

Background: V(D)J recombination, initiated by the RAG1/2 protein complex, requires two types of DNA recombination signal sequences (RSS), 12RSS and 23RSS.
Results: Improved RAG1/2 expression and purification allowed us to systematically assemble protein-DNA complexes.
Conclusion: RAG1/2 binds one copy each of 12RSS and 23RSS DNA.
Significance: Strictly enforced pairing of the 12RSS and 23RSS explains the recombination specificity.
Keywords: DNA endonuclease, DNA enzyme, immunodeficiency, mammal, protein complex, protein crystallization, V(D)J recombination, mammalian expression, metal ions, mammalian expression, DNA cleavage
Abstract
Mammalian immune receptor diversity is established via a unique restricted set of site-specific DNA rearrangements in lymphoid cells, known as V(D)J recombination. The lymphoid-specific RAG1-RAG2 protein complex (RAG1/2) initiates this process by binding to two types of recombination signal sequences (RSS), 12RSS and 23RSS, and cleaving at the boundaries of RSS and V, D, or J gene segments, which are to be assembled into immunoglobulins and T-cell receptors. Here we dissect the ordered assembly of the RAG1/2 heterotetramer with 12RSS and 23RSS DNAs. We find that RAG1/2 binds only a single 12RSS or 23RSS and reserves the second DNA-binding site specifically for the complementary RSS, to form a paired complex that reflects the known 12/23 rule of V(D)J recombination. The assembled RAG1/2 paired complex is active in the presence of Mg2+, the physiologically relevant metal ion, in nicking and double-strand cleavage of both RSS DNAs to produce a signal-end complex. We report here the purification and initial crystallization of the RAG1/2 signal-end complex for atomic-resolution structure elucidation. Strict pairing of the 12RSS and 23RSS at the binding step, together with information from the crystal structure of RAG1/2, leads to a molecular explanation of the 12/23 rule.
Introduction
During lymphocyte development, an enormous variety of immunoglobulin and T cell receptor genes can be assembled through a series of carefully orchestrated DNA breakage and rejoining events by the process called V(D)J recombination (1). V(D)J rearrangements specifically occur between gene segments flanked by recombination signal sequences (RSSs).3 An RSS typically contains conserved heptamer (5′-CACAGTG) and nonamer (5′-ACAAAAACC) elements, separated by a non-conserved spacer of 12 (12RSS) or 23 (23RSS) nucleotides (2). Recombination follows the so-called 12/23 rule, by which one gene segment has to be flanked by a 12RSS and the other has to be flanked by a 23RSS (3).
V(D)J recombination is initiated when the products of the recombination activating genes 1 and 2, the RAG1 and RAG2 proteins, bind a pair of 12RSS and 23RSS DNAs and cooperate in cleavage of DNA at the boundaries between RSSs and their neighboring gene segments (or coding flanks) (4–6). Both RAG proteins have been trimmed to smaller, more tractable species. “Core” regions of RAG1 (384–1008 out of 1040 aa) and RAG2 (1–387 out of 527 aa) are active in V(D)J recombination on episomal or integrated substrates in cell culture (7–10), and are most frequently used in biochemical characterization. Comprehensive mutational analyses have found that the catalytic center of the RAG1/2 recombinase resides in the RAG1 subunit and contains a typical DDE triad of carboxylates, in this case Asp-600, Asp-708, and Glu-962 (11–13).
Biochemical studies have shown that the DNA cleavage at an RSS-coding gene boundary occurs in two steps. The RAG1/2 complex, with the assistance of an HMGB1 protein (14), specifically interacts with a 12/23 pair of RSSs to form a paired complex (PC) and introduces site-specific nicks precisely at the boundaries between the RSS heptamer and coding flank, leaving 3′-hydroxyl groups on each coding end (4). These 3′-OH groups then attack the phosphodiester bond of the opposite strand and make double-strand breaks, with hairpin loops on the coding ends and blunt 5′-phosphorylated signal ends (4). The hairpin ends are later opened, processed, and joined together by the non-homologous end-joining machinery to form a coding joint (15, 16). Cleaved signal ends remain associated with RAG1/2 in a stable signal-end complex (SEC) (17, 18) and later are also joined by non-homologous end-joining enzymes, in most cases being lost from the cells in the form of excised circles.
Although V(D)J recombination in vivo exhibits a strong (>30-fold) preference for a 12/23 RSS pair over 12/12 and 23/23 pairs, a low level of recombination between identical sites has been demonstrated (19). The 12/23 rule can also be captured in in vitro reactions. With Mg2+ as cofactor, formation of double-strand breaks is strongly favored if both 12RSS and 23RSS are bound, whereas nicking of individual 12RSS or 23RSS is observed (20). Under less stringent conditions in Mn2+, purified RAG1/2 can generate a double-strand break on either a 12RSS or a 23RSS alone. These observations have left it unclear whether the PC can form with two RSS DNAs of the same kind (21).
The assembly pathway leading to the PC has also been unclear. Some previous work has supported an ordered assembly of functional RAG1/2-RSS complexes. A “capture” model described in several studies suggests that RAG1/2 preferentially assembles first on the 12 RSS in a single complex (12RSS-SC) and then captures the 23RSS to form a PC (22, 23). When RAG1/2 and HMGB1 were first incubated with the 12RSS, a very strong preference for acquiring and cleaving a 23RSS, versus a 12RSS partner, was observed. Although the reciprocal also appeared to be true, the adherence to the 12/23 rule was more strict if the 12RSS was the first one bound (23). A similar trend was seen on some chromatinized substrates, which is closer to the situation in cells (24). Further support of this model comes from detection of nicks of 12RSS in pre-B cells and in primary thymocytes, whereas nicks on 23RSS were not detected (22). However, an alternative model suggested that RAG1/2 binds and nicks each RSS type independently and then associates them into the final RAG1/2 complexes, with both 12 and 23 RSSs required for completing the cleavage reaction (25). In sum, it has not been clear whether RAG1/2 can simultaneously bind intact 12 and 23 RSSs to form the PC, or whether staged assembly preceded by DNA nicking is necessary to carry out the DNA cleavage that forms the SEC.
A central problem in the in vitro studies of V(D)J recombination has been to find an optimal source of active RAG proteins. Biochemical characterization has been successfully conducted using core RAG1/2 proteins expressed in insect cells (4, 12, 26, 28, 29). However, we found that only a small fraction of RAG1/2 in these preparations was active (30). To obtain homogeneous protein for structural studies, an activity-based purification was developed in our group. By immobilizing RSS DNAs on a chromatographic resin, the active subpopulation of RAG1/2 complexes in the form of SEC was isolated after cleaving the bound RSS. The SEC was produced by this activity-based purification in very low yield (∼10 μg from several liters of insect cell culture), but it was sufficient for electron microscopic analysis and was shown to contain two RAG1 and two RAG2 units, one HMGB1, and one each of cleaved 12RSS and 23RSS (30).
