

Background: The role of extracellular loop regions during substrate translocation in the serotonin transporter (SERT) is not well understood.
Results: A mutation in the extracellular loop 4 of SERT disrupts the conformational equilibrium by favoring an outward-facing conformation.
Conclusion: Extracellular loop 4 is important for conformational transitions in SERT.
Significance: New insights are gained into the coordinated conformational changes associated with substrate translocation in SERT.
Keywords: conformational change, molecular pharmacology, neurotransmitter transport, selective serotonin reuptake inhibitor (SSRI), serotonin transporter
Abstract
The serotonin transporter (SERT) terminates serotonergic neurotransmission by performing reuptake of released serotonin, and SERT is the primary target for antidepressants. SERT mediates the reuptake of serotonin through an alternating access mechanism, implying that a central substrate site is connected to both sides of the membrane by permeation pathways, of which only one is accessible at a time. The coordinated conformational changes in SERT associated with substrate translocation are not fully understood. Here, we have identified a Leu to Glu mutation at position 406 (L406E) in the extracellular loop 4 (EL4) of human SERT, which induced a remarkable gain-of-potency (up to >40-fold) for a range of SERT inhibitors. The effects were highly specific for L406E relative to six other mutations in the same position, including the closely related L406D mutation, showing that the effects induced by L406E are not simply charge-related effects. Leu406 is located >10 Å from the central inhibitor binding site indicating that the mutation affects inhibitor binding in an indirect manner. We found that L406E decreased accessibility to a residue in the cytoplasmic pathway. The shift in equilibrium to favor a more outward-facing conformation of SERT can explain the reduced turnover rate and increased association rate of inhibitor binding we found for L406E. Together, our findings show that EL4 allosterically can modulate inhibitor binding within the central binding site, and substantiates that EL4 has an important role in controlling the conformational equilibrium of human SERT.
Introduction
The human serotonin transporter (SERT)2 belongs to the solute carrier 6 (SLC6) family of transporters, that also include transporters for the neurotransmitters norepinephrine, dopamine, glycine, and γ-aminobutyric acid (GABA) (1). SERT is an integral membrane protein that facilitates sodium- and chloride-dependent reuptake of released serotonin (5-hydroxytryptamine; 5-HT) into neurons, and thereby maintains synaptic 5-HT homeostasis and controls the magnitude and duration of serotonergic neurotransmission (2).
Pharmacological modulation of the transport activity of SERT and the closely related norepinephrine transporter (NET) influences a variety of neurophysiological processes, and is used in the treatment of psychiatric diseases. In particular, inhibitors of SERT and NET are widely used in the treatment of major depressive disorder with more than 30 drugs in current clinical use (3). Additionally, SERT and NET are the major targets for psychostimulants such as amphetamine and ecstasy.
SLC6 transporters share a common topology of 12 transmembrane segments (TMs) connected by extracellular and intracellular loops. Insights into the three-dimensional SLC6 transporter architecture first came from x-ray crystal structures of the homologous prokaryotic transporter LeuT (4). In terms of transporter function, it is, however, not straightforward to correlate observations from LeuT to human SLC6 transporters, but the prokaryotic transporter has proven to be an excellent template for understanding structural aspects of SLC6 transporters, including ligand binding sites and permeation pathways (5–12).
LeuT structures have provided evidence for inhibitor binding sites located in two distinct regions: the central substrate binding site (denoted the S1 site) and a vestibular site (denoted the S2 site), which is located on the extracellular side of the central S1 site (13–16) (Fig. 1). Several residues within the equivalent S1 site in human SERT have been identified as being critical determinants for drug binding (11, 17–22). Together with the competitive binding mode of antidepressant drugs in SERT (23–25), this strongly suggests that the high affinity binding site for antidepressant drugs is overlapping with the S1 site in SERT. Recently, this notion was further supported by structures of the dopamine transporter (DAT) from Drosophila melanogaster and a LeuT/SERT hybrid protein co-crystallized with antidepressants (26, 27). The role of the S2 binding site in substrate translocation is still a matter of debate, but it has recently been suggested that this region harbors a low-affinity allosteric binding site for antidepressants in SERT (28).
FIGURE 1.
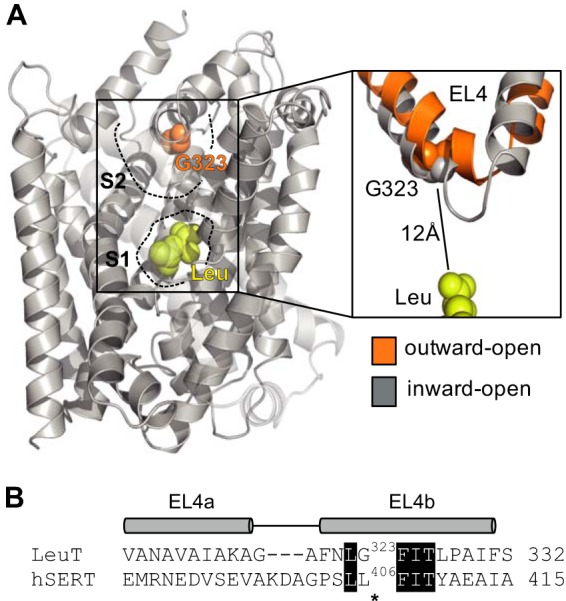
Location of the L406E mutation. A, substrate-bound x-ray crystal structure of LeuT (PDB code 2A65). The substrate (leucine) binds in the central S1 site and is shown as yellow spheres. Gly-323 (which is equivalent to Leu-406 in human SERT) is located close to the tip of EL4 in the vestibular S2 site and is shown as orange spheres. An overlay of x-ray crystal structures of LeuT in outward-facing (shown in orange; PDB code 3TT1) and inward-facing (shown in gray; PDB code 3TT3) conformations is shown on the right to illustrate the flexibility of EL4. Gly-323 is located ∼12 Å away from the central substrate binding site. B, amino acid sequence alignment of the EL4 region in LeuT and human SERT (48). Identical residues are shown as white text on black background. The segments that assume α-helical arrangement in the LeuT x-ray crystal structures (denoted EL4a and EL4b) are indicated above the sequence alignment. Asterisks indicate the position of the Leu-406 residue (SERT numbering).
Early studies utilizing chimeric constructs between SERT and NET have suggested that the extracellular loop (EL) regions are not merely passive structures connecting TMs, but important elements responsible for the conformational flexibility required for substrate translocation (29, 30). Specifically, EL4, which connects TM7 and TM8, has been proposed to adopt substantially different conformations during transport (31). LeuT structures crystallized in different conformational states corresponding to outward-facing, occluded, and inward-facing have provided structural insight into the alternating access mechanism that drives substrate translocation (32). Combined with biochemical studies of LeuT, this has confirmed the functional importance of EL4 and showed that movement of TM7 causes EL4 to dip further down into the extracellular vestibule, thereby blocking access to the central S1 binding site, when the transporter moves from the outward- to the inward-facing conformation (32–34). Furthermore, recent studies on the prokaryotic proline transporter, PutP, which shares the so-called LeuT-fold with SLC6 transporters, but is otherwise unrelated, have suggested that EL4 transmits substrate-induced conformational changes to TM domains in the core of the transporter (35).
Taken together, studies of prokaryotic transporters clearly suggest that EL4 plays an important role in the transport cycle of SLC6 transporters. However, low amino acid sequence identity between the prokaryotic transporters and their human relatives compromises the extent to which these findings can be used to generate a detailed and accurate mechanism for the role of EL4 in human SLC6 transporters. In the present study, we have identified a Leu to Glu mutation at position 406 in the EL4 region of human SERT (Fig. 1) that induces a marked gain-of-inhibitory potency for a range of different SERT inhibitors. By combining uptake experiments, ligand binding kinetics studies, site-directed mutagenesis, and the substituted cysteine accessibility method, we have investigated how L406E affects inhibitor binding and the basal transporter function of SERT. Together, our data suggest that L406E changes the equilibrium of SERT to favor an outward-facing conformation, which decreases the functional activity of SERT and increases the association rate of inhibitor binding. These findings underline that EL4 plays an important functional role in the transport cycle in human SLC6 transporters, and provide novel insight into the mechanism by which EL4 controls the conformational equilibrium of SERT.
Experimental Procedures
Chemicals
Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum, penicillin-streptomycin, and trypsin were purchased from Invitrogen. 3H-Labeled 5-HT, 125I-labeled RTI-55 ((−)-2β-carbomethoxy-3β-(4-iodophneyl)tropane), MicroScint-0, and MicroScint-20 scintillation mixtures were obtained from PerkinElmer Life Sciences. RTI-55 was purchased from ABX (Radeberg, Germany). Cocaine and 5-HT were purchased from Sigma. (2-Trimethylammonium)methanethiosulfonate (MTSET) was purchased from Toronto Research Chemicals Inc. (North York, ON, Canada) and (2-aminoethyl)methanethiosulfonate (MTSEA) was from Apollo Scientific (Stockport, UK). Ibogaine was a kind gift from Sacrament of Transition (Maribor, Slovenia). Atomoxetine, amitriptyline clomipramine, duloxetine, fluoxetine, fluvoxamine imipramine, MADAM, maprotiline, milnacipran, nisoxetine, paroxetine, escitalopram, sertraline, talopram, and venlafaxine were kindly provided by H. Lundbeck A/S (Copenhagen, Denmark).
Site-directed Mutagenesis
As expression vector, the commercially available pcDNA3.1 containing hSERT was used. Generation of point mutations in pcDNA3.1-hSERT was performed using the QuikChange site-directed mutagenesis kit (Stratagene, Carlsbad, CA), according to the manufacturer's protocol. The mutations were verified by DNA sequencing (GATC Biotech, Constance, Germany).
Cell Culturing and Expression
COS7 cells were cultured in DMEM, containing 10% fetal bovine serum, 100 units/ml of penicillin, and 100 units/ml of streptomycin, at 37 °C in a humidified 5% CO2 environment. Cells were transiently transfected using TransIT LT1 DNA transfection reagent (Mirus Bio, Madison, WI). Prior to transfection, confluent COS7 cells growing in monolayer were resuspended in DMEM, at a concentration of ∼1.3 × 106 cells/ml. Subsequently, the cell suspension was added to the transfection mixture, and immediately dispensed into poly-d-lysine-coated white 96-well plates at 50% confluence with 50 ng of plasmid DNA/well.
Functional [3H]5-HT Uptake Assay
For determination of inhibitory potencies and substrate Km and Vmax values, functional [3H]5-HT uptake assays using transiently transfected COS7 cells in 96-well plates were performed as previously described (7). All assays were carried out in triplicate and repeated at least 3 times unless otherwise noted.
