

Background: Mechanisms that drive chronic inflammation in airway disease are not well understood.
Results: We demonstrate a TGF-β-responsive enhancer element required for efficient IL-1β-dependent expression of the proinflammatory chemokine CCL20 by human lung fibroblasts.
Conclusion: Convergence of TGF-β and IL-1β signaling pathways on the CCL20 promoter are required for efficient fibroblast expression of CCL20.
Significance: These findings identify potential targets to reduce chronic inflammation in airway disease.
Keywords: inflammation, TGF-β, transcription, integrin, chemokine
Abstract
CCL20 is the only chemokine ligand for the chemokine receptor CCR6, which is expressed by the critical antigen presenting cells, dendritic cells. Increased expression of CCL20 is likely involved in the increased recruitment of dendritic cells observed in fibroinflammatory diseases such as chronic obstructive pulmonary disease (COPD). CCL20 expression is increased by the proinflammatory cytokine IL-1β. We have determined that IL-1β-dependent CCL20 expression is also dependent on the multifunctional cytokine TGF-β. TGF-β is expressed in a latent form that must be activated to function, and activation is achieved through binding to the integrin αvβ8 (itgb8). Here we confirm correlative increases in αvβ8 and IL-1β with CCL20 protein in lung parenchymal lysates of a large cohort of COPD patients. How IL-1β- and αvβ8-mediated TGF-β activation conspire to increase fibroblast CCL20 expression remains unknown, because these pathways have not been shown to directly interact. We evaluate the 5′-flanking region of CCL20 to determine that IL-1β-driven CCL20 expression is dependent on αvβ8-mediated activation of TGF-β. We identify a TGF-β-responsive element (i.e. SMAD) located on an upstream enhancer of the human CCL20 promoter required for efficient IL-1β-dependent CCL20 expression. By chromatin immunoprecipitation, this upstream enhancer complexes with the p50 subunit of NF-κB on a NF-κB-binding element close to the transcriptional start site of CCL20. These interactions are confirmed by electromobility shift assays in nuclear extracts from human lung fibroblasts. These data define a mechanism by which αvβ8-dependent activation of TGF-β regulates IL-1β-dependent CCL20 expression in COPD.
Introduction
Chronic obstructive lung disease (COPD)3 is now the third leading cause of death in the United States (1). A major physiologic component of COPD is known as airway remodeling, which is characterized by accumulation of peribronchial chronic inflammation and fibrosis leading to airway narrowing and airflow obstruction that is refractory to currently available therapies (2, 3). New therapeutic development may benefit from understanding the mechanisms driving treatment-refractory inflammation in COPD (4).
Cigarette smoke (CS) is the major cause of airway remodeling by inducing cellular injury and increasing the susceptibility to respiratory pathogens, in particular viruses (5). Rodents, when exposed to CS, viruses, or both together, demonstrate exaggerated airway remodeling (6, 7). CS and viruses engage similar host danger pathways leading to inflammasome activation and enhanced IL-1β secretion (8–10). IL-1β protein levels are increased in human COPD biospecimens (11–14), delivery of IL-1β to the airways causes experimental airway remodeling (6, 15), and perturbation of IL-1 signaling protects against experimental CS-induced airway remodeling (16, 17). The mechanisms of IL-1β-induced airway remodeling involve enhanced TGF-β1 expression (18).
TGF-β1, a multifunctional cytokine widely implicated in both pathologic immunity and fibrosis, is involved in CS-induced experimental lung pathology and is increased in COPD specimens (19, 20). Expression of TGF-β is not sufficient for its pathologic effects because it is expressed in latent form that must be activated to function. Latency of TGF-β is conferred by a noncovalent association of the homodimeric TGF-β peptide by its propeptide, latency-associated peptide (21). The latency-associated peptide of TGF-β1 and -β3 contain the integrin RGD binding motif (22–25). Integrin αvβ8 binds to the RGD sites of latency-associated peptides 1 and 3 with high affinity (21) and has been shown to be essential for TGF-β activation during development and in driving airway pathology in adult mice (6, 26–29). Integrin αvβ8 expression and αvβ8-mediated TGF-β activation is increased by IL-1β (6), suggesting that αvβ8 may be involved in CS- and viral-induced airway remodeling.
αvβ8 is implicated in the pathogenesis of airway remodeling through its increased COPD-associated expression in airway fibroblasts (6). Increased immunohistochemical staining of αvβ8 in airway fibroblasts correlates with disease severity in COPD lungs (4, 30). Intratracheal adenoviral (Ad) delivery of IL-1β to the airways of mice leads to increased αvβ8 expression (6), through a transcriptional pathway involving AP-1 and SP3 (30). The expression of αvβ8 by fibroblasts appears to be particularly important because fibroblast conditional deletion of itgb8 prevents Ad-IL-1β-induced airway inflammation and fibrosis, implicating fibroblasts as a cell type involved in driving both the inflammatory and fibrotic response during airway remodeling (6). Inhibition of αvβ8 function using a therapeutic monoclonal antibody (clone B5) prevents airway remodeling in Ad-IL-1β- or cigarette smoke-exposed mice (7). Antibody B5, an allosteric inhibitor, reduces the affinity of αvβ8 for the latency-associated peptide of TGF-β, which is sufficient to selectively block TGF-β activation without significantly affecting cell adhesion to the latency-associated peptide (7).
Fibroblasts are located in interstitial spaces surrounding the airways, and the numbers of these fibroblasts are increased during airway remodeling (31). Fibroblasts are “activated” by inflammatory stimuli such as IL-1β during the remodeling process to increase the synthesis of extracellular matrix, cytosolic smooth muscle actin, and a number of chemokines, in particular the potent dendritic cell (DC) chemoattractant, CCL20 (4, 6, 32). CCL20 is increased in COPD samples and is the only known chemokine ligand for the receptor CCR6 (33), which is expressed by DCs. DCs are critical antigen-presenting cells implicated in the pathogenesis of COPD through priming pathologic adaptive T-cell immune responses (34). DC accumulation surrounding airways correlate with COPD disease severity (33). These synthetic functions place fibroblasts at the top of a hierarchy in controlling the shape and composition of the extracellular matrix while simultaneously facilitating immune cell trafficking in response to inflammatory stimuli.
Primary human or mouse lung fibroblasts upon IL-1β stimulation increase αvβ8 expression and αvβ8-mediated TGF-β activation and CCL20 (6). Proteomic cytokine analysis of lungs from Ad-IL-1β-treated mice reveal elevated levels of CCL20 that are reduced by postnatal fibroblast-conditional deletion of itgb8, or by treatment with an affinity-matured anti-β8 antibody, B5 (6, 7). Finally, increased CCL20 levels and increased numbers of DCs are associated with airway remodeling induced by CS; these increases are efficiently blocked by B5 (7). A number of questions remain: 1) Does αvβ8 play a role in driving CCL20 expression in human COPD lungs? 2) How do IL-1β- and αvβ8-mediated TGF-β activation together drive CCL20 expression in fibroblasts? 3) How important are fibroblasts as a source of CCL20?
Here we confirm that αvβ8 and CCL20 expression levels correlate in COPD lung parenchyma; the IL-1β-dependent transcriptional regulation of CCL20 occurs through a previously unreported mechanism requiring αvβ8-depenent binding of SMAD4 to an upstream CCL20 enhancer element, which forms a long range complex with the p50 subunit of NF-κB located on the proximal CCL20 promoter; airway fibroblasts are a dominant source of CCL20 in COPD. These findings are the first report, to our knowledge, of a cooperative transcriptional complex containing SMAD4 and NF-κB.
Experimental Procedures
Human COPD Cohort
Informed consent was obtained from all study participants as part of an approved ongoing research protocol (11-05710) by the University of California San Francisco Committee on Human Research in full accordance with the declaration of Helsinki principles. Human lung tissues were collected from 152 Caucasian subjects with documented COPD and 83 with no evidence of COPD based on assessment of medical information from a prospectively maintained clinical database. Further clinical information was obtained from medical records where required. All tissue specimens were histologically normal, excised from adjacent tumor tissue, from patients undergoing lung resection for lung cancer. All tissues were snap frozen and stored in liquid nitrogen at the time of collection. Both COPD and non-COPD patient groups were either current or past smokers and approximately matched for age, gender, and smoking pack year history. Clinical information is summarized in Table 1.
TABLE 1.