In this work, a mammalian expression system has been used to generate RAG1/2 proteins that are superior in both activity and physical properties. We use these improved protein samples to show that the RAG1/2 tetramer specifically binds only a single copy of 12RSS or 23RSS DNA, but not two of the same kind. The assembled complex of RAG1/2 and a pair of intact 12/23RSS DNAs can be purified to homogeneity and can proceed to form the cleavage SEC product in good yield. We have crystallized the resulting SEC and thus paved the way to structural analysis of RAG1/2 by x-ray diffraction.
Experimental Procedures
Proteins and DNA
Several expression constructs encoding mouse core RAG1 (384–1008 aa) and RAG2 (1–387 aa) were made for co-expression in HEK293GNTI cells. A maltose-binding protein (MBP) tag was necessary for RAG2 preparation and was placed at the N terminus followed by a cleavable linker recognized by PreScission protease or a non-cleavable linker of five alanine residues. Protein yields and DNA cleavage activity were similar regardless of the type of MBP tag. For efficient removal of MBP, a 10-asparagine linker was added between the RAG1 and the PreScission cleavage site. For RAG2, seven glycines in addition to the asparagine linker were placed between the PreScission cleavage site and the RAG2. A 5′ Kozak sequence and 3′ stop codon were introduced by PCR. For MBP-tagged constructs, RAG genes were cloned between the AgeI and XhoI sites of pLEXm (31). The tagless construct of RAG1 was produced by inserting the RAG1 gene into pLEXm between its MfeI (compatible with EcoRI in pLEXm) and XhoI sites. All DNA sequences were confirmed.
For RAG1/2 co-expression, HEK293GNTI cells were cultured in suspension in FreeStyle medium supplemented with 1% FBS, 100 units/ml penicillin, and 100 μg/ml streptomycin (Gibco) to a density of 2 × 106 cells/ml. Cells were transfected by adding a mixture of 1 mg of RAG1/2 expression vector DNAs (total) and 4 mg of PEI per liter of culture. Cells were further incubated for 72 h at 37 °C, 115 rpm, 5% CO2, and 95% humidity. After 72 h, cells were pelleted at 2500 rpm, washed in PBS, and stored at −80 °C.
For protein purification, cell paste was thawed in 25 mm Tris, pH 7.5, 500 mm NaCl, 2 mm DTT (buffer A), with the addition of a Complete protease inhibitor cocktail (Roche Life Science) and then lysed by sonication. The lysate was clarified by ultracentrifugation at 35,000 rpm. The supernatant was loaded by gravity flow onto an amylose column (4 ml of resin, New England Biolabs) equilibrated in buffer A. After washing the column with 50 column volumes of buffer A, the RAG proteins were eluted with 40 mm maltose in buffer A. The heterotetrameric nature of RAG1/2 protein (30) was confirmed using a Superdex-200 GL 10/300 gel-filtration column (GE Healthcare) with molecular weight markers and SDS gel. For untagged core RAG1 and MBP-tagged core RAG2 (RAG1/M2), the protein peak fractions were pooled, and the salt concentration was lowered to 100 mm by the addition of a no-salt buffer (50 mm HEPES, pH 7.0, and 2 mm DTT). The protein solution was applied onto a 5-ml HiTrap Q HP column (GE Healthcare) equilibrated in 50 mm HEPES, pH 7.0, 100 mm KCl, 2 mm DTT and eluted with a gradient of 0.1–1 m KCl. RAG1/2 was eluted at ∼300 mm KCl. Fractions containing RAG1/2 complex were pooled, and the salt concentration was raised to 500 mm KCl before applying onto a Superdex-200 column equilibrated in 50 mm HEPES, pH 7.0, 500 mm KCl, 5% glycerol, 2 mm DTT, and 2 mm maltose. Fractions from the main eluted peak were pooled and concentrated using an Amicon Ultra-15 centrifugal filter unit (Millipore) with 100-kDa cutoff. After adjusting the glycerol concentration to 30% (w/v), RAG1/2 aliquots were stored at −20 °C and thawed only once before experiments. Human HMGB1 (1–163) (30) was expressed in BL21(DE3) cells and purified via heparin, HiTrap SP HP, HiTrap Q HP, and HiLoad 16/600 Superdex-75 columns in the final buffer (25 mm HEPES, pH 7.0, 100 mm KCl, 2 mm DTT, 0.1 mm EDTA, 10% (w/v) glycerol) and stored at −20 °C as described (30).
DNA oligonucleotides were synthesized by the NIH Facility for Biotechnology Resources and purified on Glen-Pak DNA purification cartridges according to the manufacturer's instructions, taking advantage of the hydrophobicity of the 4,4′-dimethoxytrityl protection group. Complementary strands were annealed in 10 mm Tris (pH 7.5), 50 mm NaCl, 0.2 mm EDTA and stored at −20 °C. Pre-nicked substrates were assembled in the same way from three pieces for each RSS. The 12RSS sequence was GTCTTACACAGTGATACGTACCAGAACAAAAACCCTGCAG, and the 23RSS sequence was GTCTTACACAGTGATAGAACTCCAGCTGTCAGCCAGACAAAAACCCTGCAG, where the conserved heptamer and nonamer elements are italicized and underlined.
RAG1/2-DNA Complex Assembly and Analysis
The 12RSS-SC or PC with RAG1/M2 and HMGB1 was formed in 50 mm HEPES (pH 7.0), 60 mm KCl, 5 mm CaCl2, 1 mm maltose, 0.01% n-dodecyl-β-d-maltoside (DDM), and 2 mm DTT. Protein-DNA mixtures were incubated for 20 min at room temperature followed by 5 min at 37 °C. In all experiments, the concentration of RAG1/M2 was 1 μm (as a heterotetramer), and HMGB1 was at 3 μm. The amount of 12RSS varied, depending on the experiment, from 1.2- to 2.2-fold molar excess over the RAG1/2 heterotetramer. After incubation, the complex was analyzed on a Superdex-200 GL 10/300 column, equilibrated with 50 mm HEPES (pH 7.0), 60 mm KCl, 1 mm maltose, and 2 mm DTT. Elution points of molecular mass standards (669, 443, 200, and 150 kDa) were established under the same buffer conditions. The content of the peak was analyzed by SDS-PAGE stained with Coomassie Blue and by a TBE-urea gel stained with SYBR Green.
