

Background: P450 21A2 catalyzes 21-hydroxylation of both progesterone and 17α-hydroxyprogesterone, an important step in adrenal steroidogenesis.
Results: A crystal structure of human P450 21A2 with progesterone can explain many functional variants.
Conclusion: High kinetic deuterium isotope effects show the importance of a closely spaced site for C21 hydrogen abstraction.
Significance: The structure provides insight into enzyme deficiencies in congenital adrenal hyperplasia.
Keywords: cytochrome P450, enzyme kinetics, isotope effect, steroidogenesis, x-ray crystallography
Abstract
Cytochrome P450 (P450) 21A2 is the major steroid 21-hydroxylase, and deficiency of this enzyme is involved in ∼95% of cases of human congenital adrenal hyperplasia, a disorder of adrenal steroidogenesis. A structure of the bovine enzyme that we published previously (Zhao, B., Lei, L., Kagawa, N., Sundaramoorthy, M., Banerjee, S., Nagy, L. D., Guengerich, F. P., and Waterman, M. R. (2012) Three-dimensional structure of steroid 21-hydroxylase (cytochrome P450 21A2) with two substrates reveals locations of disease-associated variants. J. Biol. Chem. 287, 10613–10622), containing two molecules of the substrate 17α-hydroxyprogesterone, has been used as a template for understanding genetic deficiencies. We have now obtained a crystal structure of human P450 21A2 in complex with progesterone, a substrate in adrenal 21-hydroxylation. Substrate binding and release were fast for human P450 21A2 with both substrates, and pre-steady-state kinetics showed a partial burst but only with progesterone as substrate and not 17α-hydroxyprogesterone. High intermolecular non-competitive kinetic deuterium isotope effects on both kcat and kcat/Km, from 5 to 11, were observed with both substrates, indicative of rate-limiting C–H bond cleavage and suggesting that the juxtaposition of the C21 carbon in the active site is critical for efficient oxidation. The estimated rate of binding of the substrate progesterone (kon 2.4 × 107 m−1 s−1) is only ∼2-fold greater than the catalytic efficiency (kcat/Km = 1.3 × 107 m−1 s−1) with this substrate, suggesting that the rate of substrate binding may also be partially rate-limiting. The structure of the human P450 21A2-substrate complex provides direct insight into mechanistic effects of genetic variants.
Introduction
Cytochrome P450 (P450)4 21A2 is the major steroid 21-hydroxylase, which catalyzes the 21-hydroxylation of progesterone and 17α-hydroxyprogesterone (17α-OH-progesterone) to form 11-deoxycorticosterone and 11-deoxycortisol, respectively (Fig. 1), primarily in the adrenal cortex (1–3). Although P450s from some other gene families (e.g. 2C and 2D) can catalyze this hydroxylation (4, 5), P450 21A2 is clearly the most important in humans, and >100 different amino acid variants have been identified in clinical settings (2, 6, 7). There are two CYP21 genes in the human genome, CYP21A1 (a pseudogene) and CYP21A2 (the functional gene) (8, 9). Both are located on chromosome 6 in the major histocompatibility locus. These two CYP21 genes have 98% exonic sequence identity. Some of the clinical variants result from recombination of the two genes, and others result from other single amino acid mutations. Deficiencies in the activity lead to a type of congenital adrenal hyperplasia, which has three clinical forms: a severe form with concurrent defects in both cortisol and aldosterone biosynthesis (“salt-wasting” type), a form with adequate aldosterone biosynthesis (“simple virilizing” type), and a mild, non-classic form that can be asymptomatic or associated with signs of postpubertal androgen excess without cortisol deficiency (2, 6). The worldwide incidence of congenital adrenal hyperplasia is 1:15,000 for the classical form and at least 1:1,000 for the non-classical form (7, 10), due primarily to gene conversion events with the adjacent CYP21A1P pseudogene; thus, congenital adrenal hyperplasia is one of the most common inborn metabolic errors known in humans. In addition, P450 21A2 is the major autoantigen in autoimmune Addison disease (11).
FIGURE 1.

Reactions catalyzed by P450 21A2.
Because of the relevance of P450 21A2 to human disease, there is considerable interest in identifying the molecular basis for loss of function in P450 21A2 variants (6, 7). Major aspects of structure-activity relationships among the deficient variants include changes in the structure, the nature of the rate-limiting steps in catalysis, and the substrate selectivity (Fig. 1). In particular, P450 21A2 is unusual in that its major 21-hydroxylase activities generate electron-deficient primary carbon radicals (-CH3 → -CH2•) in the course of the reaction (12, 13).