Patient demographics and smoking history
| COPD (n) | Non-COPD (n) | ||
|---|---|---|---|
| % | % | ||
| Age | 31–40 years | 0 (0) | 1.52 (1) |
| 41–50 years | 1.97 (3) | 4.55 (3) | |
| 51–60 years | 10.53 (16) | 13.64 (9) | |
| 61–70 years | 32.24 (49) | 34.85 (23) | |
| 71–80 years | 40.13 (61) | 28.79 (19) | |
| 81 + years | 15.13 (23) | 16.67 (11) | |
| Gender | Male | 44.08 (67) | 42.42 (28) |
| Female | 55.92 (85) | 57.58 (38) | |
| Smoking status | Current | 19.74 (30) | 15.15 (10) |
| Past | 72.37 (110) | 84.85 (56) | |
| No Information | 7.89 (12) | 0 (0) | |
| Smoking | 1–20 pack years | 9.21 (14) | 30.30 (20) |
| 21–39 pack years | 28.29 (43) | 30.30 (20) | |
| 40–59 pack years | 26.97 (41) | 25.76 (17) | |
| 60–79 pack years | 13.82 (21) | 9.09 (6) | |
| 80 + pack years | 15.13 (23) | 4.55 (3) | |
| No Information | 6.58 (10) | 0 (0) | |
Human and Mouse Lung Fibroblast Cell Culture
Leftover human explant lung samples from pulmonary pneumonectomies performed at Moffit-Long Hospital, University of California San Francisco, for lung cancer were gathered during the study period (2006–2012). Fibroblasts were cultured from the lung of humans and smad4f/f mouse parenchyma by the explant technique and used P1–P4, as previously described (4). Some fibroblasts were treated with human recombinant IL-1β (1 ng/ml) from R&D Systems. Mouse fibroblasts were either treated with Ad-LacZ or Ad-Cre to induce recombination, in vitro.
Cells and Reagents
Cell culture media and antibiotics were prepared by the University of California, San Francisco Cell Culture Facility using deionized water and analytical grade reagents. Fetal calf serum was obtained from Invitrogen, and human recombinant IL-1β and TGF-β1 were obtained from R&D Systems (Minneapolis, MN). Anti-human β8 affinity-matured clone B5, IgG2a, was produced in CHO cells as described (7). HeLa and hybridoma clone 1D11 were obtained from the American Tissue Type Collection (Manassas, VA). Clone 1D11 is a pan-TGF-β isoform monoclonal antibody that cross-reacts with TGF-β1, 2, and 3 of human, mink, and mouse origin. Anti-human MHC class I (IgG2a, ATCC) hybridomas were grown and purified using FPLC, as previously described (4). Antibodies were tested for endotoxin to confirm endotoxin levels <0.2 EU/μg as determined by the Limulus amebocyte lysate method (Genscript, Piscataway, NJ). ELISA kits for human CCL20 and IL-1β were from R&D Systems. TMLC TGF-β reporter cells were maintained in 10% FCS in DMEM (gift of John Munger, New York University Medical Center, New York, NY). P3U1 cells expressing human latent TGF-β1 were from H. Weiner (Harvard Medical School). Recombinant type 5 adenoviral vectors expressing Cre-eGFP fusion protein, eGFP, or LacZ were obtained from the Gene Transfer Vector Core (University of Iowa, Iowa City, IA).
ELISAs
Lung specimens were uniformly homogenized with stainless steel ball bearings and the TissueLyserII (Qiagen) homogenizer. Lung homogenates were obtained in phosphate-buffered saline with 1% Triton X-100 and protease inhibitors, 1× protease inhibitor mixture set I (Calbiochem), 1 mm Na3VO4 and 2 mm PMSF final concentrations. Primary human lung fibroblasts (2.5 × 106) were lysed in 1 ml of radioimmune precipitation assay buffer (Life Technologies) with 10 μl of protease inhibitor mixture (Thermo Life Technologies) and 1 mm Na3VO4 on ice for 5 min, and supernatants were collected after pelleting the insoluble material at 10,000 × g at 4 °C for 10 min. Lung homogenates and cell lysates were standardized to a working concentration of 1 mg/ml total protein using the BCA assay (Thermo Scientific Pierce). Mouse primary lung fibroblasts (3.0 × 105) from smad4 f/f mice were seeded onto 6-well dishes in complete medium, and the cells were transduced with either Ad-Cre or Ad-GFP (control). After 16 h, the medium was changed, and the cells (1 × 104) were transferred to individual wells of a 96-well plate. After 16 h, the cells were transfected with 25 nm siRNA to murine smad3 or control siRNA using Dharmafect (ON-TARGETplus Smad3 siRNA L-040706-00-0005; ON-TARGETplus nontargeting pool D-001810-10-05; Fisher Scientific). After 16 h, the cells were treated with 1 ng/ml recombinant hIL-1β (201-LB-005; R&D Systems). After 16 h, the supernatants were harvested.
Human CCL20, IL-1β, and pSMAD2/3 or mouse CCL20 sandwich ELISAs were performed using the human or murine CCL20/MIP3α (DY360, DY760), IL-1β (DY201; R&D Systems), or PathScan® Phospho-Smad2 (Ser-465/467)/Smad3 (Ser-423/425) sandwich ELISA (12001; Cell Signaling Technologies, Danvers, MA) according to the manufacturer's instructions. Briefly, 20 μg of lung homogenate or 100 μl of culture supernatant (CCL20) or 50 μg (IL-1β) or 100 μg (pSMAD2/3) of total protein lysate were loaded on the respective ELISAs. Human αvβ8 was detected by sandwich ELISA using an affinity-matured antibody (clone 42) specific to the βI domain of β8 and using biotinylated antibody (clone 6B9) specific to the β8 Psi domain as the second antibody. All ELISAs were detected using streptavidin-HRP and TMB substrate (Thermo Scientific Pierce).
Ad-Cre and siRNA-mediated gene knockdown were confirmed using SYBRGreen PCR (Applied Biosystems) with primers to smad3 or smad4 (RT2 qPCR Primer Assay, Qiagen) using murine β-actin primers as a control (forward, 5′-AGAGGGAAATCGTGCGTGAC-3′; and reverse, 5′-CAATAGTGATGACCTGGCCGT-3′).
Mice
All mice were bred and housed in specific pathogen-free housing under an institutional review board-approved protocol (institutional animal care and use committee AN098258) and in accordance with the guidelines of the Laboratory Animal Resource Center of the University of California, San Francisco. smad4f/f mice from Chuxia Deng (NIDDK, National Institutes of Health) were backcrossed eight generations to C57BL/6.
In Silico Promoter Analysis
2 kb of mouse (C57BL/6J MMCHR1_ region: 7993210 … 7995209) and human (HSCHR2 region: 78885977 … 78887976) 5′-flanking regions were interrogated using MatInspector (Genomatrix Software Suite v3.0).
Promoter Constructs
Vector PGAC −1836 + 71 hCCL20 (1.9 pGL3 (PGAC), a gift from C. D. Jun, Digestive Disease Research Institute, Wonkwang University School of Medicine, Chonbuk, Korea) (35) was used as a template for PCR (Phusion) with SBE mutant primers (forward, 5′-ctctgatatccatgCATTGGTTATTTAGGAATATGTGGTTTC-3′; and reverse, 5′-Gttctctaggataaagcatatgg-3′). The product was cloned in PGAC cut with MscI and NdeI. The NF-κB mutation was transferred from an NF-κB CCL20 mutant promoter construct into PGAC (a gift from M. Rumbo) (36).
Promoter Assays, Chromatin Immunoprecipitations, Transfections, and TFG-β Activation Assays
These assays were performed essentially, as described (4, 6, 22, 30). For ChIP, 5 μg/IP of anti-SMAD4 (Santa Cruz Biotechnology, Santa Cruz, CA) and 1 μg/IP of anti-p50 (Cell Signaling) were used. For EMSA, recombinant SMAD4 was from Fitzgerald Industries (Acton, MA), and anti-SMAD3 and anti-SMAD4 were from Santa Cruz Biotechnology (Santa Cruz, CA). Biotinylated probes corresponding to SBE2 of the CCL20 promoter were SDS-PAGE-purified (ELIM Biotechnologies, Alameda, CA).
Immunohistochemical, Immunocytochemical Staining, and Analysis
Paraffin-embedded tissue sections from 5 normal and 11 COPD patients underwent standard antigen retrieval, followed by immunohistochemical detection of CCL20, using a goat anti-human CCL20 antibody (AF360; R&D Systems) and detection using a Dako Envision kit (Dako, Carpenteria, CA). Staining of sections was graded by a pulmonary pathologist (S. L. N.) in the epithelial or mesenchymal compartment as follows: 0 to 1+, negative to indeterminate cytoplasmic/cell process staining; 2+, weak but clearly positive cytoplasmic/cell process staining; and 3+, strong cytoplasmic/cell process staining.