To determine the protein to DNA ratio, A260 and A280 were measured for protein-DNA complexes eluted from the Superdex-200 column. To investigate the ratio of the bound DNA species in the S200-purified PC, proteins were digested by proteinase K, and the DNA in the reaction mixture was 32P-labeled and analyzed on a TBE-urea gel. The 23RSS-SC or PC was formed with M1/M2 (both RAG1 and RAG2 with MBP tags) under the same condition as described above for the 12RSS-SC.
DNA Cleavage Assays
To assay coupled DNA cleavage, the bottom strands of the 12RSS and 23RSS were 32P-labeled. The complex was assembled by mixing a 1:1 molar ratio of RAG1/2 heterotetramer and 12/23RSS in 50 mm HEPES (pH 7.0), 60 mm KCl, 5 mm CaCl2, 1 mm maltose, 0.01% DDM, and 2 mm DTT. The cleavage reaction was initiated by the addition of 5 or 10 mm MgCl2, or 2, 1, or 0.5 mm MnCl2, or a combination of both metal ions. Reactions were allowed to proceed for 1–3 h at 37 °C and stopped by adding formamide solution containing 10 mm EDTA and a loading dye, heated, and resolved on TBE-urea denaturing gel. Radioactively labeled products were visualized by a PhosphorImager and quantified using ImageQuant NL (GE Healthcare).
For cleavage assays used to compare RAG1/M2 and M1/M2 complex in which both RAG1 and RAG2 contained MBP, the top strand of the 12RSS was 32P-labeled. 100 ng of RAG1/2 (as a heterotetramer), 100 ng of HMGB1, 5 mm MgCl2, and 2 nm each of labeled 12RSS and cold 23RSS were mixed in 50 mm HEPES (pH 7.0), 60 mm KCl, 0.01% DDM, 2 mm DTT, 5 mm MgCl2, and 2 mm maltose and incubated for 1 h at 37 °C. Reaction products were analyzed on a 15% TBE-urea gel and visualized by autoradiography.
SEC Preparation and Crystallization
Co-expression and purification of MBP-tagged RAG1/2 protein were conducted as described above except for omitting the HiTrap Q HP column. The PC was assembled using the following 12RSS (GTCTTACACAGTGATACGTACCAGAACAAAAACCCTGCAGCT) and 23RSS (GTCTTACACAGTGATAGAACTCCAGCTGTCAGCCAGACAAAAACCCTGCAGCT), both containing 8-bp extensions after the nonamer, in 25 mm HEPES (pH 7.0), 5 mm MgCl2, 50 mm KCl, 2 mm DTT, 1% glycerol, 2 mm DTT, and 0.01% DDM. The final concentrations in the reaction were 1 μm RAG1/2 (heterotetramer), 1.2 μm of each substrate RSS, and 3 μm HMGB1. After incubation for 20 min at room temperature, DNA cleavage was carried out for 1 h at 37 °C. PreScission protease was then added with overnight incubation at 4 °C. The cleavage mixture was loaded onto a 5-ml HiTrap Q HP column (GE Healthcare) equilibrated with 25 mm HEPES (pH 7.0), 100 mm KCl, 2 mm DTT, and 1% glycerol. Unwanted components (cleaved MBP, unreacted or aggregated RAG1/2) were eluted using a step gradient of 300 mm and 550 mm KCl. The SEC was then eluted with 650 mm KCl. After concentration using an Amicon Ultra-15 centrifugal filter unit (Millipore) with 100-kDa cutoff, the SEC complexes were loaded onto a Superdex-200 10/300 GL column equilibrated with 25 mm HEPES (pH 7.0), 500 mm KCl, 1% (v/v) glycerol, 2 mm DTT. The eluted peak was collected and concentrated to ∼10 mg/ml protein (determined by the Bradford method). Crystals were grown within a week at 4 °C using a reservoir of 0.2 m potassium citrate tribasic monohydrate and 18% (w/v) PEG 3350, by the sitting-drop diffusion method. Single crystals were cryoprotected in mother liquor supplemented with 30% (v/v) ethylene glycol, transferred, mounted in MicroLoops (MiTeGen), and flash-cooled in liquid nitrogen. Diffraction was tested on the 23-ID-D beamline at the Advanced Photon Source at Argonne National Laboratory.
Results
Expression and Purification of RAG Proteins
Because the RAG1/2 protein complex is found only in jawed vertebrates, we wondered whether expression of RAG1/2 in mammalian instead of insect cell culture could improve the protein solubility and activity. Core regions of mouse RAG1 (384–1008 aa) and RAG2 (1–387 aa), which are abbreviated as RAG1 and RAG2, were placed downstream of an MBP tag with a cleavable linker in the mammalian expression vector pLEXm (31) (see “Experimental Procedures” and Fig. 1A). MBP greatly improves the expression yield and solubility of RAGs (4). RAG1 and RAG2 co-expressed in HEK293GNTI cells were readily purified to near homogeneity by one-step amylose affinity chromatography and eluted as a heterotetramer of two RAG1s and two RAG2s in a single symmetric peak from the ensuing gel-filtration column (Fig. 1, B and C). The yield of the RAG1/2 protein expressed in mammalian suspension culture (4–8 mg/liter of cultured cells) was similar to that of insect culture. Chemically, the RAG1/2 proteins expressed in mammalian and insect cells are identical, with neither containing detectable post-translational modifications, as determined by mass spectrometry analysis.4
FIGURE 1.