Despite the clinical interest in P450 21A2, only limited work has been done directly with the human enzyme. Bovine P450 21A2 was purified from adrenal cortex (14). Arase et al. (15) successfully expressed the bovine enzyme in Escherichia coli in 2006, and in 2012, we reported the x-ray crystal structure of modified bovine P450 21A2 (6). That structure has been used to model some of the possible effects of variants observed in clinics (6, 7). Human P450 21A2 has been heterologously expressed in yeast microsomes and utilized in mutagenesis and basic kinetic isotope effect (KIE) studies (13) and also expressed in baculovirus (11) and bacterial systems (16).
In order to address several catalytic and structural issues related to the function of P450 21A2, we expressed the human enzyme in E. coli and purified the enzyme. This protein was used to obtain an x-ray crystal structure in the presence of the substrate progesterone (6) bound at the active site, with a second substrate molecule of low occupancy residing inside the access channel (in the bovine complex, the corresponding sites are fully occupied by 17α-OH-progesterone (6)). Kinetic analyses indicate that human P450 21A2 is more catalytically efficient than the bovine enzyme and may be the most catalytically efficient mammalian P450 reported (i.e. has the highest kcat/Km value) (3). In contrast to previous reports on the bovine enzyme (17), we find that the enzyme is not limited by rates of product dissociation but, at least in large part, by the rate of C–H bond breaking (Fig. 2, step 7), as revealed by high non-competitive intermolecular KIEs.
FIGURE 2.

P450 catalytic cycle.
Experimental Procedures
Chemicals
Progesterone was purchased from Sigma-Aldrich, and 17α-OH-progesterone was purchased from Steraloids (Newport, RI). All radiolabeled steroids were purchased from American Radiolabeled Chemicals (St. Louis, MO). 21,21,21-d3-progesterone and 21,21,21-d3-17α-OH-progesterone were synthesized as described previously (13).
Expression of Human P450 21A2
In the human CYP21A2 open reading frame, the region encoding the N-terminal transmembrane helix (residues 1–29) was replaced by nucleotides coding for the peptide MAKKTSSKGK (P450 2C3 N-terminal region), and the 3′-end was extended by 18 nucleotides encoding six histidines. The entire modified cDNA was synthesized by GenScript (Piscataway, NJ) and inserted into a pET17b expression vector (EMD Millipore, Billerica, MA). The pET-17b plasmid containing the gene encoding human P450 21A2 was co-transformed with a chaperone pGro12 plasmid into E. coli BL21-Gold DE(3)-competent cells. The cultures were used for inoculation in Terrific Broth medium with 100 μg/ml ampicillin and 50 μg/ml kanamycin at 37 °C and 250 rpm. When an OD600 of 1.0 was achieved (∼3.5 h), the shaker speed was lowered to 100 rpm, and δ-aminolevulinic acid and arabinose were added to final concentrations of 1.0 and 27 mm, respectively. After 60 min, the temperature was lowered to 26 °C, and 1.0 mm isopropyl 1-thio-β-d-galactopyranoside was added to induce the expression of human P450 21A2. The cells were cultured for another 40 h at 26 °C (shaking at 100 rpm) and harvested by centrifugation (2.5 × 103 g, 20 min). The cells were subsequently suspended in 50 mm Tris-HCl buffer (pH 7.4) containing 250 mm sucrose and 0.5 mm EDTA (200 ml/liter of cell culture) containing 1 mg/ml lysozyme. After incubation at 4 °C for 30 min, the cells were pelleted and stored at −80 °C.
