

Background: Modest affinity to carbohydrate targets limits the therapeutic efficacy of antibodies to tumor antigens, such as ganglioside GD2.
Results: Mutagenesis and in silico modeling were utilized to enhance the affinity of monoclonal antibody hu3F8 to GD2.
Conclusion: Alteration of electrostatic surface potential resulted in enhanced affinity and tumor cytotoxicity.
Significance: An improved clinical candidate was developed against a carbohydrate tumor antigen.
Keywords: antibody engineering, cancer therapy, electrostatics, ganglioside, immunotherapy
Abstract
Ganglioside GD2 is highly expressed on neuroectodermal tumors and an attractive therapeutic target for antibodies that have already shown some clinical efficacy. To further improve the current antibodies, which have modest affinity, we sought to improve affinity by using a combined method of random mutagenesis and in silico assisted design to affinity-mature the anti-GD2 monoclonal antibody hu3F8. Using yeast display, mutants in the Fv with enhanced binding over the parental clone were FACS-sorted and cloned. In silico modeling identified the minimal key interacting residues involved in the important charged interactions with the sialic acid groups of GD2. Two mutations, D32H (L-CDR1) and E1K (L-FR1) altered the electrostatic surface potential of the antigen binding site, allowing for an increase in positive charge to enhance the interaction with the negatively charged GD2-pentasaccharide headgroup. Purified scFv and IgG mutant forms were then tested for antigen specificity by ELISA, for tissue specificity by immunohistochemistry, for affinity by BIACORE, for antibody-dependent cell-mediated cytotoxicity (ADCC) and complement-mediated cytotoxicity in vitro, and for anti-tumor efficacy in xenografted humanized mice. The nearly 7-fold improvement in affinity of hu3F8 with a single D32H (L-CDR1) mutation translated into a ∼12-fold improvement in NK92MI-transfected CD16-mediated ADCC, a 6-fold improvement in CD32-mediated ADCC, and a 2.5-fold improvement in complement-mediated cytotoxicity while maintaining restricted normal tissue cross-reactivity and achieving substantial improvement in tumor ablation in vivo. Despite increasing GD2 affinity, the double mutation D32H (L-CDR1) and E1K (L-FR1) did not further improve anti-tumor efficacy.
Introduction
Gangliosides are sialic acid-containing glycolipids that are widely expressed in stem cells, neurons of the central nervous system, and peripheral nerve endings and represent a special class of targets in cancer immunotherapy (1, 2). Ganglioside GD2 is an abundant glycolipid found on neuroectodermal tumors, including neuroblastoma, retinoblastoma, melanoma, small cell lung cancer, brain tumors, osteosarcoma, rhabdomyosarcoma, and Ewing sarcoma in children and adolescents, but highly restricted in normal tissues (3). GD2 consists of a pentasaccharide head, which contains two sialic acids, N-acetylgalactosamine, galactose, glucose, and a ceramide tail (4). Most anti-GD2 antibodies bind to an epitope formed by sialic acids and N-acetylgalactosamine. Glycosyltransferases catalyze the synthesis of GD2 by the sequential addition of simple sugars, including sialic acid, to LacCer. GD3, GD2, GD1b, GT1b, and GQ1b are classified as belonging to the b-series gangliosides (5), whereas GM2 and GT2 are in the a- and c-series, respectively. Because of epitope similarity to GD2 in the synthetic pathways, GD3, GM2, and GD1b are potential cross-reactive epitopes during anti-GD2 antibody development (6).
Two families of therapeutic anti-GD2 antibodies have been developed and tested extensively in patients, namely the mouse immunoglobulin G3 (IgG3) antibody 3F8 (7) and its humanized version (hu3F8) (8), and the mouse IgG2a antibody 14G2a and its chimeric (ch14.18) or humanized (hu14.18) forms (9). Anti-GD2 antibodies mediate highly efficient myeloid cell-mediated antibody-dependent cell cytotoxicity (ADCC)3 via CD32 and nature killer cell-mediated ADCC via CD16 (10, 11). These antibodies have been primarily used in clinical trials for neuroblastoma, a malignancy accounting for 7% of all childhood cancers and 15% of pediatric cancer deaths (12). The use of anti-GD2 antibodies plus subcutaneous granulocyte-macrophage colony-stimulating factor (GM-CSF) has resulted in substantial increases in the survival time of patients with high-risk stage 4 neuroblastoma (7). The pain side effects, which are thought to be a consequence of complement activation (13), have limited the dose of anti-GD2 antibody and the efficacy on ablating bulky tumors. Murine 3F8 (m3F8), with a moderate affinity for GD2 (KD = 5 nm), was the first anti-GD2 antibody agent to be tested in patients (14). hu3F8 has affinity comparable with that of m3F8 and has shown low immunogenicity, despite repeated cycles in patients previously sensitized to m3F8 (8). It is often assumed for high-density antigens (e.g. GD2) that affinity may not be as important, hence the typical IgM response against such antigens. We hypothesize that affinity maturation can enhance antibody binding to GD2 that translates into improved biologic functions.
A variety of in vitro evolution strategies have proven useful to improve the affinity and specificity of antibodies obtained by display technologies (15). These strategies rely on either site-directed mutagenesis of the complementary-determining region (CDR) (16–18) or random mutagenesis of the entire variable fragment (Fv) (19, 20). The most widely adopted display technique for protein-directed evolution to date is yeast display. One of its advantages is the quantitative screening through the use of fluorescence-activated cell sorting (FACS) (21). However, it remains quite difficult to deduce which of the CDR residues directly interact with the antigen. As a rule, during in vitro affinity maturation, substituted residues involve not only the contact residues but also residues located in the periphery of the paratope (22). However, this maturation process can be accelerated by deducing the contact residues based on antibody structures or, preferably, if antibody-antigen complex structures are available (23, 24). Unlike protein antigens, glycans are generally T-cell-independent, and therefore low-affinity IgM antibodies are often produced (25). Affinity maturation of carbohydrate-specific antibodies without the loss of specificity has been attempted by incorporating limited point mutations (6, 26). A hierarchy of cross-reactivity usually accompanies increased binding affinity (6), rendering affinity maturation of anti-carbohydrate antibodies more complicated.
In the present study, we describe a strategy to improve the affinity and the anti-tumor activity of hu3F8. First, we sequence high-affinity binders from the yeast display of randomized Fv mutations. Potential residues affecting antigen binding were then identified based on the structural modeling of hu3F8. Appropriate hu3F8 variants with limited mutations were designed and tested for antigen binding, tissue immunohistochemistry, ADCC, and complement-mediated cytotoxicity (CMC) in vitro and anti-tumor effect in vivo. This is the first demonstration that anti-GD2 antibodies could be affinity-maturated by the use of yeast display and in silico analysis, achieving high specificity and high potency.
Experimental Procedures
GD2 Biotinylation
GD2-LC-LC-biotin conjugate was obtained from the Consortium for Functional Genomics. For the synthesis of GD2-PEG4-biotin, GD2-azido was conjugated to dibenzocyclooctyne-PEG4-biotin (Click Chemistry Tools) by copper-free azide-alkyne cycloaddition reactions. Briefly, the 100 μg of GD2-azido and 50 μg of dibenzocyclooctyne-PEG4-biotin in 25 μl of water reacted overnight at 4 °C with gentle rotation. On the next day, the excess dibenzocyclooctyne-PEG4-biotin was inactivated by adding 30 μg of azide-PEG3-azide (Click Chemistry Tools) and incubated for 1 h at room temperature. The product was diluted to reach a concentration of 0.5 mg/ml and stored at −80 °C.
Random and Site-directed Mutagenesis
Random mutagenesis of the entire hu3F8-scFv gene was performed by error-prone PCR with the Stratagene GeneMorph® II random mutagenesis kit as described previously (27). This introduced limited numbers of mutations into the gene by controlling the quantity of template and the number of PCR cycles. Reaction products were purified and concentrated by an ultrafilter in water for use in the yeast library construction. Site-directed mutagenesis of the hu3F8-scFv gene was performed by PCR using PfuUltra high-fidelity DNA polymerase (New England Biolabs) according to the manufacturer's instructions. Reaction products were digested by DpnI restriction enzyme (New England Biolabs) and then transformed into TOPO10-competent cells.
