

Background: RB family protein abundance is dynamic and sensitive to growth conditions.
Results: RB family turnover is mediated by C-terminal degrons in a process that is phosphorylation-sensitive.
Conclusion: The RB family degrons are important regulatory domains linking stability and transcriptional potency.
Significance: Dual control of stability and potency by RB family degrons may contribute to embryonic development.
Keywords: retinoblastoma, RB, p107, p130, E2F-DP, cyclin, CDK, protein stability, proteasome, degron, transcriptional repression.
Abstract
The retinoblastoma (RB) tumor suppressor and related family of proteins play critical roles in development through their regulation of genes involved in cell fate. Multiple regulatory pathways impact RB function, including the ubiquitin-proteasome system with deregulated RB destruction frequently associated with pathogenesis. With the current study we explored the mechanisms connecting proteasome-mediated turnover of the RB family to the regulation of repressor activity. We find that steady state levels of all RB family members, RB, p107, and p130, were diminished during embryonic stem cell differentiation concomitant with their target gene acquisition. Proteasome-dependent turnover of the RB family is mediated by distinct and autonomously acting instability elements (IE) located in their C-terminal regulatory domains in a process that is sensitive to cyclin-dependent kinase (CDK4) perturbation. The IE regions include motifs that contribute to E2F-DP transcription factor interaction, and consistently, p107 and p130 repressor potency was reduced by IE deletion. The juxtaposition of degron sequences and E2F interaction motifs appears to be a conserved feature across the RB family, suggesting the potential for repressor ubiquitination and specific target gene regulation. These findings establish a mechanistic link between regulation of RB family repressor potency and the ubiquitin-proteasome system.
Introduction
The retinoblastoma (RB)5 tumor suppressor regulates cell fate through its governance of distinct gene sets that promote cell division, differentiation, and apoptosis (1, 2). RB is related to two other family members, p107 and p130, that share substantial structural conservation along with some overlapping function in target gene regulation (3–6). Although not as tightly linked to tumor suppression as RB, tumor suppressive capacity has been assigned to these other family members in some contexts. In mouse studies, RB-deficient mice were prone to pituitary tumor formation (7), whereas mice deficient for RB and p107 or p130 developed retinoblastoma (8, 9), suggesting that p107 and p130 can influence tissue specific predisposition toward tumor development. Similarly, conditional loss of p130 in adult lung epithelial cells in a RB−/−/p53−/− null background enhanced development of small cell lung carcinoma (10). Thus, all three RB family members can act as tumor suppressors in specific contexts.
In their roles as tumor suppressors, RB family members predominately function as transcriptional repressors of target genes through their antagonism of the E2F-DP family of transcription factors (11, 12). Recent evidence suggests that RB also plays a positive role in transcriptional activation of some pro-apoptotic response genes, again in a mechanism requiring E2F activity (13), although RB may also induce apoptosis through a mechanism that is independent of transcription (14). In this scenario, RB-mediated tumor suppression is enabled through blockade of gene products necessary for cell growth with concomitant invocation of cell death pathways. As key regulators of cell fate, the RB family is tightly controlled by CDK-mediated phosphorylation in response to environmental conditions (15–17). Hypo-phosphorylated RB interacts with E2F1-DP1 (18), blocking activated transcription of cell cycle genes involved in DNA replication and S-phase progression (11, 12). In response to mitogenic signals, serial phosphorylation via cyclin D-CDK 4/6 and cyclin E-CDK2 renders RB family members inactive by disengaging their association with E2F complexes (19–22). Cyclin-CDK activity is also critically regulated during early steps in normal embryonic development (23). Rapidly dividing pluripotent embryonic stem (ES) cells of the early developing embryo employ constitutive CDK-mediated inhibition of RB proteins as a mechanism to maintain rapid cell division during blastocyst formation (24–26). As ES cells differentiate, CDK activity plummets, allowing RB family proteins to regulate E2F activity in a cell cycle-dependent manner (26, 27). Despite this unifying model for cyclin-CDK regulation, there are significant differences in the coordination of RB family member activities and steady state levels. For example, RB and p107 are active in cycling cells, whereas p130 functions predominately in quiescent cells that have exited from the cell cycle. Experiments performed with staged cells showed that steady state p130 levels peak in G0, in contrast to RB and p107, which increase as cells progress through G1 (28, 29). Consistently, CDK4 activity has opposite effects on p107 and p130 steady state levels; inhibition of the enzyme leads to diminished levels of p107 and higher levels of p130 (30). Thus, RB family member activity and stability clearly respond differently to cyclin-CDK signaling during cell cycle progression. However, the mechanisms that link regulation of RB family activity to their turnover are poorly understood.
Previous studies from our laboratory showed that the Drosophila melanogaster RB homologue Rbf1 is subjected to proteasome-mediated turnover during embryonic development (31, 32). We further demonstrated that Rbf1 turnover is influenced by an “instability element” (IE) located within its C-terminal regulatory domain. Importantly, the IE region is also critical for full repressor potency for some cell cycle-regulated genes but not for non-canonical targets whose expression is not usually integrated with the cell cycle (31, 33). Interestingly, Rbf1 ubiquitination also enhanced specific activity at select cell cycle target genes (33), suggesting that the potency of the repressor at specific genes and overall Rbf1 stability are coordinated. The IE region is well conserved within the mammalian p107 and p130 factors, and we hypothesized that the activity of mammalian RB family members may also be coordinated via integration of the cyclin-CDK signaling pathway with the ubiquitin-proteasome system. We demonstrate here that this regulatory mechanism is indeed shared among the human RB family proteins. The IE regions within the RB, p107, and p130 C-terminal domains negatively regulate repressor stability through a cyclin-CDK-responsive proteasome-dependent pathway and contribute to effective gene repression. These findings indicate that an evolutionarily conserved regulatory pathway links stability and potency for the mammalian RB family.
Materials and Methods
Expression Constructs
Expression plasmids encoding mutant forms of human RB, p107, and p130 were obtained by site-directed mutagenesis of the pCMV-GFP-RB, pCMV-GFP-p107, and pCMV-GFP-p130 parental plasmids (34). To generate GFP fusion proteins, PCR-amplified instability elements from RB (residues 786–864), p107 (residues 964–1024), and p130 (residues 1035–1095) were fused in-frame between the HindIII and KpnI sites of pEGFP-C3 (Clontech). All plasmids were verified by sequencing for the desired mutation.
ES Cell Culture, Differentiation, and Immunofluorescence
Mouse R1 ES cells were obtained from American Type Culture Collection (Manassas, VA) and cultured on mitomycin-treated mouse embryonic fibroblasts in medium containing high glucose DMEM supplemented with fetal calf serum, leukemia inhibitory factor (LIF), l-glutamine, nonessential amino acids, and β-mercaptoethanol. J1-ES cells and the RB−/−, p107−/−, p130−/− triple knock out (TKO) ES cells were a kind gift from Julien Sage (35). For ES cell differentiation, cells were plated on gelatin-coated plates to eliminate contaminating mouse embryonic fibroblasts. Differentiation was induced by growing cells in the presence of 10 μm retinoic acid (R2625, Sigma) for 72 h. Control cells were treated with DMSO for a similar time. For immunofluorescence analysis, ES cells were grown on Lab-Tek II chamber slides (Nalge Nunc International, Naperville, IL) under similar conditions, and differentiation was induced as discussed above. Cells were fixed in 3.7% freshly made paraformaldehyde for 20 min and washed 3 times in wash buffer (phosphate-buffered saline (PBS), pH 7.4, 0.1% BSA, and 0.01% Tween 20). Cells were permeabilized in PBS containing 0.1% Triton X-100 for 15 min, washed, and blocked for 1 h at room temperature in blocking solution (PBS, pH 7.4, 1% BSA, and 0.01% Tween 20). Cells were incubated in primary antibody against anti-RB (G3245, mouse monoclonal, 1:100; BD Pharmingen), anti-p107 (SC-318, rabbit polyclonal, 1:100, Santa Cruz Biotechnology), or anti-p130 (SC-317, rabbit polyclonal, 1:100, Santa Cruz Biotechnology) in blocking buffer either overnight at 4 °C (Fig. 1, A–C) or for 2 h at room temperature (Fig. 1, D–F). After three washes in wash buffer, cells were incubated in secondary antibody (Alexa Fluor 488 goat anti-rabbit A11008, Alexa Fluor 488 goat anti-mouse A11001; Life Technologies) for 1 h. After three washes in wash buffer, slides were mounted with Vectashield mounting media containing DAPI (Vector Laboratories Inc., Burlingame, CA). Images were obtained using an Olympus Fluoview 1000 filter-based laser scanning confocal microscope.
FIGURE 1.

Cellular localization of RB, p107, and p130 during mouse embryonic stem cell differentiation. A–C, confocal imaging showing localization of RB, p107, and p130 in a single section of pluripotent mouse R1-ES cell colonies before and after differentiation with RA as indicated. Cells were counterstained with DAPI to detect nuclear DNA. Three biological replicates were performed, and at least 10 ES cell colonies were imaged for each experiment. Representative examples are presented. DIC, differential interference contrast. D–F, maximal intensity projection confocal images comparing WT J1-ES and TKO cells before and after RA treatment. Samples were processed in parallel, and data were collected using identical imaging parameters. ESC, ES cells. G, Western blots were performed on whole cell extracts prepared from wild type (lane 1) and TKO ES (lane 2) cells using the indicated antibodies. Comparable amounts of p107−/− mouse embryonic fibroblast (MEF; lane 3) and U2OS (lane 4) extracts were included for comparison. Different exposures are shown for the p130 Western blot to permit visualization of the differently migrating species (labeled a–d). H, RB exhibits nuclear localization in response to RA treatment. Sections of the data presented in panel D (white boxes) were enlarged to demonstrate the predominately nuclear staining pattern for RB in WT ES cells, which was not observed for the TKO cells. Total RB staining was significantly reduced after RA treatment with a broad range of intensity noted among cells.
