the RefSeq genes, we used the DGE pipeline of HeliSphere 1.1.498.63 software using the Mouse. Txome reference files with default settings (14). To compute DGE for Repbase to deal with the fact that reads of repetitive sequence will often align to multiple locations in the genome, we used a method called Probabilistic Assessment of Sequencing Alignments that assigns weights to multiple alignments for a read. DEGseq comparisons of DGE profiles were run using the DEGexp function of version 1.0.5 of the Bioconductor DEGseq package (15). A gene or repetitive element was considered differentially expressed if it had a familywise error rate less than 0.05 and a -fold change greater than 2. For identities and genomic locations of genes, we used the knownGene and knownToLocusLink tables of the University of California Santa Cruz mm9 build of the mouse genome, and for repetitive elements we used the RepeatMasker table of that build. We performed hypergeometric gene set enrichment analysis using the gene sets from version 3.0 of MSigDB (16). See supplemental methods for details.
Results
Co-immunoprecipitation of WTX and TRIM28
The shuttling of full-length WTX protein among plasma membrane, cytoplasm, and nucleus (9) raised the possibility that it may interact with distinct protein partners within these different compartments. We used nuclear lysates from HEK293 cells expressing doxycycline-inducible FLAG-tagged, full-length WTX to immunoprecipitate WTX-bound protein complexes followed by mass spectrometry. Fourteen proteins were highly enriched (in duplicate experiments) following FLAG-immunoprecipitation of WTX-expressing nuclear lysates compared with uninduced lysates (Fig. 1A and supplemental Table S1). These proteins were enriched for components of chromatin-modifying enzyme complexes (TRIM28, DBC1, RBBP7, RUVBL2, and RUVBL1) and RNA-binding proteins (SFR14, HNRPF, DDX5, and SAM68), suggesting a potential role of WTX in chromatin regulation and RNA binding. Among these proteins, the transcriptional corepressor TRIM28 (also known as KAP1/TIF1β/KRIP1) was the most highly enriched nuclear WTX-interacting protein, and it was noteworthy in that it has been implicated in cellular differentiation as well as chromatin silencing (17–20). The co-immunoprecipitation with WTX of RBBP7, a component of the TRIM28-associated NurD complex (21), also raised the possibility of functional interaction between WTX and TRIM28. The interaction between FLAG-WTX and endogenous TRIM28 was confirmed by reciprocal co-immunoprecipitation experiments in HEK293 cells (Fig. 1B). In addition, the interaction between endogenous WTX and endogenous TRIM28 was confirmed by co-immunoprecipitation experiments in HEK293 cells, an effect that was abrogated by shRNA-targeted reduction of the endogenous WTX (Fig. 1C). Because both proteins are known to interact with p53, we also confirmed their co-immunoprecipitation using p53-null H1299 cells (Fig. 1, D and E, lanes 1).
FIGURE 1.

Co-immunoprecipitation of WTX and TRIM28. A, nuclear WTX-interacting proteins identified in HEK293 cells by duplicate FLAG immunoprecipitations followed by mass spectrometry analysis. “Peptides” indicates combined unique peptide numbers from two experiments. B, reciprocal immunoprecipitation-Western blotting analysis demonstrating co-immunoprecipitation (IP) of FLAG-WTX and endogenous TRIM28 in HEK293 cells. C, interaction between endogenous WTX and TRIM28 in HEK293 cells demonstrated by co-immunoprecipitation followed by immunoblotting. The co-immunoprecipitation is abrogated by shRNA targeting endogenous WTX. D, mapping of WTX binding domain of TRIM28 in H1299 cells expressing FLAG-WTX and truncated HA-TRIM28 proteins (schematics in upper panel) by immunoprecipitation-Western blot analysis. E, mapping of TRIM28 binding domain of WTX in H1299 cells expressing HA-TRIM28 and truncated FLAG-WTX proteins (schematics in upper panel) by immunoprecipitation-Western blot analysis. IB, immunoblotting; CCD, coiled coil domain; PR, proline-rich domain; PHD, plant homeodomain; ER, glutamic acid-rich domain; REA, arginine-glutamic acid-alanine repeats; FAM, FAM123 family homology region; RING, really interesting new gene.
Multiple domains within TRIM28 have been implicated in mediating interactions with its binding partners. The N-terminal Ring, B-box, and coiled coil domains interact with the Krüppel-associated box (KRAB) domain of KRAB-zinc finger proteins, thereby resulting in both its homo-oligomerization and its recruitment to chromatin. At the C terminus of TRIM28, the HP1 box, plant homeodomain, and bromodomains are responsible for recruiting to TRIM28-bound chromatin the silencing machinery, including HP1, the NurD complex component CHD3, and the histone methyltransferase SETDB1 (22). We used TRIM28 constructs encoding nested truncations to map the WTX-interacting domain to the N-terminal coiled coil domain, the region of TRIM28 similarly involved in binding to KRAB domains and mediating its recruitment to chromatin (Fig. 1D). Using WTX deletion constructs, we reciprocally mapped its TRIM28 interaction domain to two separate regions of WTX: the coiled coil domain 2 within the central region of WTX and its C-terminal proline-rich domain. Both of these WTX domains were required for binding to TRIM28 (Fig. 1E).