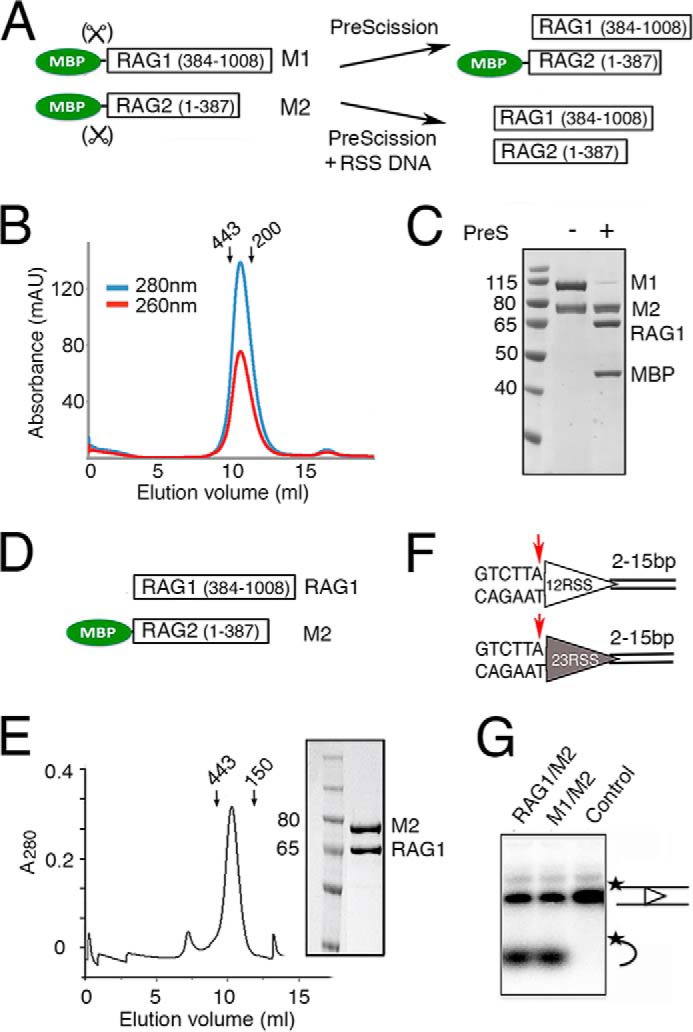
Purification and activity of RAG1 and RAG2 proteins. A, diagram of RAG1 and RAG2 constructs fused to a cleavable MBP tag (M1 and M2). The MBP tag can be efficiently removed from RAG1, but its removal from RAG2 requires binding of 12/23RSS DNAs. B, Superdex-200 elution profile of the tetrameric MBP-tagged RAG1/2 (calculated molecular mass of 406 kDa), which was co-expressed in HEK293GNTI cells and purified by amylose affinity chromatography. Arrows indicate the elution points of molecular mass markers (443 and 200 kDa). C, SDS protein gel of the size exclusion peak showing 1:1 complex of M1 and M2. MBP can be removed from M1 but not M2 by PreScission (PreS). D, diagram of the tag-free RAG1 and non-cleavable MBP-tagged RAG2 (M2) constructs used to produce RAG1/M2 for biochemical studies. E, Superdex-200 elution profile of RAG1/M2 (calculated molecular mass of 312 kDa) and the Coomassie Blue-stained SDS gel of the co-eluted RAG1/M2 protein. F, diagram of the 12RSS (open triangle) and 23RSS (filled triangle) used in paired complex formation and signal-end complex preparation for crystallization. Conserved elements are designated as triangles, and the red arrow is the RAG1/2 cleavage site. The coding flank sequence is shown, and the extensions bordering the other DNA ends were varied in crystallization trials. G, coupled cleavage of 12/23RSS in Mg2+. The 12RSS top strand was 5′-radiolabeled, allowing the detection of hairpin product by a TBE-urea gel. The results of 12/23RSS paired cleavage by RAG1/M2 and M1/M2 were compared. The control lane is DNA alone. The 32P labels on DNA strands are marked with asterisks.
From the mammalian co-expressed RAG1/2 proteins, the MBP was readily cleaved off RAG1, resulting in a RAG1 and MBP-RAG2 complex (abbreviated as RAG1/M2) (Fig. 1C). We found that the MBP tag on RAG2 was essential for obtaining good expression of the RAG1/2 complex and also allowed the use of an amylose pulldown as a first purification step. As an alternative, we created a tagless construct for RAG1 and co-expressed it with a non-cleavable MBP-tagged RAG2 (MRAG2 or M2 for short) (Fig. 1D) to avoid the overnight tag removal step (see “Experimental Procedures”). The resulting protein (Fig. 1E) was as active in coupled DNA cleavage as RAG1/2 complex with MBP tags on both subunits (Fig. 1, F–G). For the remaining experiments, unless specified, the RAG1/M2 complex was used.
Complex of RAG1/2 Tetramer and a Single 12RSS
According to the previously proposed capture model, the RAG1/2 heterotetramer preferentially assembles first on a 12RSS in a single complex (12RSS-SC), and a 23RSS then binds to form the paired complex (22, 23). To test this model, we first asked how many copies of 12RSS can be accommodated in a 12RSS-SC. The tetrameric RAG1/M2 was mixed with intact 12RSS (Figs. 1F and 2A) in a molar ratio of 1:1.2 or 1:2.2 in the presence of Ca2+ and HMGB1 protein. The small excess of DNA substrate over RAG1/2 was used to ensure that there was no free protein left in solution. After incubation and separation by size-exclusion chromatography, the major peak eluted from the sizing column, which was shifted when compared with the RAG1/M2 protein alone, contained both RAG1/M2 and 12RSS (Fig. 2, B and C) and was identical for both ratios of DNA to protein (in position and amplitude as well as the ratio of A260/A280). The later eluting peak of excess 12RSS was merely increased at the higher ratio of DNA. These observations indicated that the majority of RAG1/2 tetramer was able to accommodate only one copy of 12RSS DNA in the 12RSS-SC, although we cannot rule out that a minute fraction of RAG1/2 tetramer below our detection capability might form a complex with two copies of the same RSS DNA.
FIGURE 2.

RAG1/2 forms an SC with one copy of 12RSS or 23RSS. A, schematic representation of the RSS cleavage reaction and DNA substrate used in PC formation. B, superimposition of gel-filtration profiles of the RAG1/M2 protein complex alone and after incubation with 1.2:1 or 2.2:1 molar excess of 12RSS substrate (blue lines) or 2.2:1 molar excess of pre-nicked 12RSS (gray dashes). The elution profile of the pre-nicked 12RSS at 1.2:1 molar ratio was identical to that of the intact 12RSS at the same molar ratio and thus is omitted for clarity. The 12RSS-SC complex (355 kDa) was readily identifiable by the peak shift when compared with RAG1/M2 protein alone. Arrows indicate the elution points of molecular mass markers (443 and 150 kDa). The excess 12RSS (intact or pre-nicked) was eluted in a later peak. C, analysis of the elution peak of the 12RSS-SC by SDS-PAGE and denaturing TBE-urea gel stained by Coomassie Blue and SYBR Green, respectively. The DNA gel also shows double-strand cleavage by purified 12RSS-SC in Mn2+. D, superimposed gel-filtration profiles of the 12RSS-SC (blue) and its conversion to PC (orange, 386 kDa) after incubation with 23RSS (gray filled triangle). The excess of free DNA was eluted after SC and PC. E, analysis of components of the PC peak by SDS-PAGE and denaturing TBE-urea gel electrophoresis (stained by SYBR Green). 32P labeling of the PC peak showed that the 12RSS and 23RSS are present in 1:1 molar ratio. The 32P labels on DNA strands are marked with asterisks. F, superposition of gel-filtration profiles of 23RSS-SC formation (which did not alter by the addition of more 23RSS (green)), and its conversion to PC after the addition of 12RSS (orange). Because both RAG1 and RAG2 were MBP-tagged, the molecular masses of SC and PC were 446 and 468 kDa. Due to the flexible linkers, their apparent sizes appeared to be larger than the calculated values. The elution points of molecular mass markers (669 and 150 kDa) are indicated by arrows. G, the protein and DNA components of 23RSS-SC and PC were confirmed by SDS and TBE-urea gels. In panels C, E, and G, the open and filled triangles represent 12RSS and 23RSS, respectively, as shown in panel A.