Chromatin Immunoprecipitation
ES cells were grown on T-125 flasks and treated with either DMSO or 10 μm retinoic acid (RA; R2625, Sigma) for 3 days. Cells were washed in PBS, trypsinized, suspended in DMEM, and cross-linked with 1% formaldehyde for 18 h at 4 °C. Cells were then pelleted, washed with PBS, and flash-frozen in liquid nitrogen. Soluble chromatin was prepared as previously described (36). Chromatin-bound protein complexes were immunoprecipitated in low salt buffer (20 mm Tris HCl, pH 8.1, 2 mm EDTA, 150 mm NaCl, 0.1% SDS, 1% Triton X 100) using 5 μg of anti-RB (G3245, BD Biosciences) or anti-p107 (SC-318) or anti-p130 (SC-317) or 5 μg of rabbit nonspecific IgG (Millipore). Chromatin-antibody complexes were isolated using 50 μl of protein G Dynabeads (Life Technologies). Beads were washed once each in low salt buffer, high salt buffer (20 mm Tris HCl, pH 8.1, 2 mm EDTA, 500 mm NaCl, 0.1% SDS, 1% Triton X 100), LiCl buffer (10 mm Tris HCl, pH 8.1, 1 mm EDTA, 250 mm LiCl, 1% deoxycholate, 1% IGEPAL630), and twice with TE buffer (10 mm Tris HCl, pH 8.1, 1 mm EDTA). Chromatin-antibody complexes were eluted from the beads in 200 μl of elution buffer (100 mm NaHCO3, 1% SDS) at 65 °C for 30 min with occasional vortexing. Cross-linking was reversed by the addition of 8 μl of 5 m NaCl and incubation overnight at 65 °C. Extracts were then treated with 1 μl of RNase A (10 mg/ml) for 1 h. Subsequently, 13 μl of proteinase K buffer (8 μl of 1 m Tris, pH 6.8, 4 μl of 0.5 m EDTA and 1 μl of proteinase K (10 mg/ml) were added, and samples were incubated for an additional 1.5 h at 45 °C. Associated DNA was purified using QIAquick PCR purification kit (Qiagen, Valencia, CA). Quantitative real-time PCR was performed on input DNA and antibody-specific ChIP DNA using SYBR Green Master Mix reagents with an ABI Step one plus thermocycler (Applied Biosystems, Foster City, CA) detection system. Enrichment of RB family members at target gene promoters was examined using primers spanning known E2F binding sites at the murine CCNA2 and MCM10 loci. An intergenic region on mouse chromosome 6 was used as a negative control. Primer sequences were as follows: CCNA2-F, AATAGTCGCGGGCTACTTGA; CCNA2-R, GAGCGTAGAGCCCAGGAG; MCM10-F, AGCGTCCTCCACAAATGAAC; MCM10-R, ACCCCGTGACGCTTACCTA; Intergenic mouse chr6F, TTTTCAGTTCACACATATAAAGCAGA; Intergenic mouse chr6R, TGTTGTTGTTGTTGCTTCACTG.
RNA Extraction and Gene Expression Analysis Using Quantitative Real Time PCR
Pluripotent or differentiated ES cells were harvested, snap-frozen, and stored at −80 °C. RNA was extracted using RNeasy Mini kit (Qiagen) according to the manufacturer's instructions. cDNA was synthesized using SuperScript II Reverse Transcriptase (Invitrogen). Quantitative real-time PCR for RB, p107, and p130 was performed using gene-specific primers (10) and SYBR Green Master Mix reagents with an ABI Step one plus thermocycler (Applied Biosystems) detection system. Primers sequences were as follows: RB-F, GCTTGGCTAACTTGGGAG; RB-R, CAACTGCTGCGATAAAGATG; p107-F, CCGAAGCCCTGGATGACTT; p107-R, GCATGCCAGCCAGTGTATAACTT; p130-F, AAGGCACATGCTAACCAATGAA; p130-R, GAGCAGTTACCGCAGCATGA. Transcript levels were measured using Taqman probes (Applied Biosystems) for Pou5f1 (Mm03053917_g1), Nanog (Mm02019550_s1), and the eukaryotic elongation factor 1a1 (Mm01966122_u1) as an endogenous control. Relative gene expression was measured by the 2−ΔΔCT method (37).
Human Cell Culture, Transfection, and Drug Treatments
To determine the effect of CDK4 inhibition on endogenous RB family stability, ∼5 × 105 U2OS cells were grown for 48 h in DMEM containing 10% fetal bovine serum and penicillin-streptomycin. Cells were then treated with 1 μm PD0332991 (Selleck Chemicals, Houston, TX) and cultured for an additional 24 h with or without 1 μm MG132 for the last 6 h. Cells were harvested, and the pellet was snap-frozen in liquid nitrogen. Cell extracts were prepared in lysis buffer (50 mm Tris HCl, pH 8.0, 150 mm NaCl, 1% Triton X-100), and the total protein concentration determined using the Bradford assay. Equal amounts of whole cell extracts (50 μg) were separated by 12.5% SDS-PAGE and transferred to nitrocellulose membranes for Western analyses. Endogenous proteins were detected by using the following antibodies: anti-RB (G3245, mouse monoclonal, 1:1,000, BD Pharmingen), anti-p107 (SC-318, rabbit polyclonal, 1:1,000, Santa Cruz Biotechnology), anti-p130 (610261, mouse monoclonal, 1:1000, BD Biosciences), anti-tubulin (mouse monoclonal, 1:20,000, Iowa Hybridoma Bank), and anti-actin (A5441, mouse monoclonal, 1:10,000, Sigma). In Fig. 2B, p130 was detected using the rabbit polyclonal antibody (SC-317, rabbit polyclonal, Santa Cruz Biotechnology). All antibody incubations were performed in 5% milk in TBST (20 mm Tris HCl, pH 7.5, 120 mm NaCl, 0.1% Tween 20). Blots were developed using peroxidase-conjugated goat anti-rabbit or goat anti-mouse secondary antibodies as appropriate (1:5000, Thermo Scientific, Waltham, MA) and SuperSignal West Pico Chemiluminescent substrate (Thermo Scientific). To measure the effect of CDK4 inhibition on recombinant RB family proteins, ∼5 × 105 U2OS cells were cultured for 24 h before transfection with GFP-tagged full-length or ΔIE mutant constructs using Nanojuice transfection reagent (Novagen, EMD Chemicals, San Diego, CA). 24 h after transfection, cells were treated with 1 μm PD0332991 for an additional 24 h. Western analysis was performed as above using anti-GFP antibodies (SC-9996, mouse monoclonal, 1:1000, Santa Cruz Biotechnology).
FIGURE 2.
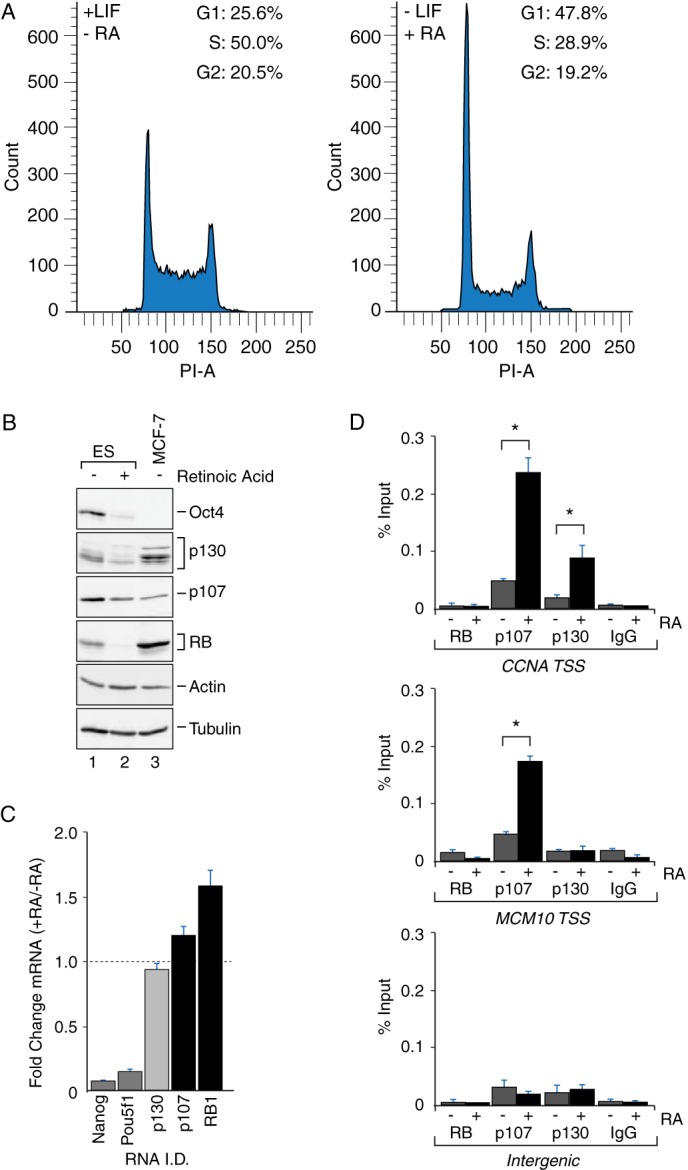
RB family protein abundance decreases during differentiation concomitant with increased engagement at target gene promoters. A, FACS analysis showing increased G1 and reduced S phase population in differentiated ES cells (−LIF, +RA) as compared with pluripotent ES cells (+LIF, −RA). B, Western blot analysis of RB, p107, and p130 in whole cell extract derived from pluripotent (−RA, lane 1) and differentiated (+RA, lane 2) mouse ES cells. RB, p107, and p130 levels in differentiated ES cells were decreased by 69, 64, and 46%, respectively, as compared with pluripotent ES cells (n = 2). Oct4 was analyzed as a positive control of differentiation and was substantially diminished in RA-treated ES cells (lane 2). Actin and tubulin were analyzed as loading controls. Whole cell extracts from MCF7 breast adenocarcinoma cells (lane 3) were analyzed as a negative control for Oct4 and as a positive control for RB, p107, and p130 detection. C, quantitative real time PCR showing relative changes in the abundance of RB, p107, and p130 mRNA transcripts upon differentiation (+RA/−RA). RB and p107 mRNA levels increased modestly after differentiation, whereas p130 levels were modestly reduced. Transcript levels of Nanog and Pou5f1 (Oct4) were reduced after RA treatment, as expected. D, p107 and p130 association at target promoters is stimulated during RA-induced differentiation. Chromatin immunoprecipitation assays were performed with the indicated antibodies to determine enrichment of RB, p107, and p130 on the CCNA2 and MCM10 promoters before and after RA treatment. After differentiation, CCNA2 start site (TSS) DNA was significantly enriched in both the p107 and p130 immunoprecipitated samples (n = 6, p < 0.05), whereas enrichment of the MCM10 promoter was observed only during p107 immunoprecipitation (n = 6, p < 0.05). Under these conditions no significant enrichment of any loci was observed for the anti-RB immunoprecipitated samples nor was enrichment observed with species-matched IgG control antibodies. Amplification of an intergenic region on mouse chromosome 6 was performed as an additional negative control.