WTX Supports TRIM28-mediated Transcriptional Repression
TRIM28 is thought to function primarily as a transcriptional corepressor for KRAB-zinc finger proteins, an effect that has been measured most consistently through TRIM28-mediated transcriptional repression of the GAL4DBS-TK-luciferase reporter in the presence of a GAL4DBD-KRAB fusion protein (11) (Fig. 2A). In this assay, recruitment of TRIM28 to the chromosomally integrated reporter is mediated through its binding to the KRAB domain, leading to repression of baseline TK-driven transcription (11). To test the consequences of endogenous WTX knockdown on TRIM28-mediated transcriptional repression, we generated U2OS cells with GAL4DBS-TK-luciferase stably integrated within a chromatin context and doxycycline-inducible GAL4DBD-KRAB. In one stable clone (clone 3) in which TK-driven luciferase activity is suppressed down to 5–10% of baseline by doxycycline-mediated induction of GAL4DBD-KRAB, shRNA knockdown of TRIM28 restored GAL4DBD-KRAB-dependent luciferase activity to 70% of baseline, whereas knockdown of WTX returned luciferase activity to 30–50% (Fig. 2, B and C). Similar results were observed in a second clone (clone 8) with 20-fold higher baseline of TK-driven luciferase activity (data not shown). Thus, suppression of either TRIM28 or WTX leads to a partial rescue of KRAB domain-mediated transcriptional repression, implicating WTX in this well characterized TRIM28 functional pathway. This functional output for the WTX/TRIM28 interaction suggests that WTX supports TRIM28-mediated transcriptional repression of its specific chromatinized target sequences.
FIGURE 2.
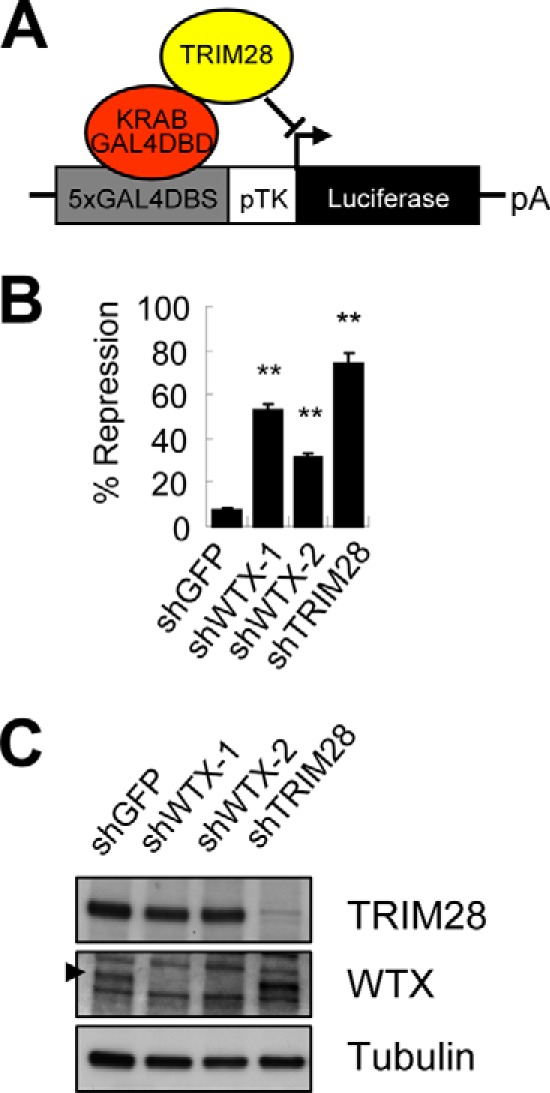
WTX supports TRIM28-mediated transcriptional repression. A, schematic illustration of GAL4DBS-TK-luciferase reporter. B, luciferase assay to measure TRIM28-mediated transcriptional repression in U2OS stable clone 3 harboring chromosomally integrated GAL4DBS-TK-luciferase and doxycycline-inducible GAL4DBD-KRAB fusion protein following knockdown of WTX or TRIM28. Luciferase activity was normalized by protein concentration of lysate. Percent repression was calculated by dividing normalized luciferase activity of doxycycline-treated by untreated sample. **, p ≤ 0.01 compared with control shGFP. C, knockdown efficiency of shWTX and shTRIM28 constructs measured by Western blot analysis. Note that the steady state level of TRIM28 protein is not changed following knockdown of WTX. The arrowhead indicates the position of WTX in the gel. Error bars represent S.E.