Purification of Human P450 21A2
Human P450 21A2 was purified from the E. coli cells as described previously for the recombinant bovine enzyme, with minor modifications (6). Briefly, the cell pellets were thawed and sonicated in 200 ml of sonication buffer (50 mm potassium phosphate buffer (pH 7.4) containing 20% glycerol (v/v), 0.5 m NaCl, 1.0% sodium cholate (w/v) (added to extract the protein from the membranes), 0.1 mm dithiothreitol (DTT), 0.1 mm EDTA, 0.1 mm phenylmethylsulfonyl fluoride (PMSF), and a protease inhibitor mixture (Roche Complete Tablets, EDTA-free, EASYpack)). Unbroken cells and cell debris were removed by centrifugation for 60 min at 105 × g. The reddish brown supernatant was applied to an Ni2+-nitrilotriacetic acid-agarose column, followed by 10 column volumes of wash buffer (50 mm potassium phosphate (pH 7.4) containing 20% glycerol (v/v), 0.5 m NaCl, 0.25% sodium cholate (w/v), 20 mm imidazole, 0.1 mm DTT, and 0.1 mm EDTA). The bound P450 21A2 was subsequently eluted with elution buffer (wash buffer containing 250 mm imidazole) and collected in 2-ml fractions. The most concentrated fractions were combined and dialyzed against 100 mm potassium phosphate buffer (pH 7.4) containing 20% glycerol (v/v), 0.3 m NaCl, and 0.1 mm EDTA, which removed the sodium cholate.
The protein for crystallization experiments was purified with a method similar to that used for purifying bovine P450 21A2, except that no detergent was used for chromatography with an SP Fast-Flow Sepharose column (GE Healthcare). The protein was eluted with 50 mm potassium phosphate buffer (pH 7.2) containing 20% glycerol (v/v), 0.1 mm DTT, 0.1 mm EDTA, and 400 mm NaCl.
Spectral Analyses of P450 21A2
Steady-state binding experiments were carried out using an Aminco DW2/OLIS spectrophotometer (On-Line Instruments, Bogart, GA) at 23 °C. In general, P450 21A2 (1.0 μm, in 0.2 m potassium phosphate (pH 7.4) buffer) was titrated with increasing concentrations of ligands (in C2H5OH) in the sample cuvette, and the same amount of C2H5OH was added to the reference cuvette. The UV-visible spectra from 350 to 500 nm were recorded. The spectral change Δ(A390 − A418), as a function of ligand concentration, was fit to a quadratic equation of the form, y = B + (A/2)*(1/E)*((Kd + E + X) − sqrt((Kd + E + X)∧2 − (4*E*X)), using Prism software (GraphPad, Inc.).
In order to obtain more accurate Kd values for the ligands with initial Kd values of <1 μm, titration experiments were repeated in a Cary 14/OLIS spectrophotometer using a 10-cm path length cylindrical cuvette (Starna Cells (Atascadero, CA), catalogue no. 34-Q-100, volume 25 ml). P450 21A2 (0.10 μm, in 0.2 m potassium phosphate (pH 7.4) buffer) was titrated with increasing concentrations of ligands (dissolved in C2H5OH). The spectral change Δ(A390 − A418), as a function of ligand concentration, was fit to a quadratic equation (see above) using GraphPad Prism software.
An OLIS RSM-1000 instrument (1.24-mm slits, 20-mm path length) was used to study pre-steady-state kinetics of ligand binding to P450 21A2. Experiments were performed in the rapid scanning mode at 23 °C in 0.2 m potassium phosphate buffer (pH 7.4) containing 60 μm l-α-1,2-dilauroyl-sn-glycero-3-phosphocholine. Briefly, 2 or 4 μm P450 was mixed with 4 μm ligand, and spectral data were collected for 1 s. The traces at 390 nm were fit to equations using the manufacturer's software (GlobalWorks). The basis of the experimental design is described by Daniels and Alberty (18). A second-order reaction 2A → A2 is kinetically equivalent to A + B → AB, if similar concentrations of A and B are mixed; the analysis is straightforward, especially if the back-reaction is slow (AB → A + B), which was the case here. This approach was used instead of trying to achieve pseudo-first-order kinetics with excess substrate, because of the low Kd values (i.e. Kd < 1–2 μm, which is a minimum for detection of changes in the kinetic binding experiments). Experiments with higher concentrations of substrate did not result in faster rates, in that the enzyme was already saturated with substrate.