We next asked whether the DNA cleavage intermediate, a pre-nicked 12RSS substrate (Fig. 2A), could be assembled with RAG1/M2. Repeating the experiments of mixing tetrameric RAG1/M2 and the pre-nicked 12RSS at 1:1.2 and 1:2.2 molar ratios, the elution profiles showed the same results, that a single copy of pre-nicked 12RSS appeared to be bound to each RAG1/M2 tetramer (Fig. 2, B and C).
To test whether single complexes (SC) of intact and pre-nicked 12RSS were active in DNA cleavage, the SC fractions eluted from the sizing column were incubated with 2 mm Mn2+. Analyses of the products showed that the SC complexes supported hairpin formation (Fig. 2C). The reaction was more complete with pre-nicked 12RSS than intact DNA. We conclude that a RAG1/M2 tetramer binds one copy of 12RSS DNA and can proceed to cleave the DNA in the presence of Mn2+.
The 12RSS-SC Reserves the Second DNA-binding Site for a 23RSS and Vice Versa
When the purified 12RSS-SC was incubated with a 1.2 molar excess of 23RSS, size-exclusion chromatography yielded a major peak, corresponding to a bona fide PC with RAG1/M2, HMGB1, and both 12RSS and 23RSS, and a small peak of excess 23RSS (Fig. 2, D and E). 32P labeling of the DNA showed that the two RSSs were present in 1:1 ratio, although by SYBR Green staining, the intensity of the 23RSS DNA in the purified PC was higher than that of the shorter 12RSS (Fig. 2E). Thus when one (and only one) site is occupied by a 12RSS in the initial 12RSS-SC, the other is reserved for a 23RSS, which can be incorporated later to form a PC.
However, assembly of an SC did not have to start from a 12RSS. The SC with 23RSS (23RSS-SC) could also be assembled by mixing M1/M2 protein (with MBP tags on both) with 23RSS (intact or pre-nicked) at 1:1.2 or 1:2.2 molar ratios in the presence of a divalent cation and HMGB1 protein. Whether the molar ratio of 23RSS to RAG1/2 protein was 1.2:1 or 2.2:1 in the incubation mixture, the protein-DNA complexes eluted from the gel-filtration column appeared to contain only one 23RSS for each RAG1/2 heterotetramer, as judged by protein and DNA gels (Fig. 2, F and G) and confirmed by their having the same A260/A280 ratios (1.35 and 1.36). The 23RSS-SC was readily converted to the PC after the addition of 12RSS (Fig. 2, F and G), with the A260/A280 ratio increased to 1.54. Therefore, the previously reported SCs (22, 23) most probably contained a single copy of either 12RSS or 23RSS DNA. SCs of 12RSS or 23RSS can be converted to a PC with the addition of the second type of RSS DNA.
The Paired Complexes Are Fully Active in DNA Cleavage
Assembly of the paired complex did not require stepwise incubation. PC was also produced by direct mixing of all components (Fig. 3, A and B). Furthermore, the PC could be assembled from pre-nicked DNA substrates in the same way, forming a pre-nicked PC (“PC-nick”) (Fig. 3, A and B). The 12/23 RSSs were present in 1:1 ratio in both the PC and PC-nick, again based on the intensity of DNAs labeled with 32P (Fig. 3, C and D).
FIGURE 3.

Activity of PC assembled from intact and pre-nicked substrates. A, scheme of PC formation using intact (PC) or pre-nicked (PC-nick) 12/23RSS substrates (open and filled triangles) in Ca2+. Subsequent DNA cleavage in the presence of Mg2+/Mn2+ led to the production of SEC. B, PC formation by simultaneous mixing of all components. Superdex-200 elution profiles of protein alone, PC, and PC-nick preparation were superimposed. Arrows indicate the elution points of molecular mass markers (443 and 150 kDa). C and D, DNA cleavage in PC assembled from intact (C) and pre-nicked 12/23RSS (D) in the presence of MgCl2 and MnCl2 for 1–2 h. The 32P labels on DNA strands are marked with asterisks. DNA cleavage products were separated on a TBE-urea gel. E, analysis of the metal ion concentrations, reaction time, and molar ratios of RAGs to DNA substrates in paired cleavage of pre-nicked RSSs by RAG1/M2. F, graphic summary of DNA cleavage reactions shown in E.
In previous studies, RSS cleavage by RAG1/2 was often incomplete even in the presence of a large excess of protein (4, 14, 20). To determine how efficient double-strand cleavage by PC or PC-nick was, both types of complexes were assembled with both 12 and 23 RSS DNAs containing 5′-32P-labeled bottom strands in the presence of Ca2+ and subjected to cleavage by adding Mg2+ or Mn2+ or a combination of both. Double-strand cleavage of a non-nicked PC did not go to completion (about 60% at best) even when all DNA was bound to RAG1/M2 (Fig. 3C). However, a PC assembled from pre-nicked substrates was more efficiently processed (Fig. 3D), presumably because the first step of DNA cleavage was bypassed. The fact that cleavage of the non-nicked PC is only ∼60% complete may suggest that the first step of the reaction is a limiting step under these conditions.
To increase the cleavage of PC-nick, we varied the metal ion composition and reaction time (Fig. 3, D and E). With Mg2+ alone, cleavage was incomplete (∼80%), whereas in 2 mm Mn2+, there was more than 90% conversion to hairpins (Fig. 3D). The best condition used a mixture of 10 mm MgCl2 and 2 mm MnCl2, which yielded nearly 100% hairpin conversion after 2 h at 37 °C (Fig. 3, E and F).
