

Background: The cardiolipin (CDL) binding and the peroxidase activity of cytochrome c (cyt c) vary with the protein source.
Results: The CDL binding and the peroxidase activity of cyt c depend on conformational heterogeneity and oligomerization.
Conclusion: Subtle variations in the surface residues of cyt c influence its conformational dynamics and stability.
Significance: Secondary functions of cyt c have been studied at single-molecule resolution.
Keywords: cardiolipin, cytochrome c, fluorescence correlation spectroscopy (FCS), protein aggregation, protein folding, single-molecule biophysics
Abstract
Although the primary function of cytochrome c (cyt c) is electron transfer, the protein caries out an additional secondary function involving its interaction with membrane cardiolipin (CDL), its peroxidase activity, and the initiation of apoptosis. Whereas the primary function of cyt c is essentially conserved, its secondary function varies depending on the source of the protein. We report here a detailed experimental and computational study, which aims to understand, at the molecular level, the difference in the secondary functions of cyt c obtained from horse heart (mammalian) and Saccharomyces cerevisiae (yeast). The conformational landscape of cyt c has been found to be heterogeneous, consisting of an equilibrium between the compact and extended conformers as well as the oligomeric species. Because the determination of relative populations of these conformers is difficult to obtain by ensemble measurements, we used fluorescence correlation spectroscopy (FCS), a method that offers single-molecule resolution. The population of different species is found to depend on multiple factors, including the protein source, the presence of CDL and urea, and their concentrations. The complex interplay between the conformational distribution and oligomerization plays a crucial role in the variation of the pre-apoptotic regulation of cyt c observed from different sources. Finally, computational studies reveal that the variation in the charge distribution at the surface and the charge reversal sites may be the key determinant of the conformational stability of cyt c.
Introduction
It has been believed generally that a protein can have only a single function. This view, however, is changing in that a growing number of proteins display multiple functions. A notable example is cytochrome c (cyt c), for which a role in many cellular processes has been amply demonstrated (1–5). Native cyt c is present in the mitochondrial intermembrane space in a loosely membrane-bound state and is responsible for the primary function of electron transfer from complex III (coenzyme Q-cytochrome c reductase) to complex IV (cytochrome c oxidase).
In addition to its role in the electron transfer processes, cyt c in its partially unfolded state can act as a death mediator by activating the apoptosome (6). This process is initiated when folded cyt c interacts with membrane cardiolipin (CDL5; 1,3-diphosphatidyl-sn-glycerol) (7–11). When the cell senses an external or internal apoptotic stimulus, such as an uncontrollable amount of reactive oxygen species production, CDL is transported into close proximity of cyt c (7, 12, 13). Cyt c, being a positively charged protein, interacts readily with the negatively charged CDL (9, 14, 15). Subsequently, the heme–Met-80 bond disruption allows cyt c to act as a peroxidase (16, 17). In a series of subsequent interconnected amplifying events, a massive release of cyt c takes place within the cytosol (5, 7, 9, 12, 18–25). In the case of higher eukaryotes, the released cyt c molecules activate the apoptosis protease-activating factor 1 (Apaf-1) in the presence of deoxy-ATP (26, 27). Once it activates Apaf-1, the cell crosses the point of no return, initiating the process of cell death (28).
The pro-apoptotic function of cyt c has been well studied in higher (for example, mammals) and lower (for example, Saccharomyces cerevisiae) eukaryotes (29–33). Although the release of cyt c in yeast is common to both proteins (34, 35), there are striking differences in the downstream mechanisms between the mammalian and yeast systems. Unlike cyt c from higher eukaryotes, the release of yeast cyt c (y-cyt c) does not activate caspases (36). Instead, the concentration of y-cyt c, once it appears in the cytosol diminishes with time (37). The mechanism of the absence of pro-apoptotic activity of the released y-cyt c in the yeast, and the observed reduction of the concentration of released y-cyt c with time is not yet understood completely.
Although the interaction of CDL with mammalian and yeast cyt c has been studied extensively (8, 9, 30, 33, 38–43), a systematic molecular level of understanding remains elusive. It should be noted that the primary amino acid sequences of mammalian horse heart cyt c (h-cyt c) and y-cyt c share almost 70% identity (44) and their structures are perfectly superimposable. Both proteins have an almost identical core composition, differing only by a few superficial amino acids at their surfaces. Nevertheless, their behavior toward the secondary functions (for example, their interactions with CDL, their peroxidase activity, and their role in the pro-apoptotic events) is remarkably different. It is also important to note that these two proteins with identical cores and a slight difference in surface residues differ in their conformational stability.
In this article, we have used the single-molecule resolution of fluorescence correlation spectroscopy (FCS) to distinguish different events that contribute to the heterogeneity of the CDL-protein complex. The results show the presence of a dynamic equilibrium between at least two conformers that differ in brightness and peroxidase activity. The involvement of a competing oligomerization further complicates these processes. Interestingly, the peroxidase activity of y-cyt c, although higher in the folded state, decreases in the presence of CDL as a result of the increase in the oligomer population. This is in contrast to h-cyt c, in which peroxidase activity increases significantly with CDL binding. This happens as a result of the increase in the population of the peroxidase-active extended conformer. The population of the fluctuating conformers and the oligomeric species depend on many factors, including the source of protein, the presence of urea and/or CDL, and their concentrations. In addition, the difference in the relative amplitudes of the participating conformers and the extent of oligomerization contributes significantly to the conformational stability of these proteins, affecting the secondary functions and their roles toward the pre-apoptotic regulations. Using molecular dynamics (MD) simulation we have shown that the change in the surface charge distribution between these two proteins and the charge reversal sites may contribute to the difference in conformational stability and their secondary functions.
Experimental Procedures
The two proteins, namely h-cyt c (catalog No. C7752-100MG) and y-cyt c (catalog No. C2436-100MG), and CDL disodium salt from bovine heart (catalog No. 21979-25-MG-F) were purchased from Sigma. Tetramethylrhodamine (TMR)-5-maleimide was obtained from Molecular Probes (Eugene, OR). All other reagents used were of the highest purity. y-cyt c contains one free cysteine (Cys-102), which was labeled with TMR using published procedures (45). h-cyt c was labeled at the N-terminal using the succinimidyl ester derivative of TMR (46). The extent of labeling was calculated to be ∼40 and 44% for y-cyt c and h-cyt c, respectively. The labeling did not have any effect on the stability of either protein as determined by independent unfolding measurements (Table 1 and Fig. 1, a and b).
TABLE 1.
The midpoint values and the change in free energy (ΔG0) obtained for different protein samples
Errors for the midpoint measurements are typically within 5 to 7%. The errors for the ΔG0 values are within 15%. All experiments were carried out using 20 mm sodium phosphate buffer, pH 7.5, at room temperature.
| Samples | Measurement methods | ΔG0 | Midpoint |
|---|---|---|---|
| kcal/mol | m | ||
| Labeled h-cyt c | Steady state fluorescence | 9 | 7 |
| Unlabeled h-cyt c | Steady state fluorescence | 9.7 | 7 |
| Labeled y-cyt c | Steady state fluorescence | 2.6 | 3.2 |
| Unlabeled y-cyt c | Steady state fluorescence | 2.9 | 3 |
| h-cyt c, no CDL | Far UV-CD | 9.9 | 7.4 |
| Peroxidase activity | 5 | 4.6 | |
| y-cyt c, no CDL | Far UV-CD | 2.7 | 3.4 |
| Peroxidase activity | 2.8 | 3.9 | |
| h-cyt c, 6 μm CDL | Far UV-CD | 9.3 | 7.5 |
| Peroxidase activity | 8.1 | 5 | |
| y-cyt c, 6 μm CDL | Far UV-CD | 3.1 | 2.1 |
| Peroxidase activity | 2.7 | 2.8 | |
| y-cyt c, 50 μm CDL | Far UV-CD | 3.9 | 3.9 |
| Peroxidase activity | 4.2 | 3.7 |
FIGURE 1.