Stability Assays
The steady state abundance of GFP-tagged full-length, ΔIE, and 4KRA proteins (Fig. 3) was determined by Western blot analyses as described above. To determine the relative stability of GFP and GFP fused to the RB family instability elements, transfected cells were treated with 100 μm cycloheximide 40 h after transfection, and samples were harvested at 3, 6, and 9 h post treatment. For proteasome inhibitor treatments in Figs. 4B and 5A, cells were treated with DMSO or 1 μm MG132 for 24 h.
FIGURE 3.

The C-terminal instability element is conserved within the mammalian RB family. A, schematic representation of the human and Drosophila RB family. The canonical instability element (magenta box) first discovered in Rbf1 (residues 728–786) is also present in the C terminus of human p107 (residues 964–1024) and p130 (residues 1035–1095). The corresponding C-terminal region in human RB that functions in E2F1-DP1 interactions, shown in red, contains two discrete regions called RBCNTer (residues 786–800) and RBCCore (residues 829–864) (40). Drosophila Rbf2 does not appear to harbor a C-terminal IE. The A and B cyclin fold domains within the central pocket domains are shown as gray boxes. Potential cyclin fold domains within the N-terminal regions are shown as blue boxes. NTD, N-terminal domain; CTD, C-terminal domain. B, sequence alignment of the C-terminal regions from RB, p107, p130, and Rbf1. Residues that are identical in at least two members are highlighted in yellow. The position of the experimentally determined instability element within Rbf1 is boxed in blue. The RBCNTer and RBCCore regions are schematically represented above the alignment. Asterisks indicate the position of CDK phosphorylation sites within RB that modulate intermolecular interactions with E2F1-DP1 and intramolecular interactions with the B domain (40). The positions of positively charged lysine and arginine residues that increase Rbf1 stability when mutated are indicated as black triangles. The position of a lysine residue within Rbf1 (K774) that induces profound developmental phenotypes when mutated (31) is indicated as an open triangle. C, Western blot (IB) analysis of whole cell extracts derived from U2OS osteosarcoma cells transfected with GFP-RBWT (lane 1), GFP-RBΔNTer (lane 2), and GFP-RBΔNTer+Core (lane 3). D, Western blot analysis of whole cell extracts derived from U2OS osteosarcoma cells transfected with wild type GFP-p107 (lane 2) or mutant GFP-107 harboring a deletion of the C-terminal instability element (GFP-p107ΔIE, lane 3) or alanine substitutions at four conserved positively charged residues (Lys-970, Arg-977, Arg-978, and Lys-991) within the instability element (GFP-p1074KR-A, lane 4). E, Western blot analysis of mutant p130 harboring a deletion of the C-terminal instability element (GFP-p130ΔIE, lane 2) or bearing alanine substitutions at four conserved positively charged residues (Arg-1041, Arg-1046, Arg-1047, and Lys-1062) within the instability element (GFP-p1304KR-A, lane 4). Total p130 levels were unaffected by deletion of the A/B pocket domain GFP-p130ΔA/B (lane 5). Actin was used as a loading control for all experiments. F, quantification of the Western data presented in C–E for IE deletion mutants. Deletion of the IE within RB (n = 5), p107 (n = 7), and p130 (n = 3) resulted in 2.4-, 2.8-, and 8.8-fold increases in abundance, respectively (*, p < 0.05).
FIGURE 4.
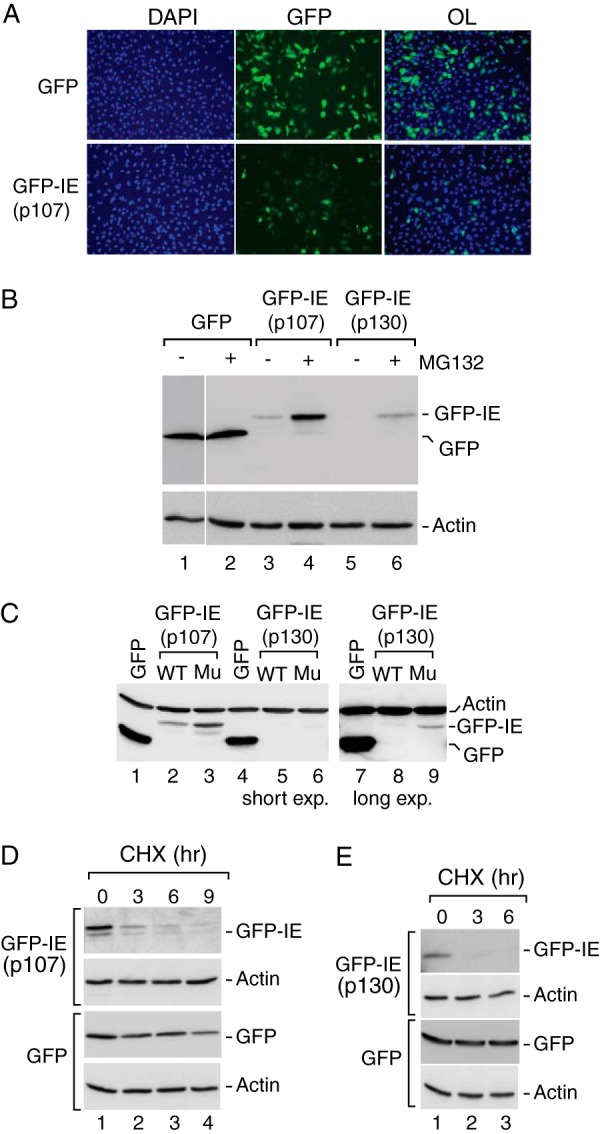
The canonical instability elements in human p107 and p130 function as autonomously acting degrons. A, the p107 IE contributes to reduced steady expression of GFP. GFP fluorescence was measured in U2OS cells transfected with GFP alone or the GFP-IEp107 chimera containing GFP fused to residues 964–1024, corresponding to the p107 instability element. Cells were counterstained with DAPI to detect cellular DNA, and overlays (OL) of the GFP and DAPI signals are shown. GFP-IEp107 showed much reduced expression as compared with GFP in most, but not all, cells. B, GFP-IEp107 and GFP-IEp130 steady state expression is enhanced by proteasome inhibition. Anti-GFP Western blot analysis was performed on whole cell extracts derived from U2OS cells transfected with GFP (lanes 1 and 2), GFP-IEp107 (lanes 3 and 4) or GFP-IEp130 containing GFP fused to residues 1035–1095 corresponding to the p130 instability element (lanes 5 and 6) in the absence or presence of MG132 as indicated. Whereas GFP remained insensitive to the proteasome inhibition, the GFP-IE fusion proteins accumulated to higher levels during MG132 treatment, suggesting the involvement of the instability element in proteasome-mediated turnover. Actin was detected as a loading control and was refractory to proteasome inhibition. C, conserved positively charged amino acids contribute to autonomous IE function. Anti-GFP Western blot analysis was performed on cells extracts transfected with either wild type GFP-IEp107 (lane 2) or mutant GFP-IEp107 containing alanine substitutions of four positively charged resides within the IE (lane 3). Similar analysis was performed for wild type GFP-IEp130 (lanes 5 and 8) and mutant GFP-IEp130 (lanes 6 and 9). In all cases the GFP chimeras containing the wild type IE sequences were expressed at lower levels than GFP alone (lanes 1, 4, and 7), whereas alanine substitution within the IE resulted in increased steady state expression. The effect of alanine substitutions on GFP-IEp130 was more evident at a higher exposure of the same blot (lane 8 versus lane 9). Actin was detected as a loading control. D and E, the p107 and p130 instability elements contribute to enhanced protein turnover. Anti-GFP Western analyses was performed on cells expressing either GFP or the wild type GFP-IE chimeras incubated in the presence of the translation inhibitor cycloheximide (CHX) for 0, 3, 6, or 9 h as indicated. GFP exhibited a half-life >9 h, whereas the half-lives of the GFP-IE chimeras were <3 h. Actin was detected as a loading control, and its levels remained unperturbed by cycloheximide treatment.
FIGURE 5.
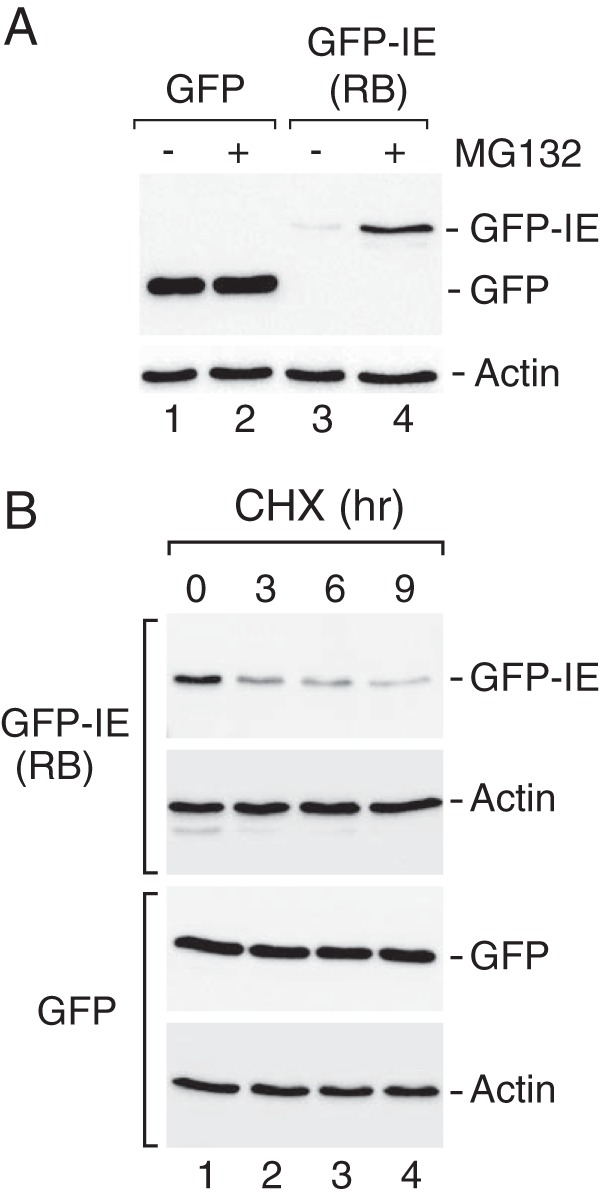
The non-canonical instability element in human RB functions as an autonomous degron. A, the RB IE contributes to reduced steady expression. Anti-GFP Western blot analysis was performed on whole cell extracts derived from U2OS cells that were transfected with GFP (lanes 1 and 2) or GFP-IERB containing GFP fused to residues 786–864 from human RB (lanes 3 and 4) in the absence or presence of the proteasome inhibitor MG132 as indicated. Actin was detected as a loading control. In these experiments the GFP-IERB chimera was expressed at much lower levels than GFP alone. GFP-IERB expression was also enhanced during proteasome inhibition. B, the RB IE region contributes to enhanced substrate turnover. Cycloheximide (CHX) experiments were performed as described previously, demonstrating that the GFP-IERB chimera exhibited diminished stability (t½ < 3 h) as compared with GFP (t½ > 9 h). Actin was used as a loading control, and its levels remained unperturbed by cycloheximide treatment during the course of this experiment.