Selection of hu3F8 Mutants (Variants) from the Yeast Libraries
The methodology for generating and isolating higher affinity hu3F8 mutants was adopted from Refs. 27 and 28 with some modifications. Appropriate GD2-PEG4-biotin was conjugated to streptavidin magnetic beads (Invitrogen) with a 1-h incubation at room temperature. Before FACS selection, induced yeast library (1 × 109 cells) was incubated with 10 μg of GD2-conjugated magnetic beads for 1 h at room temperature in PBSA buffer (0.1% BSA in PBS) in the presence of GM2, followed by the separation with a magnetic stand. The isolated beads were washed three times with PBSA buffer, put into 10 ml of SDCAA medium, and grown overnight in a 30 °C shaker with 250 rpm. The recovered yeast cells were induced in SGCAA medium for 18 h at 20 °C with 250 rpm shaking. Approximately 1 × 108 yeast cells were pelleted, washed twice with PBSA buffer, and resuspended in 1 ml of PBSA buffer with biotinylated GD2 and a 1:100 dilution of mouse anti-c-Myc antibody (Invitrogen). After incubation, yeast cells were washed three times and then resuspended in 1 ml of PBSA buffer. 1:100 dilutions of both R-phycoerythrin-conjugated streptavidin (Invitrogen) and Alexa Fluor 488-conjugated goat anti-mouse IgG antibody (Invitrogen) were added, incubated at 4 °C for 30 min, and washed three times with PBSA buffer for FACS selection. Sorting gates were determined to acquire only 0.1–0.3% population with the highest binding signal. Collected cells were grown overnight in SDCAA medium at 30 °C and induced in SGCAA for the next round of sorting. For the next two selections, ∼1–2 × 107 yeast cells were used for staining with biotinylated GD2. Yeast plasmids were isolated using the Zymoprep Yeast Plasmid Miniprep II Kit (Zymo Research) according to the manufacturer's instructions and used for templates of library construction. Plasmids from the third round were prepared, sequenced, and characterized.
Flow Cytometric Analysis
The yeast cells displaying hu3F8-scFvs were grown and induced as for flow cytometric analysis. The yeast cells (1 × 106) were incubated with 2 μg/ml biotinylated GD2 and a 1:100 dilution of mouse anti-c-Myc antibody for 30 min on ice in PBSA buffer. After one washing, cells were incubated with a 1:50 dilution of R-phycoerythrin-conjugated streptavidin and Alexa Fluor 488-conjugated goat anti-mouse antibody for 30 min on ice and then washed again and resuspended in 0.5 ml of PBSA buffer. Analysis was performed using a FACSCalibur (BD Biosciences).
Expression of scFv and IgG1
Proteins of scFvs were expressed and purified as described previously (29) with some modifications. HB2151 cells were transformed with pComb3x plasmid containing scFv sequences. Single fresh colonies were inoculated into 2YT medium containing 100 μg/ml ampicillin and 0.2% glucose. The culture was induced by isopropyl-1-thio-β-d-galactopyranoside (final concentration 0.5 mm). After overnight growth at 30 °C, the bacteria were centrifuged at 3500 × g for 15 min. Soluble proteins were released from periplasm by incubating at 30 °C for 30 min. The clear supernatant was recovered for the purification on a nickel-nitrilotriacetic acid column. Recombinant scFvs have FLAG and His tags.
IgGs were expressed in CHO suspension cells as described previously (8). The heavy and light variable regions of hu3F8 variants were cloned into a mammalian expression vector containing human IgG1 constant regions. The vector was transfected into CHO cells and selected with hygromycin B (Life Technologies). The stable cell lines were cultured in OptiCHO serum-free medium (Invitrogen), and the mature supernatant was harvested as described. The soluble IgG1 protein was purified using the Protein A affinity chromatograph medium (GE Healthcare) and subsequently concentrated using ultrafilters (Sartorius Stedim). All antibody samples were purified at >95% monomer by size exclusion HPLC analysis.
ELISA
To test cross-reactivity to gangliosides, GD2, GD1a, GD1b, and GD3 were coated on polyvinyl microtiter plates at 20 ng/well in 90% ethanol. Following air drying, wells were blocked with 0.5% BSA in PBS at 150 μl/well for 1 h at room temperature. Serials of diluted antibodies (100 μl/well) were added with duplicates in 0.5% BSA. Following incubation for 1 h at room temperature and washing with PBS, HRP-goat anti-human Fc antibody (Southern Biotech) at 1:5000 dilution for IgG antibodies or HRP-goat anti-FLAG antibody (Sigma-Aldrich) at 1:5000 dilution for scFv antibodies was added. After incubation for 1 h and further washing, the color reaction was performed, and OD was read using an ELISA plate reader at 490 nm.
Affinity Determination by Surface Plasmon Resonance
Kinetics and affinities of various antibodies were analyzed by surface plasmon resonance technology using a Biacore T100 instrument (GE Healthcare). In brief, gangliosides were directly immobilized onto the CM5 sensor chip via hydrophobic interaction. The reference surface was immobilized with GM1. The active surface was immobilized with GD2 and GM1 in a 1:1 ratio or GD1b alone. Diluted mixture of GD2 and GM1 (50 μg/ml) or GD1b was injected (300 μl) at a flow rate of 15 μl/min over 20 min. Extensive washing was followed with 10 mm NaOH (typically five washes of 20 μl at a flow rate of 5 μl/min) until a stable baseline was obtained. The association and dissociation phase data were fitted simultaneously to a 1:1 model by using BIAevaluation version 3.2.
Tumor Cell Culture
LAN-1, M14, and A4573 were obtained from Children's Hospital of Los Angeles; SK-N-CM, SK-E-PR, and SK-E-LP were obtained from Memorial Sloan Kettering Cancer Center. All other cell lines were purchased from American Type Culture Collection (ATCC) (Manassas, VA). Cells were cultured in RPMI1640 (Cellgro, Manassas, VA) supplemented with 10% fetal bovine serum (FBS) (Life Technologies, Inc.) at 37 °C in a 5% CO2-humidified incubator.
Antibody-dependent Cell-mediated Cytotoxicity (ADCC) Assay
Assays were done as described previously (8). Briefly, tumor cells were radiolabeled with 51Cr. ADCC assays were performed using NK-92MI cells stably transfected with the human Fc receptor or peripheral blood mononuclear cells (PBMCs) as effectors. The cytotoxicity was calculated by 51Cr release.
CMC Assay
Tumor cell lines were detached with trypsin-EDTA and washed in culture medium. For cytotoxicity assays, 100 μCi of 51Cr was incubated with 106 target cells in a final volume of 250 μl and incubated for 1 h at 37 °C with gentle resuspension of pellet at 15-min intervals. Cells were then pelleted, washed, and resuspended in media to ∼0.05 million cells/ml. Cells were counted, and viability was determined with trypan blue and quickly plated onto 96-well U-bottom plates. Antibodies were diluted in medium from 10 μg/ml in 10-fold dilutions. For each well, 50 μl of antibody was mixed with 100 μl of target cells and kept on ice for 45 min. 100 μl of blood serum (1:80 dilution of pooled sera) was then added to each well to initiate complement fixation. Plates were incubated in a 37 °C, 5% CO2 incubator for 4 h. Released 51Cr in the ADCC supernatant was collected for γ counting. Total release was determined using 10% SDS, and background spontaneous release was determined with medium only without serum.
Therapy of Neuroblastoma Xenograft
All animal procedures were performed in compliance with Institutional Animal Care and Use Committee guidelines. Tumor xenografts were established by subcutaneous implantation of neuroblastoma IMR-32 cells mixed with human PBMC into 5–6-week-old BALB-Rag2−/−IL-2R-γc-KO (DKO) immunodeficient mice. Mice were randomized into groups (n = 5) when tumors were 75–100 mm3. 10 or 50 μg doses of antibodies in PBS were administered intravenously twice per week for a total of four doses. An anti-Hud-peptide/HLA-A2 antibody developed at Memorial Sloan Kettering was used as a non-targeting isotype-matched control. Tumor volume (mm3) was measured once per week and was calculated as (length (mm) × width (mm)2)/2.