[125I]RTI-55 Binding Assays
Cell membranes from COS7 cells were prepared 40–50 h after transient transfection as previously described (20). Membranes were used directly for binding assays, or stored at −80 °C until use. In [125I]RTI-55 saturation binding assays (determination of Kd values), 5–30 μg/well of total membrane protein was incubated with increasing concentrations of [125I]RTI-55, diluted 1:10, 1:15, or 1:20 with unlabeled RTI-55, in phosphate-buffered saline (in mm: NaCl, 137; KCl, 2.7; Na2HPO4, 1.4, pH 7.4) containing 0.5 mm CaCl2 and 0.5 mm MgCl2 (PBSCM) and the final volume was adjusted to 150 μl/well. Binding was allowed to equilibrate for 2 h on ice. In [125I]RTI-55 competition binding assays (determination of inhibitor binding affinities), 5–30 μg/well of total membrane was incubated with 0.25 nm [125I]RTI-55 in the presence of increasing concentrations of inhibitor in PBSCM and the final volume was adjusted to 150 μl/well. Binding was allowed to equilibrate for 2 h on ice. The binding assays were terminated by rapid washing with water and membranes were collected onto 96-well glass-fiber plates (Unifilter C, PerkinElmer Life Sciences), preincubated with 0.1% polyethyleneimine, using a Packard Bell cell harvester. Filter plates were dried and soaked in MicroScint-0 scintillation mixture, followed by counting of plates in a Packard TopCounter (Packard Inc., Prospect, CT). Nonspecific binding was determined in parallel at membranes from non-transfected COS7 cells. [125I]RTI-55 binding assays were carried out in duplicates and repeated at least three times.
For calculation of turnover numbers, the maximal binding capacity (Bmax) was determined in [125I]RTI-55 saturation binding assays on whole cells. COS7 cells were transiently transfected, and plated into poly-d-lysine-coated 96-well plates (as detailed above). 40–50 h after transfection, the binding assay was carried out using the same protocol as described for the [125I]RTI-55 saturation binding assays on membranes. Binding was allowed to equilibrate for 2 h on ice before harvesting. Nonspecific binding was determined in parallel at non-transfected COS7 cells. In control experiments, we determined nonspecific binding to COS7 cells expressing SERT WT by displacing extracellular [125I]RTI-55 binding with 100 μm 5-HT as described previously (36). These experiments revealed that the level of [125I]RTI-55 binding was identical under the two background conditions (non-transfected COS7 cells and 100 μm 5-HT) (data not shown), suggesting that [125I]RTI-55 does not label intracellular SERT under the conditions used for the saturation binding assay. The kinetic Vmax values were determined in parallel on the same batches of transfected cells from [3H]5-HT uptake experiments using triplicate determinations as detailed previously (20).
Kinetic Binding Assays
Cell membranes from transiently transfected COS7 cells were prepared 40–50 h after transient transfection as previously described (20). In a 96-well plate, 5–30 μg of membrane protein was added to each well and combined with different concentrations of [3H]escitalopram or [125I]RTI-55 in PBSCM and the final volume was adjusted to 150 μl/well. Binding was allowed to proceed for 0–20 min, before it was terminated and radioactivity was quantified as described above for [125I]RTI-55 binding assays. Nonspecific binding was determined in parallel at membranes from non-transfected COS7 cells. Assays were carried out in duplicates and repeated at least three times.
Measurement of Q332C Accessibility to MTSET
COS7 cells were transiently transfected with C109A-Q332C and plated into poly-d-lysine-coated 96-well plates as described above. 40–50 h after transfection, the cells were washed and incubated with a fixed concentration of MTSET (4 mm) in the absence or presence of increasing concentrations of inhibitor at room temperature for 10 min. The cells were washed twice and incubated for 30 min in PBSCM to release bound inhibitor. Incubation of cells in PBSCM had no effect on the degree of MTSET reaction (data not shown). As control, we included cells expressing the C109A-Q332C mutant that were incubated with inhibitor alone. The residual functional activity was determined by measuring uptake of 50 nm [3H]5-HT as previously described (7). All assays were carried out in triplicate and repeated at least 3 times.
Measurement of S277C Accessibility to MTSEA
HEK293-MSR cells (Invitrogen) were transiently transfected with either SERT X5C-S277C or SERT X5C-S277C/L406E using Lipofectamine 2000 transfection reagent (Invitrogen). The SERT X5C construct (C15A/C21A/C109A/C357I/C622A) has previously been shown to have minimal sensitivity to MTS reagents (37). 48 h after transfection, cells were harvested in 50 mm Tris base buffer (50 mm Tris, 150 mm NaCl, 20 mm EDTA, pH 7.4), centrifuged at 4,700 × g, washed, and homogenized in Tris base buffer using a Ultra-turrax T25 (IKA) prior to centrifugation at a minimum 12,500 × g. Homogenization and centrifugation were performed twice. All steps were performed at 4 °C. The membrane preparations were stored in HEPES buffer (10 mm HEPES supplemented with 150 mm NaCl, pH adjusted to 8.0 with N-methyl-d-glutamine) at −20 °C until use. Membrane preparations were applied to Multiscreen HTS 96-well filtration plates (Millipore, Bedford, MA) that were pretreated with 0.1% polyethyleneimine, and the membranes were washed at least six times with HEPES buffer. Membranes were incubated with 12 increasing concentrations of MTSEA, and the reaction was terminated after 15 min by at least six washes with HEPES buffer. To quantify residual unreacted SERT, the membranes were then incubated with 0.1 nm [125I]RTI-55 for 60 min, and the membranes were subsequently washed at least five times with ice-cold HEPES buffer to remove unbound ligand. The dry filters were then dissolved in MicroScint20, and bound [125I]RTI-55 was quantified on a Packard TopCounter.
[125I]RTI-55 Dissociation Rate Assay
Dissociation rates of [125I]RTI-55 were determined using membrane preparations from HEK293-MSR cells transiently transfected with SERT WT or L406E (prepared as described above). Initially, [125I]RTI-55 was allowed to equilibrate with SERT membrane preparations for at least 1 h at room temperature. Dissociation was initiated at different time points by dilution of the SERT·[125I]RTI-55 complex into PBSCM containing either escitalopram or talopram at increasing concentrations. The dissociation was stopped by filtration through GF-B glass-fiber plates (Unifilter B, PerkinElmer) preincubated with 1% polyethyleneimine and washed twice with water using a Packard Bell cell harvester. Filter plates were soaked in MicroScint-20 scintillation mixture and captured SERT·[125I]RTI-55 complex was quantified on a Packard TopCounter. Dissociation curves were obtained by plotting residual [125I]RTI-55 binding versus time of dissociation.
Data Analysis
The experimental data from uptake and binding assays were analyzed by nonlinear regression analysis using Prism 6.0 software (GraphPad Inc., San Diego, CA). Inhibitor Ki values were calculated from the IC50 values using the equation Ki = IC50/(1 + (L/Kx), where x is either ”m“ or ”d“ whether it is an uptake or binding assay, respectively, and L is the concentration of the 3H- or 125I-labeled radioligand. For the [125I]RTI-55 dissociation assay, the dissociation rate (Koff) were determined from nonlinear regression to the Equation B = B0 × exp(−Koff × t) + b. The resulting Koff were plotted as a function of the concentration of the allosteric modulator (escitalopram or talopram) and fitted to a sigmoidal dose-response curve from which the allosteric potency was determined.
Results
L406E Mutation Increases Inhibitor Potency at Human SERT
The x-ray crystal structures of LeuT have stimulated intensive efforts into elucidating the role of S1 residues in SERT for high affinity inhibitor binding. In contrast, the role of S2 residues for high affinity inhibitor binding in SERT has attracted less attention. The S2 site is located approximately halfway across the membrane bilayer at the inner end of an extracellular cavity and likely forms part of the permeation pathway. Thus, mutation of S2 residues in SERT may affect inhibitors when they permeate this site to reach high affinity binding in the central substrate site (S1). To investigate the impact of selective perturbation in the S2 region on inhibitor binding, we selected 6 residues surrounding the S2 site in human SERT and introduced mutations into these positions. Specifically, we introduced the following mutations: Y107L (TM1), I179F (TM3), D400F (EL4), A401E (EL4), L406E (EL4), and K490T (TM10). The Y107L mutation rendered the transporter non-functional and was not studied further. The remaining five mutants retained transport activity (13–95% compared with SERT WT) and allowed determination of inhibitory potency (Ki) in a functional [3H]5-HT uptake assay. Our selection of SERT inhibitors was based on covering structural and pharmacological diversity. Specifically, three pharmacological tool compounds and 12 antidepressant drugs belonging to different drug classes, including selective 5-HT reuptake inhibitors, 5-HT and norepinephrine reuptake inhibitors, and tricyclic antidepressants, were investigated. To determine the impact on the inhibitory potency of selective perturbations of the S2 site in human SERT, we determined the Ki value for each of the 15 selected inhibitors at the five functional mutants expressed in COS7 cells in a [3H]5-HT uptake inhibition assay. The I179F, D400F, A401E, and K490T mutants generally induced differential and moderate changes in the inhibitory potency across the 15 tested compounds (data not shown). In contrast, the L406E mutation stood out with a pronounced and consistently positive effect on inhibitor potency across the 15 compounds tested. Furthermore, we tested the two recreational drugs ibogaine and cocaine, the high affinity pharmacological tool compound RTI-55, as well as the tetracyclic antidepressant maprotiline at SERT WT and L406E, and the mutation induced >10-fold gain-of-potency for 14 of the 19 inhibitors tested (Fig. 2).
FIGURE 2.
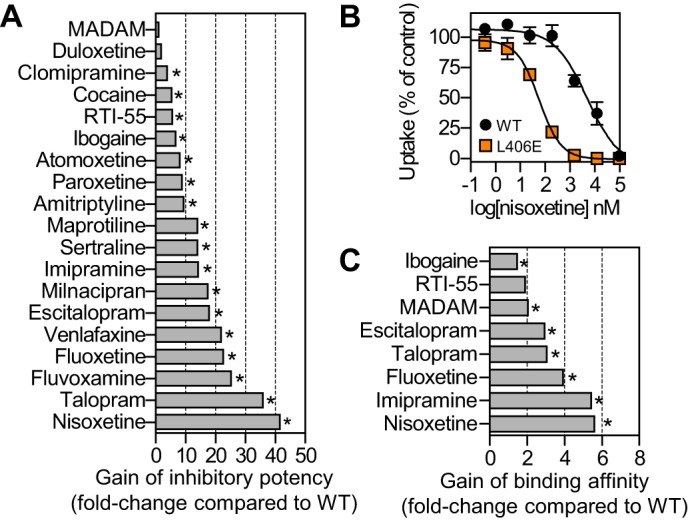
Effect of L406E in human SERT on inhibitor binding. A, the inhibitory potency of 19 structurally and pharmacologically diverse SERT inhibitors was determined at SERT WT and SERT L406E in a functional [3H]5-HT uptake inhibition assay (see “Experimental Procedures”), and the mutational induced increase in inhibitory potency (shown on x-axis) was calculated as Ki(WT)/Ki(L406E) (each Ki value was determined from at least three independent experiments each performed in duplicate). B, dose-response curves from a representative [3H]5-HT uptake inhibition experiment in COS7 cells expressing SERT WT (black circles) or SERT L406E (orange squares). Data points represent the mean ± S.E. from triplicate determinations of accumulated radioactivity in cells incubated with 50 nm [3H]5-HT in the presence of increasing concentrations of nisoxetine. Uptake has been normalized to percent uptake of cells incubated in the absence of inhibitor. C, the binding affinity of 8 selected SERT inhibitors was determined at SERT WT and SERT L406E in a competitive [125I]RTI-55 binding assay (see “Experimental Procedures”), and the mutational induced increase in binding affinity (shown on x axis) was calculated as Ki(WT)/Ki(L406E) (each Ki value was determined from at least three independent experiments each performed in duplicate). Asterisks denote that L406E induce a significant increase in inhibitory potency (panel A) or binding affinity (panel C) compared with SERT WT (p < 0.05; Student's t test).