Luciferase Reporter Assay
U2OS cells were transfected using with Nanojuice transfection reagent as described above. Typically 5 × 105 cells were transfected with 100 ng of a human cyclin A promoter-driven luciferase reporter (human cyclin A promoter (−89 to +11 (38)), 50 ng of pRL-CMV Renilla luciferase reporter (Promega), and 500 ng of plasmid expressing the GFP-tagged effector proteins. After 48 h, cells were harvested, and luciferase activity was measured using the Dual-Glo Luciferase assay system (Promega) and Veritas microplate luminometer (Turner Biosystems). Firefly luciferase activity was normalized to Renilla luciferase reading. Luciferase measurements were made in triplicate, and at least three biological experiments were performed.
Structural Homology Modeling
Structure homology modeling of the p130 IE in complex with the E2F4-DP1 was performed using SWISS-MODEL (39). The crystal structure of the RB C-terminal domain bound to an E2F1-DP1 heterodimer (PDB code 2AZE) (40) was used to generate the homology model.
Results
Regulation of RB, p107, and p130 Localization and Stability in Mouse Embryonic Stem Cells
Previous studies in Drosophila suggested that Rbf1 turnover and function are linked during embryonic development (31, 32). We, therefore, examined the behavior of mammalian RB family members in pluripotent self-renewing mouse ES cells before and after differentiation. RB function is limited in ES cells due to elevated cyclin-CDK activity but is established at the onset of differentiation in part due to down-regulation of cyclin-CDK kinase complexes (23, 27). Thus, these cells offer a useful system to examine regulation of the RB family as members are mobilized to gene promoters in response to dynamic CDK activity during development. In these experiments ES cells were cultured on mitotically inactive embryonic fibroblasts in the presence of LIF to sustain their self-renewing potential or in the presence of RA to induce differentiation, and the effect on RB, p107, and p130 was first examined by immunofluorescence analyses. In undifferentiated R1-ES cells, RB and p107 exhibited predominate cytoplasmic staining, which shifted to a stronger nuclear pattern after RA treatment (Fig. 1, A and B). In contrast, p130 was detected in the nuclear compartment both before and after RA-induced differentiation (Fig. 1C). To control for antibody specificity, RB family staining in WT J1-ES was compared with TKO ES cells under identical imaging conditions, showing that RB and p107 were preferentially detected in the WT-ES cells (Fig. 1, D and E). The pattern for RB subcellular localization was similar for both R1-ES and J1-ES cells with increased nuclear retention observed after RA challenge (see also Fig. 1H). However, the strong cytoplasmic staining for p107 was muted in J1-ES cells, and nuclear staining was observed both before and after RA treatment. Unexpectedly, p130 was equivalently detected in the nuclei of both WT and TKO ES cells (Fig. 1F). In Western blot analyses, we also detected anti-p130-reactive species in both WT and TKO cells (Fig. 1G) and using multiple antibodies that recognize distinct epitopes (not shown), whereas RB and p107 were detected only in WT but not TKO ES cells, confirming specificity for these antibodies. Based on these findings, we conclude that RB and p107 can exhibit differential subcellular localization in response to induced differentiation.
During the execution of these experiments, we frequently observed that the staining intensity of endogenous RB family members was reduced after RA treatment. These differences are not obvious in Fig. 1, A--C, because the relative luminosity of the images before and after differentiation was normalized. However, reduced RB expression after RA treatment is noticeable for the experiment presented in Fig. 1D wherein the images were acquired using identical parameters, suggesting that ES differentiation is correlated with reduced steady state expression. Variation in RB family abundance has been observed during cell cycle progression (24, 28, 29, 41); therefore, we considered that RB family levels during differentiation might be associated with the differing cell cycle profiles for pluripotent ES cells compared with cells undergoing differentiation. As shown in Fig. 2A, the proportion of cells in G1 indeed increased concomitant with a diminished S-phase population in differentiated ES cells, consistent with tight relationship between RB family levels and cell cycle progression.
Our previous studies in Drosophila further demonstrated that Rbf1 is less stable in its active state (31, 42), and we surmised that mammalian RB family proteins likewise become destabilized as they engage target genes in differentiating cells during G1 phase. To test this hypothesis, the levels of RB family proteins before and after RA treatment were compared with their genomic binding at endogenous target genes. Levels in ES cells were also compared with those in MCF-7 breast adenocarcinoma cells as a positive control for RB family members and as a negative control for the stem cell marker Oct-4. As shown in Fig. 2B, Oct-4 expression was significantly reduced by RA treatment, in line with the expected changes for this molecular marker of pluripotency. Consistent with the reduced RB family staining that we previously noted using confocal microscopy, the steady state abundances of all three family members were clearly reduced after RA-induced differentiation as observed by direct Western blot assay. In two replicates, RB levels were reduced by 69%, whereas levels of p107 and p130 were reduced by 64 and 46%, respectively. The decrease in RB family protein levels was not due to a change in transcription or RNA stability because steady state mRNA levels were either unaffected by RA treatment, such as for p130, or were modestly stimulated, such as for p107 and RB (Fig. 2C). Nanog and Pou5f1 (Oct-4) expression were significantly reduced during differentiation as expected. These results point to a post-translational mechanism for RB family regulation, such as through proteasome mediated turnover pathway. To examine this possibility we treated pluripotent ES cells and RA-differentiated cells with the proteasome inhibitor MG132. However, proteasome inhibition induced substantial cell death for both pluripotent and differentiated ES cells (not shown), precluding direct assessment of proteasome involvement for RB family turnover in these cells.
Next we determined whether the observed changes in cell cycle arrest and RB family localization during differentiation could be correlated with repressor binding at target gene promoters as one measure of function. To this end we measured RB family occupancy on a set of well characterized E2F-dependent promoters that were demonstrated to be RB family target genes (5, 6) and whose expression was affected by RB family loss in ES cells (43). As shown in Fig. 2D, increased promoter binding by p107 and p130 was correlated with RA-induced differentiation and cell cycle arrest. Interestingly, p107 and p130 exhibited distinct gene association, as both were bound to the CCNA2, TK1, and E2F1 loci (Fig. 2D and data not shown), whereas only p107 but not p130 was associated with the MCM10 locus. Finally, our data provide some evidence for differential binding by RB family members depending upon cell type. Specifically, p130 was not associated at the MCM10 locus in ES cells even though it had been detected at this locus in other cell types (6). We also did not observe RB association at any of these target genes, although we have routinely used this antibody to detect RB binding in other cell types (44), and we cannot conclude whether the lack of RB binding in these experiments is biologically relevant. The lack of RB signal at these cell cycle loci, otherwise bound by p107 and/or p130, is consistent with data previously published using T98G cells (5). Interestingly, the current observations indicate that p107 levels drop during differentiation even as there is increased residency at target genes. Unlike p107, increased p130 presence at target promoters and its diminished expression upon differentiation are independent of any changes in subcellular localization, suggesting that turnover regulation is probably not coupled to nuclear transport processes.
The RB Family C-terminal Regulatory Domains Influence Repressor Stability
We next considered a model that signaling mechanisms governing mammalian RB family activity are involved in regulation of repressor turnover. In Drosophila, the Rbf1 homolog harbors an IE within its C-terminal domain that contributes to both repressor activity and destruction (31, 33). As indicated by the alignments shown in Fig. 3, A and B, just such a sequence is clearly identifiable within the C-terminal regions of both p107 and p130. RB exhibits substantial sequence differences throughout this region. Nonetheless, previous studies demonstrated that the RB C-terminal region is structurally related to p107, and thus, this region might likewise participate in both repression and turnover. Within RB, the IE can be subdivided into two regions previously called the RBCNTer and RBCCore, which are important for specific interactions with the marked box domains of the E2F1-DP1 complex (see Fig. 3A and Ref. 40). The RBCNTer region can also interact with the MDM2 E3 ubiquitin ligase (45), suggesting that the corresponding IE region within RB similarly coordinates repressor stability. To test whether the IE regions are important for regulation of RB family turnover, we deleted these regions from RB, p107, and p130 and examined the effect on steady state expression during transient transfection in U2OS cells. These cells were chosen because we could achieve more efficient and reliable transfection in this system than in ES cells. Moreover, U2OS cells do not express the p16 CDK inhibitor (46, 47); hence, these cells exhibit unrestrained cyclin-CDK activity, analogously to ES cells (25). As shown in Fig. 3C, deletion of the RBCNTer region (Δ786–800) alone, harboring the putative MDM2 binding site, did not affect RB steady state levels. In contrast, mutant RB lacking both the RBCNTer plus RBCCore regions (RBΔ786–864) exhibited a significant elevation in steady state abundance. Thus, the IE region within RB negatively influences repressor abundance.
The C-terminal regulatory regions from p107 and p130 are less well characterized than for RB, and yet these proteins clearly exhibit the highest homology to the Drosophila Rbf1 IE region, as noted previously. Therefore, a more detailed analysis of these family members was undertaken. As shown in Fig. 3D, IE deletion from GFP-tagged p107 increased steady state expression. Similarly, GFP-p107 abundance was increased by alanine substitution of four conserved lysine and arginine residues within the p107 IE region (GFP-p1074KRA) that were previously shown to influence Rbf1 half-life. As observed for p107, deletion of the IE region in GFP-p130 resulted in an even more profound -fold increase in abundance, a result that was mirrored by the corresponding 4KR to Ala (A) substitution (Fig. 3E). Ablating E2F-DP interaction by deletion of the entire p130 A/B pocket domain did not affect steady state expression, suggesting that E2F-DP association per se does not modulate p130 abundance. Moreover, IE deletion did not affect the nuclear accumulation of either p107 or p130 as observed by immunofluorescence assay (data not shown). The quantification of the effects of IE deletion on RB family abundance is summarized in Fig. 3F. The effects of IE deletion were similar in magnitude for RB and p107, showing an increase of 2.4-fold (n = 5, p < 0.05) and 2.8-fold (n = 7, p < 0.05), respectively, whereas p130 abundance was increased 8.8-fold (n = 3, p < 0.05). Together, these data demonstrate that all three mammalian RB family members harbor C-terminal regulatory domains that contribute to their reduced steady state expression.