a and b, urea-induced unfolding transitions monitored by tryptophan fluorescence of h-cyt c (a) and y-cyt c (b), where red triangles represent labeled proteins and black triangles represent unlabeled proteins. The unfolding data are fit to a two-state unfolding model, and the fits are shown by the lines. It is clear that the labeling does not change the stability of h-cyt c and y-cyt c. c and d, absorbance spectra of h-cyt c (c) and y-cyt c (d) in the presence of 0, 6, 20, and 50 μm CDL. The binding between cyt c (y-cyt c is represented in black and h-cyt c in red). CDL was monitored by the absorbance at 410 nm (e). For h-cyt c, the absorbance decreases at the first binding site, which is then followed by an increase in absorbance for the second site. For y-cyt c, in contrast, CDL binding at both sites results in a decrease in the absorbance. f and g, far-UV CD of h-cyt c (f) and y-cyt c (g) in the presence of 0, 6, 20, and 50 μm CDL. The variation of far-UV CD at 222 nm with CDL concentration for y-cyt c (black) and h-cyt c (red) is shown in h. Far-UV CD data clearly show that the binding of CDL does not lead to any significant change in the secondary structure of h-cyt c. In contrast, the secondary structure of y-cyt c decreases considerably with the increase in CDL concentrations. i and j, steady state tryptophan fluorescence data obtained with h-cyt c (i) and y-cyt c (j) in different CDL concentrations. The variation of fluorescence intensity with CDL concentrations for y-cyt c (black) and h-cyt c (red) is shown in k, and the variation in peroxidase activity with CDL concentrations is shown in l. It is interesting to note that y-cyt c has maximal activity in the absence of CDL, which decreases as the concentration of CDL increases. In contrast, the activity of h-cyt c increases with the increase in CDL concentrations, reaching a maximum at about 35 μm. All of these experiments were carried out in 20 mm phosphate buffer, pH 7.4, and at 25 °C.
For the experiments involving CDL vesicles, the following procedure was used. A solution of CDL in chloroform was prepared. A lipid film of CDL was generated on the inside wall of a round-bottomed flask by evaporating chloroform solvent. The film of CDL was stored in a desiccator overnight under reduced pressure conditions for complete evaporation of the solvent. The film was subsequently hydrated using 20 mm sodium phosphate buffer, pH 7.5. After vigorous stirring and 10 freeze and thaw cycles, the liposome suspension was extruded 31 times using polycarbonate membranes with 100-nm pore size (38).
CD experiments were carried out using a Jasco J720 spectropolarimeter (Japan Spectroscopic Ltd.). The far-UV CD experiments (between 200 and 250 nm) were carried out using protein samples at 20 μm concentration, and each spectrum was averaged using 10 repeat scans. The path length of the cuvette used for the far-UV CD measurements was 1 mm. The value at 222 nm was plotted against urea concentration to monitor the unfolding transitions of the proteins. All of the CD experiments were carried out at 25 °C unless otherwise mentioned.
Steady state tryptophan fluorescence experiments were carried out using a PTI fluorescence spectrometer (Photon Technology Int.).The protein samples were excited at 295 nm to avoid any contributions from the tyrosine residues. The fluorescence emission spectra were recorded between 315 and 415 nm. Final concentration of the labeled cytochrome c used for the tryptophan fluorescence experiments was 2 μm.
Determination of Peroxidase Activity
The peroxidase activity of the protein samples in the absence and presence of CDL was measured at 25 °C using 200 μm H2O2 and 10 mm guaiacol. The product formation kinetics was followed using a Shimadzu 1700 Pharmaspec UV-visible spectrophotometer. The protein concentration used was 2 μm. The rate of decomposition of hydrogen peroxide (H2O2) by peroxidase using guaiacol as a hydrogen donor was determined by measuring the rate of color development spectrophotometrically at 470 nm using the extinction coefficient 2.66 × 104 m−1cm−1 (47).
Dynamic Light Scattering (DLS) Measurement
DLS experiments were performed with a Nano-ZS (Malvern Instruments) instrument (5 milliwatts, He-Ne laser, λ = 632 nm) using protein samples of 1 μm concentration in the presence of 20 mm sodium phosphate buffer at 25 °C. The experimental solutions were filtered using 0.22-μm low protein binding filters and then mixed thoroughly. The prepared samples were kept for 2 h before each experiment. The operating procedure was programmed (using the DTS software supplied with the instrument) to record the average of 10 runs, with each run collected for 30 s with an equilibration time of 3 min.
FCS Measurements and Analysis
FCS experiments were carried out using a commercial instrument, Confocor 3 LSM (Carl Zeiss, Evotec, Jena, Germany) using a ×40 water immersion objective (numerical aperture, 1.2). Approximately 400 μl of the sample (free dye or labeled protein) was placed into Nunc chambers (Nalge Nunc Int.) and excited with an argon laser at 514 nm. The fluorescence signal was separated from the excited line using a dichroic filter, and emitted fluorescence was collected using two avalanche photodiodes. The photo-current detected by the avalanche photodiode detectors was used to calculate single-color cross-correlation functions.
FCS measurements were carried out in 20 mm sodium phosphate buffer, pH 7.5, in the presence of 20 mm DTT. Typically, 50–100 nm labeled protein concentration was used. To correct for the refractive index and viscosity of urea solutions, necessary correction measurements were carried out using a microscope correction collar and height as described previously (48). In addition, control experiments were carried out with free fluorophore (TMR) under each solution condition to normalize the protein data.
For a freely diffusing system without any conformational event or chemical reaction, the diffusion time (τD) of a fluorophore and the number of particles in the observation volume (N) can be calculated by fitting the correlation function using Equation 1.
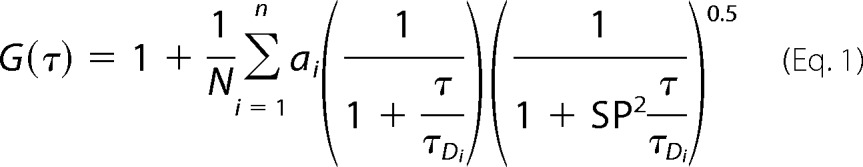 |
Here SP is the structure parameter, the depth-to-diameter ratio of the Gaussian observation volume. For a single-component system, n = 1, which corresponds the presence of only one kind of fluorophore with a diffusion time of τD. For a two-component system, n = 2, which models the presence of two diffusing species (one with the diffusion time of τD1 and an amplitude of a1 and the second with the diffusion time of τD2 and an amplitude of a2, where a1 + a2 = 1).
The correlation function for a single-component system with diffusion time τD and an associated chemical reaction or conformational change event (A ⇄ B) with a relaxation time constant of τR can be described by Equation 2.
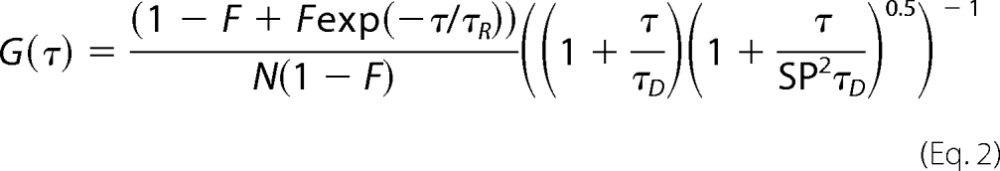 |
Equation 2 assumes that one of the states (A or B), which involves the conformational change event is non-fluorescent, and there is no difference in τD between A and B. F is the amplitude of τR, which corresponds to the average fraction of the molecule in the non-fluorescent state. The value of τR could only be measured if both the following conditions are satisfied: first, the rate of chemical reaction or conformational change event is significantly faster than the diffusion of the molecule or τR ≪ τD, and second, a large difference in fluorescence intensity between A and B exists. The diffusion coefficient (D) of the molecules can be calculated from τD using Equation 3,
 |
where ω is the beam radius of the observation volume, which can be determined by measuring the diffusion time of a dye with a known diffusion coefficient, D.
The hydrodynamic radius (rH) of a labeled molecule can be calculated from the Stokes-Einstein equation by using the value of diffusion coefficient D,
 |
where k is the Boltzmann constant, T is the temperature, and η corresponds to the viscosity of the solution.
Molecular Dynamics Simulation
Molecular dynamics simulations were carried out using Gromacs 4.5.5 with GROMOS force field 53A6 (49) at a constant temperature and pressure ensemble. The calculations were carried out using an Apple cluster equipped with operating system Darwin (release 9.6.0). Eight simultaneous processors were used for parallel processing of the mdrun program of Gromacs using Open MPI module (version 1.4.3) (50). The V-rescale coupling was employed to maintain a constant temperature of 308 K with coupling constant of 0.1 ps for both protein and non-protein molecules in the system. Parrinello-Rahman coupling was used to maintain constant semi-isotopic pressure of 1 bar with a coupling constant of 2 ps within a fixed volume of a dodecahedron box (diameter, 0.75 nm) filled with water molecules (spc216 water model). A periodic boundary condition was employed for defining the perfect three-dimensional tiling of the system. The particle mesh Ewald method with grid spacing of 0.16 nm was used for electrostatic calculations. A non-bonded cutoff of 1 nm was used for the Lennard-Jones potential. The net charge of the protein was compensated for by adding 0.1 m NaCl into the system. A steepest descent algorithm with a maximum of 50,000 steps was applied for the energy minimization of the protein. Leapfrog integrator was utilized for the molecular dynamics run.