Specificity of Leu to Glu Mutation
Leu-406 is located close to the presumed tip of EL4 in the S2 site ∼12 Å from the central substrate site (Fig. 1). Hence, it seems unlikely that the effect on inhibitor Ki induced by L406E is mediated through direct effects on high affinity inhibitor binding in the central S1 site. This prompted us to further investigate the L406E mutation, and envisioned that this could potentially uncover novel insights into the functional role of EL4 in SERT. Initially, to investigate if the L406E-induced effects in SERT could also be observed in the related transporters for norepinephrine and dopamine, we substituted the equivalent residues in NET and DAT for a Glu residue (V387E in NET; I390E in DAT). However, these mutations rendered NET and DAT functionally inactive (data not shown), which precluded us from similar examination of the corresponding mutation in NET and DAT but indicate that this EL region is involved in the function of the transporter. To investigate the site specificity of the L406E induced effects in SERT, we replaced the two adjacent positions (Leu-405 and Phe-407) with a Glu residue. However, the L405E and F407E mutants were devoid of any measurable [3H]5-HT uptake activity (data not shown), and were not studied further. Next, to investigate the specificity of the effects induced by the L406E mutation, we introduced Ala, Cys, Asp, Phe, His, Lys, or Gln into position 406 in SERT. Whereas the L406K mutant was inactive, the remaining six mutations retained 6–75% uptake activity compared with WT (Table 1). To assess if the non-functional L406K mutant was expressed at the cell surface, we examined if the mutant could bind 125I-labeled RTI-55. However, when L406K was expressed in COS7 cells we observed no measurable [125I]RTI-55 binding, suggesting that L406K inhibits protein folding and/or surface expression. We selected three inhibitors that were among the most affected by L406E (escitalopram, talopram, and nisoxetine), and determined the inhibitory potency of these compounds at L406A, L406C, L406D, L406F, L406H, and L406Q (Fig. 3). Interestingly, none of the other Leu-406 mutations affected the potency of the three inhibitors to a similar extent as L406E (which induced 18- to 41-fold gain-of-potency for the tested inhibitors). Notably, the closely related L406D mutation only induced up to 3-fold gain-of-potency, clearly showing that the effect induced by L406E is highly specific for this mutant and not simply a general charge-related effect.
TABLE 1.
Impact of SERT mutations on [3H]5-HT uptake kinetics
Relative transport activity of mutants was determined from paired uptake experiments and is expressed as percentage of mutant to WT for a 5-min uptake with 50 nm [3H]5-HT. All values are mean ± S.E. from at least 3 independent experiments.
| Mutant | Km | Transport activity |
|---|---|---|
| nm | % of WT | |
| SERT WT | 1316 ± 342 | |
| L406A | 819 ± 168 | 65 ± 4.1a |
| L406C | 756 ± 163 | 62 ± 3.2a |
| L406D | 433 ± 114 | 6.3 ± 0.8a |
| L406E | 300 ± 100a | 13 ± 0.8a |
| L406F | 2821 ± 541 | 75 ± 8.5a |
| L406H | 582 ± 74 | 13 ± 2.9a |
| L406K | NFb | NF |
| L406Q | 1273 ± 343 | 50 ± 4.7a |
a Significantl difference compared to WT (p < 0.05, Student's t test).
b NF, nonfunctional.
FIGURE 3.
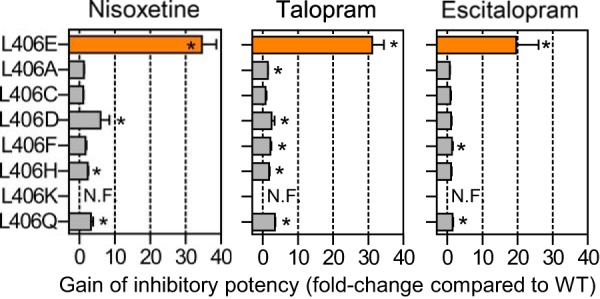
Specificity of the Leu to Glu mutation in site 406 of human SERT. The inhibitory potency of nisoxetine, talopram, and escitalopram was determined at SERT WT and seven different mutations introduced into site 406 in a functional [3H]5-HT uptake inhibition assay (see “Experimental Procedures”). The mutational induced increase in inhibitory potency (shown on x axis) was calculated as Ki(WT)/Ki(mutant) from paired experiments. Data represent mean ± S.E. from 3 to 6 independent experiments each performed in triplicate. The L406K mutation rendered the transporter non-functional (N.F.). Asterisks denote that the mutation induce a significant change in inhibitory potency compared with SERT WT (p < 0.05; one-way ANOVA followed by Dunnett's post hoc test).
The Central S1 Site Harbors the High Affinity Inhibitor Binding Site in the L406E Mutant
Next, we selected eight inhibitors that were affected differently by the L406E mutation in the functional uptake inhibition assay, and determined the binding affinity of these inhibitors by displacement of [125I]RTI-55 binding to membranes from COS7 cells expressing SERT WT or SERT L406E. Interestingly, the inhibitors were generally less affected by the L406E mutation in the competitive binding assays compared with the functional uptake assay (Fig. 2C). The most pronounced difference was observed for fluoxetine, nisoxetine, and talopram, for which the L406E mutant induced 19–41-fold gain-of-potency in the uptake inhibition assay, but only 4–6-fold gain-of-affinity in the competitive binding assay (Fig. 2). Because the large increase in inhibitory potency was not concomitant with a similar increase in binding affinity, this indicates that the effect of the mutant could arise from long-range indirect conformational perturbation of the transporter structure rather than direct modification of an inhibitor binding site. This would also be in agreement with the distinct location of Leu-406 relative to the central high affinity inhibitor binding site (Fig. 1).
To examine this further, we introduced six different S1 point mutations into the background of L406E. The selected S1 mutants (Y95A, D98E, I172M, N177S, F341Y, and S438T) have previously been shown to disrupt high affinity S1 binding of several different SERT inhibitors (21). If the inhibitors also bind to the S1 site in the L406E mutant, we would expect that these S1 mutations would disrupt binding also in the background of L406E. Initially, we tested the functional activity of the six double mutants (Fig. 4). For the two functional mutants (I172M/L406E and F341Y/L406E) we determined the inhibitory potency of four inhibitors (escitalopram, venlafaxine, talopram, and nisoxetine), for which L406E alone induced more than 10-fold gain of potency (Fig. 2). Strikingly, introduction of I172M or F341Y into the L406E background induced 15–997-fold loss of potency for all four inhibitors compared with L406E alone (Fig. 4). Hence, because the inhibitory potency of the four inhibitors could be significantly decreased by S1 mutations in the L406E background, this indicates that the inhibitors still bind to the S1 site in the L406E mutant of SERT, and that L406E affect the inhibitory potency by allosterically modulating inhibitor binding in the central S1 site. However, L406E is located close to the presumed low-affinity allosteric binding site in human SERT (28). The L406E mutation could form a high affinity inhibitor binding site in this region, which could be allosterically affected by the introduced S1 mutations. To examine if L406E increases affinity for the allosteric site, we determined the allosteric potency of escitalopram and talopram at WT SERT and the L406E mutant. Specifically, we measured the dissociation of [125I]RTI-55 binding from membranes of HEK293-MSR cells expressing SERT WT or L406E in the presence of increasing concentrations of escitalopram or talopram (Fig. 5). Consistent with previous observations (38, 39), escitalopram had a profound effect on [125I]RTI-55 dissociation from SERT WT with an allosteric potency of 102.4 ± 11.5 μm (mean ± S.E. from three independent experiments) (Fig. 5). In comparison, the allosteric potency of talopram was weaker at SERT WT (>500 μm). Importantly, L406E had no significant effect on the allosteric potency of escitalopram or talopram (Fig. 5), which further supports that L406E does not form a high affinity binding site in the extracellular vestibule of SERT, but rather allosterically modulate high affinity inhibitor binding in the central S1 site.
FIGURE 4.