For confocal microscopy, paraffin sections were prepared as above with the exception that additional antibodies were added (rabbit anti-human CD11c, Epitomics, Burlingame, CA; mouse anti-smooth muscle actin, 1A4, Cell Marque, Rocklin, CA), and anti-human CD45 clones 2B11 + PD7/26 (Cell Marque, Rocklin, CA) were detected by Alexa 647 anti-mouse or rabbit with anti-goat Alexa 488 (Life Technologies). Sections were mounted using Prolong Gold anti-fade mounting media with DAPI (Molecular Probes) and analyzed on a Yokogawa spinning disk confocal microscope.
Statistical Analysis
All data are reported as means ± S.E. Comparisons between two different groups were determined using Student's t test for parametric data or Mann-Whitney for nonparametric data. One-way analysis of variance was used for multiple comparisons and Tukey's or Bonferroni's post hoc tests used to test for statistical significance. Significance was defined as p < 0.05. Logistic regression analysis was performed using Stata (v12.1). All other statistical analyses were performed using the software package Prism 4.0b (GraphPad Software, San Diego, CA).
Results
αvβ8, CCL20, and IL-1β Expression Is Increased in COPD
A cross-sectional study was performed comparing lung parenchyma of smokers with and without COPD matched for age, sex, and cigarette pack years (cohort described in Table 1). Lung homogenates were evaluated by ELISA for αvβ8, CCL20, and IL-1β. αvβ8, CCL20, and IL-1β were significantly increased in lung parenchyma of COPD patients compared with smokers without COPD. The mid-upper level of IL-1β in the COPD cohort was 10 pg/ml (Fig. 1). We tested whether a 16-h treatment with 10 pg/ml could increase CCL20 expression in human lung fibroblasts. IL-1β (10 pg/ml) significantly induced CCL20 expression in primary human lung fibroblasts (CCL20: PBS control, 0.000 ± 0.000 versus IL-1β-treated, 36.49 ± 13.45 pg/ml supernatant, p = 0.009).
FIGURE 1.
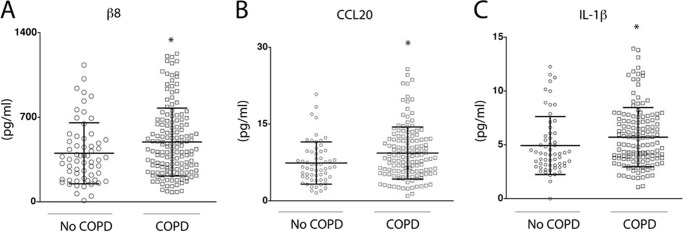
αvβ8, CCL20, and IL-1β levels in human lung parenchyma from smokers with or without COPD. Individual samples from smokers without COPD (open circles) compared with smokers with COPD (open squares) determined by ELISA (pg/ml) for integrin αvβ8 (A), CCL20 (B), or IL-1β (C). *, p < 0.05 as determined by Mann-Whitney test. n = 83 smokers without and n = 152 with COPD.
Levels of αvβ8 and CCL20 were significantly correlated, in smokers with COPD, but not those without COPD (Figs. 1 and 2 and Table 2). Levels of IL-1β and CCL20 were significantly correlated, in smokers with and without COPD (Fig. 2 and Table 3). We performed regression analysis conditioning on age, gender, and smoking duration to assess whether the increases were attributable to individual effects of these covariates independent of COPD disease status, and they were not (p ≥ 0.233). There was a nonsignificant correlation between αvβ8 and IL-1β levels (Table 3). Taken together, these data support the hypothesis that increased IL-1β levels drive CCL20 expression in smokers, and together, αvβ8 and CCL20 drive the COPD phenotype in susceptible smokers.
FIGURE 2.

CCL20 expression correlates with αvβ8 and IL-1β expression. Linear regression analysis of protein levels αvβ8, IL-1β, and CCL20 expressed as natural log (Ln) of protein (mg/ml) is shown. A–D, correlation of CCL20 expression from lung parenchymal lysates of smokers with COPD (A and C) and smokers without COPD (B and D) with αvβ8 (A and B) and IL-1β (C and D). The coefficient of determination (r2) values with associated p values are shown in each panel.
TABLE 2.
αvβ8, CCL20, and IL-1β protein detection (ELISA) in lung parenchymal lysates of smokers with and without COPD
| Protein | Disease status | n | Mean protein concentration ± S.D. | Mann-Whitney p value |
|---|---|---|---|---|
| pg/ml | ||||
| αvβ8 | Smokers, Non-COPD | 60 | 402.1 ± 252.3 | 0.0203 |
| COPD | 139 | 495.7 ± 281.4 | ||
| CCL20 | Smokers, Non-COPD | 59 | 7.398 ± 4.1 | 0.0081 |
| COPD | 137 | 9.346 ± 5.1 | ||
| IL-1β | Smokers, Non-COPD | 60 | 4.936 ± 2.7 | 0.0282 |
| COPD | 140 | 5.714 ± 2.8 |
TABLE 3.
Correlations of αvβ8, CCL20, and IL-1β protein expression in lung parenchymal lysates of smokers with and without COPD
| Correlation | Cohort | n | Pearson r | 95% confidence interval | r2 | Two-tailed p value |
|---|---|---|---|---|---|---|
| αvβ8:CCL20 | Non-COPD smokers | 53 | −0.103 | −0.363–0.172 | 0.011 | 0.464 |
| COPD smokers | 130 | 0.301 | 0.136–0.450 | 0.090 | 0.0005 | |
| αvβ8:IL-1β | Non-COPD smokers | 55 | 0.007 | −0.259–0.272 | 0.5 × 10−4 | 0.958 |
| COPD smokers | 135 | 0.056 | −0.114–0.223 | 0.003 | 0.521 | |
| IL-1β:CCL20 | Non-COPD smokers | 55 | 0.372 | 0.119–0.580 | 0.138 | 0.005 |
| COPD smokers | 137 | 0.325 | 0.116–0.468 | 0.106 | <0.0001 |
CCL20 Expression by Primary Lung Fibroblasts Is Induced by IL-1β and Is Dependent on Autocrine αvβ8-mediated TGF-β Activation
We sought to understand the mechanistic basis of interaction of αvβ8 with CCL20 in driving the COPD phenotype. We have previously determined that increased CCL20 secretion in response to IL-1β by primary human lung fibroblasts is dependent on αvβ8 and TGF-β, because anti-β8 or anti-TGF-β neutralizing antibodies significantly block IL-1β-induced CCL20 expression by both normal and COPD lung fibroblasts (6, 7). This IL-1β-dependent increase in CCL20 expression correlates with increased phosphorylation of pSMAD2/3, which is significantly blocked by anti-β8 (B5) neutralizing antibodies (Fig. 3A). To determine the SMAD dependence of IL-1β-induced CCL20 secretion by fibroblasts, smad4 was conditionally deleted from primary mouse lung fibroblasts (Fig. 3B). Addition of IL-1β to control (Ad-LacZ) treated smad4 f/f fibroblasts significantly increased murine CCL20 expression (Fig. 3B). Ad-Cre treatment of smad4 f/f fibroblasts caused a 76.2 ± 5.7% decrease (p < 0.0001) in smad4 transcript levels and inhibited 77.4 ± 11.7% of the IL-1β-induced TGF-β-dependent CCL20 expression (Fig. 3B). Thus, both human and mouse CCL20 expression is highly induced by IL-1β and significantly enhanced by autocrine SMAD4-dependent TGF-β signaling.
FIGURE 3.
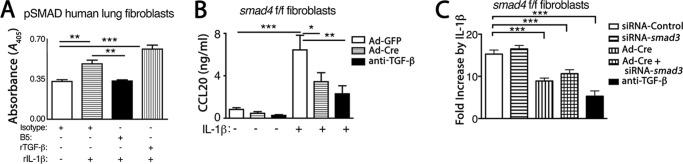
αvβ8-mediated SMAD4 signaling is required for IL-1β-mediated CCL20 expression. A, pSMAD2/3 phosphorylation as determined by ELISA (Cell Signaling) is increased by IL-1β and inhibited by antibody inhibition of αvβ8-mediated TGF-β activation. Human lung fibroblasts were treated with IL-1β, with isotype control or anti-αvβ8, clone B5 (7). As a control, recombinant active TGF-β (rTGF-β) was added to IL-1β-treated cells, which further increased pSMAD2/3 phosphorylation. B, conditional deletion of smad4 in mouse lung fibroblasts inhibits IL-1β-induced CCL20 expression. Primary cultures of lung fibroblasts from smad4 f/f mice were transduced with Ad-Cre or Ad-GFP control 24 h prior to treatment with or without recombinant human IL-1β, in the presence or absence of the pan-TGF-β blocking antibody (1D11) for 16 h. Harvested supernatants were assessed for CCL20 protein expression by ELISA. The results are expressed as ng/ml. *, p < 0.05; **, p < 0.01; ***, p < 0.001. C, siRNA knockdown of smad3 in mouse lung fibroblasts does not inhibit IL-1β-induced CCL20 expression. Primary cultures of lung fibroblasts from smad4 f/f mice were transduced with Ad-Cre or Ad-GFP control, as above. 16 h later, cells were transfected with siRNA to smad3 or siRNA control and after another 16 h were treated with or without recombinant human IL-1β (1 ng/ml). Harvested supernatants were assessed for CCL20 protein expression by ELISA, and the results are shown as fold induction over non-IL-1β-treated controls. The extent of inhibition of IL-1β-induced CCL20 expression by pan anti-TGF-β (1D11) is shown (filled bar). n = at least 4 per group. ***, p < 0.001.