RB family members lacking their IE regions exhibited increased steady state abundance; therefore, we hypothesized that the IE-containing regions function as degradation signals or degrons to direct destruction of their cognate proteins. To test whether the Rbf1-related IE regions within p107 and p130 are sufficient for autonomous recognition and target protein degradation by the ubiquitin proteasome system, these regions were appended to GFP, and the effect on chimera protein abundance was examined. As shown by the immunofluorescence data presented in Fig. 4A, the GFP-IEp107 chimera was expressed at reduced levels in most, but not all cells when directly compared with GFP. The total numbers of transfected cells were similar for both constructs, as assessed by scoring the number of GFP-positive cells independently of fluorescence intensity. These analyses indicate that the major difference in steady state abundance is likely due to reduced accumulation rather than differential transfection efficiency. The GFP-IE chimeras containing either the p107 or p130 IE regions were also markedly diminished compared with GFP alone in Western blot analyses (Fig. 4B). MG132 treatment increased the steady state abundance of both GFP-IEp107 and GFP-IEp130, whereas GFP levels were unaffected, consistent with the model that the p107 and p130 IE regions facilitate degradation by the proteasome. Significantly, alanine substitution of key lysine and arginine residues that affected p107 and p130 expression in the context of the full-length proteins also led to elevated steady state levels of the GFP-IE chimeras (Fig. 4C). These data also indicate that the cellular degradation machinery does not require additional interactions with other domains for turnover activity. Nonetheless, neither proteasome inhibition by MG132 nor IE mutation in this context restored expression to levels observed for GFP alone, suggesting that other unidentified features contribute to regulation of degron activity. We next treated cells with cycloheximide to test whether the reduced levels of GFP-IE fusion proteins were indeed a function of accelerated turnover. The measured stabilities of the GFP-IE chimeras were substantially lower, demonstrating that these effects are directed toward protein turnover (Fig. 4, D and E). Together, these studies define these canonical IE regions as independently acting degrons that are capable of directing substrate degradation by the proteasome.
The canonical IE regions from p107 and p130 differ from RB at the primary sequence level. However, structure prediction analysis suggested that these regions may be conserved at the tertiary level, and thus we next tested the ability of the non-canonical RB-IE region to function as a degron. As shown in Fig. 5A, GFP appended with the RB-IE was expressed at a substantially lower level than GFP, and levels were increased by MG132 treatment, suggesting that this region is a bona fide degron. Cycloheximide treatment demonstrated that the RB-derived IE was also destabilizing (Fig. 5B), as previously noted for the canonical p107 and p130 IE constructs. We conclude that the RBCNTer and RBCCore regions together constitute a functional degron.
RB family members are differentially expressed during cell cycle progression with low levels of RB and p107 during G0 or early G1 and increasing levels as cells progress toward late G1/S (29, 48). In contrast, p130 is typically expressed at its highest levels during G0 but at reduced levels at other stages. These differences suggest that cyclin-CDK activity may be key for regulation of RB family protein levels. At least for p130, proteasome-mediated turnover is known to contribute to cell cycle-associated changes (28, 29). To assess the potential role of the IE in this process, we first measured the effect of CDK4 inhibition by PD0332991 on endogenous RB family members in asynchronously dividing U2OS cells. As shown in Fig. 6A, p107 and RB levels were markedly diminished after CDK4 inhibition, whereas endogenous p130 levels were significantly increased. These data are consistent with the divergent regulation of the RB family during cell cycle progression in staged cells (28, 29) and with experiments testing the effect of PD0332991 in asynchronously dividing hepatocellular carcinoma cells (30). Moreover, levels of p107 and p130 were significantly increased during a short duration (6 h) of MG132 treatment, whereas RB levels were modestly diminished. These data indicate that endogenous p107 and p130 are subjected to proteasome-mediated turnover under these growth conditions. Secondly, RB is either not subjected to proteasome turnover or MG132-induced RB turnover via a proteasome-independent pathway. Interestingly, levels of p107, but not RB, were rescued by proteasome blockade during CDK4 inhibition (PD+MG132), suggesting that CDK4-mediated phosphorylation of p107 prevents its proteasome-mediated turnover. In contrast, concomitant proteasome and CDK4 inhibition did not affect p130 levels compared with that observed for PD alone, suggesting that hypophosphorylated p130 is not a substrate for ubiquitin-mediated degradation, as described previously (29).
FIGURE 6.
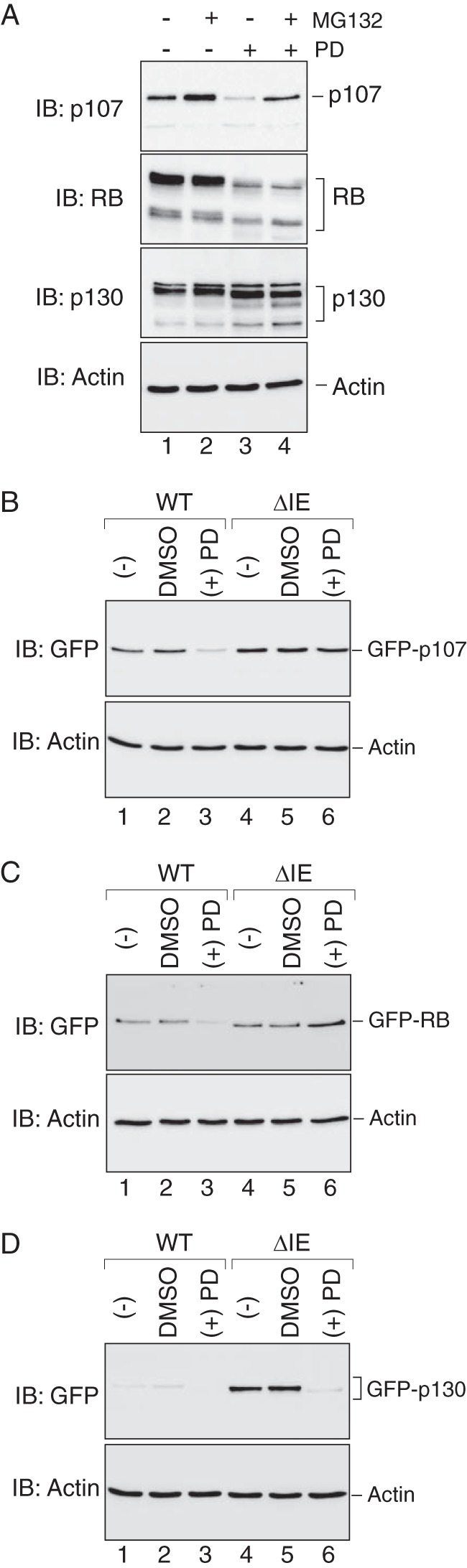
RB and p107 destabilization during CDK4 inhibition is dependent upon instability element function. A, CDK4 inhibition differentially affects steady state expression of endogenous RB, p107, and p130. Western blot (IB) analysis was performed to detect endogenous RB, p107, and p130 in whole cell extracts derived from U2OS cells treated with DMSO (lanes 1), MG132 (lane 2), the CDK4-specific inhibitor PD0332991 (PD, lane 3), or PD0332991 plus MG132 (lane 4). Proteasome inhibition by MG132 (6 h) increased steady state abundance of p107 and p130 by 90 and 30%, respectively, as compared with the DMSO-treated samples (n = 3, p < 0.05), whereas RB abundance decreased by 15% (n = 2, p < 0.05). The steady state expression of both RB and p107 were reduced by 80% during PD treatment as compared with the DMSO treated samples (n = 5, p < 0.05), whereas p130 expression was increased by 49% (n = 7, p < 0.05). The PD-induced down-regulation of p107, but not RB, could be restored by MG132 to levels comparable to the DMSO control. Actin as used as a loading control, and its levels remained unperturbed by PD or MG132 treatment. B, p107 destabilization during CDK4 inhibition requires IE function. Western blots analysis was performed on U2OS cells that expressed wild type GFP-p107 (lanes 1–3) or mutant GFP-p107ΔIE (lanes 4–6) in the absence or presence of PD0332991, as indicated. In response to CDK4 inhibition, wild type GFP-p107 levels were reduced by 68% as compared with the DMSO control (n = 3, p < 0.05), whereas the levels of p107 lacking the instability element were modestly reduced (18%) as compared with the DMSO control (n = 3, p < 0.05). C, RB destabilization during CDK4 inhibition requires IE function. Western blot analyses were performed as above, showing that wild type GFP-RB levels were reduced by 66% in response to PD as compared with the DMSO control (n = 3, p < 0.05), whereas steady state expression of GFP-RBΔIE was increased by 82% compared with the DMSO control samples (n = 3, p < 0.05). D, p130 destabilization during CDK4 inhibition does not require IE function. Western blot analyses were performed as above, showing that both wild type GFP-p130 and GFP-p130ΔIE were destabilized to similar extents during PD0332991 treatment. On average, the levels of both proteins were decreased by 92% in response to PD as compared with DMSO control (n = 3, p < 0.05). Actin was used as a loading control in panels B–D.
We next tested whether the IE is essential for the observed destabilization of p107 by examining the effect of CDK4 inhibition on wild type and mutant p107 lacking the IE in transiently transfected cells. Indeed, PD0332991 treatment destabilized the wild type but not the p107ΔIE protein, indicating that the IE is required for CDK-sensitive proteasome-mediated turnover of p107 (Fig. 6B). Wild type RB was also destabilized during CDK4 inhibition, whereas RB lacking both the RBCNTer plus RBCCore (GFP-RBΔIE) regions was refractory to PD0332991 influence (Fig. 6C), suggesting that the IE plays a similar role for CDK4 regulation of RB. In contrast to endogenous p130, recombinant GFP-p130 was destabilized, not stabilized during CDK4 inhibition, and this destabilization was observed regardless of IE status (Fig. 6D). This outcome suggests that an additional IE-independent pathway can contribute to p130 turnover. The differences in response to CDK4 inhibition for the endogenous and overexpressed p130 proteins also suggests that some turnover pathways are active only in one setting, perhaps dictated by additional regulatory phosphorylation events that affected endogenous p130 in this context.