Reduction of Chromatin-bound TRIM28 by Knockdown of WTX
We tested whether WTX expression affects either the initial recruitment of TRIM28 to chromatin or its subsequent recruitment of chromatin-silencing complexes. In chromatin immunoprecipitation (ChIP) experiments, we observed that the binding of TRIM28 to chromosomally integrated GAL4DBS in the presence of GAL4DBD-KRAB was reduced to 40–50% following knockdown of endogenous WTX (Fig. 3A). WTX knockdown also resulted in reduced binding of the histone methyltransferase SETDB1 to the GAL4DBS (Fig. 3A). SUMOylation of TRIM28, a post-translational modification critical for its binding to SETDB1 (23), was also significantly reduced following knockdown of WTX (Fig. 3B). Thus, suppression of WTX reduces the localization of TRIM28 at chromatin and its recruitment of the silencing machinery. In contrast to its previously reported effect on the stability of CBP/p300 and β-catenin proteins (7, 10), WTX knockdown did not alter the stability of TRIM28 itself (Fig. 2C). In addition to its effect at chromosomally integrated GAL4DBS loci, knockdown of WTX appears to reduce chromatin binding of TRIM28 to other silenced chromosomal loci as measured by specific loss of TRIM28 in the insoluble, histone H3-containing fraction (Fig. 3C) and increased micrococcal nuclease digestion (Fig. 3D). This effect of WTX knockdown suggests that the WTX/TRIM28 interaction may normally enhance chromatin silencing at multiple TRIM28 target sites.
FIGURE 3.

Reduction of chromatin-bound TRIM28 following WTX knockdown. A, chromatin binding of TRIM28 and SETDB1 at GAL4DBS, luciferase gene open reading frame (Luc), and control GAPDH loci determined by ChIP-quantitative PCR assay in U2OS clone 3. ns, p > 0.05; **, p ≤ 0.01; ***, p ≤ 0.001 compared with control (shGFP). B, SUMOylation of TRIM28 following knockdown of WTX in U2OS stable clone 3. Cell lysates were generated in the presence of N-ethylmaleimide and immunoprecipitated (IP) with anti-TRIM28 antibody followed by immunoblotting (IB) with anti-SUMO antibodies. C, reduced chromatin-bound TRIM28 following WTX knockdown measured by differential extraction of U2OS cells with hypotonic (C), high salt (N), and Laemmli (I) lysis buffer followed by Western blot analysis. Histone H3, lamin A/C, and Grb2 represent markers for the chromatin fractions, nucleus, and cytoplasm, respectively. Actin serves as a loading control. D, increased chromatin relaxation following knockdown of WTX determined by micrococcal nuclease digestion assay in U2OS cells. Quantification of the gel is shown in the middle panel. The density of mono- (N1), di- (N2), tri- (N3), and tetranucleosomes (N4) was measured and normalized by total density of the corresponding whole lane using ImageJ software. Knockdown efficiency of each hairpin measured by quantitative real time PCR analysis is shown in the graph. Error bars represent S.E.
Coregulation of Repetitive Sequences and Their Neighboring Coding Genes by WTX and TRIM28
Mouse ESCs provide the best model for TRIM28 transcriptional repression with the best characterized endogenous targets being the Mest gene and non-coding repetitive sequences, including the endogenous retrovirus IAP and the retrotransposon LINE1 (24). This TRIM28-dependent-, DNA methylation-independent silencing of repetitive sequences is specific to mouse ESCs (24). We therefore measured the expression of Mest, IAP, and LINE1 in mouse ESCs (ES-D3) following knockdown of WTX. Remarkably, both IAP and LINE1 transcripts were strongly induced by knockdown of WTX to a level comparable with that resulting from knockdown of TRIM28 itself (Fig. 4A). However, expression of the TRIM28-targeted Mest transcript was not affected, suggesting that WTX may contribute to transcriptional repression of just a subset of endogenous TRIM28 targets.
FIGURE 4.

Coregulation of repetitive sequences and their neighboring coding genes by WTX and TRIM28. A, expression of Trim28, Wtx, Mest, IAP, and LINE1 in the mouse embryonic stem cell line ES-D3 following knockdown of GFP, TRIM28, or WTX as measured by quantitative real time PCR. *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001 compared with control (shGFP). B, single molecule RNA sequencing analysis (Helicos platform) of ES-D3 cells following knockdown (KD) of WTX or TRIM28. Ctrl, control. Differentially expressed transcripts were identified by DEGseq analysis with a familywise error rate less than 0.05 and a -fold change greater than 2-fold. C, the overlap between differentially expressed transcripts following knockdown of TRIM28 and WTX is depicted as Venn diagrams. Red and blue circles indicate up- and down-regulated transcripts, respectively. D, genome physical proximity of RefSeq (protein-encoding) genes and Repbase (repetitive noncoding) elements that are up-regulated following knockdown of WTX. The percentage of each class of commonly up-regulated Repbase repeats located within 10 kb of commonly up-regulated RefSeq genes (red line) within the genome is compared with those of randomly selected control genes (bar graphs). E, knockdown efficiency of each hairpin is shown by immunoblotting. Error bars represent S.E. LTR, long terminal repeat.