Smad3 has been widely implicated in fibrotic responses and intensely studied in fibrotic models because smad3 (but not smad2 and smad4) knock-out mice are viable (37). To determine whether smad3, a prototypic receptor-activated SMAD (R-SMAD), was required in IL-1β-induced CCL20 expression, siRNA was used to knock down smad3 expression in smad4 f/f lung fibroblasts. siRNA to smad3 caused a 58.9 ± 18.6% (p = 0.035) decrease in smad3 transcript levels. However, smad3 knockdown had no effect on IL-1β-induced CCL20 expression (Fig. 3C). Furthermore, siRNA to smad3 when combined with smad4 Ad-Cre-mediated deletion had no additional reduction of IL-1β-induced CCL20 expression compared with smad4 deletion alone (Fig. 3C).
A TGF-β-responsive Upstream SMAD-binding Element Is Required for Efficient IL-1β-dependent CCL20 Expression
In silico examination of the CCL20 promoter including 2 kb of the 5′-flanking region from the transcriptional start site of mouse (and human) revealed canonical NF-κB binding sites located −83 and −82 bp 5′ to the respective transcriptional start sites (Fig. 4A). SMAD binding elements (SBEs) were located at −552, −779, and −1006 in the mouse and −1742 (SBE2) and −1826 (SBE1) in the human 5′ CCL20 flanking regions (Fig. 4A).
FIGURE 4.
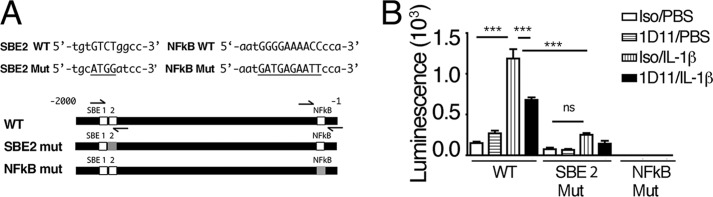
An upstream SMAD binding element is required for IL-1β-mediated CCL20 expression. A, a schematic representation of 2000 base pairs of the 5′-flanking region of human CCL20. The location of the SMAD binding elements (SBE1 and SBE2) and the NF-κB sites are shown to scale as white boxes, and the mutated sequences in the respective SBE2 and NF-κB mutations introduced into luciferase promoter constructs are shown as gray boxes. The locations of primer sequences used to amplify SBE2 and NF-κB from ChIP are indicated as half arrows above and below the bars. The nucleotide sequences of the WT and mutant (Mut) SBE2 and NF-κB probes used in EMSAs are shown. B, the WT, SBE2, and NF-κB mutant promoter constructs were transfected into HeLa cells. Transfected cells were treated for 16 h with vehicle control (PBS) or recombinant IL-1β with isotype control (Iso) or anti-TGF-β (1D11). Results from three experiments performed in duplicate are shown as luminescence in relative light units (minus background from a promoterless control construct). ***, p < 0.001; ns, not significant.
WT and mutant human promoter constructs were designed to include 1,836 bp of the human 5′-flanking region upstream from the transcriptional start site and the first 71 bp of CCL20 transcript. A truncated promoter construct lacking SBE1 was also produced. Additional constructs contained WT or a mutated SMAD4 binding element (SBE2) or a WT or mutated canonical NF-κB site (Fig. 4A). Promoter constructs were transfected into HeLa cells. The WT CCL20 promoter construct responded with a significant induction to IL-1β stimulation and was significantly inhibited by anti-TGF-β antibodies (Fig. 4B). The truncation mutant lacking SBE1 was induced nearly identically by IL-1β as the full-length construct (1.003 ± 0.13-fold induction, n = 4), indicating that it was not involved in transcriptional regulation of CCL20. In contrast, the SBE2 and the NF-κB mutants were not significantly induced by IL-1β compared with the WT construct (Fig. 4B). These data demonstrate the importance of the upstream SBE2 and the proximal NF-κB DNA-binding sites in the human CCL20 promoter as critical promoter/enhancer elements involved in autocrine TGF-β-mediated amplification of IL-1β-induced CCL20 transcriptional responses.
The TGF-β-responsive SMAD-binding Element Cooperates with a Proximal NF-κB Binding Site to Enhance IL-1β-dependent CCL20 Expression
ChIP of human lung fibroblasts stimulated with IL-1β was performed to detect an association of the NF-κB binding site with the upstream SBE2 of the CCL20 promoter region. Time courses of ChIPs with an antibody to a prototypic NF-κB complex member, NF-κB-1 (p50), revealed a significantly increased association of p50 with both the NF-κB binding consensus sequence and SBE2 that peaked 2 h after IL-1β treatment (Fig. 5, A and B). Time courses of ChIPs using antibodies to SMAD4 also revealed significant binding to SBE2 peaking at 2 h after IL-1β treatment (Fig. 5C). These results suggest that p50 forms long range complexes containing both the proximal NF-κB motif and the upstream SBE2 site on the CCL20 promoter/enhancer (Fig. 5D).
FIGURE 5.

SMAD4 and NF-κB cooperate to mediate IL-1β- and αvβ8-dependent CCL20 expression. A–E, ChIP from human lung fibroblasts using anti-p50 of the NF-κB complex (A and B) or anti-SMAD4 (C–E) demonstrating IL-1β-dependent association of p50 with the NF-κB DNA recognition motif (A) or SBE2 (B) or IL-1β-dependent association of SMAD4 with SBE2 (C and D) or the NF-κB DNA recognition motif (E). A–C, time courses after IL-1β treatment (1 ng/ml) of p50 or SMAD4 ChIPs showing amplification of the region containing the NF-κB DNA recognition motif (A) or SBE2 site (B and C) in the 5′-flanking region of human CCL20. No enrichment at any time point was seen in the absence of IL-1β, and the results are shown as fold enrichment relative to PBS treated fibroblast controls. C, an illustration of the long-range interactions of SMAD4 and p50 with their cognate recognition sequences is shown. C and D, amplifications of the SBE2 (C) or NF-κB regions (D) in anti-SMAD4 ChIPs 2 h after recombinant IL-1β (1 ng/ml) treatment of human lung fibroblasts with or without isotype or B5 (n = 3) are shown. **, p < 0.01; ***, p < 0.001.
To demonstrate that SMAD4 forms complexes with the proximal NF-κB site, we performed anti-SMAD4 ChIPs with IL-1β-stimulated human lung fibroblasts. After 2 h of IL-1β treatment, a significant enrichment of both the upstream SBE2 and the NF-κB DNA-binding sites were seen (Fig. 5, E and F). These results demonstrate that like p50, SMAD4 forms complexes with the upstream SBE2 and the proximal NF-κB motif on the CCL20 promoter/enhancer (Fig. 5F).
Integrin αvβ8-mediated TGF-β activation is critical in regulating SMAD4 interactions with the SBE2 and NF-κB sites because their enrichment in SMAD4 ChIPs was significantly inhibited by pretreatment with anti-β8 (B5) neutralizing antibody (Fig. 5, E and F). These data demonstrate that autocrine αvβ8-mediated activation of TGF-β is involved in cooperative p50/SMAD4 interactions on the CCL20 promoter.