Catalytic Activity Assays
In general, steady-state enzyme assays were done in 500-μl reaction volumes at 37 °C in 100 mm potassium phosphate buffer (pH 7.4). The reconstituted enzyme system contained 2 nm P450, 0.6 μm recombinant rat NADPH-P450 reductase (19), 15 μm l-α-1,2-dilauroyl-sn-glycero-3-phosphocholine, and varying concentrations of progesterone or 17α-OH-progesterone (0–40 μm). The reactions were initiated by the addition of an NADPH-regenerating system containing 10 mm glucose 6-phosphate, 0.5 mm NADP+, and 1 IU/ml glucose-6-phosphate dehydrogenase (20). Reactions were quenched by the addition of 2 ml of CH2Cl2, and the steroid products were extracted into the organic solvent (lower phase) by mixing with a vortex device. After centrifugation to separate the layers (3 × 103 g, 10 min), an aliquot of the organic phase was transferred and dried under an N2 stream. The extracted products were dissolved in a CH3OH/H2O mixture (70%, v/v) and injected onto an Acquity BEH octadecylsilane (C18) UPLC column (2.1 mm × 50 mm, 1.7 μm) in a Waters Acquity UPLC system connected to a UV detector set to a wavelength of 243 nm. The UPLC column was used at 40 °C. The product was identified and quantified using external standards. With progesterone as the substrate, the following CH3OH/H2O linear gradient was used, at a flow rate of 0.1 ml/min: 0–2 min, hold at 70% CH3OH (v/v); 2–6 min, linear increase from 70 to 95% CH3OH (v/v); 6–6.5 min, hold at 95% CH3OH (v/v); 6.5–7 min, linear decrease from 95 to 70% CH3OH (v/v); 7–10 min, hold at 70% CH3OH (v/v). With 17α-OH-progesterone as the substrate, the following CH3OH/H2O linear gradient was used, at a flow rate of 0.1 ml/min: 0–2 min, hold at 60% CH3OH (v/v); 2–6 min, linear increase from 60 to 85% CH3OH (v/v); 6–6.5 min, hold at 85% CH3OH (v/v); 6.5–7 min, linear decrease from 85 to 60% CH3OH (v/v); 7–10 min, hold at 60% CH3OH (v/v).
Pre-steady-state Kinetics of Product Formation
Pre-steady-state 21-hydroxylation reactions with P450 21A2 were carried out in a KinTek instrument (model RQF-3, KinTek Corp., State College, PA). All reactions were performed in 100 mm potassium phosphate buffer (pH 7.4) at 37 °C. Human P450 21A2 (2 μm) was reconstituted with 4 μm recombinant rat NADPH-P450 reductase (19), 15 μm l-α-1,2-dilauroyl-sn-glycero-3-phosphocholine, and either 20 μm progesterone ([4-14C]progesterone) or 17α-OH-progesterone (17α-OH-[1,2,6,7-3H]progesterone). The reactions were initiated by rapidly mixing with an NADPH-regenerating system (20 mm glucose 6-phosphate, 2 mm NADPH, and 2 IU/ml glucose-6-phosphate dehydrogenase (20)) and quenched with 1 m HCl as a function of time from 10 ms to 1 s. A 50-μl aliquot of each reaction mixture was spotted onto the loading zone of a channeled silica gel G TLC plate (Analtech, Newark, DE), and the plates were developed with ethyl acetate/CH2Cl2 (1:4 (v/v) for progesterone and 1:3 (v/v) for 17α-OH-progesterone). With progesterone as the substrate, the plate was exposed to a phosphor imager screen (Imaging Screen K, Bio-Rad) for 7 days and analyzed with a molecular imager system (Pharos FX Plus Molecular Imager, Bio-Rad) using the manufacturer's Quantity One software. With 17α-OH-progesterone as the substrate, the plate was scanned with an AR-2000 Radio-TLC imaging scanner (Bioscan, Poway, CA) and analyzed with the manufacturer's Winscan software. The data points were fit to a burst equation, y = A(1 − e−kpt) + ksst, where A is the burst amplitude, kp is the first-order rate of the pre-steady-state burst, and kss is the steady-state rate, using GraphPad Prism software.
Crystallization
To obtain the human P450 21A2-progesterone complex, substrate (dissolved in C2H5OH) was added in a 1:5 (P450/progesterone) molar ratio, followed by mixing and concentrating to 25 mg/ml in 50 mm potassium phosphate buffer (pH 7.2) containing 5% glycerol (v/v), 0.1 mm DTT, 0.1 mm EDTA, and 100 mm NaCl. Aliquots of the complex solution were frozen in liquid N2 and then stored at −80 °C. Human P450 21A2-progesterone complex crystals were grown using the sitting drop vapor diffusion technique by mixing equal volumes (200 nl) of complex solution and mother liquor (0.10 m sodium HEPES (pH 7.5) containing 0.20 m ammonium sulfate and 25% (w/v) polyethylene glycol 3350) and equilibrating droplets against 60 μl of reservoir solution at 20 °C. Microcrystals were obtained within 2 weeks, and in order to increase their size, one round of microseeding was performed. Wells containing microcrystals were washed with 10 μl of reservoir buffer and transferred to a microcentrifuge tube with 40 μl of mother liquor. After vortex mixing the seeds for 2 min and diluting 1:1000 with mother liquor, 200 nl of the seed solution was mixed with 200 nl of protein for each drop. Larger crystals of the complex grew from microseeded droplets in 5 days. Crystals were mounted in nylon loops and swiped through a droplet of 25% glycerol (v/v) in mother liquor for flash freezing in liquid nitrogen.