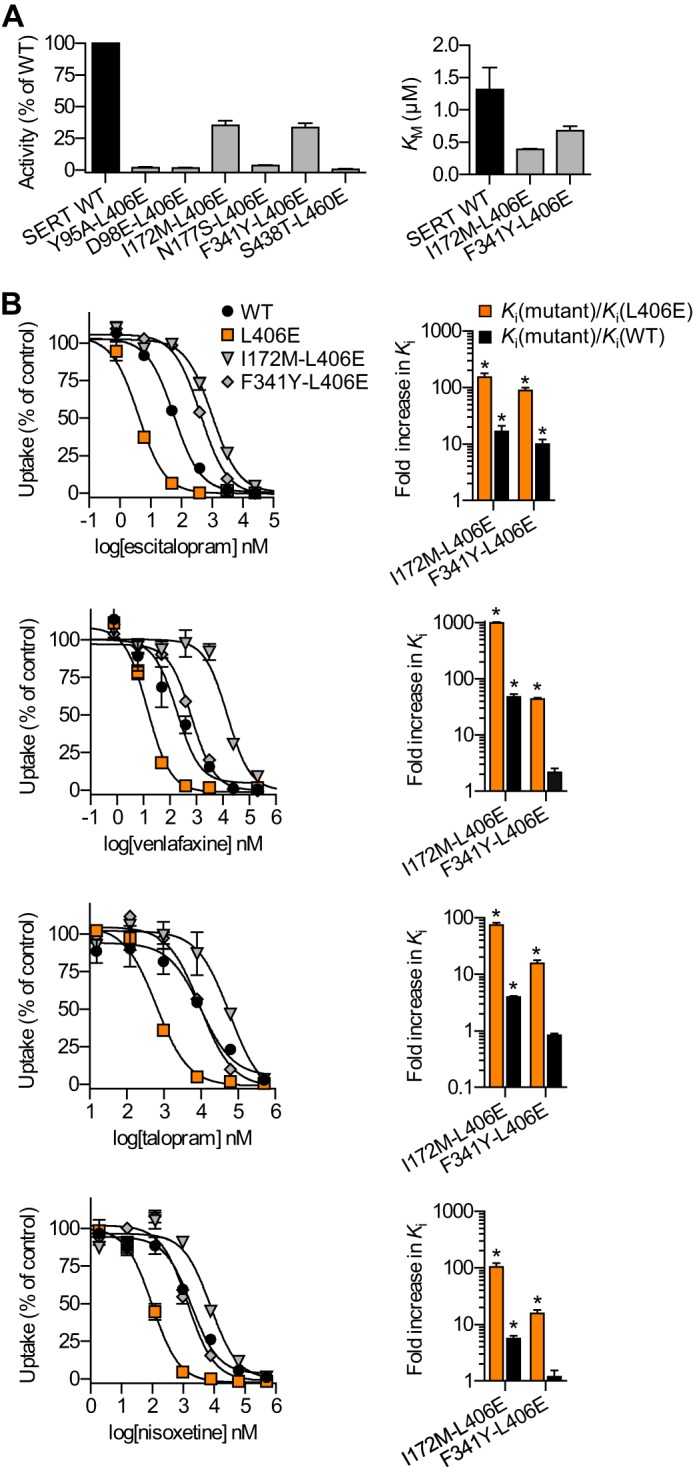
Introducing S1 mutations in the background of L406E significantly reduce inhibitor potency. A, six different S1 mutations were introduced into the background of L406E, and the mutants were assessed for functional activity by measuring uptake of 50 nm [3H]5-HT in COS7 cells expressing the double mutants. The substrate Km value was determined for the two functionally active mutants (I172M/L406E and F341Y/L406E). B, dose-response curves from representative [3H]5-HT uptake inhibition experiments in COS7 cells expressing SERT WT or the indicated mutants (left). Data points represent mean ± S.E. from triplicate determinations of accumulated radioactivity in cells incubated with 50 nm [3H]5-HT in the presence of increasing concentrations of escitalopram, venlafaxine, talopram, or nisoxetine. Uptake has been normalized to percent uptake of cells incubated in the absence of inhibitor. For I172M/L406E and F341Y/L406E, the mutational induced increase in inhibitor Ki was calculated as Ki(mutant)/Ki(WT) (black bars) or Ki(mutant)/Ki(L406E) (orange bars) from paired experiments (right). Data represent mean ± S.E. from 3 to 5 independent experiments each performed in triplicate. Asterisks denote that I172M/L406E or F341Y/L406E induce a significant increase in inhibitor Ki compared with WT (black bars) or L406E (orange bars).
FIGURE 5.
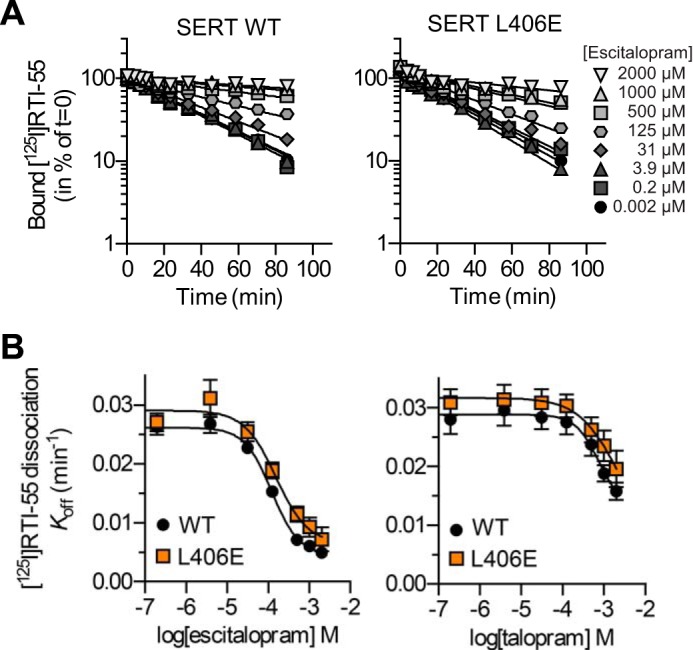
[125I]RTI-55 dissociation rates and allosteric potencies of escitalopram and talopram are unaffected by the L406E mutation. A, representative time course for dissociation of [125I]RTI-55 from HEK293-MSR membranes expressing SERT WT (left) or L406E (right) in the presence of increasing concentrations of escitalopram. The dissociation shows exponential decay and escitalopram decrease the dissociation rate of [125I]RTI-55 in a dose-dependent manner. The experiment was also conducted using increasing concentrations of talopram as allosteric modulator. B, dissociation rates are plotted as a function of the concentration of escitalopram (left) or talopram (right). From the sigmoidal dose-response curve, the allosteric potency (EC50) for escitalopram and talopram can be derived. The allosteric potency of escitalopram and talopram was found to be identical in SERT WT and L406E. Independent dissociation experiments were repeated three times.
L406E Perturbs the Conformational Equilibrium of SERT
To characterize the effect of the L406E mutant on the basal transporter function, we determined the turnover number of SERT WT and the L406E mutant. For turnover number calculations, we carried out saturation [3H]5-HT uptake assays and saturation [125I]RTI-55 binding experiments in parallel on SERT WT and SERT L406E from the same batches of transfected COS7 cells (Fig. 6A). The turnover numbers (calculated from the ratio of Vmax from [3H]5-HT uptake/Bmax from [125I]RTI-55 binding) were 15 ± 1.2 and 3.0 ± 0.4 min−1, for WT SERT and SERT L406E, respectively (values represent mean ± S.E. from at least five independent experiments each performed in duplicate). The significantly reduced turnover number for L406E compared with WT SERT (p < 0.0001, two-tailed Student's t test) clearly shows that the L406E mutation alters the kinetic properties of the transporter and that the conformational cycle underlying substrate translocation is approximately five times slower for the L406E mutant compared with the WT transporter.
FIGURE 6.
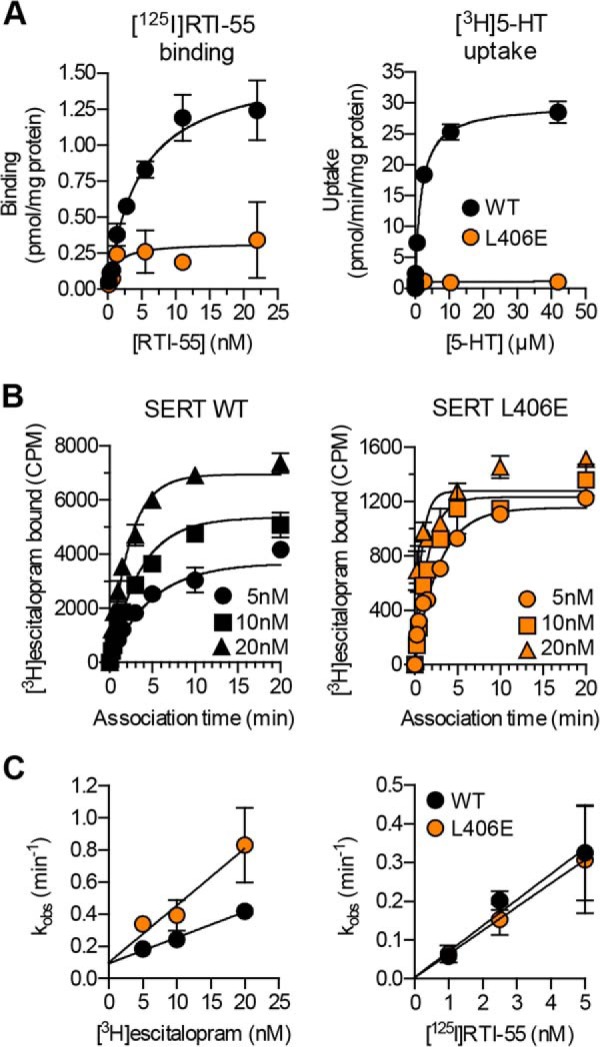
The L406E mutation affect the basal transporter function and inhibitor binding kinetics. A, dose-response curves from representative whole cell [125I]RTI-55 saturation binding (left) and [3H]5-HT saturation uptake (right) experiments, which were carried out for SERT WT and SERT L406E using transiently transfected COS7 cells (see “Experimental Procedures”). Data points represent the mean ± S.E. from triplicate determinations of accumulated radioactivity in cells incubated with increasing concentrations of RTI-55 or 5-HT. B, representative curves from a [3H]escitalopram kinetic binding experiment. Membrane preparations of COS7 cells expressing SERT WT (left) or SERT L406E (right) were incubated with the indicated concentrations (5, 10, or 20 nm) of [3H]escitalopram and the binding reaction was allowed to proceed for up to 20 min before the binding reaction was terminated by rapid washing. Data points represent the mean ± S.E. from triplicate determinations of accumulated radioactivity (CPM). Similar experiments were also carried out for [125I]RTI-55, using 1, 2.5, and 5 nm, respectively, of the radioligand (curves not shown). C, the pseudo-first order rate constant (kobs) calculated from the experiments outlined in B was plotted as a function of radioligand concentration. Data points represent the mean ± S.E. from at least three independent experiments each performed in triplicate.
L406E Increases Association Rate of Inhibitor Binding
The decreased transport function of L406E, along with the observation that the mutant induced significant gain-of-potency for inhibitors, led us to speculate that the Leu to Glu mutation might accumulate the transporter in a specific conformation that promotes inhibitor binding. To test this, we determined the kinetics of [3H]escitalopram and [125I]RTI-55 binding to SERT WT and the L406E mutant at different radioligand concentrations, which allowed us to determine the association rate (kon) and the dissociation rate (koff) (Fig. 6B). For escitalopram, L406E induced a significant (p = 0.003; two-tailed Student's t test) 2.4-fold increase in kon compared with WT SERT (16 ± 0.6 pm−1 min−1 for WT and 39 ± 6 pm−1 min−1 for L406E; values represent mean ± S.E. from five and three independent experiments, respectively, each performed in triplicate), whereas koff was unaffected (p = 0.19; two-tailed Student's t test) by the L406E mutation (0.10 ± 0.01 min−1 for WT and 0.07 ± 0.01 min−1 for L406E; values represent mean ± S.E. from five and three independent experiments, respectively, each performed in triplicate). This demonstrated that L406E indeed promotes inhibitor binding by allowing a faster association rate of escitalopram binding. In contrast, as shown on a plot of the data with kobs as a function of ligand concentration, from which kon can be derived from the slope and the koff from the intercept at the y axis, the binding kinetics of RTI-55 were not significantly affected by the L406E mutation (Fig. 6C). This is consistent with the small effect on the binding affinity of RTI-55 induced by L406E (Fig. 2). Importantly, the effects on escitalopram and RTI-55 binding induced by L406E as estimated from the kinetic binding experiments (koff/kon) were in good agreement with the effects induced by the mutation as determined in the competition binding assay (1.9- versus 2.9-fold increase in affinity for escitalopram; 1.7- versus 1.1-fold increase in affinity for RTI-55), verifying that the changes in inhibitor binding affinity at L406E were caused by a change in ligand binding kinetics. Generally, these findings indicate that L406E does not affect the dissociation rate of inhibitor binding, but for those inhibitors that are affected by the L406E mutation, the gain-of-potency is caused by an increased association rate.