To explore genome-wide transcriptional programs shared by TRIM28 and WTX, we undertook DGE profiling using Helicos single molecule sequencing (14) in ES-D3 cells subjected to knockdown of TRIM28, WTX, or control GFP. This “single molecule” sequencing platform is unique in that it avoids the need for PCR amplification of template molecules, which is required for standard next generation nucleotide sequencing, and as such, it allows direct and unbiased quantification of repetitive elements that are variably affected by PCR amplification. Digital RNA reads derived from single molecule RNA sequencing were aligned with both the standard RefSeq database of protein encoding transcripts and the Repbase database of repetitive sequences (Fig. 4B and supplemental Table S2–S5). A highly significant overlap was observed in differentially expressed transcripts following knockdown of WTX or TRIM28 in both databases (Fig. 4C). In particular, 94% (77 of 82) of repetitive transcripts (Repbase) up-regulated by knockdown of TRIM28 were also up-regulated by knockdown of WTX (p < 1e−20), suggesting that WTX plays a critical role in the physiological repression of these repetitive sequences by TRIM28. Statistically significant overlaps were also observed among protein-encoding genes that were up-regulated following knockdown of either WTX or TRIM28 (p < 1e−20), down-regulated coding genes (p < 1e−20), and down-regulated repetitive sequences (p < 4.5e−9). As a control, there was no correlation between repetitive or coding transcripts that were discordantly up-regulated or down-regulated by TRIM28 or WTX knockdown (Fig. 4C).
Transcriptional up-regulation of protein-encoding genes through derepression of neighboring repetitive elements has been reported for both LINE1 and satellite repeats (13, 25). Given the striking overlap of induced repetitive transcripts following knockdown of either TRIM28 or WTX, we tested whether the induced protein-encoding transcripts were likely to be in physical proximity to these regulated repetitive sequences. Compared with an equal number of randomly selected transcripts, the 270 TRIM28- and WTX-coregulated protein-encoding genes were highly enriched for LINE, endogenous retrovirus (long terminal repeat), and DNA transposon sequences (p < 0.005) within their 10-kb gene boundary (Fig. 4D). No such correlation was seen for simple repeats or satellite repeats. Although the presumed functional properties of the 270 TRIM28/WTX-coregulated protein-encoding genes are diverse, analysis of both DAVID and hypergeometric gene set enrichment analysis databases suggested some enrichment for pathways involving differentiation, morphogenesis, and cell motility (p < 0.05) (supplemental Tables S6 and S7). In contrast, knockdown of WTX did not mirror the well established TRIM28-mediated repression of Nanog, Oct4, and Sox2 (supplemental Table S4 and Refs. 18 and 26), indicating that WTX does not play a role in TRIM28-mediated regulation of pluripotency genes. Taken all together, these results suggest that TRIM28 and WTX together regulate the silencing of repetitive sequences, including long terminal and LINE repeats, with potential consequences for heterochromatin stability and for expression of specific protein-encoding genes that flank these repetitive sequences.
Coregulation of Adipogenic Differentiation by WTX and TRIM28
Despite significant overlap in transcriptional programs between WTX and TRIM28, knockdown of WTX in mouse ESCs did not recapitulate the known consequences of TRIM28 depletion in these cells (18, 26), including suppression of pluripotency and induction of spontaneous differentiation (data not shown). We therefore tested the functional effects of TRIM28 knockdown in the differentiation of mesenchyme-derived cells, including bone and fat, a phenotype previously established for WTX knock-out in the mouse (6). Although constitutional inactivation of WTX has a perinatal lethal phenotype, tissue-specific inactivation of WTX uncovers specific differentiation defects in lineage specification between adipocytes and osteoblasts. Its impact on β-catenin stability appears to contribute to this lineage specification step. Similarly, constitutive knock-out of TRIM28 leads to an early embryonic lethal phenotype at the postimplantation stage, but tissue-specific inactivation of TRIM28 has uncovered a role in differentiation of various hematopoietic lineages and germ cells (17, 19, 20). Thus, both WTX and TRIM28 mediate complex developmental phenotypes that may affect lineage specificity in different cell types. To test whether TRIM28 plays a role in cells that depend on WTX for osteogenic versus adipogenic differentiation, we first analyzed its effect on the murine ST2 multipotent mesenchymal stem cells, the earliest precursors in which knockdown of WTX increases osteogenic cell fate commitment at the expense of adipogenic differentiation (6). Knockdown of TRIM28 had no effect on these very early precursor cells (data not shown), suggesting that the WTX/TRIM28 interaction does not mediate initial fate specification in this setting.