X-ray Data Collection, Structure Determination, and Refinement
Diffraction data were collected on the insertion device beam line (21-ID-G) of the Life Sciences Collaborative Access Team, located at Sector 21 of the Advanced Photon Source, Argonne National Laboratory (Argonne, IL) at a wavelength of 0.9782 Å using a Mar300 CCD detector (at 100 K). Processing was done with the program HKL2000 (21), and selected crystal data and refinement statistics are summarized in Table 2. The structure of the P450 21A2-progesterone complex was determined by molecular replacement with the program MOLREP (22) (23), using the crystallographic coordinates of bovine P450 21A2 as the search model (6) (Protein Data Bank code 3QZ1). Refinement was carried out with the program Refmac5 (23, 24), and manual rebuilding was performed with Coot (25). The r.m.s.d. values from the superimposition were calculated with the Superpose routine in CCP4 (23). All structural figures were generated with the program UCSF Chimera (26).
TABLE 2.
Steady-state kinetics parameters for 21-hydroxylation catalyzed by recombinant bovine P450 21A2
| Substrate | kcat | Km | kcat/Km |
|---|---|---|---|
| min−1 | μm | μm−1 min−1 | |
| Progesterone | 47 ± 1 | 0.50 ± 0.07 | 91 ± 14 |
| 17α-OH progesterone | 40 ± 1 | 0.44 ± 0.07 | 94 ± 13 |
Results
Purification of Human P450 21A2 and Catalytic Properties
To facilitate the expression of human P450 21A2 in E. coli, the N terminus was replaced with the sequence MAKKTSSKGK to increase the solubility of this enzyme; this sequence has been utilized to improve heterologous expression of several P450s (27). P450 21A2 was stabilized in high ionic strength buffers; thus, at least 0.3 m NaCl was added in the buffers during purification. The protein was efficiently purified to >90% electrophoretic purity after one step of Ni2+-nitrilotriacetic acid affinity chromatography. The Fe2+·CO versus Fe2+-reduced difference spectra of purified human P450 21A2 displayed a typical P450 spectrum with a Soret peak at 450 nm (Fig. 3B). Only a small 420 nm peak was observed in the assays (when 1 μm safranin T was added as a mediator to increase the rate of reduction (29)), indicating that the purified human P450 21A2 is very low in the presence of the inactive form (cytochrome P420) (<5%).
FIGURE 3.

Purification of human P450 21A2. A, SDS-PAGE. The molecular weight maker (left) and purified P450 21A2 (right) are indicated. B, Fe2+·CO versus Fe2+ difference spectrum (0.38 μm P450) (28). (Safranin T (1 μm) was added as an electron mediator (29).)
Steady-state Kinetics of Hydroxylation Reactions Catalyzed by Human and Bovine P450 21A2
Steady-state kinetic parameters for both progesterone and 17α-OH-progesterone 21-hydroxylation by purified human and bovine P450 21A2 were determined (Fig. 4 and Table 1). The bovine P450 21A2 C3B21RA mutant, designed for crystallization of bovine P450 21A2, has a similar functional activity as the wild-type protein and was studied for direct comparison. Bovine P450 21A2 had comparable catalytic efficiency (∼90 min−1 μm−1) for the substrates progesterone and 17α-OH-progesterone, with similar kcat (∼40 min−1) and Km (∼0.5 μm) values (Table 2). The catalytic activities determined for this mutant were significantly higher than those reported previously (6), probably due to the lack of residual detergent (Cymal 5) used in that work. In particular, for progesterone, the kcat value is about 3-fold higher, and the Km value is about 5-fold lower, yielding a 15-fold increase in the catalytic efficiency compared with the previous kinetic data reported for the purified bovine P450 21A2 C3B21RA mutant (6). Human P450 21A2 is a more efficient enzyme than bovine P450 21A2, with catalytic efficiencies of 800 and 160 μm−1 min−1 for progesterone and 17α-OH-progesterone, respectively (i.e. 1.3 × 107 and 2.7 × 106 m−1 s−1, respectively). Although the kcat values were similar with both substrates, the 7-fold lower Km value with the substrate progesterone resulted in much higher catalytic efficiency than with the substrate 17α-OH-progesterone. Compared with other previous studies (11, 30), the significantly higher enzymatic activities of human P450 21A2 reported herein are probably due to the fact that the purified enzyme was utilized in the assays rather than whole cells or cell lysates containing expressed human P450 21A2.