L406E Renders the Cytoplasmic Permeation Pathway Less Accessible
The reduced turnover number of L406E compared with WT SERT (Fig. 6) could indicate that the mutation disrupted the conformational equilibrium of SERT by accumulating the transporter in a specific conformation. To gain further insight into which conformational state is favored by the mutation, we tested how L406E affected the accessibility of S277C. This residue is located in the cytoplasmic permeation pathway, and has previously been shown to be more accessible for MTSEA when the transporter is in an inward-facing conformation (40). The S277C and L406E mutations were introduced in the X5C background construct (C15A/C21A/C109A/C357I/C622A), which is lacking all endogenous Cys residues known to react with MTS reagents (37). In the experiments shown in Fig. 7, membranes from cells expressing SERT X5C-S277C or SERT X5C-S277C/L406E were incubated with MTSEA for 10 min, washed, and then assayed for residual binding of [125I]RTI-55. The IC50 value for MTSEA was 31-fold higher for X5C-S277C/L406E compared with X5C-S277C (mean value determined from three independent experiments where the two constructs were assayed in parallel). The significantly (p = 0.0246; two-tailed Student's t test) reduced MTSEA sensitivity strongly indicated that L406E renders the cytoplasmic pathway less accessible, implying that the mutation favors accumulation of SERT in an occluded- or outward-facing conformation.
FIGURE 7.
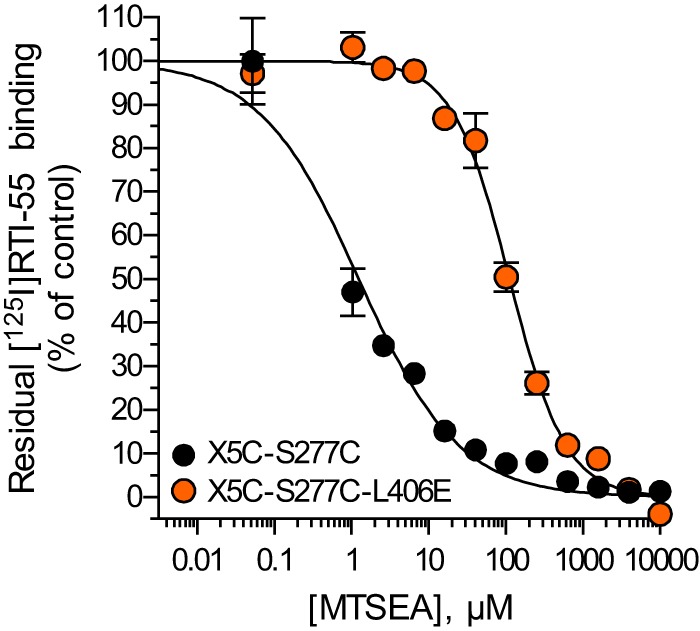
L406E confers a more outward-facing conformation of human SERT. Substituted cysteine accessibility method was used to probe the conformational change induced by the L406E mutant by measuring MTSEA inactivation of S277C in membrane preparations from HEK293-MSR cells expressing SERT X5C-S277C (black circles) or SERT X5C-S277C/L406E (orange circles). Binding of [125I]RTI-55 was used to determine residual unreacted transporter levels. Shown is the dose-response curve from a representative experiment that was repeated three times. Data points represent the mean ± S.E. of accumulated radioactivity normalized to percent of control (membrane preparations assayed in the absence of MTSEA reagent).
Examining Putative Intracellular Binding Partners to L406E
Based on structural and biophysical studies of bacterial transporters, movement of the EL4 region has been proposed to comprise a key role for the coordinated conformational changes associated with substrate translocation (32–34). We therefore examined the possibility that the negatively charged Glu residue in the L406E mutant could interact with another SERT residue in the vicinity, and thereby restrict the conformational movement of EL4, which in turn could accumulate the transporter in an occluded- or outward-facing conformation. We hypothesized that the interaction could be an attractive interaction to a positively charged residue (His, Arg, or Lys) or a repulsive interaction with a negatively charged residue (Asp or Glu). To examine this, we used a previously generated homology model of SERT (9) to guide site-directed mutagenesis of possible interaction partners of L406E. Because Leu-406 is located in the extracellular vestibule, surrounded by mobile TM and EL regions that likely adopt different orientations during the transport cycle, the L406E mutant may interact with residues that are not located in the immediate vicinity of Leu-406 in our SERT model. Therefore, we searched for His, Arg, Lys, Asp, or Glu residues within a distance of 15 Å from Leu-406, and based on the position and orientation relative to Leu-406, we selected 14 positively charged and 5 negatively charged residues for mutagenesis (Fig. 8). The 19 residues were individually mutated to Ala in the background of SERT L406E. If the mutated residue was interacting with the L406E mutant, we expected that the gain-of-inhibitory potency seen for the L406E mutant alone would be abolished by the Ala mutation. Initially, we determined the functional activity of the 19 double mutants in a [3H]5-HT uptake assay. Eleven of the mutants retained uptake activity (4–20% compared with WT), whereas eight mutants had no measurable uptake activity (Table 2). For the functionally active mutants, we determined the inhibitory potency of nisoxetine and talopram, which were largely affected by the L406E mutant (41- and 19-fold gain-of-potency, respectively). However, none of the Ala mutants significantly affected the gain-of-potency observed at L406E alone (one-way ANOVA with a post hoc Dunnett multiple comparison test) (Fig. 8A). To examine if a putative interaction partner to L406E could be found among the non-functional Ala mutants, we determined their ability to bind [125I]RTI-55 in membrane preparations from transfected COS7 cells (Fig. 8B). Three of the eight non-functional double mutants (L406E/E230A, L406E/H240A, and L406E/E493A) could not bind [125I]RTI-55, indicating that these mutations either inhibit protein folding and/or surface expression or severely decrease the ability of SERT to transport 5-HT and bind RTI-55. The binding affinity of talopram was determined at the five double mutants that could bind [125I]RTI-55 in a competition binding assay. However, none of the Ala mutants significantly affected the gain-of-affinity observed at L406E alone (one-way ANOVA with a post hoc Dunnett multiple comparison test) (Fig. 8B). Because the L406E-induced effects were not significantly affected by any of the Ala mutants, we conclude that a putative interaction partner of L406E was not among the 16 residues that we investigated (Fig. 8).
FIGURE 8.

Examining putative intracellular interaction partners to the L406E mutant. A, the inhibitory potency of nisxoetine (upper) and talopram (lower) was determined in a functional [3H]5-HT uptake inhibition assay (see “Experimental Procedures”) at SERT WT (black bars), SERT L406E (orange bars), and at 19 different mutations where a charged residue was mutated to Ala in the background of L406E (gray bars). Data represent the mean ± S.E. from at least three independent experiments each performed in triplicate. The stippled line indicates inhibitor Ki at L406E. Eight of the double mutants rendered the transporter non-functional (N.F.). Asterisks denote that the mutation induce a significant change in inhibitory potency compared with SERT WT (p < 0.05; one-way ANOVA followed by Dunnett's post hoc test). None of the Ala mutations in the L406E background induce a significant change in inhibitory potency compared with L406E alone (p > 0.05; one-way ANOVA followed by Dunnett's post hoc test). B, the binding affinity of talopram was determined in a competitive [125I]RTI-55 binding assay (see “Experimental Procedures”) at SERT WT (black bars), SERT L406E (orange bars), and at 14 different mutations where a charged residue was mutated to Ala in the background of L406E (gray bars). Data represent the mean ± S.E. from at least three independent experiments each performed in duplicate. The stippled line indicates the talopram binding affinity at L406E. Three of the double mutants showed no binding (N.B.) activity. Asterisks denote that the mutation induce a significant change in binding affinity compared with SERT WT (p < 0.05; one-way ANOVA followed by Dunnett's post hoc test). None of the Ala mutations in the L406E background induce a significant change in binding affinity compared with L406E alone (p > 0.05; one-way ANOVA followed by Dunnett's post hoc test). C, location of the Cα atoms (shown as yellow spheres) of the mutated charged residues (outlined in A) mapped onto a homology model of SERT (9). For clarity, only the extracellular part of the transporter is depicted, and Leu-406 is shown as orange spheres.
TABLE 2.
Impact of SERT mutations on [3H]5-HT uptake kinetics and [125I]RTI-55 binding
Relative transport activity of mutants was determined from paired uptake experiments and is expressed as percentage of mutant to WT from a 5-min uptake with 50 nm [3H]5-HT. All values are mean ± S.E. from at least 3 independent experiments.
| Mutant | [3H]5-HT uptake assay |
[125I]RTI-55 binding assay |
||
|---|---|---|---|---|
| Km | Transport activity | KD | Bmax | |
| nm | % of WT | nm | % of WT | |
| SERT WT | 1316 ± 342 | 0.63 ± 0.06 | (100 ± 29) | |
| L406E | 300 ± 100a | 13 ± 0.8a | 0.38 ± 0.11 | 23 ± 1.8a |
| L406E/W103A | NFb | 0.46 ± 0.09 | 5.5 ± 1.0a,c | |
| L406E/R104A | NF | 3.1 ± 1.0a | 5.2 ± 3.1a,c | |
| L406E/K201A | 341 ± 32a | 9.2 ± 1.9a | ND | |
| L406E/D216A | 519 ± 36a,c | 20 ± 3.5a | ND | |
| L406E/H223A | 281 ± 37a | 5.2 ± 0.9a | 0.52 ± 0.10 | 12 ± 2.1a,c |
| L406E/E229A | NF | 0.37 ± 0.06a | 1.7 ± 0.1a,c | |
| L406E/E230A | NF | NB | ||
| L406E/R234A | 333 ± 36a | 8.2 ± 2.3a | 1.0 ± 0.3 | 5.8 ± 2.1a, |
| L406E/H235A | 338 ± 57a | 16 ± 2.5a | 1.6 ± 0.4a,c | 25 ± 10a |
| L406E/H240A | NF | NB | ||
| L406E/R241A | 227 ± 24a | 3.4 ± 0.6a | ND | |
| L406E/K243A | 342 ± 46a | 18 ± 3.3a | ND | |
| L406E/R298A | NF | 4.1 ± 0.6a,c | 2.1 ± 1.6a,c | |
| L406E/R307A | NF | 1.1 ± 0.3 | 4.1 ± 1.4a,c | |
| L406E/K314A | 316 ± 26a | 5.0 ± 0.9a | 1.1 ± 0.3a | 17 ± 8.1a |
| L406E/K319A | 271 ± 13a | 14 ± 1.5a | 1.3 ± 0.5 | 29 ± 14a |
| L406E/R390A | NF | 1.6 ± 0.4a,c | 7.2 ± 3.2a,c | |
| L406E/K399A | 537 ± 54a | 3.8 ± 0.4a | 0.97 ± 0.46 | 21 ± 11a |
| L406E/E493A | NF | NB | ||
| L406E/E494A | 697 ± 127 | 15 ± 2.7a | ND | |
a Indicates significant difference compared to WT (p < 0.05, Student's t test).
b NF, nonfunctional; NB, no binding; ND, not determined.
c Indicates significant difference compared to L406E (p < 0.05, Student's t test).