Immunohistochemistry
Human tumors and normal tissues were obtained at Memorial Sloan Kettering Cancer Center with institutional review board approval. 5–7-μm sections of snap-frozen tissues were fixed in acetone for 30 min at −20 °C. Endogenous biotin-binding activity was blocked by sequential treatment with avidin and biotin (Vector avidin-biotin blocking kit; Invitrogen) for 20 min each. Sections were incubated with 3 μg/ml scFv at room temperature for 1 h. Following washing, sections were incubated with HRP anti-FLAG antibodies for 30 min at room temperature and subsequent incubation with 3,3-diaminobenzidine for 5 min. H&E staining was also performed.
Simulated Models of hu3F8 Fv Structure and GD2 Docking
Molecular modeling and energy calculations were done using Discovery Studio version 4.1 (Dassault Systems, Waltham, MA). We previously solved the crystal of murine 3F8 to 1.65 Å resolution (Protein Data Bank 3VFG) and created a docked model of bound GD2 using CDOCKER (30). A structural model of hu3F8-GD2 was generated by using the murine 3F8-GD2 model generated previously as a template using MODELER followed by CHARMm energy minimization on Discovery Studio. Effects of point mutations were calculated from the difference between the folding free energies of the mutated structure and the parental protein. Generalized Born approximation was used to account for the effect of the solvent, and all electrostatic terms were calculated as a sum of coulombic interactions and polar contributions to the solvation energy. A weighted sum of the van der Waals, electrostatic, entropy, and non-polar terms was calculated for each point mutation. Electrostatic surface potentials were generated using DelPhi on Discovery Studio. Additional image renderings were done using PyMOL (Schroedinger, LLC, New York).
Results
Biotinylated GD2 with a Polyethylene Glycol (PEG) Spacer
Biotinylated glycan targets are valuable carbohydrate probes during aqueous selection of antibodies in yeast or phage display systems, where fluorescent streptavidin provides the optical signal. Hydrophobic GD2 was azidolated at the glucose residue and subsequently coupled with biotin spacer by copper-free azide-alkyne cycloaddition reactions. A 4-unit polyethylene glycol spacer (PEG4) was introduced between GD2 and biotin to avoid steric hindrance when streptavidin tried to capture biotinylated GD2 bound to hu3F8-scFv displayed on the yeast cell surface. A Myc tag was used to track the scFv protein itself. By flow cytometric analysis, GD2 binding to yeast and scFv expression were detected as the x- and y-axis, respectively. The PEG4 spacer was necessary for successful streptavidin capture when compared with GD2 probes with the alkyl spacer (LC-LC) (Fig. 1). Contrary to traditional alkyl spacers, which are hydrophobic, hydrophilic PEG is a stable spacer that offsets the hydrophobic nature of biotin. The PEG spacer potentially eliminates nonspecific binding that causes aggregation and precipitation problems.
FIGURE 1.

Evidence of improved biotinylated GD2 with flexible PEG spacer. Yeast cells displayed low- and high-affinity hu3F8 scFv with c-Myc tag on the cell surface, which were used to bind biotinylated GD2 conjugates with PEG or LC-LC Spacer. GD2 binding onto scFv was detected by dual-color flow cytometric analysis. ScFv expression is indicated on the x axis. GD2 binding is indicated on the y axis.
Affinity Maturation of hu3F8 by Random Mutagenesis and Yeast Display
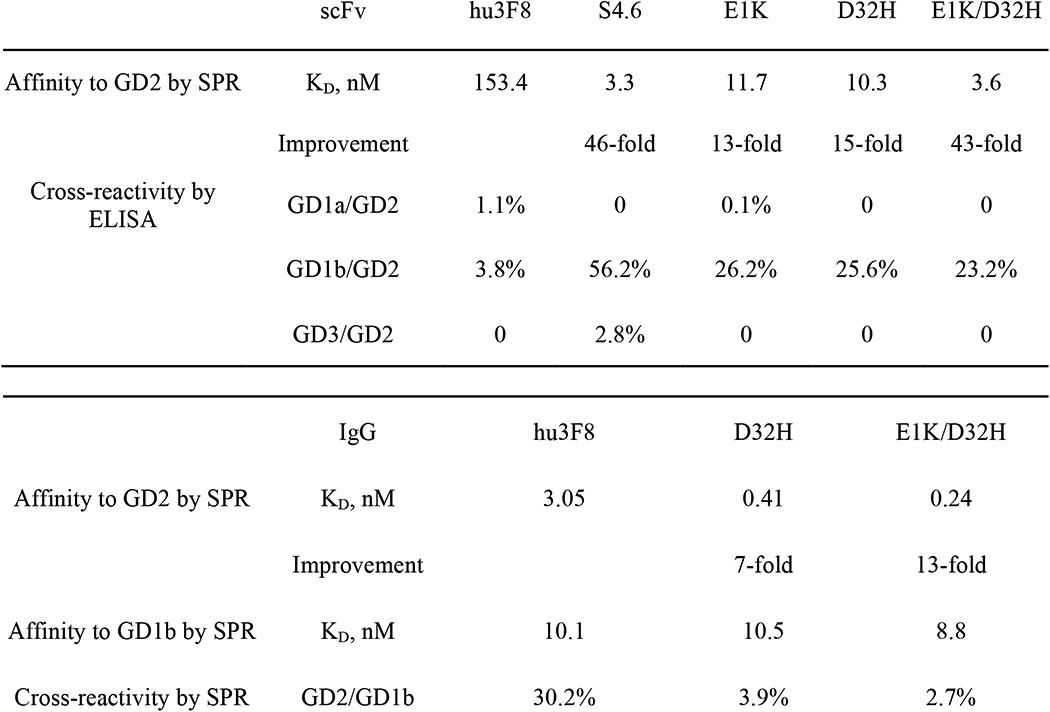
A yeast scFv library of 5 × 108 transforms was constructed by randomizing the entire hu3F8-scFv after introducing low (<5/1000 bp) and moderate (5–9/1000 bp) mutations. GM2 was used as the negative selector because cross-reactivity was consistently detected during phase display affinity maturation of antibodies to GD2 (6). Clones from the yeast library were selected for high and selective binding to GD2-PEG4-biotin conjugate. By repeating FACS three times using stringent conditions, scFvs with increased binding to GD2 were enriched (Fig. 2A). By sequencing, a recurrent mutant (S4.6) and two unique clones (S4.10 and S4.13) were selected. The scFvs of these three mutants were expressed, purified, and tested for GD2 binding by ELISA, which showed a substantial (∼10-fold) increase in binding efficiency relative to hu3F8-scFv (Fig. 2B). Analysis by surface plasmon resonance showed that all mutants had a better kon than the parental hu3F8-scFv (Fig. 2C). The most improved S4.6 scFv exhibited a 46-fold increase in affinity over hu3F8-scFv (Table 1).
FIGURE 2.

Affinity maturation of hu3F8 scFv by random mutagenesis. A, FACS for yeast display selection. The yeast library was labeled with mouse anti-c-Myc antibody followed by goat anti-mouse dye as well as biotinylated GD2 followed by streptavidin dye. During three FACS selections, yeast cells were stained with concentrations of biotinylated GD2 at 100 μm (Sort 1), 100 μm (Sort 2), and 25 μm (Sort 3), respectively. The 0.1–0.3% cells were selected from sort gates. B, ELISA of yeast-selected hu3F8 scFv variants against GD2. Three scFvs (S4.6, S4.10, and S4.13) and parental hu3F8 scFv were serially diluted and bound with GD2, followed by measurement as optical densities (OD) at 490 nm. C, comparative binding kinetics of yeast-selected hu3F8 scFv variants on solid phase GD2 when measured by surface plasmon resonance. An overlay plot of association and dissociation curves was obtained against immobilized GD2. RU, resonance units.