Previous biochemical and structural studies of the C-terminal domains of the human RB family showed that these regions are important for interactions with the marked box domains of E2F-DP complexes (40, 49–51). A model of the p130 IE region in a complex with the marked box domains of E2F4 and DP1 was generated by homology modeling using the RB C-terminal domain bound to a heterodimer of E2F1 and DP1 (Fig. 7A). In this model, the C-terminal portion of the IE harbors a sheet-turn-helix motif that contacts the E2F4-DP1 complex, consistent with a potential role for the IE in repression. Interestingly, we noted that p107 (RBL1) and p130 (RBL2) harbor low frequency somatic mutations in cancer patients that map within the IE regions, as documented in the TCGA and COSMIC databases (cancergenome.nih.gov) (52). These missense and nonsense mutations are found in carcinomas of the ovary, large intestine, endometrium, and pancreas. An additional independent study focusing on RBL2 found that mutations within the p130 IE were frequently observed in a cohort of lung adenocarcinomas (53), including missense mutations affecting lysine 1083. It is notable that comparable lesions have strong biological effects in Drosophila wherein Rbf1 bearing a homologous substitution at K1083 (K774R) caused severe developmental defects (31, 54), suggesting that some mutations may play important roles in vivo. As also shown in Fig. 7A, the locations of cancer-associated RBL2 mutations map to different regions of the p130 structure, suggesting that these mutations may generate different effects, including modulation of E2F-DP interactions or potential E3 ligase association. We first tested whether cancer-associated point mutations can affect p130 stability by expression of the proteins in U2OS cells. Substitutions within the C-terminal portion of the α helix (S1090I, I1092M) significantly enhanced p130 levels, comparable with the effect of mutating four conserved lysine/arginine residues in the adjacent but unstructured region of the IE. Other point mutations tested, including R1070G, N1079F, K1083R, and K1083T that map more proximal to the E2F4-DP1 dimer interface, had no effect on p130 steady state levels (Fig. 7B). Thus, some IE-associated cancer mutations result in enhanced expression of p130 potentially due to disrupted E3 ligase binding.
FIGURE 7.

p130 IE activity in repressor potency and stability are biochemically separable functions. A, model of the human p130 IE in a complex with E2F4-DP1. Homology model of the p130 C terminus (residues 1035–1113, red) in complex with the coiled coil-marked box domains from E2F4 (residues 94–198, light green) and DP1 (residues 199–350, dark green) was generated using the crystal structure of the RB C-terminal domain bound to an E2F1-DP1 heterodimer as an template (PDB code 2AZE; Ref. 40). The N-terminal portion of the p130 IE is unstructured in this model (dashed red line), whereas the C-terminal portion of the IE harbors a sheet (residues 1071–1077)-turn-helix (1083–1093) motif. The positions of some amino acid residues altered in human cancer patients are indicated (documented in COSMIC-S1090I, TCGA-23-1118-01; I1092M, TCGA-AA-3864-01; R1093H, TCGA-AA-A01Q-01; R1093C, TCGA-D1-A15W-01A-11D-A122-09). In all cases, these mutations are rare, occurring at a frequency if ≤0.3%. Additional mutants (R1070G, 2/14 cases; N1079F, 2/14 cases; K1083R, 4/14 cases; K1083T, 1/14 cases) are based on the study presented in Ref. 53. Residues highlighted in yellow (Asn-1079, Lys-1083) are located toward the N-terminal region of the α-helix, whereas residues highlighted in blue (Ser-1090, Ile-1092, Arg-1093) are located within the C-terminal region of this helix. B, cancer-associated IE mutations have variable effects on p130 levels. Anti-GFP Western blot (IB) analysis was performed on U2OS cells transfected with plasmids expressing either wild type GFP-p130 or mutant versions harboring single point substitutions, as indicated. In two replicates, GFP-130 containing the S1090I and I1092M substitutions was expressed 2.2- and 1.7-fold greater than the wild type (n = 2). Vertical dashed lines indicate positions where the image had been cut to rearrange the order of lanes. C, the p130 IE is required for full repression potency. Luciferase reporter assays were performed in the presence of wild type or mutant versions of GFP-130, as indicated, testing repression of the human CCNA2 luciferase reporter. Wild type GFP-p130 repressed transcription by 63% as compared with GFP alone. GFP-p1304KR-A, GFP-p130ΔIE, and GFP-p130ΔC were significantly less effective than wild type GFP-p130 (n = 8; *, p < 0.05). None of the IE mutations reported in human cancers statistically altered the repression of CCNA2 reporter by GFP-p130. D, the p107 IE is required for full repression potency. Luciferase reporter assays were performed in the presence of wild type or mutant versions of GFP-107, as indicated, testing repression of the human CCNA2 luciferase reporter. Wild GFP-p107 repressed transcription by 50% as compared with GFP alone. Both GFP-p1074KR-A and GFP-p107ΔIE were significantly less effective than wild type GFP-p107 (n = 5; *, p < 0.05).
We next tested whether deletion of the entire IE or amino acid substitutions within the IE regions from p107 and p130 can impact repression potency (Fig. 7, C and D). Both wild type p107 and p130 repressed transcription driven by the CCNA2 reporter to levels ∼50–63% that observed for the control. This magnitude of repression is consistent with previously reported activities for p107 and p130 (34) but is not as profound as that reported for Drosophila Rbf1 (31, 33). Removal of the IE or substitution of four conserved lysines/arginines with alanine significantly impaired repression activity for both p107 and p130, indicating that the IE in these mammalian homologs contributes to full repression potential. Consistently, truncation of the entire C terminus (p130-ΔC) significantly impaired repression, consistent with the role of the C terminus in nuclear localization and in mediating contacts with E2F4/DP (55). In contrast, none of the missense mutations within the p130-IE, as reported in human cancer samples, was significantly altered for repression of either the cell cycle regulated CCNA2 gene or the apoptotic TP73 reporter (not shown). We conclude that these particular point mutations are unlikely to critically affect transcriptional regulation of canonical E2F-DP target genes.
Discussion
The RB tumor suppressor family governs key steps in cellular proliferation through the transcriptional regulation of distinct genes associated with growth control (3, 4). Phosphorylation of RB proteins by cyclin-CDK complexes in mouse and human ES cells was previously demonstrated to inhibit RB/E2F interaction with consequent effects on gene expression and cell proliferation (25, 26, 43, 56). In this study we report that early steps during developmental regulation of the RB family in ES cells are additionally rendered through governance of subcellular localization. Specifically, RB and p107 accumulated in the nucleus during mouse ES cell differentiation mediated by concomitant LIF withdrawal and retinoic acid addition. This transition was concurrent with enhanced p107 and p130 association at select target genes. Promoter association by p107 and p130, but not RB in this developmental context, is similar to that observed in tissue culture experiments performed using T98G glioblastoma cells (5). It is also interesting that p107 and p130 exhibited different behavior for promoter binding with some genes harboring only p107 and others harboring both p107 and p130. However, the functional significance of differential promoter association during ES differentiation remains to be determined. It should be noted that ES cells can undergo differentiation independently of RB family control (43), and thus consequences for promoter-specific binding may depend upon additional cues, such as cell type and developmental context. These data point to two mechanisms governing activity of some RB family members during early development, one involving post-translational regulation by the cyclin-CDK system and another involving control of subcellular localization.
Our data further indicate that RB family function in transcriptional repression is linked to increased repressor turnover. This connection was first suggested by observations that steady state levels of RB family members in ES cells were diminished during RA-induced differentiation and cell cycle attenuation. Our observations are also consistent with previous studies in mouse ES cells where total RB levels dropped in early G1 after release from nocodazole blockade (24). Taken together these observations suggest an intimate connection between cell cycle progression and RB protein levels in development. In response to RA, cellular mRNA levels encoding RB, p107, and p130 were either unaffected or were slightly increased, suggesting that changes in protein abundance during differentiation are influenced at a post transcriptional level, potentially involving regulated protein turnover. To understand the mechanism underlying RB family abundance, we tested the effect of proteasome inhibition on repressor levels in ES cells. However, these cells were extremely sensitive to MG132 treatment precluding direct assessment of RB family member half-lives. We, therefore, performed a biochemical structure-function study in a human osteosarcoma cell line that also maintains elevated CDK activity, analogously to that observed in ES cells. In this context, p107 and p130 turnover were indeed directed via a proteasome-mediated pathway that involved the evolutionarily conserved instability elements located within their C-terminal regulatory domains. The primary sequences of the mammalian p107 and p130 IE regions are most similar, and both of these are clearly related to the prototypical IE initially identified within Drosophila Rbf1 (31). However, RB differs substantially in primary sequence throughout the IE. Nonetheless, structural studies have indicated that pocket proteins maintain secondary and tertiary conservation throughout this region (40). We show herein that the corresponding region within RB also functions as an autonomously acting degron, suggesting that regulation of repressor stability via these C-terminal degrons represents an important and evolutionarily conserved component of global RB family control.
Mammalian RB family members are differentially expressed during cell cycle progression (28, 29, 48) with rapid degradation of hyperphosphorylated p130 correlated with G0 exit and cell cycle reentry. In contrast, p107 and RB levels tend to increase as cells progress toward S phase. Our studies showed that both wild type p107 and RB, but not mutant versions lacking the IE, are diminished by CDK4 inhibition and are consistent with a role of IE-mediated degron function in these cell cycle fluctuations. These findings are consistent with earlier observations wherein mouse ES cells ablated for the CDK2 inhibitor CDK2AP1 exhibited enhanced RB phosphorylation concomitant with increased RB abundance (57). Together, these studies demonstrate a clear linkage between the onset of CDK regulatory activity in ES cells and inversely correlated changes in RB family activity and abundance.
Although the IE regions of RB, p107, and p130 share similar function in turnover control, the notable primary sequence divergence within these regions suggests that different E3 ligases participate in RB family turnover. We propose that these distinct IE regions provide regulatory flexibility for RB family responses to distinct cell signaling events. Such events may include differential responses to DNA damage or during regulated cell cycle progression. For example, the RBCNTer region can bind to the Mdm2 E3 ligase for targeted destruction of hypophosphorylated RB (45, 58, 59). As noted, corresponding regions in p107 and p130 are only minimally conserved with RB, and these proteins are refractory to Mdm2 expression (59). In contrast, proteolysis of p130, but not that of RB or p107, is dependent on the ubiquitin ligase activity of SCFSkp2 (28, 29), which is minimally expressed during G0 but peaks during S phase (60–62), suggesting that E3 availability also plays a significant role in turnover of specific family members. Although SCFSkp2 and Mdm2 have been suggested as E3 ligases for cell cycle and DNA damage-associated degradation of RB family proteins, the involvement of these ligases in turnover during ES cell differentiation remains to be established. It is interesting that the RB C-terminal domain is also sufficient for F-box protein Skp2 association, but in this case the RB-Skp2 interaction is mainly implicated in the regulation of p27 turnover (63), suggesting a role for the RB-IE in non-autonomous protein turnover. We consider it likely that multiple E3 ligases participate in RB family regulation through differential contacts with the IE regions of the different RB family members.