We ran several molecular dynamics simulations corresponding to different temperature conditions, starting with the crystal structures of the proteins. Two 300-ns simulations were performed with y-cyt c and h-cyt c at 25 and 40 °C. The sulfur atoms of Cys-14 and -17 of both proteins are bond-restrained with nitrogen atoms of the pyrrole ring to hold the heme at the proper position. The Gromacs in-built option for coupling between His-18 and the iron atom of heme was employed to create better heme geometry at the protein core. All trajectory analysis was done after a satisfactorily equilibrated period of 100 ns. The Adaptive Poisson-Boltzmann Solver (APBS) (51) program was used on the end simulation structure of both proteins to calculate surface macromolecular electrostatic properties. PyMOL, version 1.5.0.3, was used for the visualization of protein structure.
Results
Although the Binding of CDL Takes Place through Two Binding Sites, the Nature of the Transitions May be Different for h-cyt c and y-cyt c
The absorbance spectra of h-cyt c and y-cyt c in the presence of different concentrations of CDL are shown in Fig. 1, c and d, respectively. The presence of an iron porphyrin ring as the prosthetic group in a heme protein (for example, cyt c) is responsible for the absorbance around 410 nm (the Soret band) (52). For both proteins, the change in the Soret absorbance at 410 nm with increasing concentrations of CDL show two CDL binding sites (Fig. 1e), supporting the observations of Rytömaa and Kinnunen (14) and Sinibaldi et al. (17). However, the nature of the transitions seems to be different for the two proteins (Fig. 1e). For h-cyt c, a decrease in the absorbance at 410 nm takes place, which is then followed by an increase in the intensity (Fig. 1e, red). In contrast, the binding of CDL with y-cyt c leads to a biphasic decrease in the absorbance at 410 nm (Fig. 1e, black).
Fig. 1, f and 1g, shows the far-UV CD spectra of h-cyt c and y-cyt c, respectively, in the absence and presence of 6, 20, and 50 μm CDL at pH 7.5. Fig. 1h shows the variation of the ellipticity at 222 nm with the increase in CDL concentrations for both proteins. Far-UV CD data, which monitor the change in the secondary structure of a protein, clearly show that the binding of CDL with y-cyt c leads to a large decrease in the ellipticity at 222 nm (Fig. 1h, black). The secondary structure of h-cyt c, in contrast, remains intact even in the presence of the highest concentration of CDL studied (50 μm) (Fig. 1h, red).
The tryptophan fluorescence of cyt c is generally quenched in the native state of cyt c (Fig. 1, i and j). Any conformational alteration or unfolding generally results in an increase in tryptophan fluorescence emission. This is because the unfolding increases the distance between tryptophan and the heme group. Steady state tryptophan fluorescence intensity at 350 nm increases for both proteins as a result of the addition of CDL (Fig. 1k). The nature of the increase in fluorescence intensity with CDL concentrations also suggests the presence of two binding transitions for both the proteins. Although these two transitions are well separated in y-cyt c, they are somewhat overlapping in h-cyt c (Fig. 1k).
The above data suggest that the binding of CDL with cyt c is biphasic, which agrees with previous reports (14, 17). For y-cyt c, the concentrations of CDL where half of the binding site is occupied are 3 and 19 μm for the first and second binding, respectively (Fig. 1e). For h-cyt c, the respective values are 3 and 26 μm (Fig. 1e). In addition, Scatchard analyses suggested that the binding constants for these two sites for h-cyt c are 0.09 and 0.03 μm−1, respectively. The binding constant for the first binding of y-cyt c was found to be very similar. In contrast, the binding constant for the second site is higher (0.075 μm−1) for y-cyt c.
Binding with CDL results in a conformational change of cyt c, although the extent of structural perturbation is significantly higher in the case of y-cyt c. The conformation and coordination geometry of the heme active site become perturbed as a result of CDL binding. It has been shown that, in the presence of CDL, the native iron–methionine bond breaks, and as a result heme iron either remains unbound or forms axial bond with other residues (53–55).
We measured the peroxidase activity of h-cyt c and y-cyt c in the absence and presence of different concentrations of CDL, and the results are shown in Fig. 1l. The peroxidase activity data indicate interesting differences between these two proteins. The data obtained in the absence of CDL show greater peroxidase activity for y-cyt c compared with h-cyt c (Fig. 1l). In the presence of CDL, the peroxidase activity of h-cyt c increases initially and then decreases slightly above 35 μm CDL (Fig. 1l, red). The effect of CDL is very different on the peroxidase activity of y-cyt c. The peroxidase activity of y-cyt c is at its maximum in the folded condition (in the absence of CDL) and decreases continuously with the increase in the concentration of CDL (Fig. 1l, black). It is interesting to note that although both proteins have similar value of maximum peroxidase activity, they reach these maxima at different concentrations of CDL (no CDL for y-cyt c, but 35 μm CDL for h-cyt c).
FCS Measurements Distinguish Different Conformers Present in cyt c CDL Interaction Pathways
FCS measures fluctuations in fluorescence intensity occurring at a small observation volume. The observation volume can be defined by using a number of pinholes or by using a tightly focused multi-photon laser line. Intensity fluctuations may occur due to the diffusion of the molecule (with diffusion time τD) in or out of the observation volume (Equation 1) or due to the kinetics of a conformational change (with the time constant τR), which rate is faster than the molecular diffusion (τR ≪ τD (Equation 2)). Analysis of the diffusion component (τD) provides the hydrodynamic radii (rH) of the folded, unfolded, and intermediate states of a protein. Analysis of the kinetic component provides information about the time constant (τR) of the conformational fluctuations in the μs time scale.
The correlation functions obtained from the FCS experiments on TMR-labeled y-cyt c and h-cyt c in their folded states and in the absence of CDL are shown in Fig. 2a. Cyt c in its folded state is characterized by a conformational fluctuation with the time constant in the μs time scale (τR ∼30 μs) (56). The residual distributions of the fits are shown as an inset in Fig. 2a. This conformational fluctuation takes place between a fluorescently dark conformer (N, in which the attached fluorophore and the quencher heme are close to each other) and a bright conformer (E, in which they are separated) of cyt c. Further characterization of this fluctuation has been discussed in our previous article (56). Correlation functions (Fig. 2a) show that y-cyt c has a slightly larger rH, and hence it takes more time to diffuse through the observation volume compared with h-cyt c. The amplitude of the bright state (E) is significantly higher in the case of y-cyt c (85% population in equilibrium between N and E), as compared with h-cyt c (less than 5%). This observation is in line with the steady state fluorescence data, which show that the fluorescence emission intensity is significantly higher in the case of y-cyt c, compared with that of h-cyt c.
FIGURE 2.

a, the autocorrelation functions of TMR-labeled h-cyt c and y-cyt c in the native folded state of the proteins and in the absence of CDL. The lines through the data represent their fits using a one diffusion-one exponential model. This model assumes a conformational fluctuation between a quenched and a bright conformer. The goodness of the fits was determined using the randomness of the residual distributions (shown in the inset). b, the correlation function obtained using the TMR-labeled y-cyt c in the presence of 50 μm CDL. The long tail effect in the correlation functions suggests the presence of oligomers. c, the presence of oligomers was also noted by the observance of large spikes in the intensity fluctuations in FCS data. This experiment was carried out using TMR-labeled y-cyt c in the presence of 50 μm CDL. d and e, the variation of the populations of the compact (N), extended (E), and oligomerized (O) forms of h-cyt c (d) and y-cyt c (e) with CDL concentrations. f, the variation of rH with CDL concentrations for y-cyt c (black) and h-cyt c (red). The values of rH were calculated from the values of τD1 (the diffusion time of the fast component) using Stokes-Einstein approximation. g, comparison between the FCS and DLS results. The population of the oligomerized component obtained using FCS (blue) with y-cyt c is compared with the values of the average diameter obtained using DLS (green). The left and right y axes represent the population values of the oligomerized component and average diameter, respectively. h, the average diameters of y-cyt c (black) and h-cyt c (red) in the presence of different CDL concentrations as monitored by DLS. A comparison between the population of E and peroxidase activity is shown for h-cyt c (i) and y-cyt c (j).
In the presence of relatively high concentrations of CDL, the proteins are found to form large particles, as indicated by large spikes in the intensity fluctuations data and by the long tail in the correlation functions. This effect is more prominent with y-cyt c. The representative correlation function observed with y-cyt c in the presence of 50 μm CDL is shown in Fig. 2b. The associated intensity fluctuation and the presence of spikes are shown in Fig. 2c. The aggregation and pore formation of cyt c in the presence of CDL has been noted previously (25, 57). The presence of a long tail in the correlation function and large spikes has been shown in the aggregation of the intestinal fatty acid-binding proteins (58). In addition, the quantification of the number of spikes in the intensity distribution data has been used as a diagnostics tool to monitor the aggregation of β-amyloid in Alzheimer's patients (59).
FCS data observed with the TMR-labeled cyt c in the presence of different concentrations of CDL have been analyzed using the following model (Reaction 1).