Recent studies on bacterial transporters have suggested that interactions between a hydrophobic residue on TM1 (equivalent to Trp103 in human SERT) and a Phe residue on EL4 (equivalent to Phe407 in human SERT) is important for the transition from outward- to inward-facing conformation (35, 41). To test if the L406E-induced effects were caused by disruption of the proposed TM1-EL4 interaction, we substituted Trp-103 for Ala in the background of L406E in SERT. This double mutant was devoid of any uptake activity, but could bind [125I]RTI-55 (Table 2). Determination of the binding affinity of talopram at SERT W103A/L406E showed that the W103A mutation did not significantly affect the increased talopram affinity seen at L406E alone (63.7 ± 7.4 nm at L406E versus 60.4 ± 5.5 nm at W103A/L406E), thus suggesting that Trp-103 is not involved in the L406E-induced effects.
Inhibitors That Are Sensitive to L406E Decrease Accessibility of Q332C
Inhibitors of SERT block the functional activity by binding to the transporter and stabilize a specific conformational state, thereby obstructing substrate translocation. Cocaine and ibogaine have been shown to stabilize outward- and inward-facing SERT conformations, respectively, as demonstrated by their ability to increase or decrease accessibility of positions in the extracellular or cytoplasmic permeation pathways (12, 40, 42–44). However, much less is known about the conformational state of SERT that is stabilized by most other SERT inhibitors. We have shown that the L406E mutant affected the conformational equilibrium of SERT (Fig. 7), and speculated that there could be a correlation between the conformational state stabilized by an inhibitor and the gain in inhibitory potency induced by L406E. We therefore selected four inhibitors, atomoxetine, escitalopram, nisoxetine, and talopram, that were highly sensitive to the L406E mutation (>18-fold gain-of-potency) in addition to ibogaine and cocaine, which were less sensitive to the mutation (<6-fold gain-of-potency), and determined the ability of the inhibitors to modify the reactivity of Q332C in the extracellular permeation pathway. The Q332C mutant (generated in the C109A background that is insensitive to externally applied MTS reagents (45)) has previously been shown to be accessible to MTSET only when the transporter resides in an outward-facing conformation (44). COS7 cells expressing the C109A/Q332C mutant were preincubated with MTSET in the absence or presence of increasing inhibitor concentrations. In the absence of inhibitor, a concentration of 4 mm MTSET reduced the activity of C109A/Q332C by ∼50% (data not shown), and this MTSET concentration was therefore used in these studies. The residual functionality after preincubation with MTSET and inhibitor was normalized to the residual activity of cells preincubated with MTSET alone and plotted against increasing concentrations of the inhibitors (Fig. 9). A decrease in functionality indicates that the inhibitor increases the accessibility of Q332C, whereas an increase in functionality indicates that Q332C becomes less accessible when the inhibitor is present. Cocaine has been shown to stabilize an outward-facing conformation of SERT, and thus increases accessibility of residues in the extracellular permeation pathway. Accordingly, and consistent with previous findings (44), cocaine increased the accessibility of Q332C (Fig. 9). In contrast, ibogaine decreased the accessibility of Q332C, which is consistent with the idea that ibogaine stabilizes an inward-facing conformation of SERT in which access to the extracelllular permeation pathway is restricted (12). Interestingly, the four compounds that were highly sensitive to L406E (atomoxetine, escitalopram, nisoxetine and talopram), all decreased the accessibility of Q332C in a dose-dependent manner. This indicates that the compounds stabilize a conformation in which access to the extracellular pathway is hindered; i.e. an occluded- or inward-facing conformation. An alternative explanation could be that the inhibitors bind in proximity of Q332C and thereby sterically block the MTSET reaction. However, escitalopram, talopram, and atomoxetine have been shown to be competitive inhibitors of SERT (8, 23, 46), indicating that their binding site overlaps the central substrate binding site. Thus, rather than blocking access to Q332C, which is located outside the central S1 site (44), it seems more likely that the inhibitors decrease the reactivity of this residue by stabilizing an occluded- or inward-facing conformation.
FIGURE 9.
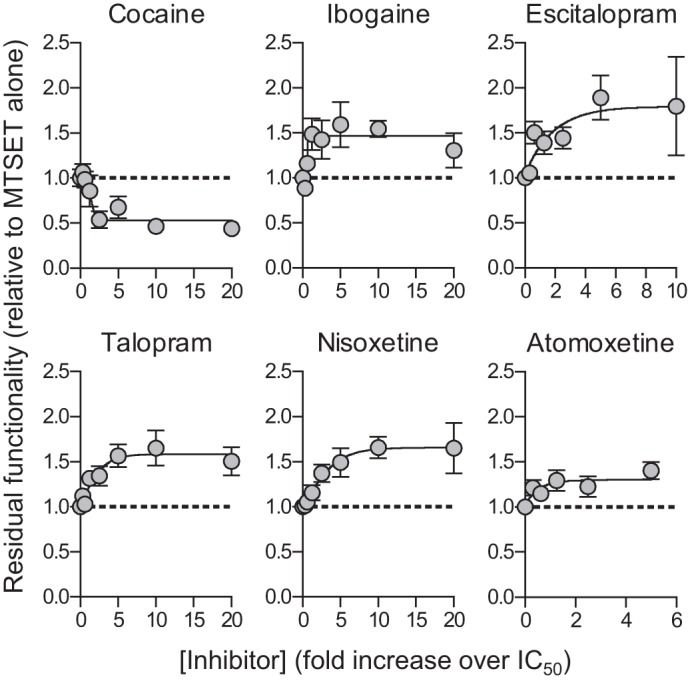
Inhibitors alter MTSET-induced inactivation of Q332C. COS7 cells expressing C109A/Q332C were incubated with MTSET (4 mm) and increasing concentrations of cocaine, ibogaine, escitalopram, talopram, nisoxetine, or atomoxetine for 10 min. After extensive washing, the residual functionality was determined by measuring uptake of [3H]5-HT (see “Experimental Procedures”). Shown are composite dose-response curves from at least three independent experiments each performed in triplicate. Data points represent the mean ± S.E. from 9 to 12 determinations of accumulated radioactivity in cells incubated with 50 nm [3H]5-HT. Uptake was normalized to cells incubated with MTSET alone.
Discussion
Neurotransmitter transporters are believed to mediate substrate translocation via an alternating access mechanism, implying that the transporter protein shuttles through at least three conformational states: 1) an outward-facing conformation, 2) an occluded conformation, and 3) an inward-facing conformation. The first x-ray crystal structure of LeuT revealed a pseudo symmetry, in which two structurally similar repeats (TMs 1–5 and TMs 6–10) are inverted with respect to the plane of the membrane (4). This structural symmetry has been used to generate experimentally validated structural models describing the alternating access mechanism of SERT (12, 47). Specifically, the transporter can be divided into two domains, a four-helix bundle (TMs 1, 2, 6, and 7) and a scaffold domain (TMs 3–5 and 8–10). Movement of the flexible four-helix bundle relative to the more static scaffold domain allow alternating access from the central substrate site to either the extracellular side or the cytoplasm, respectively (12). At present, we therefore have a good impression of how the TM domains are orientated relative to each other during the transport cycle. Much less is known about the role and importance of the extra- and intracellular loop regions in the transport cycle. Structural and biophysical studies of LeuT have suggested a critical role of EL4 in the conformational cycle of the bacterial transporter (32–34). However, the loop regions of SLC6 transporters are only poorly conserved and contain numerous insertions and deletions compared with LeuT (48). For the loop regions, it is therefore difficult to correlate findings directly from bacterial to human transporters, and further experiments are thus warranted to examine an equivalent important role of EL4 in SERT.
Here, we have identified a Leu to Glu mutation (L406E) close to the tip of EL4 in human SERT that disrupts the conformational equilibrium and accumulates the transporter in a more outward-facing conformation, which is also reflected in the 5-fold decrease in the turnover number for the mutant compared with the WT transporter (Figs. 6 and 7). This implies that EL4 is indeed involved in the transition between inward- and outward-facing conformations in SERT, which corroborates previous substituted cysteine accessibility method analysis of SERT and the GABA transporter GAT-1, showing that substrate translocation is associated with relative movement around the EL4 region (31, 49).
By probing the accessibility of S277C, we found that L406E rendered the cytoplasmic pathway less accessible (Fig. 7), but from these experiments we cannot determine whether the mutation favors accumulation of SERT in an occluded- or an outward-facing conformation. However, this may be inferred by the effect on inhibitor binding by L406E. We found that escitalopram reduced MTSET accessibility to Q332C in the extracellular pathway, indicating that the inhibitor stabilizes either an occluded or an inward-facing conformation (Fig. 9). Together with previous experiments, showing that citalopram inhibits copper(II)(1,10-phenanthroline)3-mediated cross-linking between Cys residues inserted in TMs 1 and 3, supposedly because the inhibitor restricts SERT in an occluded- or outward-facing conformation (50), this suggests that escitalopram stabilizes an occluded conformation of SERT. If L406E increases the binding affinity of escitalopram by stabilizing the inhibitor-bound occluded conformation, we would expect that the mutation reduced the dissociation rate of escitalopram binding. However, from our binding kinetics experiments we found that the dissociation rate for escitalopram was unaffected, whereas the association rate was significantly increased (Fig. 4). Together with the decreased accessibility of S277C induced by L406E, this is consistent with a model where the Leu to Glu mutation stabilizes an outward-open conformation of SERT.
Rather than affecting inhibitor binding allosterically by disrupting the conformational equilibrium of SERT, the marked increase in inhibitory potency could instead be mediated through direct interactions between the inhibitors and L406E. Specifically, all tested inhibitors have an amino group, and it is thus tempting to speculate that this positively charged moiety could interact with the negatively charged L406E mutant, and thereby capture the inhibitors within the extracellular vestibule. However, we have shown that introduction of S1 mutations in the background of L406E severely decrease the potency of inhibitors (up to 997-fold compared with L406E alone) (Fig. 4). Combined with the findings that inhibitor binding to the allosteric low affinity binding pocket in the S2 site is unaffected by the L406E mutation (Fig. 5), this strongly suggest that L406E allosterically affect high affinity inhibitor binding in the central S1 site.