We then tested the lineage-committed preadipocyte 3T3-L1 cells, which represent a later stage in the differentiation cascade. Adipogenic differentiation of 3T3-L1 cells was significantly inhibited by shRNA knockdown of either WTX or TRIM28 (Fig. 5A). Consistent with this phenotype, induction of both Pparγ and C/ebpα, the two master regulators of adipocyte differentiation (27), was dramatically suppressed by knockdown of either WTX or TRIM28 under adipogenic differentiation conditions (Fig. 5B). WNT signaling has been implicated as a negative regulator of adipocyte differentiation, and PNU-74654, a small molecule inhibitor specifically targeting the β-catenin/T cell factor interaction without affecting cytoplasmic/plasma membrane β-catenin (28), accelerates adipogenic differentiation of 3T3-L1 cells (Fig. 5A, shGFP). However, this inhibitor only partially rescued the suppressed adipogenic differentiation mediated by knockdown of WTX or TRIM28 (Fig. 5A). Thus, the functional effect of WTX and TRIM28 is distinct from the proposed WTX-dependent destabilization of β-catenin. Interestingly, we observed that TRIM28 associates with Pparγ and C/ebpα promoters under non-induced condition, suggesting that these two master regulators of adipogenic differentiation are direct targets of TRIM28. TRIM28 binding to Pparγ promoter under non-induced condition, but not to C/ebpα or Mest promoter, was significantly reduced following knockdown of WTX (Fig. 5C). Pparγ expression is regulated by multiple factors (27, 29), and these observations suggest that the TRIM28/WTX interaction contributes to this regulatory network.
FIGURE 5.

WTX and TRIM28 regulate adipogenic differentiation. A, adipogenic differentiation of preadipocyte 3T3-L1 cells following knockdown of WTX or TRIM28. Lipid formation was visualized with oil red-O staining after culture under adipogenic differentiation conditions for 12 days. Where indicated, PNU-74654 (PNU) was added to 100 μm final concentration. Magnified images of each well in the upper panel are shown in the lower panel with hematoxylin counter staining. B, expression of adipogenic marker genes Pparγ and C/ebpα in 3T3-L1 cells cultured under adipogenic condition for 8 days. Control cultures were maintained under non-induced (NI) condition. **, p ≤ 0.01; ***, p ≤ 0.001; ****, p ≤ 0.0001 compared with control (shGFP). C, chromatin binding of TRIM28 on Pparγ, C/ebpα, and Mest promoters in 3T3-L1 cells cultured under non-induced (NI) or adipogenic (Adipo) conditions determined by ChIP-quantitative PCR following knockdown of WTX or control GFP. ns, p > 0.05; *, p ≤ 0.05 compared with control (shGFP). Error bars represent S.E. IP, immunoprecipitation.
We also studied the effects of TRIM28 and WTX on bone differentiation, another abnormal phenotype observed in WTX-null mice (6). Terminal osteogenic differentiation of murine preosteoblast MC3T3-E1 cells was also suppressed by knockdown of either WTX or TRIM28 (data not shown). In contrast to its stimulatory effect on adipogenesis, the β-catenin inhibitor PNU-74654 suppressed osteogenic differentiation in vitro. This inhibitory effect was further enhanced by knockdown of either WTX or TRIM28 (data not shown). Taken together, these results suggest that the WTX/TRIM28 interaction may have a role in terminal differentiation of both preadipocytes and preosteoblasts but not on initial fate specification, an effect that is distinct from the effect of cytoplasmic WTX on β-catenin stability.
Discussion
We have identified TRIM28 as a major protein interaction partner for the nuclear pool of the WTX tumor suppressor, a protein whose function has previously been linked to the modulation of cytoplasmic WNT signaling. The WTX/TRIM28 interaction was identified using an unbiased immunoprecipitation-mass spectrometry screen for WTX partners in cell nuclear lysates. Of note, our analysis made use of full-length WTX, including the C-terminal 350 amino acids that had been lacking in the protein bait used to pull down cytoplasmic β-catenin (7). This C-terminal 350-amino acid domain of WTX, initially missing from exon predictions derived from genomic databases, was identified through the cloning and characterization of full-length WTX (1). It is specifically responsible for WTX binding to nuclear proteins such as WT1 (9) and p53 (10), and it is also critical for WTX binding to TRIM28. In addition to the lead interacting protein TRIM28, our immunoprecipitation-mass spectrometry screen also identified the NurD complex component RBBP7 (21, 30) and the TIP60, INO80, and SWR1 chromatin-modifying complex components RUVBL1 and RUVBL2 (31). We have previously reported interaction of WTX with the histone acetyltransferase CBP (10). Thus, nuclear WTX appears to associate directly or indirectly with a number of chromatin-regulatory complexes, including TRIM28. Of note, WTX interacts with the N-terminal coiled coil domain of TRIM28 that is required for its homo-oligomerization, its binding to KRAB domains of KRAB-zinc finger proteins, and hence its recruitment to chromatin. Consistent with this domain mapping, our results using the synthetic GAL4-TK promoter-reporter and the endogenous Pparγ promoter suggest that the knockdown of WTX destabilizes the chromatin recruitment of TRIM28. Further studies will be required to define whether this effect results from destabilization of the TRIM28/KRAB domain interaction alone, from destabilization of TRIM28 homo-oligomerization, or from its binding to and stabilization of multiple distinct components of the TRIM28 complex. Such interactions are consistent with the postulated scaffolding role of WTX in the cytoplasm and plasma membrane where it appears to modulate interactions between β-catenin and components of its destruction complex (7) and phosphorylation of LRP6 by tethering CK1γ, GSK3β, AXIN, and LRP6 to the plasma membrane (8). Notably, we cannot exclude the possibility that WTX and TRIM28 interact indirectly or as part of a larger protein complex that is disrupted by knockdown of either component.