Preparation and Crystallization of SEC
The efficient DNA cleavage of our assembled PC produced an ample amount of the SEC, a stable complex that is known to resist challenge by high salt or heparin (18). To obtain homogeneous tagless SEC for x-ray crystallography, we prepared RAG1 and RAG2 both containing a cleavable MBP tag on the N terminus (Fig. 1A). The amylose affinity-purified fully tagged RAG1/2 (Fig. 1A) and RSS DNAs (Fig. 1F) were assembled under the DNA cleavage conditions to allow PC formation and its conversion to the SEC (Fig. 4A). The MBP tags were removed later by overnight incubation with PreScission protease (Fig. 4A). When the entire DNA and protein cleavage reaction mixture was loaded on a HiTrap Q column equilibrated in the low salt reaction buffer, the HMGB1 protein, which was likely displaced by the positively charged column resin, dissociated from the SEC and flowed through the column (Fig. 4, B and C). The SEC was separated from MBP, PreScission protease, free RAG1/2, and DNA with a step gradient of KCl, and then eluted alone at 650 mm KCl as peak 2 (Fig. 4C). The HMGB1-free SEC was purified to homogeneity over a Superdex-200 column (Fig. 4D). SDS-PAGE and TBE-urea gels confirmed that it contained the cleaved 12 and 23 RSS as well as RAG1/2 proteins (Fig. 4E).
FIGURE 4.
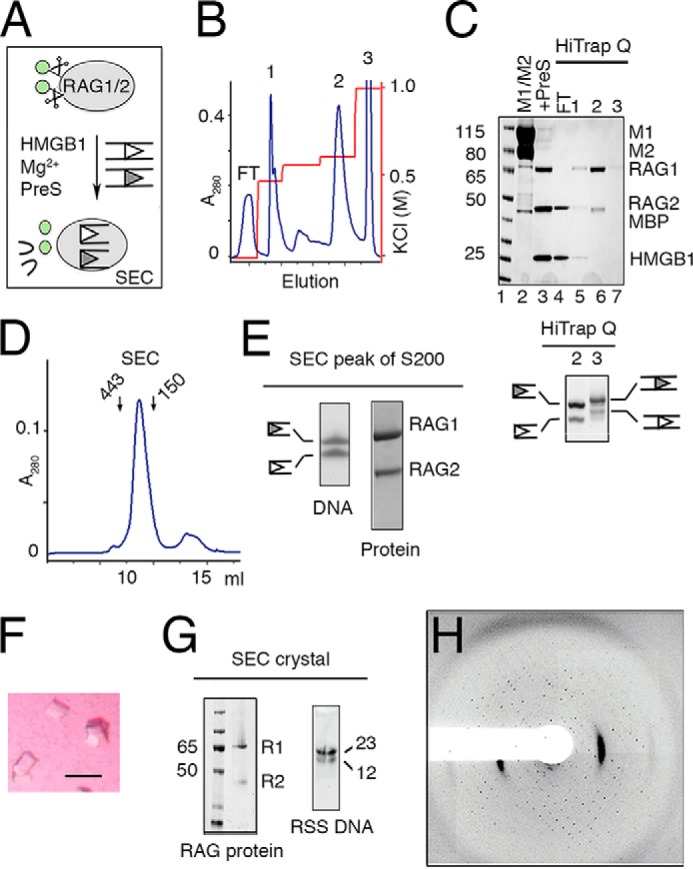
Preparation and crystallization of SEC. A, schematic of SEC production. RAG1/2 (gray oval) with cleavable MBP (green circles) was incubated with RSS DNAs (open and filled triangles) and HMGB1. After cleavage of RSS DNAs in the presence of Mg2+, MBP tags were removed by PreScission protease (PreS). B, elution profile of the reaction mixture applied onto a HiTrap Q ion-exchange column. HMGB1, MBP, unreacted RAG1/2, and free DNA were eluted at different concentrations of KCl. SEC was eluted in a sharp peak at 650 mm KCl (peak 2). C, upper panel, SDS gel of M1/M2 protein (lane 2), assembled SEC after PreScission cleavage (lane 3), flow-through (FT, lane 4), and the peak fractions eluted from HiTrap Q (B). Lower panel, a SYBR Green-stained TBE-urea gel of peaks 2 (SEC) and 3 (free and intact DNA) eluted from HiTrap Q (B). D, Superdex-200 elution profile of the SEC (281 kDa) after the ion-exchange step. Arrows indicate the elution points of molecular mass markers (443 and 150 kDa). E, TBE-urea and SDS protein gels of the Superdex-200 peak fraction. F, tetragonal and square-shaped crystals of the SEC complex obtained after optimization of the DNA lengths. G, SDS-PAGE and TBE-urea gel (stained with SYBR Green) of dissolved SEC crystals after thorough wash. The crystals contained tag-free RAG1/2 (R1 and R2) and 12/23RSS DNAs. H, diffraction image of a typical SEC crystal.
The SEC complex was easily concentrated to 5–10 mg/ml and screened for crystallization. Crystals of the SEC were obtained in 0.2 m tri-potassium citrate and 18% (w/v) PEG 3350 (see “Experimental Procedures” and Fig. 4F) and were verified to contain RAG1/2 proteins as well as 12/23RSS DNAs (Fig. 4G). To optimize the SEC crystals, the extensions downstream of the nonamer on 12RSS and 23RSS were varied from 2 to 15 bp in parallel. An 8-bp extension on both RSS DNAs (Fig. 1F) gave the best crystals. These SEC crystals diffracted x-rays up to 4.5Å, but the diffraction pattern indicated that these SEC crystals were mosaic and consisted of clusters of thin plates (Fig. 4H). Although not resulting in a three-dimensional structure immediately, the readiness of SEC to crystallize has inspired continuous efforts and led to the eventual crystal structure of RAG1/2 (34), for which the SEC was prepared from pre-cleaved RSS DNAs.
Discussion
Using HEK293 cells and a mammalian expression vector, we were able to produce ample RAG1/2 protein in the active tetrameric form. By showing that this protein complex can be quantitatively converted to the PC and then SEC after DNA cleavage, we found that RAG1/2 in this preparation is essentially 100% active. This contrasts with RAG1/2 expressed in insect cells, which forms amorphous soluble aggregates and contains only a small fraction of active species (30). Because RAG1/2 produced by both insect and human are not post-translationally modified according to mass spectrometry analysis, it may be that protein folding of RAG1/2 in insect cells is defective. We also found that MBP tags on RAG1, RAG2, or both do not alter the biochemical properties of the RAG1/2 recombinase, which is consistent with previous characterization of the SEC complex (30). However, the stability of RAG2 depends on the MBP fusion until RAG1/2 is associated with DNA. The MBP tags on RAG2 can be removed in either the PC or the SEC complex but not from the tetrameric RAG1/2 proteins alone. Apparently, RSS DNA binding stabilizes the RAG1/2 recombinase.