TABLE 1.
Affinity and cross-reactivity of hu3F8 scFv and IgG variants
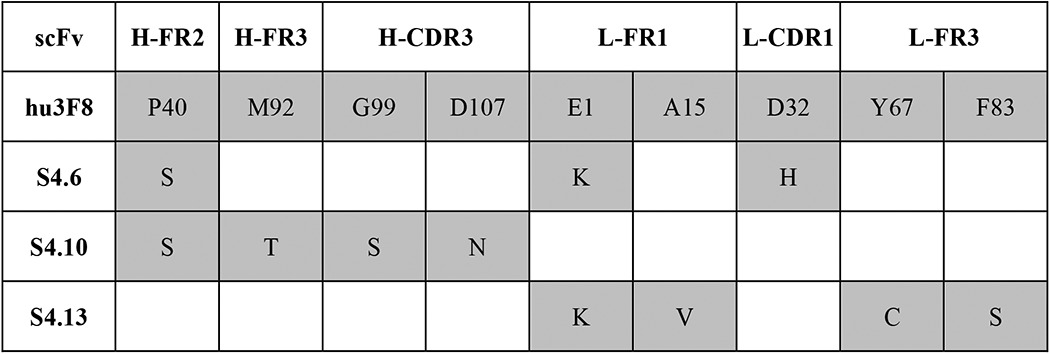
The alignment of the amino acid mutations with the parental hu3F8 is shown in Table 2, identifying the framework regions (FRs) and CDRs where these mutations occurred. Of the nine mutations in the variable regions of the heavy chain and light chains, only three occurred in the CDR. The most improved variant (S4.6) carried three amino acid mutations (VH-FR2 P40S, VL-FR1 E1K, and VL-CDR1 D32H). In two variants (S4.10 and S4.13) with moderate affinity improvement, we observed a glutamic acid-to-lysine substitution at residue 1 (E1K, VL-FR1) and a proline-to-serine at residue 40 (P40S, VH-FR2). Two other mutations in the VH-CDR3 of S4.10 (G99S and D107N) did not appear to affect affinity improvement but were later analyzed by structural modeling.
TABLE 2.
Mutated residues of hu3F8 scFv identified by yeast selection
Blank, amino acid positions where no mutation was observed. Gray, amino acid positions that were mutated during the random maturation process.
In Silico-assisted Affinity Maturation of hu3F8 by Site-directed Mutagenesis
Because parental hu3F8 is not immunogenic in patients,4 we wanted to limit the number of mutations to avoid neoantigens and epitope spread (6). The coordinates of the antibody Fv domains of hu3F8 were generated by homology modeling based on the crystal structure of m3F8 (Protein Data Bank code 3VFG). m3F8 carried a binding cavity formed by CDR loops that had only two charged residues protruding from the H-CDR3 loop (30). Positively charged side chains of amino acid residues in the binding cavity were good candidates for the negatively charged sialic acid groups of GD2 (−2 formal charge). Several potential mutants were considered and analyzed on the basis of the energy predictions from docking hu3F8 to GD2 (Fig. 3A). Two particular mutations stood out: E1K and D32H of the VL chain. A Lys in VL position 1 faced away from the binding cavity but could interact with GD2 via long range electrostatic forces. VL position 32 was a CDR interface residue of the antigen-binding region. Substitution of the negatively charged aspartic acid with a positively charged residue histidine was expected to directly strengthen the interaction with the negatively charged GD2. Neither the E1K nor the D32H mutation was expected to directly interact with GD2 in the docked hu3F8-GD2 model via hydrogen bonds, but CHARMm force field-based energy calculations did predict that the resulting mutations would increase the interaction energy of hu3F8-GD2 by ∼0.5 kcal/mol. Electrostatic surface potentials were calculated for parental hu3F8, a single D32H mutation, and the double E1K/D32H mutations (Fig. 3, B–D). In parental hu3F8 (Fig. 3B), the GD2 molecule was predicted to predominantly interact with the VH CDR loops due to the large area of positive charge shown in blue. The single D32H mutation (shown in Fig. 3C) dramatically altered the charge distribution of the VL domain from negative to positive. The addition of the E1K mutation (Fig. 3D) also contributed to this shift in charge, although to a lesser degree. Consideration was also given to VH positions 99 and 107, which were located in the CDR loops, but structural analysis showed that these residues were fully buried and excluded from interacting with GD2. We therefore chose two mutations, Lys at position 1 and/or His at position 32, to evaluate antibody-antigen interactions.
FIGURE 3.

Structural models of hu3F8 variants interacting with pentasaccharide headgroup of GD2. A, top down view of the antigen binding site of the hu3F8 homology model, based on the solved crystal structure of murine 3F8 (Protein Data Bank code 3VFG) and docked GD2. LC residues Glu-1 and Asp-32 are shown in a stick rendering. B, electrostatic surface map of hu3F8 with docked GD2. Surface potential was calculated using DelPhi. C, electrostatic potential surface of hu3F8 D32H showing a large shift toward partially positive charge relative to hu3F8. D, electrostatic potential surface of hu3F8 E1K/D32H showing additional positive charge from the double point mutations.
Three scFv mutants, E1K (Lys at residue 1), D32H (His at residue 32), and E1K/D32H (Lys at residue 1 and His at residue 32), were produced in bacteria and affinity-purified. They showed 13–43-fold higher affinity than parental hu3F8-scFv (Table 1). Similar to parental hu3F8, scFv mutants showed cross-reactivity with GD1b and no cross-reactivity with GD3 or GD1a. Of interest, the designed mutant scFvs (E1K, D32H, and E1K/D32H) all showed less cross-reactivity with GD1b when compared with the random mutant S4.6, which carried additional FR mutations.
For further functional characterization, we converted these affinity-maturated scFvs to full-length human IgG1 formats. Two different IgG1 variants, D32H (VL D32H mutation) and E1K/D32H (VL E1K and D32H mutations), were constructed. To evaluate the impact of E1K and D32H mutations in IgG on affinity, the KD for each IgG1 was determined by surface plasmon resonance using Biacore (Table 1). The KD values of the two affinity-maturated IgG1s reached 0.41 and 0.24 nm, respectively. The increases in affinities of the D32H and E1K/D32H mutants were 7- and 13-fold, respectively, compared with 3 nm for the parental IgG1. Interestingly, when tested against GD1b by surface plasmon resonance, neither D32H nor E1K/D32H IgG1 showed a substantial improvement in KD value, suggesting that the increased affinity for GD2 was preferential.