To demonstrate the direct interaction of the upstream SBE2 with SMAD4, EMSAs were performed using recombinant SMAD4 and biotinylated oligonucleotides matching the sequence of the upstream SBE2 (Fig. 4A). Recombinant SMAD4 bound to the labeled oligonucleotide and gave three different bands likely representing different oligomerization states of SMAD4. These bands were specific to interactions to SMAD4 with SBE2 because they competed with an excess of WT oligonucleotides but not oligonucleotides with a mutant SBE2 (Fig. 6A). The binding of SBE2 to oligomeric SMAD4 containing complexes occurred in intact human cells because the labeled SBE2 oligonucleotide bound to primary fibroblast nuclear extracts with a similar banding pattern as seen with recombinant SMAD4 (Fig. 6B). The intensity of several of these bands was slightly increased by treatment with IL-1β, consistent with the enrichment of this DNA region seen in ChIP (Fig. 5, A–C). Finally, supershift assays with SMAD3 or SMAD4 antibodies revealed a diminution in band intensity relative to control IgG treated cells, with weak shifts seen with SMAD3 antibodies and a near complete absence of signal with SMAD4 antibodies (Fig. 6C), suggesting that SMAD3 and SMAD4 antibodies interfere with complex assembly or stability required for DNA binding. Taken together, these data demonstrate the existence of higher order IL-1β- and TGF-β-dependent NF-κB-SMAD4 complexes on the human CCL20 promoter.
FIGURE 6.

Direct binding of SMAD4 to the CCL20 SMAD binding element (SBE2). A, EMSA with a biotinylated probe containing SBE2 of the human CCL20 promoter and recombinant SMAD4 (lane 1) in the presence of WT (lane 2) or SBE2 mutant oligonucleotides (lane 3) or anti-SMAD4 (lane 4). The positions of three bands and a fourth supershifted band with anti-SMAD4 are shown. B, EMSA using nuclear extracts of human lung fibroblasts treated with vehicle control (lanes 1–3) or recombinant IL-1β (lanes 3–6) in the presence of WT (lanes 2 and 5) or SBE2 mutant oligonucleotides (lanes 3 and 6). The positions of the three lower bands are similar to those seen in A. C, EMSA using nuclear extracts of human lung fibroblasts treated with recombinant IL-1β with no antibody (lane 1), control IgG (lane 2), anti-SMAD3 (lane 3), or anti-SMAD4 (lane 4). Supershifts with anti-SMAD3 (lane 4) or competition with anti-SMAD4 (lane 4) are shown. Mut, mutant; oligo, oligonucleotide.
Fibroblasts Are a Major Source of Increased CCL20 Secretion in COPD
We sought to confirm that airway fibroblasts were an important CCL20-expressing cell type in COPD airways, as a step in understanding the translational relevance of our findings. We performed CCL20 immunofluorescent confocal microscopy of COPD airways and observed two patterns of staining: the first on the luminal border of epithelial cells and the second as linear spindle cell-like extensions located on the edge of autofluorescent collagen bundles in the lamina propria below the airway epithelium. These spindle cell-like extensions most likely represent fibroblast cell processes because they are located in the lamina propria where fibroblasts are enriched, are located next to collagen fibers, and do not colocalize with smooth muscle actin, a pan-leukocyte marker (CD45), or a dendritic cell and macrophage marker (CD11c) (Fig. 7, A–C). The intensity of staining in airway epithelial cells and the fibroblast-like cells in normal controls and COPD airways was determined using immunohistochemistry. We determined that staining in the subepithelial fibroblast-like cells from COPD patients was highly enriched and significantly increased from subepithelial fibroblast-like cells from patients without COPD (Fig. 7, D–F). Airway epithelial cells had a similar high staining intensity in COPD and normal patients (Fig. 7, D–F). To determine the relative cell type contribution to CCL20 expression, primary cultures of human lung fibroblasts, bronchial epithelial cells, and astrocytes, all cell types known to express CCL20 (6, 38), were evaluated. IL-1β robustly increased CCL20 expression in all cell types; IL-1β-treated fibroblasts secreted the highest levels of CCL20 on a per cell basis (Fig. 7G).
FIGURE 7.

Increased expression of CCL20 by human airway fibroblasts. A–C, immunofluorescence localization of CCL20 in COPD airway samples. Coimmunofluorescence staining and detection by confocal microscopy of CCL20 with a smooth muscle cell marker, smooth muscle actin (SMA) (A), a pan-leukocyte marker, CD45 (B), or a macrophage and dendritic cell marker (CD11c) (C). CCL20 staining is green; smooth muscle actin, CD45, and CD11c are in red; and nuclear counterstain and autofluorescent collagen are blue. Scale bar, 15 μm. Arrowheads point to CCL20 staining in airway epithelial cells, arrows point to CCL20 staining of fibroblast-like cell processes, and asterisks indicate smooth muscle actin staining (A), CD45-stained cells (B), and CD11c-stained cells (C). D and E, immunohistochemical staining for CCL20 from normal (D) or COPD (E) airway samples reveals increased CCL20 expression in subepithelial fibroblasts in COPD. AE, airway epithelium; Fib, fibroblasts. Arrowheads point to apical staining of CCL20 on epithelial cells; arrows point to CCL20 staining on mesenchymal cell processes. F, semiquantitative analysis of CCL20 expression by fibroblasts, as well as airway epithelial cells reveals increased staining of fibroblasts in COPD (n = 11) compared with normal (n = 5). ***, p < 0.001 by Student's t test. G, primary human COPD lung fibroblasts (n = 3) show increased CCL20 expression in response to IL-1β stimulation. Human bronchial epithelial cells (HBEC) and astrocytes secrete less CCL20 on a per cell basis compared with lung fibroblasts. ***, p < 0.001 by analysis of variance and Tukey's post-test.
Discussion
This study has advanced the mechanistic understanding of the complex regulation of CCL20 in COPD. Here we have determined that αvβ8, CCL20, and IL-1β are increased in COPD lung parenchyma compared with smokers without COPD and/or nonsmoking controls. We have elucidated a pathway where TGF-β activated via a αvβ8-dependent mechanism synergistically increases IL-1β-dependent CCL20 expression by lung fibroblasts. Finally, we have determined that airway fibroblasts are an important source of increased CCL20 in COPD patients. These results and the demonstrated efficacy of inhibiting αvβ8-mediated TGF-β activation in prevention of experimental airway remodeling (7), together with the mechanistic insights provided here, provide compelling evidence that CCL20 is a crucial downstream mediator of the fibroinflammatory effects of αvβ8.
Fibroblasts Are an Important Cellular Source of CCL20 in COPD
The cell types relevant to airway remodeling that express αvβ8 and IL-1β have been previously suggested to be fibroblasts (6) and airway epithelial cells (38), respectively, with the major source of CCL20 open to debate. We have use confocal colocalization studies to identify CCL20 positive fibroblast-like cell processes on the edges of collagen fibers in the lamina propria of COPD airways. These cell processes are not from smooth muscle or immune cells, because they do not colocalize with immune and smooth muscle cell markers. Thus, the evidence of the CCL20-expressing cells in the lamina propria being fibroblasts remains indirect, because there are no specific markers for fibroblasts. We have studied primary cells in vitro to determine that on a per cell basis, CCL20 is expressed most highly by IL-1β-stimulated fibroblasts, which supports the contention that the CCL20-positive cells in the COPD airway lamina propria are indeed fibroblasts.
Increased IL-1β expression in COPD lungs provides a mechanism for the observed increased CCL20 expression by airway fibroblasts in remodeled airways in COPD patients. Local production of IL-1β by airway epithelial cells acts on adjacent fibroblasts to increase both αvβ8 expression and CCL20 production (4, 6). Increased DC numbers detected in COPD airway-adjacent zones (6) could be plausibly attributed to local increases in fibroblast CCL20 secretion because DCs express CCR6, the only receptor for CCL20 (39). An increase in CCL20 through this mechanism could contribute to increased immune cell priming and the chronic inflammation that characterizes COPD (40).
A Novel SMAD-p50 Complex Regulates CCL20 Expression
The basis of the αvβ8 and TGF-β dependence for IL-1β-induced CCL20 production by fibroblasts was initially unclear, because the TGF-β and IL-1β pathways have not to our knowledge been shown to directly interact. In fact, the TGF-β and IL-1β pathways are generally thought to indirectly antagonize each other through such mechanisms as the IL-1β-mediated induction of the inhibitory SMAD7 (41). A potential mechanism for TGF-β and IL-1β interaction became immediately clear by investigating the 5′ noncoding region of CCL20 where a number of SBEs were identified upstream of the canonical NF-κB DNA-binding site in the CCL20 promoter.