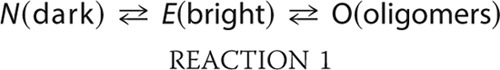 |
The values of the diffusion time of the species N (or E) and O are denoted as τD1 and τDO, respectively. It may be noted that FCS analyses did not differentiate between N and E in terms of the values in diffusion time. Hence the observed diffusion time (τD1) of N and E is an average estimate depending on their relative populations in equilibrium. The time constant of interconversion between N and E and that between E and O are denoted as τR and τO, respectively. τR is faster than the diffusion time of either N or E (τR ≪ τDN ∼ τDE). Because O represents the oligomeric species, the diffusion of O is significantly slower than that of either N or E (τDO ≫ τDN ∼ τDE ∼ τD1). The time constant of the formation of species O (τO) is also large compared with the τD values corresponding to E (or N) and O (τO ≫ τDO > τD1). Consequently, our model contains two diffusional components (τD1 and τDO) in addition to one exponential component (τR). One of these two diffusion components (with the diffusion time of τD1) corresponds to the population-weighted average of N and E, whereas the second component (with the diffusion time of τDO) represents the oligomeric species (O). The exponential component (with the time constant of τR and an amplitude of ar) represents the conformational fluctuation between N and E. The amplitude, ar, is proportional to the population of the fluorescently dark state, N.
The variations in the equilibrium population of N (the dark component), E (the bright component), and O (the oligomeric species) with CDL concentrations were determined from the above model and plotted for h-cyt c and y-cyt c in Fig. 2, d and e, respectively. For h-cyt c in its completely folded state (in the absence of CDL), the component N was found to be the predominant species, with the populations of E and O making very low contributions (Fig. 2d). With the increase in CDL concentrations, the population of E increases, which takes place mostly at the expense of the population of N. The population of E is maximal at about 35 μm CDL concentration, beyond which the population of E decreases slowly as a result of the increase in the population of O, the oligomeric species (Fig. 2d).
The result is different with y-cyt c (shown in Fig. 2e). In the folded state (in the absence of CDL), the population of E is significantly large (about 85%). This is in contrast to its almost negligible concentration for h-cyt c. The population of the dark conformer, N, increases slightly in the presence of a low concentration of CDL, maximizes at concentrations between 4 and 8 μm, and then decreases when a higher concentration of CDL is added. In contrast, the population of E decreases systematically with the increase in CDL concentration. Additionally, the increase in the concentration of CDL results in a large increase in the population of O. A comparison of the FCS data for y-cyt c and h-cyt c suggests identical increase in τD1 (the diffusion time of the first component) for both h-cyt c and y-cyt c. This is shown in Fig. 2f using the values of rH determined from the values of τD1. As suggested in our model for FCS data analyses, the variation of the first component with CDL concentration probes the change in the size of the protein-CDL complex as CDL molecules bind. The present data suggest that the binding of CDL leads to similar increase in the size for both proteins.
In contrast, a greater extent of aggregation for y-cyt c is observed when compared with h-cyt c (Fig. 2, d and e). This was substantiated further by dynamic light scattering data (Fig. 2, g and h). For h-cyt c and y-cyt c in the presence of 50 μm CDL, the values of the average diameter obtained by DLS experiments were found to be 49.2 nm (polydispersity index (PdI), 0.3) and 480 nm (PdI, 0.5), respectively.
Fig. 2i shows the comparison between the changes in peroxidase activity and the population of E in the case of h-cyt c. The corresponding data for y-cyt c is shown in Fig. 2j. It is interesting to note that a direct correlation exists between these two experimental results, which suggests that the bright component, E, plays a profound role in the peroxidase activity for both h-cyt c and y-cyt c. For h-cyt c, the population of E is very low in the absence of CDL, resulting in low peroxidase activity. The interaction of h-cyt c with CDL leads to an increase in the population of E, resulting in an increase in the peroxidase activity. The higher peroxidase activity of y-cyt c in the absence of CDL comes from the higher population of E in its native folded state. The drop in peroxidase activity for y-cyt c in the presence of CDL is the result of the decrease in the population of E, primarily because of the formation of the large oligomeric species, O.
Unfolding of cyt c Monitored by Far-UV CD, Peroxidase Activity, and FCS Suggests Higher Conformational Stability of h-cyt c Compared with y-cyt c
The results discussed above clearly show that the equilibrium population of the extended conformer, E, is significantly larger in the folded state of y-cyt c as compared with h-cyt c. The data also show that the component E may play a direct role in the peroxidase activity of the protein. In addition, the protein y-cyt c has been found to have greater propensities for oligomer formation. All of these results suggest that the ability of these proteins to sample the more extended conformer (E) may have a direct relevance to their peroxidase activity. In the following sections, we asked whether oligomerization has any role in peroxidase activity and also how the different components of the cyt c secondary function (peroxidase activity and CDL binding) are modulated by structural and conformational factors, including the conformational stability and oligomer forming propensities.
To obtain a detailed insight into the conformational stability of h-cyt c and y-cyt c in the absence and presence of different concentrations of CDL, urea-induced unfolding transitions were carried out using far-UV CD, FCS, and peroxidase activity measurements. Fig. 3 shows the variations of ellipticity at 222 nm and peroxidase activity for h-cyt c and y-cyt c with urea concentrations in the absence and presence of 6 and 50 μm CDL. The two chosen concentrations (6 and 50 μm) represent the end points of the first and second CDL binding transitions (as discussed above). The representative fit parameters obtained from the unfolding experiments are shown in Table 1.
FIGURE 3.

Urea-induced unfolding transitions were monitored by far-UV CD at 222 nm (black) and peroxidase activity (blue) for h-cyt c (a) and y-cyt c (b) in the absence of CDL, h-cyt c (c) and y-cyt c (d) in the presence of 6 μm CDL, and h-cyt c (e) and y-cyt c (f) in the presence of 50 μm CDL. The unfolding transitions (except those shown in e) were fit to a two-state unfolding model, and the thermodynamic parameters are given in Table 1. The transitions shown in Fig. 3e could not be fit to a two-state model.
For both proteins, unfolding transitions monitored by far-UV CD can be fit perfectly using a two-state transition model. In the absence of CDL, the fits of the far-UV CD data yield midpoints of 7.4 and 3.4 m urea for h-cyt c (Fig. 3a) and y-cyt c (Fig. 3b), respectively. The CD results clearly suggest that the secondary structure of h-cyt c is significantly more stable compared with that of y-cyt c. Although the qualitative nature of the unfolding transitions of both the proteins is similar when monitored by far-UV CD (both proteins show two-state behavior), their differences become very prominent when the transitions monitored by far-UV CD and peroxidase activity are compared.
In the case of h-cyt c, unfolding transitions monitored by far-UV CD and peroxidase activity are not superimposable (Fig. 3, a, c, and e). The above conclusion holds both in the absence and presence of CDL. In the case of h-cyt c, the increase in peroxidase activity (supposedly representing the formation of an intermediate with local unfolding of the heme pocket) takes place at low urea concentration. This is followed by a decrease in the far-UV CD, representing the unfolding of the secondary structure. In the absence and presence of low concentrations of CDL, the formation of the intermediate state could not be detected directly by spectroscopy and was identifiable only by comparing the far-UV CD and peroxidase data. However, direct proof of intermediate formation was obtained in the presence of 50 μm CDL, where the presence of the intermediate was observed prominently in the far-UV CD measurement (Fig. 3e). The formation of an intermediate in this condition is accompanied by a significant decrease in the secondary structure (about 35% of the total change in ellipticity) (Fig. 3e). The rest of the secondary structure unfolds in a second unfolding event, which takes place with a midpoint of 7.5 m urea (Fig. 3e). In addition, Fig. 3e clearly shows that the increase in peroxidase activity takes place mostly as a result of the first unfolding transition (the formation of the early intermediate) and the second transition (the global unfolding of the secondary structure) does not contribute significantly to the increase in the peroxidase activity.
In contrast, the unfolding transitions monitored by far-UV CD and peroxidase activity measurements are superimposable in the case of y-cyt c (Fig. 3, b, d, and f). In addition, both in the absence and presence of different concentrations of CDL, these transitions can be fit perfectly using a two-state transition model (Fig. 3, b, d, and f). A large decrease in the conformational stability was observed for y-cyt c in the presence of 6 μm CDL, evident by the midpoints of 3.4 and 2.1 m as determined from the unfolding transitions in the absence and presence of 6 μm CDL, respectively.
FCS experiments were carried out with h-cyt c and y-cyt c in the presence of different concentrations of CDL and urea, and the relative populations of the conformational states were determined. The FCS data were modeled using a compact native state (N), an intermediate state (I), and an unfolded state (U). The far-UV CD and peroxidase activity data described above have suggested the presence of I in the unfolding equilibrium. In addition, we incorporated the oligomeric species (O) in FCS analyses, the presence of which was confirmed by independent FCS and DLS experiments.