Tumor Cell Killing Properties of Affinity-maturated hu3F8 IgG1
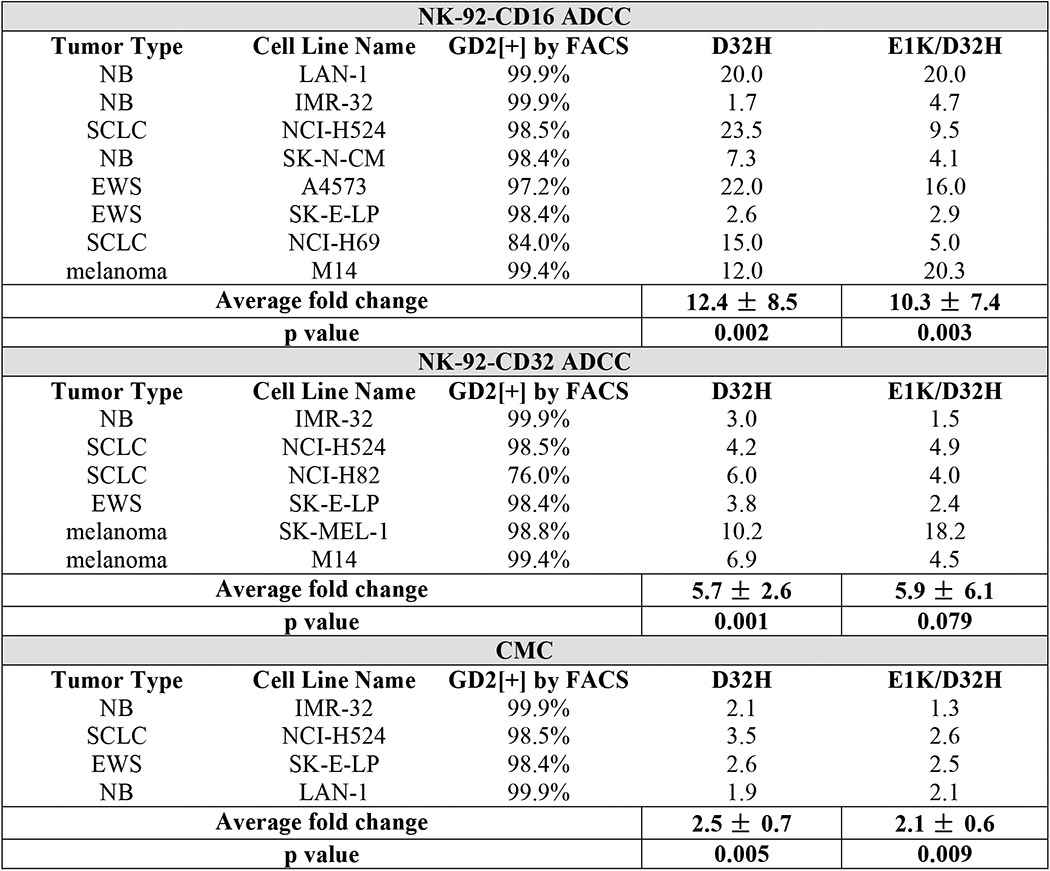
ADCC activity was a critical mechanism of hu3F8-mediated tumor cytotoxicity in vitro and in patients (31). Using human neuroblastoma LAN-1 as targets in the presence of human PBMC (Fig. 4A) or NK92-CD16 (Fig. 4B) as effectors, both D32H-IgG1 and E1K/D32H-IgG1 were ∼7–20-fold stronger by left shifting in the EC50. PMBC-ADCC is primarily an NK-mediated, CD16-dependent cytotoxicity, although monocytes that expressed CD32 could also mediate low levels of cytotoxicity. By transfecting a FcR(−) natural killer cell line NK92-MI with CD16 or CD32, we separated CD16 (FcγRIII)-mediated cytotoxicity from CD32 (FcγRII)-mediated cytotoxicity. In Table 3, NK92-CD16-ADCC, NK92-CD32-ADCC, and CMC were compared in a panel of human tumor cell lines that included neuroblastoma, Ewing sarcoma, melanoma, and small cell lung cancer; potencies were computed as the ratio, (EC50 for variant)/(EC50 for parental). When averaged across cell lines, the -fold increase in potency for D32H variant was 12, 6, and 2.5, respectively, all highly significant (p = 0.001 to p = 0.005) compared with parental hu3F8. For the double mutant E1K/D32H, these -fold changes were 10, 6, and 2, respectively (Table 3). These results demonstrated that mutant-mediated ADCC and CMC against GD2(+) tumor cells were substantially enhanced, but the differences between single D32H versus double E1K/D32H mutations were not statistically significant.
FIGURE 4.
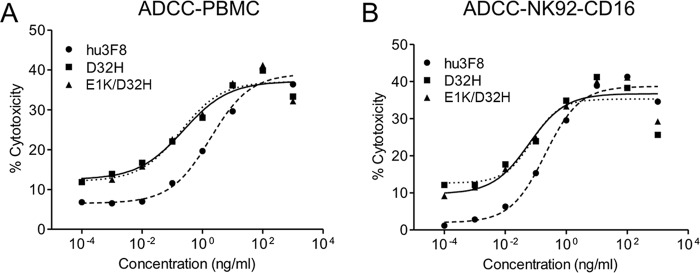
Antibody potency of mature hu3F8 IgG1 in ADCC against neuroblastoma. A and B, three hu3F8 variants mediate in vitro ADCC on LAN-1 neuroblastoma cells with human PBMC (A) and NK92-CD16 (B) effector cells.
TABLE 3.
ADCC and CMC assays of hu3F8 IgG1 variants
Antibody potencies for variants D32H and E1K/D32H are shown as the fold change in EC50 using parental hu3F8 as a reference. NB, neuroblastoma; SCLC, small cell lung cancer; EWS, Ewing sarcoma.
Immunohistochemistry of Affinity-maturated hu3F8
The three scFvs (parental hu3F8, D32H, and E1K/D32H mutants) were tested for tissue specificity by immunohistochemistry on neuroblastoma, osteosarcoma, rhabdomyosarcoma, Ewing sarcoma, desmoplastic small round cell tumors, adult sarcoma, and normal human tissues. Staining intensity scores are shown in Table 4. All neuroblastoma, 60% of osteosarcoma, 60% of desmoplastic small round cell tumors, 50% of rhabdomyosarcoma, 33% of Ewing sarcoma, and 72% of adult sarcoma stained positive. 12 normal human tissues were also tested (Table 5). Compared with parental hu3F8-scFv, affinity-maturated scFvs showed the reactivity in GD2(+) tissues expected to be positive (including frontal lobe, pons, cerebellum, and spinal cord), whereas all other normal tissues had negative staining.
TABLE 4.
Immunohistochemistry of hu3F8 scFv variants on different tumor samples
The strength is defined as follows: negative (−); weak, heterogeneous membrane staining (+); weak, homogeneous membrane staining (++); strong, heterogeneous membrane staining (+++); strong, homogeneous membrane staining (++++).
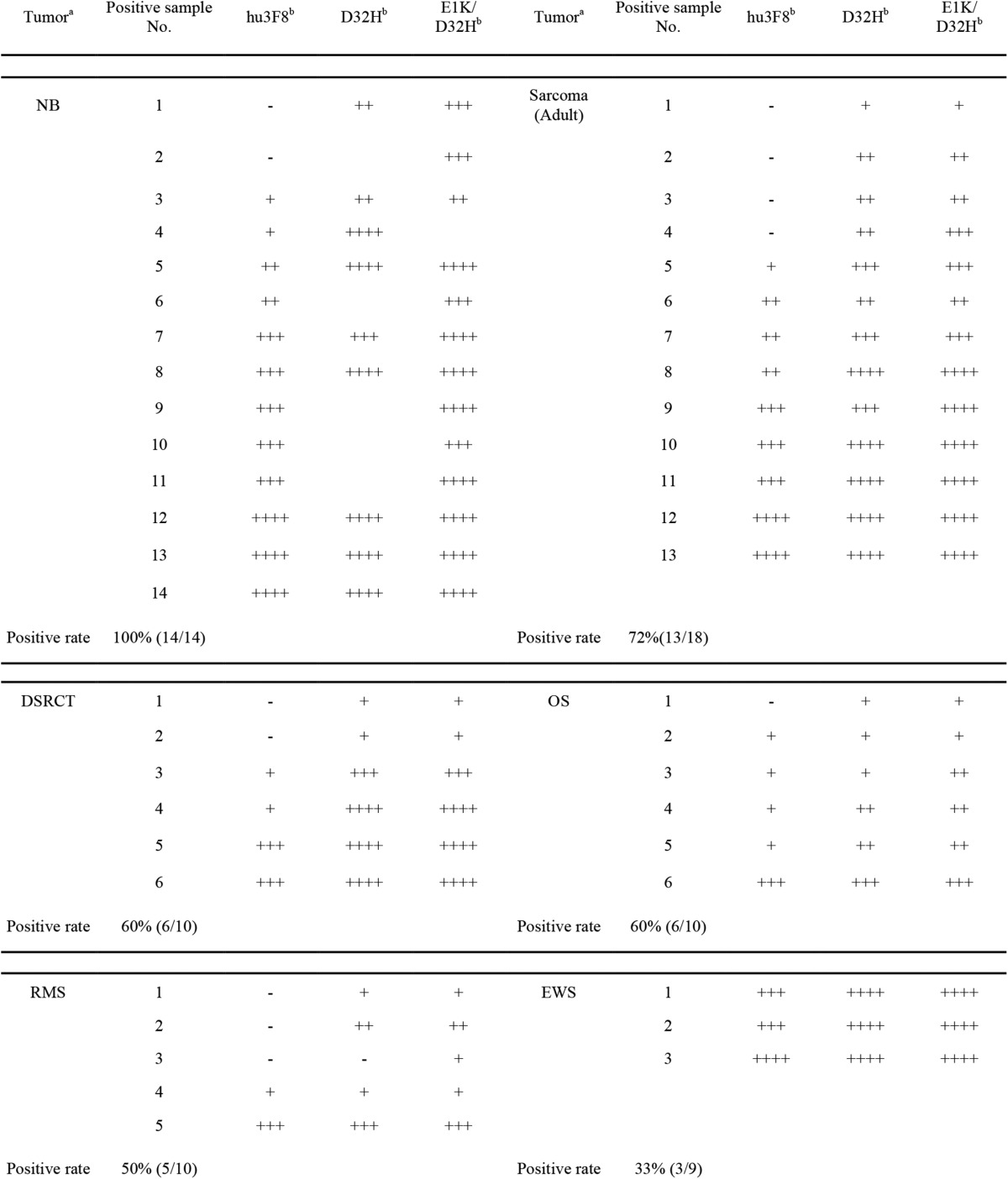
a NB, neuroblastoma; OS, osteosarcoma; RMS, rhabdomyosarcoma; Ewings, Ewing sarcoma; DSRCT, desmoplastic small round cell tumor.