Ad-Cre-mediated deletion of smad4 inhibited IL-1β-mediated induction of CCL20 ∼77%, as well as a pan-isoform TGF-β blocking antibody. The ∼23% of enhanced IL-1β-mediated induction of CCL20 that was not inhibited by loss of smad4 was likely due to incomplete deletion because Ad-Cre only reduced smad4 transcript levels by ∼76%. Alternatively, it is possible that SMAD-independent TGF-β signaling is able to partially compensate for loss of smad4 (42). It has previously been reported that smad4-deficient mouse embryo fibroblasts retain some responsiveness to TGF-β (43). Whatever the case, in adult lung fibroblasts, smad4 represents the major common mediator of TGF-β signaling involved in enhancing IL-1β-mediated induction of CCL20.
TGF-β typically signals through an ALK5 (activin-linked kinase 5) receptor-containing complex to induce phosphorylation and activation of SMAD2 and SMAD3, which form heterotrimers and heterodimers with SMAD4 before translocating to the nucleus to initiate gene transcription (42). SMAD2 and SMAD3 share many overlapping functions, but SMAD2 differs from SMAD3 and SMAD4 by lacking an essential DNA-binding domain (42). We attempted to address R-SMAD signaling mediators upstream of SMAD4 using a knockdown approach. We were not able to verify that SMAD3 was required for efficient IL-1β-mediated induction of CCL20 by lung fibroblasts despite having identified SMAD3 bound to SBE2 in EMSA assays. Failure to detect a functional role for SMAD3 may be due to incomplete knockdown and/or redundant involvement of other R-SMADs. The most likely candidate is SMAD2, but TGF-β has also been reported to signal through a receptor complex containing ALK1 in mesenchymal cells via SMAD1 and/or SMAD5 (44, 45). Therefore, the individual roles of the various R-SMADs in enhancing IL-1β-mediated induction of CCL20 remain to be determined.
We verified that SMAD4 binds to SBE2, and the interaction peaked 2 h after IL-1β treatment. This increase overlapped with the peak of p50 binding to the proximal NF-κB site, which in turn also overlapped with the peak of detection of SBE2 in p50 ChIPs. The temporal IL-1β-dependent increase of SBE2 and NF-κB DNA-binding sites detected in p50 ChIPs was transient and not sustained, consistent with the known fast association and disassociation rates of NF-κB from DNA (46–48).
The magnitude of the IL-1β-dependent increases in the SBE2 and the proximal NfκB sites detected in p50 ChIPs (∼2.5-fold increases) were less than the corresponding increases in SMAD4 ChIPs (∼10–20-fold increases). The reason for this difference may be rapid dissociation of NF-κB from DNA, differences in amounts in antibody used (i.e. 1 μg versus 5 μg/IP for p50 versus SMAD4), the immunoprecipitation efficiencies of the respective antibodies, or the relative affinities of p50 or SMAD4 for their DNA substrates.
Our study represents the first evidence of a SMAD-p50 complex. Convergence of TGF-β and IL-1β signaling via higher order transcription factor complexes provides a mechanism by which αvβ8-dependent TGF-β activation amplifies inflammatory responses. However, we have not explored the exact mechanism of interaction of p50 with SMAD4. The interaction is likely to be indirect and complex, requiring multiple other protein components such as R-SMADs and other NF-κB family members (i.e. pSMAD2/3, RelA, RelB, or c-Rel).
TGF-β in Amplifying Pathologic Immune Responses
TGF-β has complex roles in immune regulation with an extensive literature documenting both proinflammatory and anti-inflammatory roles (49). Recent toxicity studies using pan-TGF-β antibody inhibitors fail to reveal evidence of systemic autoimmunity (50). To the contrary, antibody inhibition or condition genetic deletion of αvβ8-mediated TGF-β activation significantly inhibits lung inflammation, fibroinflammatory responses, and airway smooth muscle hyper-responsiveness in several preclinical models of asthma or COPD (6, 7). The role of TGF-β function in pathology appears to be highly context- and cell type-dependent. It appears that in the lung, αvβ8-mediated TGF-β activation plays important roles in the recruitment and immune priming functions of DCs, which are required for adaptive immune responses (6, 7). Therefore, the TGF-β-dependent regulation of CCL20 is likely to play a central and crucial role in fibroinflammatory responses in the lung.
The αvβ8/IL-1β/CCL20 Network in COPD
Previous studies have investigated increased expression of αvβ8, CCL20, or IL-1β in small cohorts of COPD patients, using a variety of specimen sources, but none have used quantitative measures of these proteins from lung parenchyma, arguably the most relevant biospecimen. In COPD patients, increased αvβ8 in lung sections is suggested by semiquantitative immunohistochemistry (6), increased CCL20 levels by ELISAs of sputum (33), and increased protein levels of IL-1β by ELISA of human serum (11), exhaled breath condensate (12), and sputum (13, 14). Our current study represents a comprehensive analysis of these proteins in a large COPD case control cohort. This cohort was sufficiently powered to find significant increases of each of these proteins in COPD samples that were independent of smoking history and other covariates.
Elevations in IL-1β and CCL20 were relatively small in COPD compared with non-COPD patients, which might be expected because the cohort contained only stable COPD patients. It is likely that the cytokine and chemokine levels would be significantly increased during episodes of acute exacerbation (51). Even with this caveat, the slight IL-1β elevations of 10–15 pg/ml are sufficient to significantly induce CCL20 expression in human fibroblasts. Furthermore, this amount is in the same range of IL-1β secreted by the synovium of patients with rheumatoid arthritis, a disease where IL-1β targeted therapies result in patient benefit (52, 53).
The threshold of biologic activity for CCL20 in vitro is ∼20 pg/ml (54). The upper range of levels of CCL20 that we find in COPD patients is ∼25 pg/mg in lung tissue homogenates. The relatively low level of CCL20 that we find in human lung tissue homogenates likely reflects the relative paucity of airways in the random lung parenchymal samples that were collected for this study because CCL20 expression is concentrated around airways. Furthermore, the elevations of CCL20 in human COPD lung tissue (on the basis of CCL20/mg lung tissue) is close to what we find in lung homogenates of cigarette-exposed mice (7). This level of CCL20 induction is physiologically significant because it correlates with an increase in adaptive T-cell immunity and airway remodeling in CS-exposed mice. These increases are not seen in ccr6-deficient mice (40). Finally, induced sputum from COPD patients (which represent concentrated large airway washings) contains ∼2 ng/ml CCL20, reinforcing the idea that CCL20 is concentrated in and around the airways in physiologically significant amounts in COPD (33). Individual correlations between αvβ8 or IL-1β with CCL20 expression in smokers with COPD validates a hypothetical framework where IL-1β- and αvβ8-mediated TGF-β activation cooperate to drive CCL20 expression; αvβ8 and CCL20 together drive the COPD phenotype.
We have previously determined that IL-1β increases the expression of αvβ8 by fibroblasts, yet here we failed to find a significant correlation between IL-1β and αvβ8 levels (30). It is possible that our relatively large cohort was still underpowered to find a correlation. Alternatively, it is possible that the IL-1β levels were not sufficiently elevated in our cohort to affect ITGB8 transcription. Therefore, it is possible that IL-1β-driven increased αvβ8 expression occurs mainly during COPD exacerbations.
How Do Increased CCL20 Levels Affect the COPD Phenotype?
CCR6 is the only known receptor for CCL20 and is highly expressed by immature DCs, which accumulate around COPD airways (33). DCs are critical to prime adaptive immunity but have not been shown to directly influence airway remodeling. We have recently found that DC depletion and deficiency in the crucial DC chemokine receptor, CCR6, both protect from Ad-IL-1β-induced airway adaptive T-cell immune responses, and airway fibrosis (40). These results provide direct evidence that DCs are critical to airway fibrosis. We have further determined that ccr6 and fibroblast expression of αvβ8 are crucial for lung dendritic cell motility (chemokinesis), which causes DCs to accumulate around the airways of cigarette-smoke exposed mice.4 Taken together, our findings suggest an important functional role for αvβ8 and CCL20 in the chronic inflammation that contributes to airway remodeling in COPD.
Acknowledgments
We thank John Munger (New York University) for TMLC reporter cells. Special thanks to M. Rumbo and C. D. Jun for CCL20 promoter constructs and Chuxia Deng for smad4+/− mice.
This work was supported, in whole or in part, by National Institutes of Health Grants HL113032, HL063993, HL090662, and NS044155; University of California-Center for Accelerated Innovation Grant U54HL119893; University of California Proof of Concept Award; University of California San Francisco Liver Center Grant P30DK026743 (to S. L. N. and J. L. B.); and by funds from the University of California Tobacco Related Disease Research Program, the University of California San Francisco Academic Senate, the Nina Ireland Lung Disease Program (to P. W.), the Leon Fredericq Foundation, the Belgian Respiratory Society, the University of Liege, and the European Union Seventh Framework Programme for Research (FP7), Co-funding of regional, national, and international programmes (COFUND)-BeIPD Project (to C. M.). J. L., J. D. M., J. L. B., and S. N. have received royalty payments from the Regents of the University of California for β8 neutralizing antibodies used in this work.