RAG1/2 isolated from HEK293 cells is a preformed tetramer without DNA (Fig. 1B) (34). Our step-by-step assembly and characterization of single and paired complexes unequivocally show that the tetrameric RAG1/2 binds and synapses one 12RSS and one 23RSS into a paired complex without change of oligomeric composition, whether the RSS DNAs are intact or pre-nicked. The tetrameric RAG1/2 within PC is fully active in cleaving 12/23RSS DNAs, as shown previously (30, 32). Characterization of the RSS DNA cleavage product SEC by EM and atomic force microscopy analyses (30) also gives no evidence of species other than RAG1/2 tetramer. These observations are in contrast to the report, largely based on atomic force microscopy measurements, that a RAG PC assembled on a long coding DNA contains a pair of RAG1/2 tetramers in the form of a heterooctamer (33).
The detectable single complexes of RAG1/2 tetramer with 12RSS or 23RSS DNA contain only one copy of either RSS, and the second DNA-binding site is reserved for binding the complementary RSS DNA. Although this conclusion was also drawn from earlier work (20), it then relied on gel electrophoresis, with the possibility of confounding gel-specific effects. This specificity in DNA selection could arise in two ways. The first possibility is that each tetrameric RAG1/2 initially contains two equivalent sites that can bind either a 12RSS or a 23RSS DNA, and the protein undergoes a conformational change upon binding of one RSS, becoming asymmetric and selecting specifically for the absent RSS DNA (Fig. 5). The alternative, that two non-equivalent DNA-binding sites are present in RAG1/2 before any single complex is formed, is less likely than the first scenario because the two copies of RAG1/2 in the tetramer are intrinsically identical.
FIGURE 5.
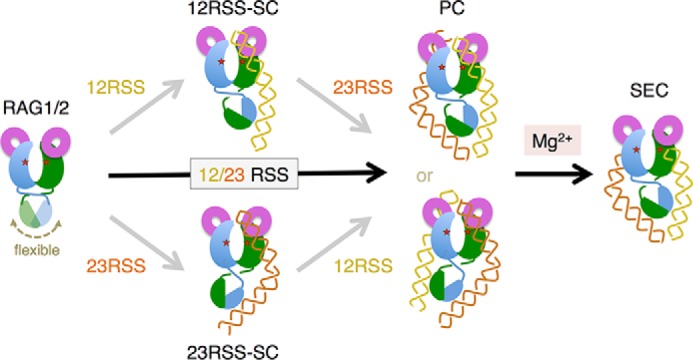
A model of SC, PC and SEC complex formation. The RAG1/2 heterotetramer has a pair of NBDs, which are attached to the bulk of the two RAG1 subunits by a flexible link (motion indicated by a double arrowhead). Once bound to either a 12RSS or a 23RSS to form an SC, the NBD becomes tilted asymmetrically relative to the two active sites (marked by red stars), and the second DNA-binding site can only bind the complementary RSS to form a PC. The PC can also assemble from 12RSS and 23RSS DNAs added simultaneously, whether the DNAs are intact or pre-nicked. DNA cleavage in the PC occurs in the presence of Mg2+, leading to SEC formation. HMGB1, which facilitates both SC and PC formation, is omitted in the diagram for clarity.
Recently, following the procedure described here, we have determined the crystal structure of RAG1/2 without RSS DNA (PDB: 4WWX) (34). Interestingly, the nonamer-binding domains (NBDs) of RAG1 subunits are dimerized as reported (27) and appear to be connected to the bulk of the protein by a flexible link. In the crystals, they are positioned asymmetrically relative to the two catalytic centers, presumably due to crystal packing forces. We thus hypothesize that the NBDs are mobile relative to the bulk of the RAG1/2 tetramer and to the catalytic domains that bind the heptamers of the RSS DNAs (Fig. 5). Upon binding of either 12RSS or 23RSS, the RAG1/2 tetramer becomes asymmetric, with the NBD tilted such that only the other type of RSS DNA can bind (Fig. 5). The 12/23 rule for V(D)J recombination is thereby established at the paired complex formation and is re-enforced by the efficiency of DNA cleavage only in the paired and not single complex (20).
- RSS
- recombination signal sequence
- RAG
- recombination activation gene
- SC
- single complex
- PC
- paired complex
- SEC
- signal-end complex
- MBP
- maltose-binding protein
- HMGB1
- high mobility group protein B1
- HEK293GNTI
- N-acetylglucosaminyltransferase I-negative human embryonic kidney 293 cells
- TBE
- Tris, boric acid, and EDTA
- DDM
- n-dodecyl-β-d-maltoside
- aa
- amino acids
- NBD
- nonamer-binding domain.