b scFv concentration is 3 μg/ml; blank values indicate no data collected.
TABLE 5.
Organ cross-reactivity of hu3F8 scFv variants by immunohistochemistry
The strength is defined as follows: negative (−); weak, heterogeneous membrane staining (+); weak, homogeneous membrane staining (++); strong, heterogeneous membrane staining (+++); strong, homogeneous membrane staining (++++).
| Organ | hu3F8a | D32Ha | E1K/D32Ha |
|---|---|---|---|
| NBb | ++++ | ++++ | ++++ |
| Ileum | − | − | − |
| Skeletal muscle | − | − | − |
| Cerebellum | − | + | + |
| Frontal lobe | − | + | + |
| Pons | − | + | + |
| Stomach | − | − | − |
| Spinal cord | − | + | + |
| Lung | − | − | − |
| Spleen | − | − | − |
| Thyroid | − | − | − |
| Kidney | − | − | − |
| Testes | − | − | − |
a scFv concentration is 3 μg/ml.
b Stage 4 neuroblastoma tumor.
Anti-tumor Effect of hu3F8 Variants in Vivo
We next tested the efficacy of affinity-maturated variants in vivo using human neuroblastoma IMR-32 xenografted together with human PBMC subcutaneously in DKO mice. Intravenous injections of antibodies were given twice weekly for 2 weeks. In the first experiment, D32H antibody (at 10- and 50-μg doses) was compared with parental hu3F8 (at 50-μg doses). Parental hu3F8 was able to reduce tumor volume area under the curve by 51% compared with an isotype-matched non-targeting control antibody (Fig. 5A) although statistically insignificant (p = 0.2). The D32H-IgG1 antibody (at 10- and 50-μg doses) had much higher tumor suppression, reducing tumor volume area under the curve by 83 and 89%, respectively, compared with control (p = 0.029 and 0.035). A second experiment was done to compare 50-μg doses of both variants D32H and E1K/D32H, compared with parental hu3F8 (Fig. 5B). In the non-treatment group, none of the mice survived through the end of the experiment, whereas all five mice in the D32H and E1K/D32H groups survived, with similar reductions in tumor growth.
FIGURE 5.
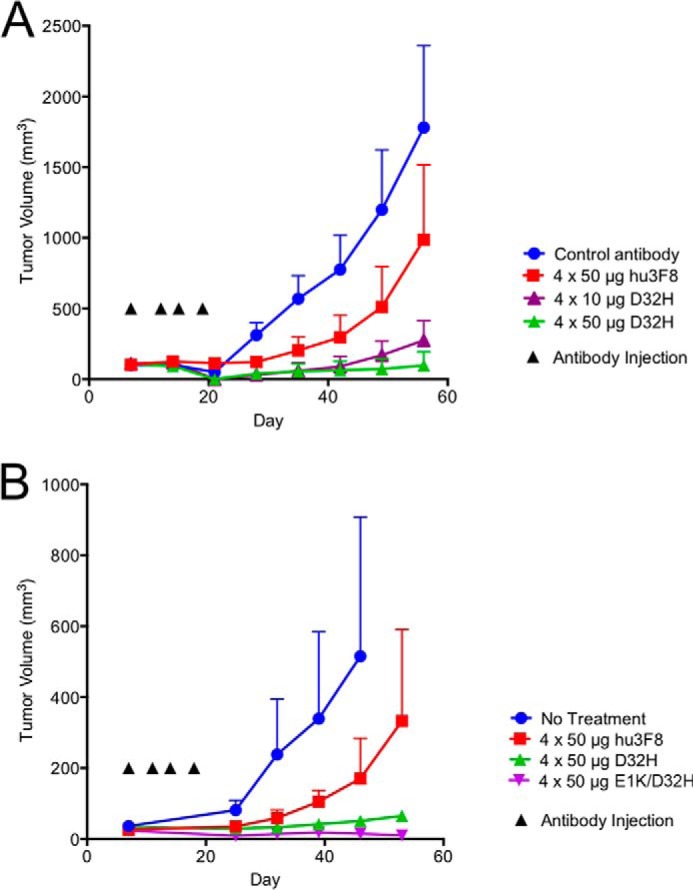
Affinity-maturated hu3F8 IgG1 exhibits potent in vivo anti-tumor activity toward neuroblastoma xenografts. Xenografts were established by implanting IMR-32 neuroblastoma cells mixed with human PBMC subcutaneously into immunodeficient mice. Antibodies were administered intravenously twice weekly for a total of four doses. Tumor volumes were measured weekly and shown as the mean ± S.E. (error bars). A, five mice per group were treated with isotype-matched control antibody, 4 × 50 μg of parental hu3F8, 4 × 10 μg of D32H antibody, or 4 × 50 μg of D32H antibody. One mouse died unexpectedly in the 4 × 50 μg of D32H group with no measurable tumor. B, five mice per group were given no treatment, 4 × 50 μg of parental hu3F8, 4 × 50 μg of D32H, or 4 × 50 μg E1K/D32H. All mice survived in the D32H and E1K/D32H antibody groups. Four of five mice in the hu3F8 group survived. No mice survived in the non-treatment group at the end of the experiment.
Discussion
Carbohydrate antigens play important roles in cancer biology. Antibodies targeting tumor-associated carbohydrate antigens have enormous potential as cancer therapeutics (32). Because the immune response to oligosaccharides is T-cell-independent, antibodies generated toward carbohydrate antigens are usually low-affinity IgM antibodies (25). To increase the therapeutic index of anti-carbohydrate antibodies, affinity improvement while preserving antigen specificity is an attractive approach (12). However, the structural similarities of gangliosides can result in a hierarchy of cross-reactivity during affinity maturation (6) and remains a challenge in creating effective antibody therapeutics. One such antigen is the ganglioside GD2, which is targeted by monoclonal antibodies for the clinical treatment of children with high-risk neuroblastoma. Effective affinity maturation approaches are needed to create next generation candidates with higher therapeutic potential.
Humanized 3F8 has so far shown minimal immunogenicity in Phase I trials, although its affinity is slightly lower than that of murine 3F8 (3). To recover affinity and to improve these antibodies, we described a combined yeast display and computational approach to accelerate and direct the affinity maturation process of anti-GD2 antibodies, exploiting whole Fv random mutation and structure-guided specific mutagenesis of key residues. Although directly introducing mutations into CDR loops has theoretical advantages over random mutagenesis of the entire variable regions, we considered that framework mutations could indirectly impact antibody-antigen interactions by affecting the antibody conformation (33) or by the positioning of contact residue side chains (16).