M. Hashimoto, submitted for publication.
- COPD
- chronic obstructive pulmonary disease
- CS
- cigarette smoke
- Ad
- adenoviral
- DC
- dendritic cell
- SBE
- SMAD binding element.
References
- 1. Mininño A. M., Xu J. Q., Kochanek K. D. (2010) Deaths: preliminary data for 2008. National Vital Statistics Reports 59, 1–52 [PubMed] [Google Scholar]
- 2. Ito K., Ito M., Elliott W. M., Cosio B., Caramori G., Kon O. M., Barczyk A., Hayashi S., Adcock I. M., Hogg J. C., Barnes P. J. (2005) Decreased histone deacetylase activity in chronic obstructive pulmonary disease. N. Engl. J. Med. 352, 1967–1976 [DOI] [PubMed] [Google Scholar]
- 3. Postma D. S., Timens W. (2006) Remodeling in asthma and chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc. 3, 434–439 [DOI] [PubMed] [Google Scholar]
- 4. Araya J., Cambier S., Markovics J. A., Wolters P., Jablons D., Hill A., Finkbeiner W., Jones K., Broaddus V. C., Sheppard D., Barzcak A., Xiao Y., Erle D. J., Nishimura S. L. (2007) Squamous metaplasia amplifies pathologic epithelial-mesenchymal interactions in COPD patients. J. Clin. Invest. 117, 3551–3562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Papi A., Bellettato C. M., Braccioni F., Romagnoli M., Casolari P., Caramori G., Fabbri L. M., Johnston S. L. (2006) Infections and airway inflammation in chronic obstructive pulmonary disease severe exacerbations. Am. J. Respir. Crit. Care Med. 173, 1114–1121 [DOI] [PubMed] [Google Scholar]
- 6. Kitamura H., Cambier S., Somanath S., Barker T., Minagawa S., Markovics J., Goodsell A., Publicover J., Reichardt L., Jablons D., Wolters P., Hill A., Marks J. D., Lou J., Pittet J. F., Gauldie J., Baron J. L., Nishimura S. L. (2011) Mouse and human lung fibroblasts regulate dendritic cell trafficking, airway inflammation, and fibrosis through integrin αvβ8-mediated activation of TGF-β. J. Clin. Invest. 121, 2863–2875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Minagawa S., Lou J., Seed R. I., Cormier A., Wu S., Cheng Y., Murray L., Tsui P., Connor J., Herbst R., Govaerts C., Barker T., Cambier S., Yanagisawa H., Goodsell A., Hashimoto M., Brand O. J., Cheng R., Ma R., McKnelly K. J., Wen W., Hill A., Jablons D., Wolters P., Kitamura H., Araya J., Barczak A. J., Erle D. J., Reichardt L. F., Marks J. D., Baron J. L., Nishimura S. L. (2014) Selective targeting of TGF-β activation to treat fibroinflammatory airway disease. Sci. Transl. Med. 6, 241ra79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lucattelli M., Cicko S., Müller T., Lommatzsch M., De Cunto G., Cardini S., Sundas W., Grimm M., Zeiser R., Dürk T., Zissel G., Sorichter S., Ferrari D., Di Virgilio F., Virchow J. C., Lungarella G., Idzko M. (2011) P2X7 receptor signaling in the pathogenesis of smoke-induced lung inflammation and emphysema. Am. J. Respir. Cell Mol. Biol. 44, 423–429 [DOI] [PubMed] [Google Scholar]
- 9. Geraghty P., Dabo A. J., D'Armiento J. (2011) TLR4 protein contributes to cigarette smoke-induced matrix metalloproteinase-1 (MMP-1) expression in chronic obstructive pulmonary disease. J. Biol. Chem. 286, 30211–30218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kang M. J., Lee C. G., Lee J. Y., Dela Cruz C. S., Chen Z. J., Enelow R., Elias J. A. (2008) Cigarette smoke selectively enhances viral PAMP- and virus-induced pulmonary innate immune and remodeling responses in mice. J. Clin. Invest. 118, 2771–2784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Singh B., Arora S., Khanna V. (2010) Association of severity of COPD with IgE and interleukin-1β: Monaldi archives for chest disease. Secondo Ateneo 73, 86–87 [DOI] [PubMed] [Google Scholar]
- 12. Gessner C., Scheibe R., Wötzel M., Hammerschmidt S., Kuhn H., Engelmann L., Hoheisel G., Gillissen A., Sack U., Wirtz H. (2005) Exhaled breath condensate cytokine patterns in chronic obstructive pulmonary disease. Respir. Med. 99, 1229–1240 [DOI] [PubMed] [Google Scholar]
- 13. Pauwels N. S., Bracke K. R., Dupont L. L., Van Pottelberge G. R., Provoost S., Vanden Berghe T., Vandenabeele P., Lambrecht B. N., Joos G. F., Brusselle G. G. (2011) Role of IL-1α and the Nlrp3/caspase-1/IL-1β axis in cigarette smoke-induced pulmonary inflammation and COPD. Eur. Respir. J. 38, 1019–1028 [DOI] [PubMed] [Google Scholar]
- 14. Sapey E., Ahmad A., Bayley D., Newbold P., Snell N., Rugman P., Stockley R. A. (2009) Imbalances between interleukin-1 and tumor necrosis factor agonists and antagonists in stable COPD. J. Clin. Immunol. 29, 508–516 [DOI] [PubMed] [Google Scholar]
- 15. Lappalainen U., Whitsett J. A., Wert S. E., Tichelaar J. W., Bry K. (2005) Interleukin-1β causes pulmonary inflammation, emphysema, and airway remodeling in the adult murine lung. Am. J. Respir. Cell Mol. Biol. 32, 311–318 [DOI] [PubMed] [Google Scholar]
- 16. Churg A., Zhou S., Wang X., Wang R., Wright J. L. (2009) The role of interleukin-1β in murine cigarette smoke-induced emphysema and small airway remodeling. Am. J. Respir. Cell Mol. Biol. 40, 482–490 [DOI] [PubMed] [Google Scholar]
- 17. Doz E., Noulin N., Boichot E., Guénon I., Fick L., Le Bert M., Lagente V., Ryffel B., Schnyder B., Quesniaux V. F., Couillin I. (2008) Cigarette smoke-induced pulmonary inflammation is TLR4/MyD88 and IL-1R1/MyD88 signaling dependent. J. Immunol. 180, 1169–1178 [DOI] [PubMed] [Google Scholar]
- 18. Kolb M., Margetts P. J., Anthony D. C., Pitossi F., Gauldie J. (2001) Transient expression of IL-1β induces acute lung injury and chronic repair leading to pulmonary fibrosis. J. Clin. Invest. 107, 1529–1536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Podowski M., Calvi C., Metzger S., Misono K., Poonyagariyagorn H., Lopez-Mercado A., Ku T., Lauer T., McGrath-Morrow S., Berger A., Cheadle C., Tuder R., Dietz H. C., Mitzner W., Wise R., Neptune E. (2012) Angiotensin receptor blockade attenuates cigarette smoke-induced lung injury and rescues lung architecture in mice. J. Clin. Invest. 122, 229–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Takizawa H., Tanaka M., Takami K., Ohtoshi T., Ito K., Satoh M., Okada Y., Yamasawa F., Nakahara K., Umeda A. (2001) Increased expression of transforming growth factor-β1 in small airway epithelium from tobacco smokers and patients with chronic obstructive pulmonary disease (COPD). Am. J. Respir. Crit. Care Med. 163, 1476–1483 [DOI] [PubMed] [Google Scholar]
- 21. Shi M., Zhu J., Wang R., Chen X., Mi L., Walz T., Springer T. A. (2011) Latent TGF-β structure and activation. Nature 474, 343–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mu D., Cambier S., Fjellbirkeland L., Baron J. L., Munger J. S., Kawakatsu H., Sheppard D., Broaddus V. C., Nishimura S. L. (2002) The integrin α(v)β8 mediates epithelial homeostasis through MT1-MMP-dependent activation of TGF-β1. J. Cell Biol. 157, 493–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Munger J. S., Harpel J. G., Giancotti F. G., Rifkin D. B. (1998) Interactions between growth factors and integrins: latent forms of transforming growth factor-β are ligands for the integrin αvβ1. Mol. Biol. Cell 9, 2627–2638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wipff P. J., Rifkin D. B., Meister J. J., Hinz B. (2007) Myofibroblast contraction activates latent TGF-β1 from the extracellular matrix. J. Cell Biol. 179, 1311–1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Munger J. S., Huang X., Kawakatsu H., Griffiths M. J., Dalton S. L., Wu J., Pittet J. F., Kaminski N., Garat C., Matthay M. A., Rifkin D. B., Sheppard D. (1999) The integrin αvβ6 binds and activates latent TGF β1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell 96, 319–328 [DOI] [PubMed] [Google Scholar]