Fig. 4 shows the variations in the populations of individual components with urea concentrations for both proteins in the absence and presence of different concentrations of CDL. For h-cyt c, the population of I initially increases with urea concentrations, reaches a maximum, and then decreases at high urea concentration. The decrease in the population of I occurs simultaneously with the increase in the population of U. As both U and I would sample the extended state (E), the population of E is also calculated and shown in Fig. 4, which suggests an increase in the urea concentration followed by saturation. This trend is similar also in the presence of CDL. However, both the population and the nature of the intermediate species change as a result of CDL binding.
FIGURE 4.

Variations in the populations of E (green), N (black), I (red), and U (blue) are shown with urea concentration for h-cyt c (a) and y-cyt c (b) in the absence of CDL, h-cyt c (c) and y-cyt c (d) in the presence of 6 μm CDL, and h-cyt c (e) and y-cyt c (f) in the presence of 50 μm CDL. The insets show the variation of oligomerized components (black) of y-cyt c (d) in the presence of 6 μm CDL and h-cyt c (e) and y-cyt c (f) in the presence of 50 μm CDL with urea concentrations.
For y-cyt c, however, the protein under native-like conditions (in the absence of CDL and urea) contains a significant population of I. With the increase in the urea concentration, the population of I decreases, which occurs simultaneously with the increase in the population of U. The calculated population of E (which combines the populations of I and U) dips at low urea concentrations, which is then followed by an increase in its population. A similar trend was obtained also in the presence of different concentrations of CDL (Fig. 4).
A comparison among FCS, CD, and peroxidase activity data for both proteins is shown in Fig. 5. The data clearly show that the increase in the population of E occurs simultaneously with the increase in the peroxidase activity. The increase in the value of rH, on the other hand, corresponds to the change in the far-UV CD, representing the formation of U. For both proteins, the extent of oligomerization decreases significantly in the presence of high urea concentrations, an effect that is particularly prominent in the case of y-cyt c (Fig. 4, d–f).
FIGURE 5.

Variations in the peroxidase activity (blue) and the population of E (red) are shown for h-cyt c (a) and y-cyt c (b) in the absence of CDL, h-cyt c (c) and y-cyt c (d) in the presence of 6 μm CDL, and h-cyt c (e) and y-cyt c (f) in the presence of 50 μm CDL. The insets show the variations in ellipticity at 222 nm (black) and rH (blue) with urea concentrations.
Although h-cyt c and y-cyt c Have Identical Structural Cores, They Have Different Extents of Conformational Fluctuation
As discussed above, cyt c experiences conformational fluctuations occurring in the μs time scale. This fluctuation takes place between a relatively compact conformer (N) and an extended conformer (E). The protein y-cyt c samples the conformer, E, effectively in its native solution condition. This is not the case for h-cyt c, which needs the presence of CDL or urea for the effective sampling of the E conformer. We have shown that E is responsible for the increase in the peroxidase activity of both proteins. We have also shown that y-cyt c has significantly less conformational stability and is prone to aggregation.
To obtain further insight into the difference in stability observed for these two proteins in their native conditions, computational methods were used. Fig. 6a shows the structural superimposition of h-cyt c and y-cyt c, which was obtained using available crystal structures (Protein Data Bank codes 1HRC and 1YCC for h-cyt c and y-cyt c, respectively). The proteins are superimposable with a very low Cα root mean square deviation (0.54 Å). They have identical core regions surrounding their heme cofactors (amino acids present within an 8 Å sphere with an iron atom at the center) with a root mean square deviation of 0.05 Å. Only a 2% difference in the amino acid sequence (due to the substitution by similar amino acids) was observed in the core region. In contrast, these two proteins differ slightly in their surface-exposed regions. A molecular surface view, showing the difference in the solvent-exposed amino acids of the superimposed y-cyt c and h-cyt c, is presented in Fig. 6b.
FIGURE 6.

a, structural superimposition of h-cyt c (orange color schematic of backbone with red stick representation of heme cofactor and ball representation of the iron atom at the center) with y-cyt c (light green color schematic of backbone with dark green stick representation of heme cofactor and ball representation of the iron atom at the center). The red color surface representation of dissimilar regions in y-cyt c from h-cyt c is shown in b, where the rest of the similar regions are represented by dots; the heme cofactor is represented by pink and blue sticks, and the iron atom is represented by an orange sphere. The RMSF of h-cyt c and y-cyt c are plotted against the residue numbers at 25 °C (c) and 40 °C (d), and the increase in RMSF of y-cyt over h-cyt c is shown by the gray area. e, a bar diagram of ΔΔRMSF is plotted against the residue numbers. ΔΔRMSF is defined under “Results.” APBS electrostatic calculation shows a continuous positive charge (blue) for region 1 for y-cyt c (f) and an even distribution of the positive charge surface (blue) and negative charge surface (red) for h-cyt c (g). h and i, region 2 showing the continuous negatively charged surface in y-cyt c (h) and even distribution of charge in h-cyt c (i).
The equilibrated structure of h-cyt c in aqueous solution shows minimal thermal fluctuations as determined by the root mean square fluctuations (RMSF). However, the extent of fluctuation is much larger for y-cyt c, which was observed using 200-ns MD simulations after the equilibration period (Fig. 6c). Additionally, the calculations were repeated at 40 °C, because the increase in the thermal energy at elevated temperature allows loosely bound and unstable amino acids to fluctuate more, providing a better understanding of the unstable regions of the protein. 200-ns MD simulations after an equilibrated period with the system temperature of 40 °C also showed a greater degree of fluctuation for y-cyt c compared with h-cyt c (Fig. 6e). To identify which regions of y-cyt c fluctuate more than the rest compared with h-cyt c, we plotted the values of ΔΔRMSF with the residue numbers in Fig. 6e. The parameter ΔΔRMSF can be defined as follows,
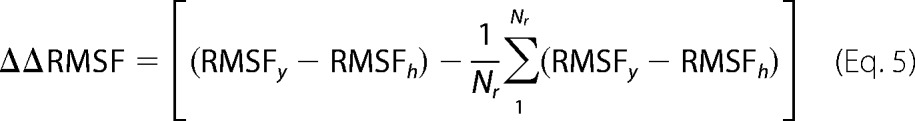 |
where RMSFy and RMSFh correspond to the values of RMSF of y-cyt c and h-cyt c, respectively, and Nr is the number of residues.
Positive values of ΔΔRMSF over a region of amino acids would hence indicate large fluctuations in that region with respect to the average change in fluctuation between y-cyt c and h-cyt c. In contrast, the negative values would suggest fewer fluctuations with respect to the average. The zero value indicates average fluctuations.
The simulation results shown in Fig. 6e indicate that the extent of fluctuation is not identical in the different regions of y-cyt c and some regions of y-cyt c fluctuate more than others. The highly fluctuating regions of y-cyt c are composed of surface-exposed residues, which are clustered into two distinct exposed regions. Region 1 is composed of residues 2–4, 10–13, and 79–88, and region 2 contains residues 23, 36–53, and 101–103. Region 1 can be classified as the cluster of positive charges and region 2 as the negative charge center, as shown by the equilibrated structure of y-cyt c obtained using a 300 ns simulation (Fig. 6, f and g). In contrast, h-cyt c shows a comparatively even distribution of charge at these two regions (Fig. 6, f and g). Interestingly, a virtually mutated h-cyt c with the core made identical to y-cyt c, differing only at the surface residues, shows no significant difference in RMSF compared with that observed with h-cyt c (data not shown).
As mentioned above, positively charged residues of y-cyt c are clustered together (Lys-4, -5, -54, -55, -72, -73, -79, -86, -87, and -89 and Arg-13) to form region 1, whereas in h-cyt c, Asp-2, Glu-4, Asn-54, Thr-89, and Glu-90 break the network of these positively charged residues (Fig. 6f). This in turn results in an evenly charged distribution of this region for h-cyt c. Among these residues, the changes from Ser-2 (in y-cyt c) to Asp-2 (in h-cyt c) and from Lys-11 to Val-11 constitute a reduction of the positive charge, whereas the changes at positions 4 (Lys to Glu), 54 (Lys to Asn), and 89 (Lys to Thr) are charge reversals. In addition, y-cyt c also has a continuously negatively charged region (region 2) composed of Glu-21, -44, -61, -66, and -103 and Asp-50 and -60. For h-cyt c, Lys-39, -53, -55, and -79 and Pro-39 break the continuity of a negative surface, generating a relatively evenly charged distribution. In this case, the charge reversal sites from y-cyt c to h-cyt c are Glu-44 to Pro and Ile-53 to Lys.