Our study has additionally uncovered an intriguing aspect of mammalian RB regulation, namely that the sequences guiding repressor instability physically overlap with regions that are important for transcriptional repression. The inability of mutant p107 and p130 lacking the IE to fully engage in transcriptional repression is consistent with biochemical studies that have demonstrated a role for the IE in intermolecular contacts with the coiled coil-marked box regions of E2F1-DP1 complexes (Ref. 40; see also Fig. 7). In this regard our observations with the RB family of repressor proteins are similar to the intimate association of degrons within the activation domains of potent transactivator proteins, such as E2F1 and c-Myc (64), regulatory factors that control critical steps in cellular proliferation. A common theme emerges from these studies that key activators and repressors governing cell fate outcomes are inherently engineered. with limitations on their life span through turnover by the ubiquitin-proteasome system. Depending upon how the RB family interacts with distinct E2F-DP complexes, the interesting possibility arises that IE-E2F-DP engagement may reciprocally influence interactions with E3 ubiquitin ligases. In one model, E2F-DP complexes compete with E3 ligase for access to IE surfaces. In an alternative cooperative model, IE interactions with E2F complexes may portend engagement with E3 ubiquitin ligases. This latter model is supported, in part, by our data showing that the steady state levels of p107 and RB are diminished by cyclin D-CDK4 inhibition, a process that also licenses these pocket proteins for E2F-DP interactions and target gene engagement. Moreover, some cancer-associated p130 mutants tested in the current study showed increased steady state expression without significant effects on CCNA2 repression in vitro, suggesting that E3 binding and E2F-DP engagement are biochemically separable. Although ineffectual for perturbation of p130-mediated repression in this context, it remains possible that in vivo these mutations are associated with deregulation of other, as yet uncharacterized classes of target genes with significant effects on cellular physiology (6, 65). We note that in the developing Drosophila embryo, Rbf1 associates with many genes involved in cell signaling and metabolism (66), and similar categories of genes may likewise become deregulated during human cancer progression in cells lacking IE function. In flies, expression of Rbf1 lacking the IE enhanced DNA replication in vitro (67) and drove increased organ size when expressed in a tissue-specific manner during development (54), suggesting a critical role for IE function in developmental and proliferative pathways.
Acknowledgments
We are grateful to Julien Sage and Jenny Hsu for providing the wild type J1-ES cells, triple knock out ES cells, and p107−/− mouse embryonic fibroblasts. We also thank Erik Knudsen and Fred Dick for kindly sharing plasmids and Seth Rubin for providing a mouse monoclonal p130 antibody. We also acknowledge the assistance of Melinda Frame at the Michigan State University Center for Advanced Microscopy for help with confocal imaging.
This work was supported, in whole or in part, by National Institutes of Health Grants R01GM095347 (to J. G. K.) and R01GM079098 (to D. N. A. and R. W. H.).
- RB
- retinoblastoma
- CDK
- cyclin-dependent kinase
- ES
- embryonic stem
- IE
- instability element
- LIF
- leukemia inhibitory factor
- TKO
- triple knock out
- RA
- retinoic acid.
References
- 1. Burkhart D. L., Sage J. (2008) Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat. Rev. Cancer 8, 671–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dick F. A., Rubin S. M. (2013) Molecular mechanisms underlying RB protein function. Nat. Rev. Mol. Cell Biol. 14, 297–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hurford R. K., Jr., Cobrinik D., Lee M. H., Dyson N. (1997) pRB and p107/p130 are required for the regulated expression of different sets of E2F responsive genes. Genes Dev. 11, 1447–1463 [DOI] [PubMed] [Google Scholar]
- 4. Black E. P., Huang E., Dressman H., Rempel R., Laakso N., Asa S. L., Ishida S., West M., Nevins J. R. (2003) Distinct gene expression phenotypes of cells lacking Rb and Rb family members. Cancer Res. 63, 3716–3723 [PubMed] [Google Scholar]
- 5. Takahashi Y., Rayman J. B., Dynlacht B. D. (2000) Analysis of promoter binding by the E2F and pRB families in vivo: distinct E2F proteins mediate activation and repression. Genes Dev. 14, 804–816 [PMC free article] [PubMed] [Google Scholar]
- 6. Chicas A., Wang X., Zhang C., McCurrach M., Zhao Z., Mert O., Dickins R. A., Narita M., Zhang M., Lowe S. W. (2010) Dissecting the unique role of the retinoblastoma tumor suppressor during cellular senescence. Cancer Cell 17, 376–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jacks T., Fazeli A., Schmitt E. M., Bronson R. T., Goodell M. A., Weinberg R. A. (1992) Effects of an Rb mutation in the mouse. Nature 359, 295–300 [DOI] [PubMed] [Google Scholar]
- 8. Ajioka I., Martins R. A., Bayazitov I. T., Donovan S., Johnson D. A., Frase S., Cicero S. A., Boyd K., Zakharenko S. S., Dyer M. A. (2007) Differentiated horizontal interneurons clonally expand to form metastatic retinoblastoma in mice. Cell 131, 378–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. MacPherson D., Conkrite K., Tam M., Mukai S., Mu D., Jacks T. (2007) Murine bilateral retinoblastoma exhibiting rapid-onset, metastatic progression and N-myc gene amplification. EMBO J. 26, 784–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schaffer B. E., Park K. S., Yiu G., Conklin J. F., Lin C., Burkhart D. L., Karnezis A. N., Sweet-Cordero E. A., Sage J. (2010) Loss of p130 accelerates tumor development in a mouse model for human small-cell lung carcinoma. Cancer Res. 70, 3877–3883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dyson N. (1998) The regulation of E2F by pRB-family proteins. Genes Dev. 12, 2245–2262 [DOI] [PubMed] [Google Scholar]
- 12. Trimarchi J. M., Lees J. A. (2002) Sibling rivalry in the E2F family. Nat. Rev. Mol. Cell Biol. 3, 11–20 [DOI] [PubMed] [Google Scholar]
- 13. Ianari A., Natale T., Calo E., Ferretti E., Alesse E., Screpanti I., Haigis K., Gulino A., Lees J. A. (2009) Proapoptotic function of the retinoblastoma tumor suppressor protein. Cancer Cell 15, 184–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hilgendorf K. I., Leshchiner E. S., Nedelcu S., Maynard M. A., Calo E., Ianari A., Walensky L. D., Lees J. A. (2013) The retinoblastoma protein induces apoptosis directly at the mitochondria. Genes Dev. 27, 1003–1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Buchkovich K., Duffy L. A., Harlow E. (1989) The retinoblastoma protein is phosphorylated during specific phases of the cell cycle. Cell 58, 1097–1105 [DOI] [PubMed] [Google Scholar]
- 16. Chen P. L., Scully P., Shew J. Y., Wang J. Y., Lee W. H. (1989) Phosphorylation of the retinoblastoma gene product is modulated during the cell cycle and cellular differentiation. Cell 58, 1193–1198 [DOI] [PubMed] [Google Scholar]
- 17. DeCaprio J. A., Ludlow J. W., Lynch D., Furukawa Y., Griffin J., Piwnica-Worms H., Huang C. M., Livingston D. M. (1989) The product of the retinoblastoma susceptibility gene has properties of a cell cycle regulatory element. Cell 58, 1085–1095 [DOI] [PubMed] [Google Scholar]
- 18. Chellappan S. P., Hiebert S., Mudryj M., Horowitz J. M., Nevins J. R. (1991) The E2F transcription factor is a cellular target for the RB protein. Cell 65, 1053–1061 [DOI] [PubMed] [Google Scholar]
- 19. Hinds P. W., Mittnacht S., Dulic V., Arnold A., Reed S. I., Weinberg R. A. (1992) Regulation of retinoblastoma protein functions by ectopic expression of human cyclins. Cell 70, 993–1006 [DOI] [PubMed] [Google Scholar]
- 20. Lundberg A. S., Weinberg R. A. (1998) Functional inactivation of the retinoblastoma protein requires sequential modification by at least two distinct cyclin-cdk complexes. Mol. Cell. Biol. 18, 753–761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Narasimha A. M., Kaulich M., Shapiro G. S., Choi Y. J., Sicinski P., Dowdy S. F. (2014) Cyclin D activates the Rb tumor suppressor by mono-phosphorylation. Elife 10.7554/eLife.02872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Weinberg R. A. (1995) The retinoblastoma protein and cell cycle control. Cell 81, 323–330 [DOI] [PubMed] [Google Scholar]
- 23. White J., Dalton S. (2005) Cell cycle control of embryonic stem cells. Stem Cell Rev. 1, 131–138 [DOI] [PubMed] [Google Scholar]
- 24. Savatier P., Huang S., Szekely L., Wiman K. G., Samarut J. (1994) Contrasting patterns of retinoblastoma protein expression in mouse embryonic stem cells and embryonic fibroblasts. Oncogene 9, 809–818 [PubMed] [Google Scholar]
- 25. Stead E., White J., Faast R., Conn S., Goldstone S., Rathjen J., Dhingra U., Rathjen P., Walker D., Dalton S. (2002) Pluripotent cell division cycles are driven by ectopic Cdk2, cyclin A/E, and E2F activities. Oncogene 21, 8320–8333 [DOI] [PubMed] [Google Scholar]
- 26. White J., Stead E., Faast R., Conn S., Cartwright P., Dalton S. (2005) Developmental activation of the Rb-E2F pathway and establishment of cell cycle-regulated cyclin-dependent kinase activity during embryonic stem cell differentiation. Mol. Biol. Cell 16, 2018–2027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sage J. (2012) The retinoblastoma tumor suppressor and stem cell biology. Genes Dev. 26, 1409–1420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bhattacharya S., Garriga J., Calbó J., Yong T., Haines D. S., Graña X. (2003) SKP2 associates with p130 and accelerates p130 ubiquitylation and degradation in human cells. Oncogene 22, 2443–2451 [DOI] [PubMed] [Google Scholar]