References
- 1. Sakano H., Hüppi K., Heinrich G., Tonegawa S. (1979) Sequences at the somatic recombination sites of immunoglobulin light-chain genes. Nature 280, 288–294 [DOI] [PubMed] [Google Scholar]
- 2. Lewis S. M. (1994) The mechanism of V(D)J joining: lessons from molecular, immunological, and comparative analyses. Adv. Immunol. 56, 27–150 [DOI] [PubMed] [Google Scholar]
- 3. Sakano H., Maki R., Kurosawa Y., Roeder W., Tonegawa S. (1980) Two types of somatic recombination are necessary for the generation of complete immunoglobulin heavy-chain genes. Nature 286, 676–683 [DOI] [PubMed] [Google Scholar]
- 4. McBlane J. F., van Gent D. C., Ramsden D. A., Romeo C., Cuomo C. A., Gellert M., Oettinger M. A. (1995) Cleavage at a V(D)J recombination signal requires only RAG1 and RAG2 proteins and occurs in two steps. Cell 83, 387–395 [DOI] [PubMed] [Google Scholar]
- 5. Oettinger M. A., Schatz D. G., Gorka C., Baltimore D. (1990) RAG-1 and RAG-2, adjacent genes that synergistically activate V(D)J recombination. Science 248, 1517–1523 [DOI] [PubMed] [Google Scholar]
- 6. Schatz D. G., Oettinger M. A., Baltimore D. (1989) The V(D)J recombination activating gene, RAG-1. Cell 59, 1035–1048 [DOI] [PubMed] [Google Scholar]
- 7. Cuomo C. A., Oettinger M. A. (1994) Analysis of regions of RAG-2 important for V(D)J recombination. Nucleic Acids Res. 22, 1810–1814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sadofsky M. J., Hesse J. E., Gellert M. (1994) Definition of a core region of RAG-2 that is functional in V(D)J recombination. Nucleic Acids Res. 22, 1805–1809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sadofsky M. J., Hesse J. E., McBlane J. F., Gellert M. (1993) Expression and V(D)J recombination activity of mutated RAG-1 proteins. Nucleic Acids Res. 21, 5644–5650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Silver D. P., Spanopoulou E., Mulligan R. C., Baltimore D. (1993) Dispensable sequence motifs in the RAG-1 and RAG-2 genes for plasmid V(D)J recombination. Proc. Natl. Acad. Sci. U.S.A. 90, 6100–6104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kim D. R., Dai Y., Mundy C. L., Yang W., Oettinger M. A. (1999) Mutations of acidic residues in RAG1 define the active site of the V(D)J recombinase. Genes Dev. 13, 3070–3080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Landree M. A., Wibbenmeyer J. A., Roth D. B. (1999) Mutational analysis of RAG1 and RAG2 identifies three catalytic amino acids in RAG1 critical for both cleavage steps of V(D)J recombination. Genes Dev. 13, 3059–3069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fugmann S. D., Villey I. J., Ptaszek L. M., Schatz D. G. (2000) Identification of two catalytic residues in RAG1 that define a single active site within the RAG1/RAG2 protein complex. Mol. Cell 5, 97–107 [DOI] [PubMed] [Google Scholar]
- 14. van Gent D. C., Hiom K., Paull T. T., Gellert M. (1997) Stimulation of V(D)J cleavage by high mobility group proteins. EMBO J. 16, 2665–2670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Boboila C., Alt F. W., Schwer B. (2012) Classical and alternative end-joining pathways for repair of lymphocyte-specific and general DNA double-strand breaks. Adv. Immunol. 116, 1–49 [DOI] [PubMed] [Google Scholar]
- 16. Deriano L., Roth D. B. (2013) Modernizing the nonhomologous end-joining repertoire: alternative and classical NHEJ share the stage. Annu. Rev. Genet. 47, 433–455 [DOI] [PubMed] [Google Scholar]
- 17. Agrawal A., Schatz D. G. (1997) RAG1 and RAG2 form a stable postcleavage synaptic complex with DNA containing signal ends in V(D)J recombination. Cell 89, 43–53 [DOI] [PubMed] [Google Scholar]
- 18. Jones J. M., Gellert M. (2001) Intermediates in V(D)J recombination: a stable RAG1/2 complex sequesters cleaved RSS ends. Proc. Natl. Acad. Sci. U.S.A. 98, 12926–12931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hesse J. E., Lieber M. R., Mizuuchi K., Gellert M. (1989) V(D)J recombination: a functional definition of the joining signals. Genes Dev. 3, 1053–1061 [DOI] [PubMed] [Google Scholar]
- 20. Hiom K., Gellert M. (1997) A stable RAG1–RAG2–DNA complex that is active in V(D)J cleavage. Cell 88, 65–72 [DOI] [PubMed] [Google Scholar]
- 21. Gellert M. (2002) V(D)J recombination: RAG proteins, repair factors, and regulation. Annu. Rev. Biochem. 71, 101–132 [DOI] [PubMed] [Google Scholar]
- 22. Curry J. D., Geier J. K., Schlissel M. S. (2005) Single-strand recombination signal sequence nicks in vivo: evidence for a capture model of synapsis. Nat. Immunol. 6, 1272–1279 [DOI] [PubMed] [Google Scholar]
- 23. Jones J. M., Gellert M. (2002) Ordered assembly of the V(D)J synaptic complex ensures accurate recombination. EMBO J. 21, 4162–4171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kwon J., Morshead K. B., Guyon J. R., Kingston R. E., Oettinger M. A. (2000) Histone acetylation and hSWI/SNF remodeling act in concert to stimulate V(D)J cleavage of nucleosomal DNA. Mol. Cell 6, 1037–1048 [DOI] [PubMed] [Google Scholar]
- 25. Swanson P. C. (2004) The bounty of RAGs: recombination signal complexes and reaction outcomes. Immunol. Rev. 200, 90–114 [DOI] [PubMed] [Google Scholar]
- 26. Akamatsu Y., Oettinger M. A. (1998) Distinct roles of RAG1 and RAG2 in binding the V(D)J recombination signal sequences. Mol. Cell. Biol. 18, 4670–4678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yin F. F., Bailey S., Innis C. A., Ciubotaru M., Kamtekar S., Steitz T. A., Schatz D. G. (2009) Structure of the RAG1 nonamer binding domain with DNA reveals a dimer that mediates DNA synapsis. Nat. Struct. Mol. Biol. 16, 499–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li W., Swanson P., Desiderio S. (1997) RAG-1 and RAG-2-dependent assembly of functional complexes with V(D)J recombination substrates in solution. Mol. Cell. Biol. 17, 6932–6939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. van Gent D. C., McBlane J. F., Ramsden D. A., Sadofsky M. J., Hesse J. E., Gellert M. (1995) Initiation of V(D)J recombination in a cell-free system. Cell 81, 925–934 [DOI] [PubMed] [Google Scholar]
- 30. Grundy G. J., Ramón-Maiques S., Dimitriadis E. K., Kotova S., Biertümpfel C., Heymann J. B., Steven A. C., Gellert M., Yang W. (2009) Initial stages of V(D)J recombination: the organization of RAG1/2 and RSS DNA in the postcleavage complex. Mol. Cell 35, 217–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Aricescu A. R., Lu W., Jones E. Y. (2006) A time- and cost-efficient system for high-level protein production in mammalian cells. Acta Crystallogr. D Biol. Crystallogr. 62, 1243–1250 [DOI] [PubMed] [Google Scholar]
- 32. Swanson P. C. (2002) A RAG-1/RAG-2 tetramer supports 12/23-regulated synapsis, cleavage, and transposition of V(D)J recombination signals. Mol. Cell. Biol. 22, 7790–7801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shlyakhtenko L. S., Gilmore J., Kriatchko A. N., Kumar S., Swanson P. C., Lyubchenko Y. L. (2009) Molecular mechanism underlying RAG1/RAG2 synaptic complex formation. J. Biol. Chem. 284, 20956–20965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kim M.-S., Lapkouski M., Yang W., Gellert M. (2015) Crystal structure of the V(D)J recombinase RAG1-RAG2. Nature 518, 507–511 [DOI] [PMC free article] [PubMed] [Google Scholar]