FIGURE 4.

Steady-state kinetics of 21-hydroxylation of progesterone and 17α-hydroxyprogesterone by human and bovine P450 21A2. The curves represent the best non-linear fits to the Michaelis-Menten equation. A, human P450 21A2 with the substrate progesterone; B, human P450 21A2 with the substrate 17α-OH-progesterone. C, bovine P450 21A2 with the substrate progesterone; D, bovine P450 21A2 with the substrate 17α-OH-progesterone.
TABLE 1.
Steady-state kinetics parameters for 21-hydroxylation catalyzed by recombinant human P450 21A2
| Substrate | kcat | Km | kcat/Km | DV | D(V/K) |
|---|---|---|---|---|---|
| min−1 | μm | μm−1 min−1 | |||
| d0-Progesterone | 168 ± 4 | 0.21 ± 0.03 | 800 ± 120 | 6.5 ± 0.3 | 11 ± 2 |
| 21,21, 21-d3- Progesterone | 26 ± 1 | 0.37 ± 0.01 | 70 ± 2 | 6.5 ± 0.3 | 11 ± 2 |
| d0-17α-OH progesterone | 240 ± 5 | 1.5 ± 0.1 | 160 ± 11 | 8.0 ± 1.0 | 5.3 ± 0.6 |
| 21,21, 21-d3-17α-OH progesterone | 30 ± 1 | 1.0 ± 0.1 | 30 ± 3 | 8.0 ± 1.0 | 5.3 ± 0.6 |
Binding of Substrates to Human P450 21A2
The binding of the substrates progesterone and 17α-OH-progesterone to P450 21A2 produces a classic Type I binding spectrum (Fig. 5), indicating the effects of substrate binding on the heme iron spin state equilibrium. High affinities of P450 21A2 for the steroid substrates progesterone and 17α-OH-progesterone were observed, with Kd values of ∼10 ± 3 and 30 ± 3 nm, respectively. In order to estimate the rate constants (kon) for substrate binding, rates of spectral changes at 390 nm were monitored using stopped-flow spectroscopy, and the kon values were determined by fitting. The estimated kon values were 2.4 × 107 and 2.2 × 107 m−1 s−1 for progesterone and 17α-OH-progesterone, respectively. The dissociation rate constants (koff) were estimated using the relationship Kd = koff/kon and were 0.24 and 0.66 s−1 for progesterone and 17α-OH-progesterone, respectively. These studies indicated that substrate binding and release are relatively fast for P450 21A2 with both substrates. The monophasic nature of the binding of both substrates differs from the apparent biphasic binding of 17α-OH-progesterone to bovine P450 21A2 (6).
FIGURE 5.

Binding of steroid substrates to human P450 21A2. Steady-state titrations of P450 21A2 (1.0 μm) were conducted with varying concentrations of steroids in 0.2 m potassium phosphate buffer (pH 7.4) (shown in A (progesterone) and B (17α-OH-progesterone)). The plots in C (progesterone) and D (17α-OH-progesterone) show quadratic fits of spectral changes (Δ(A390 − A418)) when the titrations were repeated with 0.10 μm P450 21A2 (progesterone Kd = 0.010 ± 0.003 μm; 17α-OH-progesterone Kd = 0.030 ± 0.003 μm). Stopped-flow absorbance changes (ΔA390) for binding of different substrates (2 μm) to P450 21A2 (1 μm) are shown in E and F. The absorbance changes at 390 nm were fit using the manufacturer's software (OLIS GlobalWorks): binding of progesterone to human P450 21A2, kon = 2.4 × 107 m−1 s−1; binding of 17α-OH-progesterone to human P450 21A2, kon = 2.2 × 107 m−1 s−1). The residuals analysis is shown in the insets in E and F.