Discussion
The interaction between cyt c and CDL has been studied and reviewed extensively in the (5, 30, 38, 60, 61). The data presented here are described schematically in Fig. 7, which depicts the CDL binding and stability landscapes of h-cyt c and y-cyt c proteins. As discussed above, one the most important differences between these two proteins lies in the position of the intermediate states in the reaction coordinate. Although y-cyt c has a significant population of intermediate states in the native solution condition, h-cyt c requires the presence of CDL or urea to sample these peroxidase active intermediates. Consequently, the increase in peroxidase activity and the unfolding of the secondary structure do not superimpose in the case of h-cyt c, whereas they do for y-cyt c. It is interesting to note that peroxidase activity is higher in the presence of urea compared with that in the presence of CDL. Because the extent of E is maximal in both cases, the present data do not explain this difference. However, we emphasize that the partially unfolded intermediates formed in the presence of CDL and in the presence of urea are not necessarily the same species, although they sample the E state equally. Consequently, they have been denoted in Fig. 7 using different names. It may be noted that the enhancement of peroxidase activity in urea with respect to that in the presence of CDL is relatively less for y-cyt c compared with h-cyt c, and this small increase may possibly result from urea-induced deaggregation of y-cyt c along with its global unfolding.
FIGURE 7.
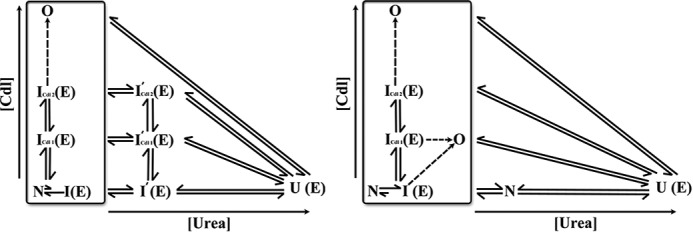
CDL binding (vertical axis) and urea-induced unfolding (horizontal axis) of h-cyt c (left) and y-cyt c (right) are shown. The designations N and E in parentheses indicate whether a particular state samples the compact (N) or extended conformer (E). The h-cyt c intermediates I′cdl1 and I′cdl2 (left) are defined based on urea unfolding data probed by far-UV CD and peroxidase activity (see Fig. 3, a, c, and e). Although the FCS data were analyzed using a generic intermediate state (I), this figure uses different symbols for the intermediates depending on the solution conditions.
The importance of positively charged residues for h-cyt c, particularly Lys-72, Lys-73, and Lys-79, is appreciated. It has been shown that Lys-72 and Lys-79, but not Lys-73, play a direct role in the molecular recognition of CDL (55). Interestingly, unlike h-cyt c, Lys-72 of y-cyt c is methylated, and methylation in Lys-72 aggravates the pro-apoptotic role of cyt c (36). It has been shown that the mutation of trimethyl Lys-72 to Ala in y-cyt c allows the displacement of the Met-80 ligand (which forms a coordinate bond with iron atom) from the heme crevice and is replaced by a water molecule (62). This conformational change increases the peroxidase activity of cyt c and has been reported previously for h-cyt c (63, 64). Pletneva and co-workers (39) have used a time-resolved FRET technique to show that CDL-attached cyt c has a mixed population of compact as well as massively unfolded conformers, a result that agrees with the present FCS data. They have suggested that the unfolding process starts with the unfolding of a low stability foldon followed by a massive distortion in structure, exposing the heme part, and penetration of the C-terminal portion of the protein into a CDL-containing membrane (39). Moreover, it has been shown that there exists a conformational exchange between the closed and extended conformers, which also is in agreement with the present findings (40). It has been proposed that the C-terminal insertion may have a direct correlation with the pore formation and release of cyt c into the cytosol (25). Confocal time lapse imaging has been used to monitor the crossing of fluorescent cyt c through synthetic CDL containing membranes in the absence of any other membrane permeabilizing pro-apoptitic factors (25). Green and co-workers (31) have suggested that the machinery of cyt c release and the activation of caspases is an ancient one, lost during evolution.
The core region of the protein, which houses the heme active site and is responsible for the primary function of electron transfer, is essentially conserved between different species. In contrast, the secondary functions of cyt c are fine-tuned by small changes in the surface residues. We show here that the secondary functions of cyt c, which include CDL binding and peroxidase activity, are governed by two important factors: first, a conformational fluctuation between N and E and then the CDL-induced oligomerization. For h-cyt c, the aggregation component (O) is generally low, and hence the increase in the peroxidase activity is more or less directly correlated to the population of E. For y-cyt c, however, the contribution of O is substantial, particularly in the presence of CDL. Consequently, the oligomer formation plays a major role in the peroxidase activity of y-cyt c.
This finding on the formation of oligomers may be important, as this could rationalize the observed differences between h-cyt c and y-cyt c during the initial stage of apoptosis. The partially unfolded cyt c from higher eukaryotes interacts and activates Apaf-1 (65), forming apoptosome, and initiating the apoptosis process. Interestingly, cyt c from yeast (and other invertebrates) does not interact with Apaf-1 (in fact yeast cells do not have any Apaf-1-homologous protein), acting as a reactive oxygen species scavenger in the cytosol. Studies have also shown that the released and dissociated y-cyt c becomes degraded over time. Our study shows that CDL-induced partial unfolding of y-cyt c leads to the formation of a large fraction of oligomeric species resulting in a decrease in peroxidase activity. It can be speculated that the oligomerization of a large fraction of partially unfolded y-cyt c may have direct relevance to the previous findings that y-cyt c lacks pro-apoptotic activity and decreases in its cytosolic concentration with time upon release from the mitochondria. Conversely, h-cyt c may experience an insignificant loss in its pro-apoptotic functionality due to lesser amounts of oligomeric species formation. It should be noted that the present study has been carried out completely under in vitro solution conditions, and hence a detailed investigation in a more complex cellular environment would be needed for further validation.
For h-cyt c, with the increase in the CDL concentration, the equilibrium between N and E initially shifts toward E followed by a slight decrease in the E population due to the formation of oligomeric species. These data along with the increase in the tryptophan fluorescence suggest that CDL induces the h-cyt c core (containing tryptophan) to open up for peroxidase activity while the overall fold remains the same as indicated by CD and FCS studies. In contrast, in the case of y-cyt c the protein does not require CDL induction for its peroxidase activity, but rather it relies on the fluctuation in regions 1 and 2 for the presence of a high fraction of E in the native condition. Those fluctuating regions allow the core of y-cyt c to open up for its peroxidase activity. As a result y-cyt c has slightly a larger rH value compared with h-cyt c.
There could be several reasons for the high degree of fluctuation sampled by the native state of y-cyt c. We hypothesized that the presence of local charge clusters in the surface region of the protein may be one of the responsible factors. The confinement of a similar charge (positive or negative) in a local surface cluster (observed in y-cyt c) may provide greater instability and structural movements in the protein, whereas charge units separated by an opposite charge (in the case of h-cyt c) may result in greater stability. These fluctuations are essential not only for its function but also for its stability, and hence any perturbation of the surface charge (for example, the binding of CDL) results in aggregation. The extent of fluctuation is significantly less in the case of h-cyt c and hence the extent of aggregation also. In the case of y-cyt c, a higher degree of fluctuation takes place in the structure around the exposed charge reversal sites. Surface-exposed charge reversal sites generate a different distribution of charges on the surface.
Although the fluctuations sampled by the MD simulations are in the ns time range and the dynamics observed in FCS is in the order of μs, their agreement may not be coincidental. It is possible that the faster fluctuations sampled by the MD simulations trigger slower and longer range fluctuations, which FCS can monitor. With the advancements in computational techniques and processor speeds, μs simulations are now possible. It would be exciting to carry out a direct comparison between the experiments and simulations in identical time regions.
Acknowledgments
We thank the Director of CSIR IICB for his encouragement as well as Prof. Carl Frieden of Washington University School of Medicine for help with this manuscript. We also thank the anonymous reviewers for their excellent critical suggestions.
This study was funded by a network project grant (UNSEEN) from the Council of Scientific and Industrial Research (CSIR), India.
- CDL
- cardiolipin
- cyt c
- cytochrome c
- y-
- yeast
- h-
- horse heart
- MD
- molecular dynamics
- TMR
- tetramethylrhodamine
- DLS
- dynamic light scattering
- RMSF
- root mean square fluctuation.