We first introduced random mutations into the entire Fv of humanized 3F8, including both CDR and the framework regions, displayed them on yeast cells, and competitively selected by FACS sorting. This is the first report on improving the affinity of carbohydrate-specific antibodies using yeast display in solution. The use of stepwise decrease in antigen concentration in FACS, in addition to negative selection against GM2, resulted in the enrichment of high-affinity binders. The mutations in high binders were scattered over CDRs and FRs of heavy and light chains, some of which were considered as irrelevant for binding. Using in silico mutagenesis analysis as a second step, focusing on the key interacting residues of humanized 3F8, we observed that single point mutations could dramatically change the electrostatic potential surfaces of the antigen-binding site, changing the charge distribution to more favorably interact with the negatively charged GD2-carbohydrate moiety. Using these approaches, we confirmed the importance of two mutations (E1K in FR1-VL and D32H in CDR1-VL), which had emerged in a single mutant of the yeast display. The side chain of histidine 32 was positioned at the bottom of the binding cavity and was a likely candidate for coordinating the negative charge of a sialic acid group. Lysine 1, on the other hand, was not positioned at a CDR loop, but it also increased the electrostatic interaction of hu3F8 with GD2 through long range interactions. Although not tested here, it is likely that inserting other positively charged residues at VL positions 1 and 32 could increase GD2 affinity, but these would need to be experimentally verified with particular attention to the loss of specificity. We did consider combining the D32H and E1K mutations with our previously identified heavy chain G54L mutation, which was identified solely by in silico scanning mutagenesis (30), but the resulting variants had enhanced aggregation properties, which is not therapeutically useful (data not shown).
We showed that the D32H and E1K/D32H variants led to enhancement of binding and created new energetically favorable interactions with the antigen. The improvement in affinity for the optimized IgG was found to be 7- to 13-fold compared with the parental hu3F8 IgG, giving rise to subnanomolar affinity, generally accepted as the desirable affinity ceiling for therapeutic antibodies (34). More importantly, despite nearly 3-fold enhancement in affinity when the second mutation, to Lys-1, was added to His-32, there was no substantial advantage in ADCC or CMC in vitro and no difference in anti-tumor effect in vivo. We conclude that the His-32 mutation was the minimally sufficient change to achieve a substantial improvement in affinity and biological effects for potential clinical application.
The use of immunohistochemistry as accompanying diagnostics for patient selection can be critical in validating the clinical utility of therapeutic antibodies. For trastuzumab, immunohistochemical screening has proven to be useful in selecting responsive patients who can benefit from antibody treatment (35). In this study, immunohistochemistry-based assays were used to confirm tissue specificity as the antibody affinity was being maturated. These results confirmed their ELISA specificity against known gangliosides in the epitope neighborhood. Epitope spread has clinical implications for ganglioside-specific antibodies. Anti-GD2 antibodies are known to induce significantly painful side effects with occasional reports of cranial or peripheral neuropathy (7, 36). Mutating antibodies to increase the affinity of anti-ganglioside antibodies can result in clones with epitope spread. Cross-reactivity with GD1a or GD1b can potentially cause significant motor (or axonal) or sensory (or ataxic) neuropathies (6). 3F8 has inherent low-level cross-reactivity with GD1b. These engineered IgG antibodies showed unexpectedly low reactivity with GD1b even when their affinities for GD2 were so much higher. In the future, with the advent of glycan arrays, the issue of cross-reactivity can be mapped with more granularity and precision (37).
In summary, affinity-maturated humanized 3F8 with a single mutation (D32H in CDR1-VL) achieved log-fold improvements in binding and in vitro and in vivo tumor cytotoxicity while maintaining tissue specificity. Our results also suggest an affinity ceiling for carbohydrate antibodies beyond which the gain in anti-tumor effect will diminish. This affinity maturation strategy could provide a rational platform for functional enhancement of other anti-carbohydrate therapeutic proteins.
Acknowledgments
We thank Dr. Mamoru Ito (Central Institute for Experimental Animals, Kawasaki, Japan) for providing the DKO mice and the MSK Small-Animal Imaging Core Facility and Molecular Cytology Core Facility for providing technical services. Technical services provided by the MSK Small-Animal Imaging Core Facility and Molecular Cytology Core Facility were supported in part by National Institutes of Health Cancer Center Core Support Grant P30 CA008748-48. We thank the Consortium for Functional Genomics, Emory University (Atlanta, GA), and Dr. Steven Robert of the Scripps Research Institute (La Jolla, CA) for providing biotin-GD2. We thank Yi Feng and Hoa Tran for excellent technical support and Drs. Ming Cheng and Hong Xu for reviewing the manuscript.
This study was supported in part by grants from the Band of Parents, the Kids Walk for Kids with Cancer NYC, and the Robert Steel Foundation. Q. Z., M. A., and N. K. C. were named as inventors for a patent application filed by Memorial Sloan Kettering Cancer Center related to high-affinity anti-GD2 monoclonal antibodies.
Cheung, I. Y., Basu, E. M., Modak, M., Kushner, B., Roberts, S., Y., F., Tran, H., Enero, C., Gregoria, L., O'Neill, T., and Cheung, N. K. (2014) Pharmacokinetics of humanized anti-GD2 monoclonal antibody Hu3F8 in patients with metastatic GD2-positive tumors. Poster presented at Advances in Neuroblastoma Research 2014, Cologne, Germany.
- ADCC
- antibody-dependent cell-mediated cytotoxicity
- CDR
- complementarity-determining region
- CMC
- complement-mediated cytotoxicity
- PBMC
- peripheral blood mononuclear cell
- FR
- framework region.