Elucidation of the molecular details underlying accumulation of the L406E mutant in an outward-facing conformation could uncover novel information about the role of EL4 in the transport cycle. The x-ray crystal structure of LeuT captured in an inward-facing conformation show that EL4 occludes the extracellular pathway by tightly packing with TMs 1, 3, 7, and 8 in addition to EL2 (32). The L406E mutation could possibly impair interactions to these domains and thereby hinder SERT from entering the inward-facing conformation. A recent study of the bacterial LeuT-fold transporter, PutP, has suggested that interactions between residues on TM1 and EL4 (equivalent to Trp103 (TM1) and Phe407 (EL4), respectively, in human SERT) are important for transporter function (35). Furthermore, mutation of Trp33 in the bacterial SLC6 homologue MhsT (equivalent to Trp103 in human SERT) reduced functional uptake activity, supposedly by enhancing the transition to the inward-facing conformation (41). Hence, Trp103 on TM1 in SERT might have a critical role for the conformational equilibrium, possibly through interactions with Phe407 on EL4. Mutation of the adjacent Leu-406 could possibly disrupt the proposed Trp-103/Phe-407 interaction. Leu-406 is also located close to the GPSL motif (from Gly-402 to Leu-405) at the tip of EL4 in SERT, which has previously been proposed to form a dynamic hinge important for the conformational changes associated with substrate translocation (31). Mutation of the proximate Leu-406 could possibly affect the flexibility of the proposed EL4 hinge motif, which in turn could affect the conformational equilibrium of SERT. However, other mutations in position 406 would probably also affect the proposed Trp-103/Phe-407 interaction as well as the dynamic GPSL hinge motif, and these possibilities are therefore difficult to reconcile with the high specificity we found for L406E (Fig. 3).
We examined the possibility that L406E could stabilize an outward-facing conformation through interactions with negatively or positively charged residues localized in the vicinity of Leu-406. From x-ray crystal structures of LeuT and Drosophila DAT it is evident that EL2 is located on the extracellular side of EL4 (4, 26). In SERT, Leu-406 is located on the cytoplasm facing side of EL4 with the side chain pointing toward the core of the transporter (Fig. 8), and it may therefore not seem obvious that the L406E mutation affect interactions between EL2 and EL4. However, there are likely several conformational intermediates that are transitory in the transport cycle that are not portrayed in the currently available x-ray crystal structures, and the importance of the L406E mutation may reside in one of the presumed intermediate states. Furthermore, a recent study revealed a close proximity of EL2 and EL4 in GAT-1 when the transporter resided in an outward-facing conformation (51), suggesting an intimate relationship between these loop regions in SLC6 transporters. Thus, in our search for a putative interaction partner to L406E we included 10 EL2 residues (Lys-201, Asp-216, His-223, Glu-229, Glu-230, Arg-234, His-235, His-240, Arg-241, and Lys-243) that were not obvious candidates for directly interacting with L406E, but which may adopt a transient alternate conformation that could affect the functional role of EL4. In total, we mutated 14 positively and 5 negatively charged residues in the background of L406E. None of the tested mutations significantly changed the effects on inhibitor binding induced by L406E alone, suggesting that none of the charged residues contribute to the L406E-induced effects (Fig. 8). Mutation of Glu-230 (EL2), His-240 (EL2), or Glu-493 (TM10) to Ala in the background of L406E rendered the transporter devoid of any uptake or binding activity (Tables 1 and 2), and we can therefore not rule out that the L406E-induced effects are mediated through interactions with these residues.
The L406E mutation could also disrupt the conformational equilibrium of SERT by interacting with the protein backbone. Inspection of our SERT homology model (9) showed that the side chain of L406E is located close to backbone of Val-102 and Trp-103 on TM1 (Fig. 10). The backbone NH-groups are pointing away from L406E, but the backbone carbonyl group of Val-102 is pointing directly toward L406E and is located within 2 Å of the carboxylate group on the side chain of L406E (Fig. 10). The close proximity of the negatively charged carboxylate group of L406E and the partially negative backbone carbonyl oxygen could induce a repulsive effect between TM1 and EL4. Such an interaction would presumably have high spatiotemporal requirements, thus substantiating the high specificity of the L406E-induced effects compared with other mutations in the same site, including the closely related L406D mutation (Fig. 3). Repulsion between L406E and a backbone carbonyl on TM1 could restrict the flexibility of the 4-helix bundle (consisting of TMs 1, 2, 6, and 7), thereby hindering extracellular packing of the 4-helix bundle against the scaffold domain, leading to accumulation of SERT in outward-facing conformations. This model therefore seems to be in good agreement with the proposed movements of TM domains during the alternating access mechanism, and substantiates the idea that a downward movement of EL4 toward the central binding site is a key regulator for the conformational transition between outward- and inward-facing conformations. Together, our findings thus substantiate that EL4 has an important role in controlling the conformational equilibrium of SERT, and we believe that our findings will be important for a complete understanding of the molecular mechanism defining the transport cycle in human neurotransmitter transporters.
FIGURE 10.
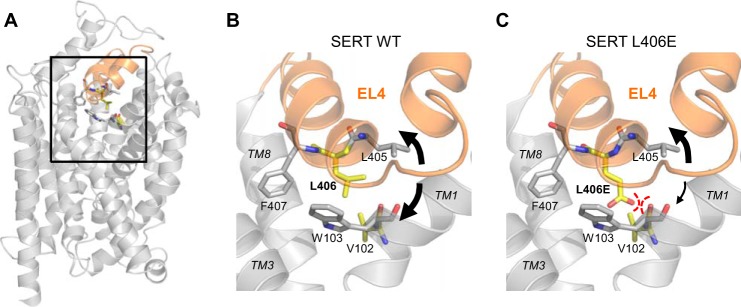
Putative interaction between L406E and a backbone carbonyl on TM1. A, location of EL4 (shown in orange) depicted on a homology model of SERT (9). B, close-up view of the EL4 (boxed area in A) and the surrounding TMs (TM 1, 3, and 8). Val-102 (TM1) and Leu-406 (EL4) are shown as yellow sticks, and Trp-103 (TM1) and Phe-407 (EL4) are shown as gray sticks. The presumed flexibility of EL4 is indicated by black arrows. C, the L406E mutation was constructed using PyMOL Molecular Graphics System, version 1.3 (Schrödinger, LLC) using its backbone-dependent rotamer library. The close proximity between the negatively charged carboxylate group on the side chain of L406E and the backbone carbonyl group of Val-102 could possibly induce a repulsive interaction that would restrict the flexibility of EL4 and TM1.
Acknowledgments
We thank Dr. Benny Bang-Andersen for providing compounds from the H. Lundbeck A/S compound collection, Sacrament of Transition (Maribor, Slovenia) for the kind gift ibogaine, and Dr. Claus J. Loland for the kind gift of [3H]escitalopram.
This work was supported by the Lundbeck Foundation.
- SERT
- serotonin transporter
- 5-HT
- serotonin
- DAT
- dopamine transporter
- EL
- extracellular loop
- LeuT
- amino acid transporter from Aquifex aeolicus
- MTSEA
- (2-aminoethyl)methanethiosulfonate
- MTSET
- (2-trimethylammonium)methanethiosulfonate
- NET
- norepinephrine transporter
- RTI-55
- (−)-2β-carbomethoxy-3β-(4-iodophneyl)tropane
- SLC6
- solute carrier 6
- TM
- transmembrane segments
- ANOVA
- analysis of variance.
References
- 1. Kristensen A. S., Andersen J., Jørgensen T. N., Sørensen L., Eriksen J., Loland C. J., Strømgaard K., Gether U. (2011) SLC6 neurotransmitter transporters: structure, function, and regulation. Pharmacol. Rev. 63, 585–640 [DOI] [PubMed] [Google Scholar]
- 2. Rudnick G. (1998) Bioenergetics of neurotransmitter transport. J. Bioenerg. Biomembr. 30, 173–185 [DOI] [PubMed] [Google Scholar]
- 3. Andersen J., Kristensen A. S., Bang-Andersen B., Strømgaard K. (2009) Recent advances in the understanding of the interaction of antidepressant drugs with serotonin and norepinephrine transporters. Chem. Commun. 3677–3692 [DOI] [PubMed] [Google Scholar]
- 4. Yamashita A., Singh S. K., Kawate T., Jin Y., Gouaux E. (2005) Crystal structure of a bacterial homologue of Na+/Cl−-dependent neurotransmitter transporters. Nature 437, 215–223 [DOI] [PubMed] [Google Scholar]
- 5. Beuming T., Kniazeff J., Bergmann M. L., Shi L., Gracia L., Raniszewska K., Newman A. H., Javitch J. A., Weinstein H., Gether U., Loland C. J. (2008) The binding sites for cocaine and dopamine in the dopamine transporter overlap. Nat. Neurosci. 11, 780–789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Celik L., Sinning S., Severinsen K., Hansen C. G., Møller M. S., Bols M., Wiborg O., Schiøtt B. (2008) Binding of serotonin to the human serotonin transporter. Molecular modeling and experimental validation. J. Am. Chem. Soc. 130, 3853–3865 [DOI] [PubMed] [Google Scholar]
- 7. Andersen J., Olsen L., Hansen K. B., Taboureau O., Jørgensen F. S., Jørgensen A. M., Bang-Andersen B., Egebjerg J., Strømgaard K., Kristensen A. S. (2010) Mutational mapping and modeling of the binding site for (S)-citalopram in the human serotonin transporter. J. Biol. Chem. 285, 2051–2063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Andersen J., Stuhr-Hansen N., Zachariassen L., Toubro S., Hansen S. M., Eildal J. N., Bond A. D., Bøgesø K. P., Bang-Andersen B., Kristensen A. S., Strømgaard K. (2011) Molecular determinants for selective recognition of antidepressants in the human serotonin and norepinephrine transporters. Proc. Natl. Acad. Sci. U.S.A. 108, 12137–12142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Andersen J., Stuhr-Hansen N., Zachariassen L. G., Koldsø H., Schiøtt B., Strømgaard K., Kristensen A. S. (2014) Molecular basis for selective serotonin reuptake inhibition by the antidepressant agent fluoxetine (Prozac). Mol. Pharmacol. 85, 703–714 [DOI] [PubMed] [Google Scholar]
- 10. Severinsen K., Koldsø H., Thorup K. A., Schjøth-Eskesen C., Møller P. T., Wiborg O., Jensen H. H., Sinning S., Schiøtt B. (2014) Binding of mazindol and analogs to the human serotonin and dopamine transporters. Mol. Pharmacol. 85, 208–217 [DOI] [PubMed] [Google Scholar]