- 29. Tedesco D., Lukas J., Reed S. I. (2002) The pRb-related protein p130 is regulated by phosphorylation-dependent proteolysis via the protein-ubiquitin ligase SCF(Skp2). Genes Dev. 16, 2946–2957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rivadeneira D. B., Mayhew C. N., Thangavel C., Sotillo E., Reed C. A., Graña X., Knudsen E. S. (2010) Proliferative suppression by CDK4/6 inhibition: complex function of the retinoblastoma pathway in liver tissue and hepatoma cells. Gastroenterology 138, 1920–1930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Acharya P., Raj N., Buckley M. S., Zhang L., Duperon S., Williams G., Henry R. W., Arnosti D. N. (2010) Paradoxical instability-activity relationship defines a novel regulatory pathway for retinoblastoma proteins. Mol. Biol. Cell 21, 3890–3901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ullah Z., Buckley M. S., Arnosti D. N., Henry R. W. (2007) Retinoblastoma protein regulation by the COP9 signalosome. Mol. Biol. Cell 18, 1179–1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Raj N., Zhang L., Wei Y., Arnosti D. N., Henry R. W. (2012) Ubiquitination of retinoblastoma family protein 1 potentiates gene-specific repression function. J. Biol. Chem. 287, 41835–41843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Stengel K. R., Thangavel C., Solomon D. A., Angus S. P., Zheng Y., Knudsen E. S. (2009) Retinoblastoma/p107/p130 pocket proteins: protein dynamics and interactions with target gene promoters. J. Biol. Chem. 284, 19265–19271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sage J., Mulligan G. J., Attardi L. D., Miller A., Chen S., Williams B., Theodorou E., Jacks T. (2000) Targeted disruption of the three Rb-related genes leads to loss of G1 control and immortalization. Genes Dev. 14, 3037–3050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wang K., Sengupta S., Magnani L., Wilson C. A., Henry R. W., Knott J. G. (2010) Brg1 is required for Cdx2-mediated repression of Oct4 expression in mouse blastocysts. PloS ONE 5, e10622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Livak K. J., Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔΔCT) method. Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]
- 38. Philips A., Huet X., Plet A., Le Cam L., Vié A., Blanchard J. M. (1998) The retinoblastoma protein is essential for cyclin A repression in quiescent cells. Oncogene 16, 1373–1381 [DOI] [PubMed] [Google Scholar]
- 39. Arnold K., Bordoli L., Kopp J., Schwede T. (2006) The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22, 195–201 [DOI] [PubMed] [Google Scholar]
- 40. Rubin S. M., Gall A. L., Zheng N., Pavletich N. P. (2005) Structure of the Rb C-terminal domain bound to E2F1-DP1: a mechanism for phosphorylation-induced E2F release. Cell 123, 1093–1106 [DOI] [PubMed] [Google Scholar]
- 41. Beijersbergen R. L., Carlée L., Kerkhoven R. M., Bernards R. (1995) Regulation of the retinoblastoma protein-related p107 by G1 cyclin complexes. Genes Dev. 9, 1340–1353 [DOI] [PubMed] [Google Scholar]
- 42. Zhang L., Wei Y., Pushel I., Heinze K., Elenbaas J., Henry R. W., Arnosti D. N. (2014) Integrated stability and activity control of the Drosophila Rbf1 retinoblastoma protein. J. Biol. Chem. 289, 24863–24873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wirt S. E., Adler A. S., Gebala V., Weimann J. M., Schaffer B. E., Saddic L. A., Viatour P., Vogel H., Chang H. Y., Meissner A., Sage J. (2010) G1 arrest and differentiation can occur independently of Rb family function. J. Cell Biol. 191, 809–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hirsch H. A., Jawdekar G. W., Lee K. A., Gu L., Henry R. W. (2004) Distinct mechanisms for repression of RNA polymerase III transcription by the retinoblastoma tumor suppressor protein. Mol. Cell. Biol. 24, 5989–5999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sdek P., Ying H., Zheng H., Margulis A., Tang X., Tian K., Xiao Z. X. (2004) The central acidic domain of MDM2 is critical in inhibition of retinoblastoma-mediated suppression of E2F and cell growth. J. Biol. Chem. 279, 53317–53322 [DOI] [PubMed] [Google Scholar]
- 46. McConnell B. B., Gregory F. J., Stott F. J., Hara E., Peters G. (1999) Induced expression of p16(INK4a) inhibits both CDK4- and CDK2-associated kinase activity by reassortment of cyclin-CDK-inhibitor complexes. Mol. Cell. Biol. 19, 1981–1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Stott F. J., Bates S., James M. C., McConnell B. B., Starborg M., Brookes S., Palmero I., Ryan K., Hara E., Vousden K. H., Peters G. (1998) The alternative product from the human CDKN2A locus, p14(ARF), participates in a regulatory feedback loop with p53 and MDM2. EMBO J. 17, 5001–5014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Classon M., Harlow E. (2002) The retinoblastoma tumour suppressor in development and cancer. Nat. Rev. Cancer 2, 910–917 [DOI] [PubMed] [Google Scholar]
- 49. Cecchini M. J., Dick F. A. (2011) The biochemical basis of CDK phosphorylation-independent regulation of E2F1 by the retinoblastoma protein. Biochem. J. 434, 297–308 [DOI] [PubMed] [Google Scholar]
- 50. Julian L. M., Palander O., Seifried L. A., Foster J. E., Dick F. A. (2008) Characterization of an E2F1-specific binding domain in pRB and its implications for apoptotic regulation. Oncogene 27, 1572–1579 [DOI] [PubMed] [Google Scholar]
- 51. Zhu L., Enders G., Lees J. A., Beijersbergen R. L., Bernards R., Harlow E. (1995) The pRB-related protein p107 contains two growth suppression domains: independent interactions with E2F and cyclin/cdk complexes. EMBO J. 14, 1904–1913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Forbes S. A., Bindal N., Bamford S., Cole C., Kok C. Y., Beare D., Jia M., Shepherd R., Leung K., Menzies A., Teague J. W., Campbell P. J., Stratton M. R., Futreal P. A. (2011) COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 39, D945–D950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Claudio P. P., Howard C. M., Pacilio C., Cinti C., Romano G., Minimo C., Maraldi N. M., Minna J. D., Gelbert L., Leoncini L., Tosi G. M., Hicheli P., Caputi M., Giordano G. G., Giordano A. (2000) Mutations in the retinoblastoma-related gene RB2/p130 in lung tumors and suppression of tumor growth in vivo by retrovirus-mediated gene transfer. Cancer Res. 60, 372–382 [PubMed] [Google Scholar]
- 54. Elenbaas J. S., Mouawad R., Henry R. W., Arnosti D. N., Payankaulam S. (2015) Role of Drosophila retinoblastoma protein instability element in cell growth and proliferation. Cell cycle 14, 589–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chestukhin A., Litovchick L., Rudich K., DeCaprio J. A. (2002) Nucleocytoplasmic shuttling of p130/RBL2: novel regulatory mechanism. Mol. Cell. Biol. 22, 453–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Conklin J. F., Baker J., Sage J. (2012) The RB family is required for the self-renewal and survival of human embryonic stem cells. Nat. Commun. 3, 1244. [DOI] [PubMed] [Google Scholar]
- 57. Kim Y., Deshpande A., Dai Y., Kim J. J., Lindgren A., Conway A., Clark A. T., Wong D. T. (2009) Cyclin-dependent kinase 2-associating protein 1 commits murine embryonic stem cell differentiation through retinoblastoma protein regulation. J. Biol. Chem. 284, 23405–23414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sdek P., Ying H., Chang D. L., Qiu W., Zheng H., Touitou R., Allday M. J., Xiao Z. X. (2005) MDM2 promotes proteasome-dependent ubiquitin-independent degradation of retinoblastoma protein. Mol. Cell 20, 699–708 [DOI] [PubMed] [Google Scholar]
- 59. Uchida C., Miwa S., Kitagawa K., Hattori T., Isobe T., Otani S., Oda T., Sugimura H., Kamijo T., Ookawa K., Yasuda H., Kitagawa M. (2005) Enhanced Mdm2 activity inhibits pRB function via ubiquitin-dependent degradation. EMBO J. 24, 160–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wei W., Ayad N. G., Wan Y., Zhang G. J., Kirschner M. W., Kaelin W. G., Jr. (2004) Degradation of the SCF component Skp2 in cell-cycle phase G1 by the anaphase-promoting complex. Nature 428, 194–198 [DOI] [PubMed] [Google Scholar]
- 61. Wirbelauer C., Sutterlüty H., Blondel M., Gstaiger M., Peter M., Reymond F., Krek W. (2000) The F-box protein Skp2 is a ubiquitylation target of a Cul1-based core ubiquitin ligase complex: evidence for a role of Cul1 in the suppression of Skp2 expression in quiescent fibroblasts. EMBO J. 19, 5362–5375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zhang H., Kobayashi R., Galaktionov K., Beach D. (1995) p19Skp1 and p45Skp2 are essential elements of the cyclin A-CDK2 S phase kinase. Cell 82, 915–925 [DOI] [PubMed] [Google Scholar]
- 63. Ji P., Jiang H., Rekhtman K., Bloom J., Ichetovkin M., Pagano M., Zhu L. (2004) An Rb-Skp2-p27 pathway mediates acute cell cycle inhibition by Rb and is retained in a partial-penetrance Rb mutant. Mol. Cell 16, 47–58 [DOI] [PubMed] [Google Scholar]
- 64. Salghetti S. E., Muratani M., Wijnen H., Futcher B., Tansey W. P. (2000) Functional overlap of sequences that activate transcription and signal ubiquitin-mediated proteolysis. Proc. Natl. Acad. Sci. U.S.A. 97, 3118–3123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Cam H., Balciunaite E., Blais A., Spektor A., Scarpulla R. C., Young R., Kluger Y., Dynlacht B. D. (2004) A common set of gene regulatory networks links metabolism and growth inhibition. Mol. Cell 16, 399–411 [DOI] [PubMed] [Google Scholar]
- 66. Acharya P., Negre N., Johnston J., Wei Y., White K. P., Henry R. W., Arnosti D. N. (2012) Evidence for autoregulation and cell signaling pathway regulation from genome-wide binding of the Drosophila retinoblastoma protein. G3 2, 1459–1472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Raj N., Zhang L., Wei Y., Arnosti D. N., Henry R. W. (2012) Rbf1 degron dysfunction enhances cellular DNA replication. Cell Cycle 11, 3731–3738 [DOI] [PMC free article] [PubMed] [Google Scholar]