Our shRNA knockdown followed by single molecule RNA sequencing approach revealed that WTX and TRIM28 share a significant fraction of their transcriptional programs in mouse ESCs. These shared transcriptional targets include both non-coding repetitive sequences commonly associated with TRIM28 suppression and a subset of protein-encoding genes. Among the shared WTX and TRIM28 targets, the overlapping subset of non-coding repetitive sequences was particularly significant with virtually all up-regulated transcripts derepressed by TRIM28 knockdown also induced by WTX knockdown. Given the evidence for the role of WTX in enhancing TRIM28 recruitment to chromatin, these observations suggest a model whereby a subset of TRIM28-silenced repeats requires WTX for transcriptional repression. The consequences of derepression of repetitive elements in cancer are poorly understood. They may be an incidental result of reduced heterochromatin silencing, or they may be specific to subsets of repeats. For instance, using single molecule RNA sequencing, we previously noted the massive and highly specific derepression of human satellite II (HSATII) pericentromeric repeats in multiple epithelial cancers (13). Similarly, re-expression of endogenous retroviral elements has been reported in several tumor types (32, 33), and satellite repeats are aberrantly expressed in BRCA1 mutant breast cancers (34, 35). Activation of retroelements may trigger chromosomal insertions and rearrangements, but they may also lead to the aberrant expression of protein-encoding genes whose regulatory sequences are normally corepressed with the repeat sequences. This phenomenon was observed following derepression of human satellite II (HSATII) pericentromeric repeats (13), and it is also evident here given the highly significant fraction of protein-encoding genes induced following knockdown of either TRIM28 or WTX, both of which have promoters within 10 kb of a derepressed repetitive sequence.
Finally, we note that beyond their role in repeat silencing in embryonic stem cells, WTX and TRIM28 may coregulate additional target genes in different cell types. Both of these genes are involved at multiple stages of embryonic and tissue differentiation. Constitutional inactivation of WTX in mice has a neonatal lethal phenotype, and its tissue-specific inactivation has uncovered specific differentiation defects in lineage specification of adipocytes and osteoblasts. The effect of WTX on β-catenin stability is most evident in its contribution to early stage lineage specification (6). Similarly, constitutive knock-out of TRIM28 in mice leads to an early embryonic lethal phenotype at the postimplantation stage, but tissue-specific inactivation is associated with differentiation defects in various hematopoietic lineages and in germ cells (17, 19, 20). Our results indicate that the nuclear interaction of WTX and TRIM28 modulates a later stage in mesenchymal cell differentiation. While this manuscript was under revision, Singh et al. (36) reported that TRIM28 modulates the function of MyoD, a master regulator of mesenchyme to skeletal muscle differentiation. Further tissue-specific analyses using conditional inactivation models will be required to fully dissect the potential functional interactions between WTX and TRIM28 in vivo. Finally, the fact that WTX, the most commonly deleted gene in Wilms tumor, interacts with TRIM28, a bona fide epigenetic regulator implicated in heterochromatin silencing, provides new insight into potential mechanisms by which WTX inactivation leads to the expansion of embryonic renal precursors, the cell of origin for pediatric kidney cancers.
Supplementary Material
Acknowledgments
We thank Drs. Yun Sok Lee, Jungwoo Lee, Mingzhu Liu, Miguel Rivera, Ryan Walsh, David Schultz, Nabeel Bardeesy, and Nick Dyson for reagents, technical help, thoughtful discussions, and critical comments.
Note Added in Proof
Information about the bioinformatic analysis was missing from the version of the article that was published as a Paper in Press on April 16, 2015. That information is now available as a supplement.
This work was supported by the Howard Hughes Medical Institute (to D. A. H.) and an American Cancer Society institutional research grant (to W. J. K.).

This article contains supplemental methods, supplemental Refs. 1–6, and supplemental Tables S1–S7.
- CBP
- cAMP-response element-binding protein (CREB)-binding protein
- KRAB
- Krüppel-associated box
- ESC
- embryonic stem cell
- TK
- thymidine kinase
- SUMO
- small ubiquitin-like modifier
- DGE
- digital gene expression
- DBS
- DNA binding sequence
- DBD
- DNA binding domain
- LINE
- long interspersed element
- IAP
- intercisternal A-type particles.