- 26. Aluwihare P., Mu Z., Zhao Z., Yu D., Weinreb P. H., Horan G. S., Violette S. M., Munger J. S. (2009) Mice that lack activity of αvβ6- and αvβ8-integrins reproduce the abnormalities of Tgfb1- and Tgfb3-null mice. J. Cell Sci. 122, 227–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kudo M., Melton A. C., Chen C., Engler M. B., Huang K. E., Ren X., Wang Y., Bernstein X., Li J. T., Atabai K., Huang X., Sheppard D. (2012) IL-17A produced by αβ T cells drives airway hyper-responsiveness in mice and enhances mouse and human airway smooth muscle contraction. Nat. Med. 18, 547–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Melton A. C., Bailey-Bucktrout S. L., Travis M. A., Fife B. T., Bluestone J. A., Sheppard D. (2010) Expression of αvβ8 integrin on dendritic cells regulates Th17 cell development and experimental autoimmune encephalomyelitis in mice. J. Clin. Invest. 120, 4436–4444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Travis M. A., Reizis B., Melton A. C., Masteller E., Tang Q., Proctor J. M., Wang Y., Bernstein X., Huang X., Reichardt L. F., Bluestone J. A., Sheppard D. (2007) Loss of integrin α(v)β8 on dendritic cells causes autoimmunity and colitis in mice. Nature 449, 361–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Markovics J. A., Araya J., Cambier S., Somanath S., Gline S., Jablons D., Hill A., Wolters P. J., Nishimura S. L. (2011) Interleukin-1β induces increased transcriptional activation of the transforming growth factor-β-activating integrin subunit β8 through altering chromatin architecture. J. Biol. Chem. 286, 36864–36874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jeffery P. K. (2004) Remodeling and inflammation of bronchi in asthma and chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc. 1, 176–183 [DOI] [PubMed] [Google Scholar]
- 32. Araya J., Cambier S., Morris A., Finkbeiner W., Nishimura S. L. (2006) Integrin-mediated transforming growth factor-β activation regulates homeostasis of the pulmonary epithelial-mesenchymal trophic unit. Am. J. Pathol. 169, 405–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Demedts I. K., Bracke K. R., Van Pottelberge G., Testelmans D., Verleden G. M., Vermassen F. E., Joos G. F., Brusselle G. G. (2007) Accumulation of dendritic cells and increased CCL20 levels in the airways of patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 175, 998–1005 [DOI] [PubMed] [Google Scholar]
- 34. Lambrecht B. N., Hammad H. (2009) Biology of lung dendritic cells at the origin of asthma. Immunity 31, 412–424 [DOI] [PubMed] [Google Scholar]
- 35. Wu S., Metcalf J. P., Wu W. (2011) Innate immune response to influenza virus. Curr. Opin. Infect. Dis. 24, 235–240 [DOI] [PubMed] [Google Scholar]
- 36. Rumbo M., Sierro F., Debard N., Kraehenbuhl J. P., Finke D. (2004) Lymphotoxin β receptor signaling induces the chemokine CCL20 in intestinal epithelium. Gastroenterology 127, 213–223 [DOI] [PubMed] [Google Scholar]
- 37. Flanders K. C. (2004) Smad3 as a mediator of the fibrotic response. Int. J. Exp. Pathol. 85, 47–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Reibman J., Hsu Y., Chen L. C., Bleck B., Gordon T. (2003) Airway epithelial cells release MIP-3α/CCL20 in response to cytokines and ambient particulate matter. Am. J. Respir. Cell Mol. Biol. 28, 648–654 [DOI] [PubMed] [Google Scholar]
- 39. Greaves D. R., Wang W., Dairaghi D. J., Dieu M. C., Saint-Vis B., Franz-Bacon K., Rossi D., Caux C., McClanahan T., Gordon S., Zlotnik A., Schall T. J. (1997) CCR6, a CC chemokine receptor that interacts with macrophage inflammatory protein 3α and is highly expressed in human dendritic cells. J. Exp. Med. 186, 837–844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hashimoto M., Yanagisawa H., Minagawa S., Sen D., Goodsell A., Ma R., Moermans C., McKnelly K. J., Baron J. L., Krummel M. F., Nishimura S. L. (2015) A critical role for dendritic cells in the evolution of IL-1β-mediated murine airway disease. J. Immunol. 194, 3962–3969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Saika S., Kono-Saika S., Ohnishi Y., Sato M., Muragaki Y., Ooshima A., Flanders K. C., Yoo J., Anzano M., Liu C. Y., Kao W. W., Roberts A. B. (2004) Smad3 signaling is required for epithelial-mesenchymal transition of lens epithelium after injury. Am. J. Pathol. 164, 651–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Derynck R., Zhang Y. E. (2003) Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature 425, 577–584 [DOI] [PubMed] [Google Scholar]
- 43. Sirard C., Kim S., Mirtsos C., Tadich P., Hoodless P. A., Itié A., Maxson R., Wrana J. L., Mak T. W. (2000) Targeted disruption in murine cells reveals variable requirement for Smad4 in transforming growth factor β-related signaling. J. Biol. Chem. 275, 2063–2070 [DOI] [PubMed] [Google Scholar]
- 44. Larrivée B., Prahst C., Gordon E., del Toro R., Mathivet T., Duarte A., Simons M., Eichmann A. (2012) ALK1 signaling inhibits angiogenesis by cooperating with the Notch pathway. Dev. Cell 22, 489–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Muñoz-Félix J. M., Perretta-Tejedor N., Eleno N., López-Novoa J. M., Martínez-Salgado C. (2014) ALK1 heterozygosity increases extracellular matrix protein expression, proliferation and migration in fibroblasts. Biochim. Biophys. Acta 1843, 1111–1122 [DOI] [PubMed] [Google Scholar]
- 46. Alverdi V., Hetrick B., Joseph S., Komives E. A. (2014) Direct observation of a transient ternary complex during IκBα-mediated dissociation of NF-κB from DNA. Proc. Natl. Acad. Sci. U.S.A. 111, 225–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bosisio D., Marazzi I., Agresti A., Shimizu N., Bianchi M. E., Natoli G. (2006) A hyper-dynamic equilibrium between promoter-bound and nucleoplasmic dimers controls NF-κB-dependent gene activity. EMBO J. 25, 798–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Siegel R., Eskdale J., Gallagher G. (2011) Regulation of IFN-λ1 promoter activity (IFN-λ1/IL-29) in human airway epithelial cells. J. Immunol. 187, 5636–5644 [DOI] [PubMed] [Google Scholar]
- 49. Li M. O., Flavell R. A. (2008) TGF-β: a master of all T cell trades. Cell 134, 392–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Vitsky A., Waire J., Pawliuk R., Bond A., Matthews D., Lacasse E., Hawes M. L., Nelson C., Richards S., Piepenhagen P. A., Garman R. D., Andrews L., Thurberg B. L., Lonning S., Ledbetter S., Ruzek M. C. (2009) Homeostatic role of transforming growth factor-β in the oral cavity and esophagus of mice and its expression by mast cells in these tissues. Am. J. Pathol. 174, 2137–2149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Caramori G., Adcock I. M., Di Stefano A., Chung K. F. (2014) Cytokine inhibition in the treatment of COPD. Int. J. Chron. Obstruct. Pulmon. Dis. 9, 397–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chomarat P., Vannier E., Dechanet J., Rissoan M. C., Banchereau J., Dinarello C. A., Miossec P. (1995) Balance of IL-1 receptor antagonist/IL-1β in rheumatoid synovium and its regulation by IL-4 and IL-10. J. Immunol. 154, 1432–1439 [PubMed] [Google Scholar]
- 53. Mertens M., Singh J. A. (2009) Anakinra for rheumatoid arthritis: a systematic review. J. Rheumatol. 36, 1118–1125 [DOI] [PubMed] [Google Scholar]
- 54. Varona R., Zaballos A., Gutiérrez J., Martín P., Roncal F., Albar J. P., Ardavín C., Márquez G. (1998) Molecular cloning, functional characterization and mRNA expression analysis of the murine chemokine receptor CCR6 and its specific ligand MIP-3α. FEBS Lett. 440, 188–194 [DOI] [PubMed] [Google Scholar]