Binding of Products to Human P450 21A2
Titration of P450 21A2 with the 21-OH products deoxycorticosterone and 11-deoxycortisol also yielded Type I binding spectra (Fig. 6). The Kd values were 1.2 and 2.9 μm for deoxycorticosterone and 11-deoxycortisol, respectively, which are 2 orders of magnitude higher than those for the substrates. Pre-steady-state binding studies using stopped-flow spectroscopy yielded kon values of 2.2 × 107 and 1.9 × 107 m−1 s−1 for deoxycorticosterone and 11-deoxycortisol, respectively, which are comparable with the rate constants for binding of substrates. The calculated koff rates were 26 s−1 for deoxycorticosterone and 55 s−1 for 11-deoxycortisol. The lower affinities of products for P450 21A2 are probably due to fast product dissociation from P450 21A2, as is the case with most proteins (31).
FIGURE 6.

Binding of steroid products to human P450 21A2. Steady-state titrations of P450 21A2 (1 μm) were conducted with varying concentrations of steroid products in 0.2 m potassium phosphate buffer (pH 7.4) (shown in A and B). The plots show quadratic fits of spectral changes (ΔA390 − A418), and insets show spectral changes. C, titration of human P450 21A2 with 11-deoxycorticosterone (Kd = 1.2 ± 0.2 μm). D, titration of human P450 21A2 with 11-deoxycortisol (Kd = 2.9 ± 0.7 μm). Stopped-flow absorbance changes (ΔA390) for binding of different steroids (2 μm) to P450 21A2 (2 μm) are shown in E and F. The absorbance changes at 390 nm were fit using the manufacturer's software (OLIS GlobalWorks). E, kinetics of binding of 11-deoxycorticosterone to human P450 21A2 (kon = 1.1 × 107 m−1 s−1), F, kinetics of binding of 11-deoxycortisol to human P450 21A2 (kon = 9.5 × 106 m−1 s−1). The residuals analysis is shown in the insets in E and F.
Analysis of Burst Kinetics
In order to determine whether steps following product formation are rate-limiting for 21-hydroxylation by P450 21A2, pre-steady-state kinetic analysis was performed (Fig. 7). Product formation was monitored over the range of 10 ms to 1 s using a rapid quench flow instrument. A small kinetic burst within the initial 80 ms (20%) was followed by a slower product formation with the substrate progesterone, whereas an almost linear curve was observed with 17α-OH-progesterone as the substrate.
FIGURE 7.
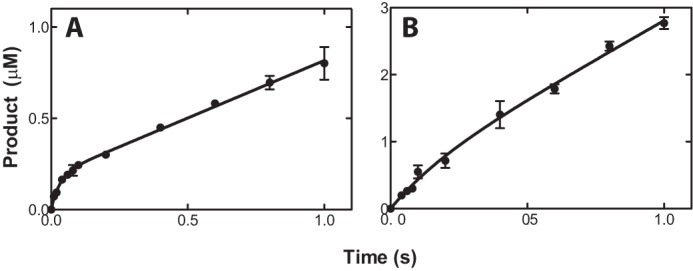
Pre-steady-state-kinetics for 21-hydroxylation catalyzed by human P450 21A2. The final P450 concentration was 1.0 μm. The curves represent the best fits to the equation, y = A(1 − e−kpt) + ksst, where A represents the burst amplitude, kp represents the first-order rate of the pre-steady-state burst, and kss represents steady-state rate. A, formation of deoxycorticosterone from progesterone. A = 0.19 ± 0.02 μm, kp = 32 ± 10 s−1, and kss = 0.63 ± 0.03 s−1. B, formation of 11-deoxycortisol from 17α-OH-progesterone. The parameters were not calculated due to the lack of burst reaction.
Kinetic Isotope Effects
In order to determine the kinetic contribution of step 7 (C–H bond breaking; Fig. 2) in the catalytic cycle of P450 21A2, KIE approaches were used to analyze the rate-limiting nature of this step. The steady-state kinetic parameters were determined with the deuterium-labeled substrates 21,21,21-d3-progesterone and 17α-OH-21,21,21-d3-progesterone. Comparison of the kcat and Km values between non-deuterated (d0) and deuterated (d3) substrates yielded non-competitive intermolecular KIEs, designated DV and D(V/K) following the convention of Northrop (32). DV and D(V/K) values determined for progesterone were 6.5 and 11, respectively. DV and D(V/K) values determined for 17α-OH-progesterone were 8.0 and 5.3, respectively (Fig. 8 and Table 1). The relatively high non-competitive intermolecular isotope effects indicate that C–H bond cleavage is at least partially rate-limiting in both 21-hydroxylation reactions catalyzed by human P450 21A2.