- 11. Sinning S., Musgaard M., Jensen M., Severinsen K., Celik L., Koldsø H., Meyer T., Bols M., Jensen H. H., Schiøtt B., Wiborg O. (2010) Binding and orientation of tricyclic antidepressants within the central substrate site of the human serotonin transporter. J. Biol. Chem. 285, 8363–8374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Forrest L. R., Zhang Y. W., Jacobs M. T., Gesmonde J., Xie L., Honig B. H., Rudnick G. (2008) Mechanism for alternating access in neurotransmitter transporters. Proc. Natl. Acad. Sci. U.S.A. 105, 10338–10343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Singh S. K., Yamashita A., Gouaux E. (2007) Antidepressant binding site in a bacterial homologue of neurotransmitter transporters. Nature 448, 952–956 [DOI] [PubMed] [Google Scholar]
- 14. Singh S. K., Piscitelli C. L., Yamashita A., Gouaux E. (2008) A competitive inhibitor traps LeuT in an open-to-out conformation. Science 322, 1655–1661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhou Z., Zhen J., Karpowich N. K., Goetz R. M., Law C. J., Reith M. E., Wang D. N. (2007) LeuT-desipramine structure reveals how antidepressants block neurotransmitter reuptake. Science 317, 1390–1393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhou Z., Zhen J., Karpowich N. K., Law C. J., Reith M. E., Wang D. N. (2009) Antidepressant specificity of serotonin transporter suggested by three LeuT-SSRI structures. Nat. Struct. Mol. Biol. 16, 652–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Barker E. L., Perlman M. A., Adkins E. M., Houlihan W. J., Pristupa Z. B., Niznik H. B., Blakely R. D. (1998) High affinity recognition of serotonin transporter antagonists defined by species-scanning mutagenesis: an aromatic residue in transmembrane domain I dictates species-selective recognition of citalopram and mazindol. J. Biol. Chem. 273, 19459–19468 [DOI] [PubMed] [Google Scholar]
- 18. Barker E. L., Moore K. R., Rakhshan F., Blakely R. D. (1999) Transmembrane domain I contributes to the permeation pathway for serotonin and ions in the serotonin transporter. J. Neurosci. 19, 4705–4717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Henry L. K., Field J. R., Adkins E. M., Parnas M. L., Vaughan R. A., Zou M. F., Newman A. H., Blakely R. D. (2006) Tyr-95 and Ile-172 in transmembrane segments 1 and 3 of human serotonin transporters interact to establish high affinity recognition of antidepressants. J. Biol. Chem. 281, 2012–2023 [DOI] [PubMed] [Google Scholar]
- 20. Andersen J., Taboureau O., Hansen K. B., Olsen L., Egebjerg J., Strømgaard K., Kristensen A. S. (2009) Location of the antidepressant binding site in the serotonin transporter: importance of Ser-438 in recognition of citalopram and tricyclic antidepressants. J. Biol. Chem. 284, 10276–10284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sørensen L., Andersen J., Thomsen M., Hansen S. M., Zhao X., Sandelin A., Strømgaard K., Kristensen A. S. (2012) Interaction of antidepressants with the serotonin and norepinephrine transporters: mutational studies of the S1 substrate binding pocket. J. Biol. Chem. 287, 43694–43707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Koldsø H., Severinsen K., Tran T. T., Celik L., Jensen H. H., Wiborg O., Schiøtt B., Sinning S. (2010) The two enantiomers of citalopram bind to the human serotonin transporter in reversed orientations. J. Am. Chem. Soc. 132, 1311–1322 [DOI] [PubMed] [Google Scholar]
- 23. Apparsundaram S., Stockdale D. J., Henningsen R. A., Milla M. E., Martin R. S. (2008) Antidepressants targeting the serotonin reuptake transporter act via a competitive mechanism. J. Pharmacol. Exp. Ther. 327, 982–990 [DOI] [PubMed] [Google Scholar]
- 24. Graham D., Esnaud H., Habert E., Langer S. Z. (1989) A common binding site for tricyclic and nontricyclic 5-hydroxytryptamine uptake inhibitors at the substrate recognition site of the neuronal sodium-dependent 5-hydroxytryptamine transporter. Biochem. Pharmacol. 38, 3819–3826 [DOI] [PubMed] [Google Scholar]
- 25. Koe B. K., Lebel L. A., Welch W. M. (1990) [3H]Sertraline binding to rat brain membranes. Psychopharmacology 100, 470–476 [DOI] [PubMed] [Google Scholar]
- 26. Penmatsa A., Wang K. H., Gouaux E. (2013) X-ray structure of dopamine transporter elucidates antidepressant mechanism. Nature 503, 85–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang H., Goehring A., Wang K. H., Penmatsa A., Ressler R., Gouaux E. (2013) Structural basis for action by diverse antidepressants on biogenic amine transporters. Nature 503, 141–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Plenge P., Shi L., Beuming T., Te J., Newman A. H., Weinstein H., Gether U., Loland C. J. (2012) Steric hindrance mutagenesis in the conserved extracellular vestibule impedes allosteric binding of antidepressants to the serotonin transporter. J. Biol. Chem. 287, 39316–39326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Smicun Y., Campbell S. D., Chen M. A., Gu H., Rudnick G. (1999) The role of external loop regions in serotonin transport: loop scanning mutagenesis of the serotonin transporter external domain. J. Biol. Chem. 274, 36058–36064 [DOI] [PubMed] [Google Scholar]
- 30. Stephan M. M., Chen M. A., Penado K. M., Rudnick G. (1997) An extracellular loop region of the serotonin transporter may be involved in the translocation mechanism. Biochemistry 36, 1322–1328 [DOI] [PubMed] [Google Scholar]
- 31. Mitchell S. M., Lee E., Garcia M. L., Stephan M. M. (2004) Structure and function of extracellular loop 4 of the serotonin transporter as revealed by cysteine-scanning mutagenesis. J. Biol. Chem. 279, 24089–24099 [DOI] [PubMed] [Google Scholar]
- 32. Krishnamurthy H., Gouaux E. (2012) X-ray structures of LeuT in substrate-free outward-open and apo inward-open states. Nature 481, 469–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Claxton D. P., Quick M., Shi L., de Carvalho F. D., Weinstein H., Javitch J. A., McHaourab H. S. (2010) Ion/substrate-dependent conformational dynamics of a bacterial homolog of neurotransmitter:sodium symporters. Nat. Struct. Mol. Biol. 17, 822–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kazmier K., Sharma S., Quick M., Islam S. M., Roux B., Weinstein H., Javitch J. A., McHaourab H. S. (2014) Conformational dynamics of ligand-dependent alternating access in LeuT. Nat. Struct. Mol. Biol. 21, 472–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Raba M., Dunkel S., Hilger D., Lipiszko K., Polyhach Y., Jeschke G., Bracher S., Klare J. P., Quick M., Jung H., Steinhoff H.-J. (2014) Extracellular loop 4 of the proline transporter PutP controls the periplasmic entrance to ligand binding sites. Structure 22, 769–780 [DOI] [PubMed] [Google Scholar]
- 36. Zhu C. B., Hewlett W. A., Feoktistov I., Biaggioni I., Blakely R. D. (2004) Adenosine receptor, protein kinase G, and p38 mitogen-activated protein kinase-dependent up-regulation of serotonin transporters involves both transporter trafficking and activation. Mol. Pharmacol. 65, 1462–1474 [DOI] [PubMed] [Google Scholar]
- 37. Sato Y., Zhang Y. W., Androutsellis-Theotokis A., Rudnick G. (2004) Analysis of transmembrane domain 2 of rat serotonin transporter by cysteine scanning mutagenesis. J. Biol. Chem. 279, 22926–22933 [DOI] [PubMed] [Google Scholar]
- 38. Chen F., Larsen M. B., Sánchez C., Wiborg O. (2005) The S-enantiomer of R,S-citalopram, increases inhibitor binding to the human serotonin transporter by an allosteric mechanism: comparison with other serotonin transporter inhibitors. Eur. Neuropsychopharmacol. 15, 193–198 [DOI] [PubMed] [Google Scholar]
- 39. Neubauer H. A., Hansen C. G., Wiborg O. (2006) Dissection of an allosteric mechanism on the serotonin transporter: a cross-species study. Mol. Pharmacol. 69, 1242–1250 [DOI] [PubMed] [Google Scholar]
- 40. Zhang Y. W., Rudnick G. (2006) The cytoplasmic substrate permeation pathway of serotonin transporter. J. Biol. Chem. 281, 36213–36220 [DOI] [PubMed] [Google Scholar]
- 41. Malinauskaite L., Quick M., Reinhard L., Lyons J. A., Yano H., Javitch J. A., Nissen P. (2014) A mechanism for intracellular release of Na by neurotransmitter/sodium symporters. Nat. Struct. Mol. Biol. 21, 1006–1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jacobs M. T., Zhang Y. W., Campbell S. D., Rudnick G. (2007) Ibogaine, a noncompetitive inhibitor of serotonin transport, acts by stabilizing the cytoplasm-facing state of the transporter. J. Biol. Chem. 282, 29441–29447 [DOI] [PubMed] [Google Scholar]
- 43. Henry L. K., Adkins E. M., Han Q., Blakely R. D. (2003) Serotonin and cocaine-sensitive inactivation of human serotonin transporters by methanethiosulfonates targeted to transmembrane domain I. J. Biol. Chem. 278, 37052–37063 [DOI] [PubMed] [Google Scholar]
- 44. Torres-Altoro M. I., Kuntz C. P., Nichols D. E., Barker E. L. (2010) Structural analysis of the extracellular entrance to the serotonin transporter permeation pathway. J. Biol. Chem. 285, 15369–15379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chen J. G., Liu-Chen S., Rudnick G. (1997) External cysteine residues in the serotonin transporter. Biochemistry 36, 1479–1486 [DOI] [PubMed] [Google Scholar]
- 46. Tsuruda P. R., Yung J., Martin W. J., Chang R., Mai N., Smith J. A. (2010) Influence of ligand binding kinetics on functional inhibition of human recombinant serotonin and norepinephrine transporters. J. Pharmacol. Toxicol. Methods 61, 192–204 [DOI] [PubMed] [Google Scholar]
- 47. Forrest L. R., Rudnick G. (2009) The rocking bundle: a mechanism for ion-coupled solute flux by symmetrical transporters. Physiology 24, 377–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Beuming T., Shi L., Javitch J. A., Weinstein H. (2006) A comprehensive structure-based alignment of prokaryotic and eukaryotic neurotransmitter/Na+ symporters (NSS) aids in the use of the LeuT structure to probe NSS structure and function. Mol. Pharmacol. 70, 1630–1642 [DOI] [PubMed] [Google Scholar]
- 49. Zomot E., Kanner B. I. (2003) The interaction of the γ-aminobutyric acid transporter GAT-1 with the neurotransmitter is selectively impaired by sulfhydryl modification of a conformationally sensitive cysteine residue engineered into extracellular loop IV. J. Biol. Chem. 278, 42950–42958 [DOI] [PubMed] [Google Scholar]
- 50. Tao Z., Zhang Y. W., Agyiri A., Rudnick G. (2009) Ligand effects on cross-linking support a conformational mechanism for serotonin transport. J. Biol. Chem. 284, 33807–33814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hilwi M., Dayan O., Kanner B. I. (2014) Conformationally sensitive proximity of extracellular loops 2 and 4 of the γ-aminobutyric acid (GABA) transporter GAT-1 inferred from paired cysteine mutagenesis. J. Biol. Chem. 289, 34258–34266 [DOI] [PMC free article] [PubMed] [Google Scholar]