References
- 1. Fredman P., Hedberg K., Brezicka T. (2003) Gangliosides as therapeutic targets for cancer. BioDrugs 17, 155–167 [DOI] [PubMed] [Google Scholar]
- 2. Durrant L. G., Noble P., Spendlove I. (2012) Immunology in the clinic review series; focus on cancer: glycolipids as targets for tumour immunotherapy. Clin. Exp. Immunol. 167, 206–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cheung N. K., Dyer M. A. (2013) Neuroblastoma: developmental biology, cancer genomics and immunotherapy. Nat. Rev. Cancer 13, 397–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yu R. K., Tsai Y. T., Ariga T., Yanagisawa M. (2011) Structures, biosynthesis, and functions of gangliosides: an overview. J. Oleo Sci. 60, 537–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chester M. A. (1998) IUPAC-IUB Joint Commission on Biochemical Nomenclature (JCBN): nomenclature of glycolipids: recommendations 1997. Eur. J. Biochem. 257, 293–298 [DOI] [PubMed] [Google Scholar]
- 6. Hu J., Huang X., Ling C. C., Bundle D. R., Cheung N. K. (2009) Reducing epitope spread during affinity maturation of an anti-ganglioside GD2 antibody. J. Immunol. 183, 5748–5755 [DOI] [PubMed] [Google Scholar]
- 7. Cheung N. K., Cheung I. Y., Kushner B. H., Ostrovnaya I., Chamberlain E., Kramer K., Modak S. (2012) Murine anti-GD2 monoclonal antibody 3F8 combined with granulocyte-macrophage colony-stimulating factor and 13-cis-retinoic acid in high-risk patients with stage 4 neuroblastoma in first remission. J. Clin. Oncol. 30, 3264–3270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cheung N. K., Guo H., Hu J., Tassev D. V., Cheung I. Y. (2012) Humanizing murine IgG3 anti-GD2 antibody m3F8 substantially improves antibody-dependent cell-mediated cytotoxicity while retaining targeting in vivo. Oncoimmunology 1, 477–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shusterman S., London W. B., Gillies S. D., Hank J. A., Voss S. D., Seeger R. C., Reynolds C. P., Kimball J., Albertini M. R., Wagner B., Gan J., Eickhoff J., DeSantes K. B., Cohn S. L., Hecht T., Gadbaw B., Reisfeld R. A., Maris J. M., Sondel P. M. (2010) Antitumor activity of hu14.18-IL2 in patients with relapsed/refractory neuroblastoma: a Children's Oncology Group (COG) phase II study. J. Clin. Oncol. 28, 4969–4975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tarek N., Le Luduec J. B., Gallagher M. M., Zheng J., Venstrom J. M., Chamberlain E., Modak S., Heller G., Dupont B., Cheung N. K., Hsu K. C. (2012) Unlicensed NK cells target neuroblastoma following anti-GD2 antibody treatment. J. Clin. Invest. 122, 3260–3270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cheung I. Y., Hsu K., Cheung N. K. (2012) Activation of peripheral-blood granulocytes is strongly correlated with patient outcome after immunotherapy with anti-GD2 monoclonal antibody and granulocyte-macrophage colony-stimulating factor. J. Clin. Oncol. 30, 426–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ahmed M., Cheung N. K. (2014) Engineering anti-GD2 monoclonal antibodies for cancer immunotherapy. FEBS Lett. 588, 288–297 [DOI] [PubMed] [Google Scholar]
- 13. Navid F., Santana V. M., Barfield R. C. (2010) Anti-GD2 antibody therapy for GD2-expressing tumors. Curr. Cancer Drug Targets 10, 200–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cheung N. K., Lazarus H., Miraldi F. D., Abramowsky C. R., Kallick S., Saarinen U. M., Spitzer T., Strandjord S. E., Coccia P. F., Berger N. A. (1987) Ganglioside GD2 specific monoclonal antibody 3F8: a phase I study in patients with neuroblastoma and malignant melanoma. J. Clin. Oncol. 5, 1430–1440 [DOI] [PubMed] [Google Scholar]
- 15. Bradbury A. R., Sidhu S., Dübel S., McCafferty J. (2011) Beyond natural antibodies: the power of in vitro display technologies. Nat. Biotechnol. 29, 245–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Garcia-Rodriguez C., Levy R., Arndt J. W., Forsyth C. M., Razai A., Lou J., Geren I., Stevens R. C., Marks J. D. (2007) Molecular evolution of antibody cross-reactivity for two subtypes of type A botulinum neurotoxin. Nat. Biotechnol. 25, 107–116 [DOI] [PubMed] [Google Scholar]
- 17. Nakanishi T., Maru T., Tahara K., Sanada H., Umetsu M., Asano R., Kumagai I. (2013) Development of an affinity-matured humanized anti-epidermal growth factor receptor antibody for cancer immunotherapy. Protein Eng. Des. Sel. 26, 113–122 [DOI] [PubMed] [Google Scholar]
- 18. Votsmeier C., Plittersdorf H., Hesse O., Scheidig A., Strerath M., Gritzan U., Pellengahr K., Scholz P., Eicker A., Myszka D., Coco W. M., Haupts U. (2012) Femtomolar Fab binding affinities to a protein target by alternative CDR residue co-optimization strategies without phage or cell surface display. MAbs 4, 341–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhao Q., Feng Y., Zhu Z., Dimitrov D. S. (2011) Human monoclonal antibody fragments binding to insulin-like growth factors I and II with picomolar affinity. Mol. Cancer Ther. 10, 1677–1685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang Y., Keck Z. Y., Saha A., Xia J., Conrad F., Lou J., Eckart M., Marks J. D., Foung S. K. (2011) Affinity maturation to improve human monoclonal antibody neutralization potency and breadth against hepatitis C virus. J. Biol. Chem. 286, 44218–44233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Boder E. T., Raeeszadeh-Sarmazdeh M., Price J. V. (2012) Engineering antibodies by yeast display. Arch. Biochem. Biophys. 526, 99–106 [DOI] [PubMed] [Google Scholar]
- 22. Valjakka J., Hemminki A., Niemi S., Söderlund H., Takkinen K., Rouvinen J. (2002) Crystal structure of an in vitro affinity- and specificity-matured anti-testosterone Fab in complex with testosterone. Improved affinity results from small structural changes within the variable domains. J. Biol. Chem. 277, 44021–44027 [DOI] [PubMed] [Google Scholar]
- 23. Inoue H., Suganami A., Ishida I., Tamura Y., Maeda Y. (2013) Affinity maturation of a CDR3-grafted VHH using in silico analysis and surface plasmon resonance. J. Biochem. 154, 325–332 [DOI] [PubMed] [Google Scholar]
- 24. Barderas R., Desmet J., Timmerman P., Meloen R., Casal J. I. (2008) Affinity maturation of antibodies assisted by in silico modeling. Proc. Natl. Acad. Sci. U.S.A. 105, 9029–9034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Heimburg-Molinaro J., Rittenhouse-Olson K. (2009) Development and characterization of antibodies to carbohydrate antigens. Methods Mol. Biol. 534, 341–357 [DOI] [PubMed] [Google Scholar]
- 26. Thomas R., Patenaude S. I., MacKenzie C. R., To R., Hirama T., Young N. M., Evans S. V. (2002) Structure of an anti-blood group A Fv and improvement of its binding affinity without loss of specificity. J. Biol. Chem. 277, 2059–2064 [DOI] [PubMed] [Google Scholar]
- 27. Zhao Q., Zhu Z., Dimitrov D. S. (2012) Yeast display of engineered antibody domains. Methods Mol. Biol. 899, 73–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chao G., Lau W. L., Hackel B. J., Sazinsky S. L., Lippow S. M., Wittrup K. D. (2006) Isolating and engineering human antibodies using yeast surface display. Nat. Protoc. 1, 755–768 [DOI] [PubMed] [Google Scholar]
- 29. Zhao Q., Chan Y. W., Lee S. S., Cheung W. T. (2009) One-step expression and purification of single-chain variable antibody fragment using an improved hexahistidine tag phagemid vector. Protein Expr. Purif. 68, 190–195 [DOI] [PubMed] [Google Scholar]
- 30. Ahmed M., Goldgur Y., Hu J., Guo H. F., Cheung N. K. (2013) In silico driven redesign of a clinically relevant antibody for the treatment of GD2 positive tumors. PLoS One 8, e63359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kushner B. H., Kramer K., Cheung N. K. (2001) Phase II trial of the anti-G(D2) monoclonal antibody 3F8 and granulocyte-macrophage colony-stimulating factor for neuroblastoma. J. Clin. Oncol. 19, 4189–4194 [DOI] [PubMed] [Google Scholar]
- 32. Magnani J. L., Ernst B. (2009) Glycomimetic drugs: a new source of therapeutic opportunities. Discov. Med. 8, 247–252 [PubMed] [Google Scholar]
- 33. Foote J., Milstein C. (1994) Conformational isomerism and the diversity of antibodies. Proc. Natl. Acad. Sci. U.S.A. 91, 10370–10374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wittrup K. D., Thurber G. M., Schmidt M. M., Rhoden J. J. (2012) Practical theoretic guidance for the design of tumor-targeting agents. Methods Enzymol. 503, 255–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Leyland-Jones B. (2001) Maximizing the response to Herceptin therapy through optimal use and patient selection. Anticancer Drugs 12, S11–S17 [DOI] [PubMed] [Google Scholar]
- 36. Yu A. L., Gilman A. L., Ozkaynak M. F., London W. B., Kreissman S. G., Chen H. X., Smith M., Anderson B., Villablanca J. G., Matthay K. K., Shimada H., Grupp S. A., Seeger R., Reynolds C. P., Buxton A., Reisfeld R. A., Gillies S. D., Cohn S. L., Maris J. M., Sondel P. M., and Children's Oncology, G. (2010) Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N. Engl. J. Med. 363, 1324–1334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang L., Cummings R. D., Smith D. F., Huflejt M., Campbell C. T., Gildersleeve J. C., Gerlach J. Q., Kilcoyne M., Joshi L., Serna S., Reichardt N. C., Parera Pera N., Pieters R. J., Eng W., Mahal L. K. (2014) Cross-platform comparison of glycan microarray formats. Glycobiology 24, 507–517 [DOI] [PMC free article] [PubMed] [Google Scholar]