References
- 1. Rivera M. N., Kim W. J., Wells J., Driscoll D. R., Brannigan B. W., Han M., Kim J. C., Feinberg A. P., Gerald W. L., Vargas S. O., Chin L., Iafrate A. J., Bell D. W., Haber D. A. (2007) An X chromosome gene, WTX, is commonly inactivated in Wilms tumor. Science 315, 642–645 [DOI] [PubMed] [Google Scholar]
- 2. Wegert J., Wittmann S., Leuschner I., Geissinger E., Graf N., Gessler M. (2009) WTX inactivation is a frequent, but late event in Wilms tumors without apparent clinical impact. Genes Chromosomes Cancer 48, 1102–1111 [DOI] [PubMed] [Google Scholar]
- 3. Cancer Genome Atlas Network (2012) Comprehensive molecular characterization of human colon and rectal cancer. Nature 487, 330–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Akhavanfard S., Vargas S. O., Han M., Nitta M., Chang C. B., Le L. P., Fazlollahi L., Nguyen Q., Ma Y., Cosper A., Dias-Santagata D., Han J. Y., Bergethon K., Borger D. R., Ellisen L. W., Pomeroy S. L., Haber D. A., Iafrate A. J., Rivera M. N. (2014) Inactivation of the tumor suppressor WTX in a subset of pediatric tumors. Genes Chromosomes Cancer 53, 67–77 [DOI] [PubMed] [Google Scholar]
- 5. Jenkins Z. A., van Kogelenberg M., Morgan T., Jeffs A., Fukuzawa R., Pearl E., Thaller C., Hing A. V., Porteous M. E., Garcia-Miñaur S., Bohring A., Lacombe D., Stewart F., Fiskerstrand T., Bindoff L., Berland S., Adès L. C., Tchan M., David A., Wilson L. C., Hennekam R. C., Donnai D., Mansour S., Cormier-Daire V., Robertson S. P. (2009) Germline mutations in WTX cause a sclerosing skeletal dysplasia but do not predispose to tumorigenesis. Nat. Genet. 41, 95–100 [DOI] [PubMed] [Google Scholar]
- 6. Moisan A., Rivera M. N., Lotinun S., Akhavanfard S., Coffman E. J., Cook E. B., Stoykova S., Mukherjee S., Schoonmaker J. A., Burger A., Kim W. J., Kronenberg H. M., Baron R., Haber D. A., Bardeesy N. (2011) The WTX tumor suppressor regulates mesenchymal progenitor cell fate specification. Dev. Cell 20, 583–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Major M. B., Camp N. D., Berndt J. D., Yi X., Goldenberg S. J., Hubbert C., Biechele T. L., Gingras A. C., Zheng N., Maccoss M. J., Angers S., Moon R. T. (2007) Wilms tumor suppressor WTX negatively regulates WNT/β-catenin signaling. Science 316, 1043–1046 [DOI] [PubMed] [Google Scholar]
- 8. Tanneberger K., Pfister A. S., Brauburger K., Schneikert J., Hadjihannas M. V., Kriz V., Schulte G., Bryja V., Behrens J. (2011) Amer1/WTX couples Wnt-induced formation of PtdIns(4,5)P2 to LRP6 phosphorylation. EMBO J. 30, 1433–1443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rivera M. N., Kim W. J., Wells J., Stone A., Burger A., Coffman E. J., Zhang J., Haber D. A. (2009) The tumor suppressor WTX shuttles to the nucleus and modulates WT1 activity. Proc. Natl. Acad. Sci. U.S.A. 106, 8338–8343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kim W. J., Rivera M. N., Coffman E. J., Haber D. A. (2012) The WTX tumor suppressor enhances p53 acetylation by CBP/p300. Mol. Cell 45, 587–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sripathy S. P., Stevens J., Schultz D. C. (2006) The KAP1 corepressor functions to coordinate the assembly of de novo HP1-demarcated microenvironments of heterochromatin required for KRAB zinc finger protein-mediated transcriptional repression. Mol. Cell. Biol. 26, 8623–8638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Meerbrey K. L., Hu G., Kessler J. D., Roarty K., Li M. Z., Fang J. E., Herschkowitz J. I., Burrows A. E., Ciccia A., Sun T., Schmitt E. M., Bernardi R. J., Fu X., Bland C. S., Cooper T. A., Schiff R., Rosen J. M., Westbrook T. F., Elledge S. J. (2011) The pINDUCER lentiviral toolkit for inducible RNA interference in vitro and in vivo. Proc. Natl. Acad. Sci. U.S.A. 108, 3665–3670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ting D. T., Lipson D., Paul S., Brannigan B. W., Akhavanfard S., Coffman E. J., Contino G., Deshpande V., Iafrate A. J., Letovsky S., Rivera M. N., Bardeesy N., Maheswaran S., Haber D. A. (2011) Aberrant overexpression of satellite repeats in pancreatic and other epithelial cancers. Science 331, 593–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lipson D., Raz T., Kieu A., Jones D. R., Giladi E., Thayer E., Thompson J. F., Letovsky S., Milos P., Causey M. (2009) Quantification of the yeast transcriptome by single-molecule sequencing. Nat. Biotechnol. 27, 652–658 [DOI] [PubMed] [Google Scholar]
- 15. Wang L., Feng Z., Wang X., Wang X., Zhang X. (2010) DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 26, 136–138 [DOI] [PubMed] [Google Scholar]
- 16. Subramanian A., Tamayo P., Mootha V. K., Mukherjee S., Ebert B. L., Gillette M. A., Paulovich A., Pomeroy S. L., Golub T. R., Lander E. S., Mesirov J. P. (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102, 15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cammas F., Mark M., Dollé P., Dierich A., Chambon P., Losson R. (2000) Mice lacking the transcriptional corepressor TIF1β are defective in early postimplantation development. Development 127, 2955–2963 [DOI] [PubMed] [Google Scholar]