References
- 1. Ow Y. P., Green D. R., Hao Z., Mak T. W. (2008) Cytochrome c: functions beyond respiration. Nat. Rev. Mol. Cell Biol. 9, 532–542 [DOI] [PubMed] [Google Scholar]
- 2. Winge D. R. (2012) Sealing the Mitochondrial Respirasome. Mol. Cell. Biol. 32, 2647–2652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mueller G. P., Driscoll W. J. (2007) In vitro synthesis of oleoylglycine by cytochrome c points to a novel pathway for the production of lipid signaling molecules. J. Biol. Chem. 282, 22364–22369 [DOI] [PubMed] [Google Scholar]
- 4. Driscoll W. J., Chaturvedi S., Mueller G. P. (2007) Oleamide synthesizing activity from rat kidney: identification as cytochrome c. J. Biol. Chem. 282, 22353–22363 [DOI] [PubMed] [Google Scholar]
- 5. Hüttemann M., Pecina P., Rainbolt M., Sanderson T. H., Kagan V. E., Samavati L., Doan J. W., Lee I. (2011) The multiple functions of cytochrome c and their regulation in life and death decisions of the mammalian cell: from respiration to apoptosis. Mitochondrion 11, 369–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Liu X., Kim C. N., Yang J., Jemmerson R., Wang X. (1996) Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell 86, 147–157 [DOI] [PubMed] [Google Scholar]
- 7. Kagan V. E., Tyurin V. A., Jiang J., Tyurina Y. Y., Ritov V. B., Amoscato A. A., Osipov A. N., Belikova N. A., Kapralov A. A., Kini V., Vlasova I. I., Zhao Q., Zou M., Di P., Svistunenko D. A., Kurnikov I. V., Borisenko G. G. (2005) Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat. Chem. Biol. 1, 223–232 [DOI] [PubMed] [Google Scholar]
- 8. Basova L. V., Kurnikov I. V., Wang L., Ritov V. B., Belikova N. A., Vlasova I. I., Pacheco A. A., Winnica D. E., Peterson J., Bayir H., Waldeck D. H., Kagan V. E. (2007) Cardiolipin switch in mitochondria: shutting off the reduction of cytochrome c and turning on the peroxidase activity. Biochemistry 46, 3423–3434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Belikova N. A., Vladimirov Y. A., Osipov A. N., Kapralov A. A., Tyurin V. A., Potapovich M. V., Basova L. V., Peterson J., Kurnikov I. V., Kagan V. E. (2006) Peroxidase activity and structural transitions of cytochrome c bound to cardiolipin-containing membranes. Biochemistry 45, 4998–5009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pinheiro T. J. (1994) The interaction of horse heart cytochrome c with phospholipid bilayers: structural and dynamic effects. Biochimie 76, 489–500 [DOI] [PubMed] [Google Scholar]
- 11. Nantes I. L., Zucchi M. R., Nascimento O. R., Faljoni-Alario A. (2001) Effect of heme iron valence state on the conformation of cytochrome c and its association with membrane interfaces: A CD and EPR investigation. J. Biol. Chem. 276, 153–158 [DOI] [PubMed] [Google Scholar]
- 12. Tyurin V. A., Tyurina Y. Y., Osipov A. N., Belikova N. A., Basova L. V., Kapralov A. A., Bayir H., Kagan V. E. (2007) Interactions of cardiolipin and lyso-cardiolipins with cytochrome c and tBid: conflict or assistance in apoptosis. Cell Death Differ. 14, 872–875 [DOI] [PubMed] [Google Scholar]
- 13. Liu J., Dai Q., Chen J., Durrant D., Freeman A., Liu T., Grossman D., Lee R. M. (2003) Phospholipid scramblase 3 controls mitochondrial structure, function, and apoptotic response. Mol. Cancer Res. 1, 892–902 [PubMed] [Google Scholar]
- 14. Rytömaa M., Kinnunen P. K. (1994) Evidence for two distinct acidic phospholipid-binding sites in cytochrome c. J. Biol. Chem. 269, 1770–1774 [PubMed] [Google Scholar]
- 15. Rytömaa M., Kinnunen P. K. (1995) Reversibility of the binding of cytochrome c to liposomes: implications for lipid-protein interactions. J. Biol. Chem. 270, 3197–3202 [DOI] [PubMed] [Google Scholar]
- 16. Kapralov A. A., Kurnikov I. V., Vlasova I. I., Belikova N. A., Tyurin V. A., Basova L. V., Zhao Q., Tyurina Y. Y., Jiang J., Bayir H., Vladimirov Y. A., Kagan V. E. (2007) The hierarchy of structural transitions induced in cytochrome c by anionic phospholipids determines its peroxidase activation and selective peroxidation during apoptosis in cells. Biochemistry 46, 14232–14244 [DOI] [PubMed] [Google Scholar]
- 17. Sinibaldi F., Fiorucci L., Patriarca A., Lauceri R., Ferri T., Coletta M., Santucci R. (2008) Insights into cytochrome c-cardiolipin interaction: role played by ionic strength. Biochemistry 47, 6928–6935 [DOI] [PubMed] [Google Scholar]
- 18. Miyamoto S., Nantes I. L., Faria P. A., Cunha D., Ronsein G. E., Medeiros M. H., Di Mascio P. (2012) Cytochrome c-promoted cardiolipin oxidation generates singlet molecular oxygen. Photochem. Photobiol. Sci. 11, 1536–1546 [DOI] [PubMed] [Google Scholar]
- 19. Joshi A. S., Zhou J., Gohil V. M., Chen S., Greenberg M. L. (2009) Cellular functions of cardiolipin in yeast. Biochim. Biophys. Acta 1793, 212–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Brustovetsky N., Klingenberg M. (1996) Mitochondrial ADP/ATP carrier can be reversibly converted into a large channel by Ca2+. Biochemistry 35, 8483–8488 [DOI] [PubMed] [Google Scholar]
- 21. Petrosillo G., Ruggiero F. M., Di Venosa N., Paradies G. (2003) Decreased complex III activity in mitochondria isolated from rat heart subjected to ischemia and reperfusion: role of reactive oxygen species and cardiolipin. FASEB J. 17, 714–716 [DOI] [PubMed] [Google Scholar]
- 22. Genova M. L., Baracca A., Biondi A., Casalena G., Faccioli M., Falasca A. I., Formiggini G., Sgarbi G., Solaini G., Lenaz G. (2008) Is supercomplex organization of the respiratory chain required for optimal electron transfer activity? Biochim. Biophys. Acta 1777, 740–746 [DOI] [PubMed] [Google Scholar]
- 23. Gogvadze V., Orrenius S., Zhivotovsky B. (2006) Multiple pathways of cytochrome c release from mitochondria in apoptosis. Biochim. Biophys. Acta 1757, 639–647 [DOI] [PubMed] [Google Scholar]
- 24. Turrens J. F. (2003) Mitochondrial formation of reactive oxygen species. J. Physiol. 552, 335–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bergstrom C. L., Beales P. A., Lv Y., Vanderlick T. K., Groves J. T. (2013) Cytochrome c causes pore formation in cardiolipin-containing membranes. Proc. Natl. Acad. Sci. U.S.A. 110, 6269–6274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yu T., Wang X., Purring-Koch C., Wei Y., McLendon G. L. (2001) A mutational epitope for cytochrome c binding to the apoptosis protease activation factor-1. J. Biol. Chem. 276, 13034–13038 [DOI] [PubMed] [Google Scholar]
- 27. Olteanu A., Patel C. N., Dedmon M. M., Kennedy S., Linhoff M. W., Minder C. M., Potts P. R., Deshmukh M., Pielak G. J. (2003) Stability and apoptotic activity of recombinant human cytochrome c. Biochem. Biophys. Res. Commun. 312, 733–740 [DOI] [PubMed] [Google Scholar]
- 28. Sukhotnik I., Bernshteyn A., Mogilner J. G. (2005) The basic biology of apoptosis and its implications for pediatric surgery. Eur. J. Pediatr. Surg. 15, 229–235 [DOI] [PubMed] [Google Scholar]
- 29. Carmona-Gutierrez D., Eisenberg T., Büttner S., Meisinger C., Kroemer G., Madeo F. (2010) Apoptosis in yeast: triggers, pathways, subroutines. Cell Death Differ. 17, 763–773 [DOI] [PubMed] [Google Scholar]
- 30. Kagan V. E., Bayir H. A., Belikova N. A., Kapralov O., Tyurina Y. Y., Tyurin V. A., Jiang J., Stoyanovsky D. A., Wipf P., Kochanek P. M., Greenberger J. S., Pitt B., Shvedova A. A., Borisenko G. (2009) Cytochrome c/cardiolipin relations in mitochondria: a kiss of death. Free Radic. Biol. Med. 46, 1439–1453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bender C. E., Fitzgerald P., Tait S. W., Llambi F., McStay G. P., Tupper D. O., Pellettieri J., Sánchez Alvarado A., Salvesen G. S., Green D. R. (2012) Mitochondrial pathway of apoptosis is ancestral in metazoans. Proc. Natl. Acad. Sci. U.S.A. 109, 4904–4909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Oberst A., Bender C., Green D. R. (2008) Living with death: the evolution of the mitochondrial pathway of apoptosis in animals. Cell Death Differ. 15, 1139–1146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Guaragnella N., Zdraleviæ M., Antonacci L., Passarella S., Marra E., Giannattasio S. (2012) The role of mitochondria in yeast programmed cell death. Front. Oncol. 2, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ludovico P., Rodrigues F., Almeida A., Silva M. T., Barrientos A., Côrte-Real M. (2002) Cytochrome c release and mitochondria involvement in programmed cell death induced by acetic acid in Saccharomyces cerevisiae. Mol. Biol. Cell 13, 2598–2606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Manon S., Chaudhuri B., Guérin M. (1997) Release of cytochrome c and decrease of cytochrome c oxidase in Bax-expressing yeast cells, and prevention of these effects by coexpression of Bcl-xL. FEBS Lett. 415, 29–32 [DOI] [PubMed] [Google Scholar]