- 18. Hu G., Kim J., Xu Q., Leng Y., Orkin S. H., Elledge S. J. (2009) A genome-wide RNAi screen identifies a new transcriptional module required for self-renewal. Genes Dev. 23, 837–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Barde I., Rauwel B., Marin-Florez R. M., Corsinotti A., Laurenti E., Verp S., Offner S., Marquis J., Kapopoulou A., Vanicek J., Trono D. (2013) A KRAB/KAP1-miRNA cascade regulates erythropoiesis through stage-specific control of mitophagy. Science 340, 350–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Weber P., Cammas F., Gerard C., Metzger D., Chambon P., Losson R., Mark M. (2002) Germ cell expression of the transcriptional co-repressor TIF1β is required for the maintenance of spermatogenesis in the mouse. Development 129, 2329–2337 [DOI] [PubMed] [Google Scholar]
- 21. Schultz D. C., Friedman J. R., Rauscher F. J., 3rd. (2001) Targeting histone deacetylase complexes via KRAB-zinc finger proteins: the PHD and bromodomains of KAP-1 form a cooperative unit that recruits a novel isoform of the Mi-2α subunit of NuRD. Genes Dev. 15, 428–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Iyengar S., Farnham P. J. (2011) KAP1 protein: an enigmatic master regulator of the genome. J. Biol. Chem. 286, 26267–26276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ivanov A. V., Peng H., Yurchenko V., Yap K. L., Negorev D. G., Schultz D. C., Psulkowski E., Fredericks W. J., White D. E., Maul G. G., Sadofsky M. J., Zhou M. M., Rauscher F. J., 3rd. (2007) PHD domain-mediated E3 ligase activity directs intramolecular sumoylation of an adjacent bromodomain required for gene silencing. Mol. Cell 28, 823–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rowe H. M., Jakobsson J., Mesnard D., Rougemont J., Reynard S., Aktas T., Maillard P. V., Layard-Liesching H., Verp S., Marquis J., Spitz F., Constam D. B., Trono D. (2010) KAP1 controls endogenous retroviruses in embryonic stem cells. Nature 463, 237–240 [DOI] [PubMed] [Google Scholar]
- 25. Rodić N., Burns K. H. (2013) Long interspersed element-1 (LINE-1): passenger or driver in human neoplasms? PLoS Genet. 9, e1003402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fazzio T. G., Huff J. T., Panning B. (2008) An RNAi screen of chromatin proteins identifies Tip60-p400 as a regulator of embryonic stem cell identity. Cell 134, 162–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tontonoz P., Spiegelman B. M. (2008) Fat and beyond: the diverse biology of PPARγ. Annu. Rev. Biochem. 77, 289–312 [DOI] [PubMed] [Google Scholar]
- 28. Trosset J. Y., Dalvit C., Knapp S., Fasolini M., Veronesi M., Mantegani S., Gianellini L. M., Catana C., Sundström M., Stouten P. F., Moll J. K. (2006) Inhibition of protein-protein interactions: the discovery of druglike β-catenin inhibitors by combining virtual and biophysical screening. Proteins 64, 60–67 [DOI] [PubMed] [Google Scholar]
- 29. Farmer S. R. (2006) Transcriptional control of adipocyte formation. Cell Metab. 4, 263–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang Y., Ng H. H., Erdjument-Bromage H., Tempst P., Bird A., Reinberg D. (1999) Analysis of the NuRD subunits reveals a histone deacetylase core complex and a connection with DNA methylation. Genes Dev. 13, 1924–1935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Baek S. H. (2008) When ATPases pontin and reptin met telomerase. Dev. Cell 14, 459–461 [DOI] [PubMed] [Google Scholar]
- 32. Hohn O., Hanke K., Bannert N. (2013) HERV-K(HML-2), the best preserved family of HERVs: endogenization, expression, and implications in health and disease. Front. Oncol. 3, 246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Katoh I., Kurata S. (2013) Association of endogenous retroviruses and long terminal repeats with human disorders. Front. Oncol. 3, 234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Filipponi D., Muller J., Emelyanov A., Bulavin D. V. (2013) Wip1 controls global heterochromatin silencing via ATM/BRCA1-dependent DNA methylation. Cancer Cell 24, 528–541 [DOI] [PubMed] [Google Scholar]
- 35. Zhu Q., Pao G. M., Huynh A. M., Suh H., Tonnu N., Nederlof P. M., Gage F. H., Verma I. M. (2011) BRCA1 tumour suppression occurs via heterochromatin-mediated silencing. Nature 477, 179–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Singh K., Cassano M., Planet E., Sebastian S., Jang S. M., Sohi G., Faralli H., Choi J., Youn H. D., Dilworth F. J., Trono D. (2015) A KAP1 phosphorylation switch controls MyoD function during skeletal muscle differentiation. Genes Dev. 29, 513–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.