- 36. Kluck R. M., Ellerby L. M., Ellerby H. M., Naiem S., Yaffe M. P., Margoliash E., Bredesen D., Mauk A. G., Sherman F., Newmeyer D. D. (2000) Determinants of cytochrome c pro-apoptotic activity: the role of lysine 72 trimethylation. J. Biol. Chem. 275, 16127–16133 [DOI] [PubMed] [Google Scholar]
- 37. Giannattasio S., Atlante A., Antonacci L., Guaragnella N., Lattanzio P., Passarella S., Marra E. (2008) Cytochrome c is released from coupled mitochondria of yeast en route to acetic acid-induced programmed cell death and can work as an electron donor and a ROS scavenger. FEBS Lett. 582, 1519–1525 [DOI] [PubMed] [Google Scholar]
- 38. Sinibaldi F., Droghetti E., Polticelli F., Piro M. C., Di Pierro D., Ferri T., Smulevich G., Santucci R. (2011) The effects of ATP and sodium chloride on the cytochrome c-cardiolipin interaction: the contrasting behavior of the horse heart and yeast proteins. J. Inorg. Biochem. 105, 1365–1372 [DOI] [PubMed] [Google Scholar]
- 39. Hanske J., Toffey J. R., Morenz A. M., Bonilla A. J., Schiavoni K. H., Pletneva E. V. (2012) Conformational properties of cardiolipin-bound cytochrome c. Proc. Natl. Acad. Sci. U.S.A. 109, 125–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hong Y., Muenzner J., Grimm S. K., Pletneva E. V. (2012) Origin of the conformational heterogeneity of cardiolipin-bound cytochrome c. J. Am. Chem. Soc. 134, 18713–18723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Muenzner J., Toffey J. R., Hong Y., Pletneva E. V. (2013) Becoming a peroxidase: cardiolipin-induced unfolding of cytochrome c. J. Phys. Chem. B 117, 12878–12886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Muenzner J., Pletneva E. V. (2014) Structural transformations of cytochrome c upon interaction with cardiolipin. Chem. Phys. Lipids 179, 57–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Patriarca A., Polticelli F., Piro M. C., Sinibaldi F., Mei G., Bari M., Santucci R., Fiorucci L. (2012) Conversion of cytochrome c into a peroxidase: inhibitory mechanisms and implication for neurodegenerative diseases. Arch. Biochem. Biophys. 522, 62–69 [DOI] [PubMed] [Google Scholar]
- 44. Louie G. V., Brayer G. D. (1990) High-resolution refinement of yeast iso-1-cytochrome c and comparisons with other eukaryotic cytochromes c. J. Mol. Biol. 214, 527–555 [DOI] [PubMed] [Google Scholar]
- 45. Chattopadhyay K., Saffarian S., Elson E. L., Frieden C. (2002) Measurement of microsecond dynamic motion in the intestinal fatty acid binding protein by using fluorescence correlation spectroscopy. Proc. Natl. Acad. Sci. U.S.A. 99, 14171–14176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sen Mojumdar S., Chowdhury R., Chattoraj S., Bhattacharyya K. (2012) Role of ionic liquid on the conformational dynamics in the native, molten globule, and unfolded states of cytochrome c: a fluorescence correlation spectroscopy study. J. Phys. Chem. B 116, 12189–12198 [DOI] [PubMed] [Google Scholar]
- 47. Diederix R. E., Ubbink M., Canters G. W. (2002) Peroxidase activity as a tool for studying the folding of c-type cytochromes. Biochemistry 41, 13067–13077 [DOI] [PubMed] [Google Scholar]
- 48. Chattopadhyay K., Saffarian S., Elson E. L., Frieden C. (2005) Measuring unfolding of proteins in the presence of denaturant using fluorescence correlation spectroscopy. Biophys. J. 88, 1413–1422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Oostenbrink C., Villa A., Mark A. E., van Gunsteren W. F. (2004) A biomolecular force field based on the free enthalpy of hydration and solvation: the GROMOS force-field parameter sets 53A5 and 53A6. J. Comput. Chem. 25, 1656–1676 [DOI] [PubMed] [Google Scholar]
- 50. Haldar S., Paul S., Joshi N., Dasgupta A., Chattopadhyay K. (2012) The presence of the iron-sulfur motif is important for the conformational stability of the antiviral protein, Viperin. PLoS One 7, e31797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Baker N. A., Sept D., Joseph S., Holst M. J., McCammon J. A. (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Blauer G., Sreerama N., Woody R. W. (1993) Optical activity of hemoproteins in the Soret region: circular dichroism of the heme undecapeptide of cytochrome c in aqueous solution. Biochemistry 32, 6674–6679 [DOI] [PubMed] [Google Scholar]
- 53. Nantes I. L., Kawai C., Pessoto F. S., Mugnol K. C. (2010) Study of respiratory cytochromes in liposomes. Methods Mol. Biol. 606, 147–165 [DOI] [PubMed] [Google Scholar]
- 54. Godoy L. C., Muñoz-Pinedo C., Castro L., Cardaci S., Schonhoff C. M., King M., Tórtora V., Marín M., Miao Q., Jiang J. F., Kapralov A., Jemmerson R., Silkstone G. G., Patel J. N., Evans J. E., Wilson M. T., Green D. R., Kagan V. E., Radi R., Mannick J. B. (2009) Disruption of the M80-Fe ligation stimulates the translocation of cytochrome c to the cytoplasm and nucleus in nonapoptotic cells. Proc. Natl. Acad. Sci. U.S.A. 106, 2653–2658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sinibaldi F., Howes B. D., Droghetti E., Polticelli F., Piro M. C., Di Pierro D., Fiorucci L., Coletta M., Smulevich G., Santucci R. (2013) Role of lysines in cytochrome c-cardiolipin interaction. Biochemistry 52, 4578–4588 [DOI] [PubMed] [Google Scholar]
- 56. Haldar S., Mitra S., Chattopadhyay K. (2010) Role of protein stabilizers on the conformation of the unfolded state of cytochrome c and its early folding kinetics: investigation at single molecular resolution. J. Biol. Chem. 285, 25314–25323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Haldar S., Sil P., Thangamuniyandi M., Chattopadhyay K. (2014) Conversion of amyloid fibrils of cytochrome c to mature nanorods through a honeycomb morphology. Langmuir DOI: 10.1021/la5029993 [DOI] [PubMed] [Google Scholar]
- 58. Sarkar S., Chattopadhyay K. (2014) Studies of early events of folding of a predominately β-sheet protein using fluorescence correlation spectroscopy and other biophysical methods. Biochemistry 53, 1393–1402 [DOI] [PubMed] [Google Scholar]
- 59. Pitschke M., Prior R., Haupt M., Riesner D. (1998) Detection of single amyloid β-protein aggregates in the cerebrospinal fluid of Alzheimer's patients by fluorescence correlation spectroscopy. Nat. Med. 4, 832–834 [DOI] [PubMed] [Google Scholar]
- 60. Ascenzi P., Polticelli F., Marino M., Santucci R., Coletta M. (2011) Cardiolipin drives cytochrome c proapoptotic and antiapoptotic actions. IUBMB Life 63, 160–165 [DOI] [PubMed] [Google Scholar]
- 61. Santucci R., Sinibaldi F., Patriarca A., Santucci D., Fiorucci L. (2010) Misfolded proteins and neurodegeneration: role of non-native cytochrome c in cell death. Expert Rev. Proteomics 7, 507–517 [DOI] [PubMed] [Google Scholar]
- 62. McClelland L. J., Mou T. C., Jeakins-Cooley M. E., Sprang S. R., Bowler B. E. (2014) Structure of a mitochondrial cytochrome c conformer competent for peroxidase activity. Proc. Natl. Acad. Sci. U.S.A. 111, 6648–6653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hirota S., Hattori Y., Nagao S., Taketa M., Komori H., Kamikubo H., Wang Z., Takahashi I., Negi S., Sugiura Y., Kataoka M., Higuchi Y. (2010) Cytochrome c polymerization by successive domain swapping at the C-terminal helix. Proc. Natl. Acad. Sci. U.S.A. 107, 12854–12859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wang Z., Matsuo T., Nagao S., Hirota S. (2011) Peroxidase activity enhancement of horse cytochrome c by dimerization. Org. Biomol. Chem. 9, 4766–4769 [DOI] [PubMed] [Google Scholar]
- 65. Jemmerson R., Liu J., Hausauer D., Lam K. P., Mondino A., Nelson R. D. (1999) A conformational change in cytochrome c of apoptotic and necrotic cells is detected by monoclonal antibody binding and mimicked by association of the native antigen with synthetic phospholipid vesicles. Biochemistry 38, 3599–3609 [DOI] [PubMed] [Google Scholar]