FIGURE 8.
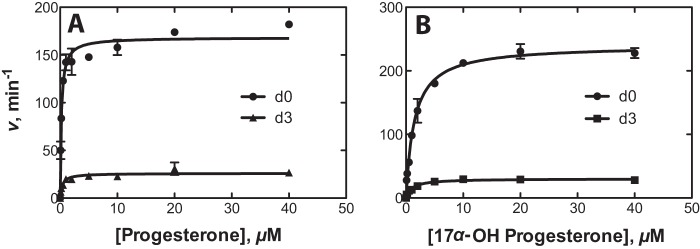
Kinetic isotope effects for 21-hydroxylation reactions catalyzed by human P450 21A2. A, progesterone; B, 17α-OH-progesterone.
Crystal Structure of Human P450 21A2
The structure of the complex between human P450 21A2 and progesterone was phased by molecular replacement using the bovine P450 21A2 protein portion from the previously determined structure of the complex of the latter with 17α-OH-progesterone as the search model (6). The monoclinic crystal of human P450 21A2 contained three molecules of the complex with progesterone per asymmetric unit, and the structure was refined to a resolution of 2.64 Å (Table 3 and Fig. 9A). The conformations adopted by the independent complexes exhibit close similarity (Fig. 9B), with 440, 440, and 442 amino acids traced for complexes 1, 2, and 3, respectively, in the electron density map. The sequence identity between the human and bovine P450 21A2 proteins is 80% (Fig. 10A). However, the initial model of the human P450 21A2·progesterone complex based on the molecular replacement solution obtained from the search with the coordinates of the 3 Å resolution structure of the bovine P450 21A2·17α-OH-progesterone complex required extensive rebuilding to arrive at the final structure. The degree of deviation between the superimposed human P450 21A2·progesterone and bovine P450 21A2·17α-OH-progesterone complexes (Fig. 11A) is indicated by an r.m.s.d. of 4.68 Å for main chain atoms (C, O, N, Cα; residue range 29–485; complex A). Residue by residue r.m.s.d. values for main chain atoms along the entire chain based on an overall superimposition of the human and bovine P450 21A2 complex structures are depicted in Fig. 10B. The computed average values for r.m.s.d. regarding main chain atoms and based on separate superimpositions of individual segments of the human and bovine P450 21A2 proteins are 0.85 Å (aa 29–100), 2.38 Å (aa 101–200), 1.50 Å (aa 201–300), 1.05 Å (aa 301–400), and 3.21 Å (aa 401–485). This analysis provides a clear indication that the N-terminal portions of the two crystal structures exhibit the most similar conformations among the analyzed segments. On the other hand, even when just main chain atoms are included, the comparison reveals significant deviations between the tertiary structures of the human and bovine P450 21A2 proteins.
TABLE 3.
Selected crystal data, data collection, and refinement parameters
| P450 21A2·progesterone | |
|---|---|
| Crystal data | |
| Space group | C2 |
| Unit cell constants a, b, c (Å), and β (degrees) | 150.38, 86.86, 108.92, and 102.1 |
| Data collection | |
| Wavelength (Å) | 0.9782 |
| Temperature | 110 K |
| Resolution (outer shell) (Å) | 30.00-2.64 (3.42-2.64)a |
| Unique reflections | 40,299 (3,979) |
| Completeness (%) | 100 (100) |
| I/σ(I) | 10.2 (1.4) |
| Rpim | 0.455 (0.081) |
| Redundancy | 7.5 (7.1) |
| Refinement | |
| Phasing method | Molecular replacement (Protein Data Bank code 3QZ1 model) |
| Rwork | 0.230 (0.225)b |
| Rfree | 0.281 (0.281)b |
| No. of amino acids | 1,322 (440,440,442)c |
| No. of protein atoms | 10,537 |
| No. of heme atoms | 129 |
| No. of ligand atoms (3 progesterones) | 69 |
| No. of water molecules/sulfate ions | 81/4 |
| Average B-factor, protein atoms (Å2) | 43.1 |
| Average B-factor, ligand/heme atoms (Å2) | 32.5/26.6 |
| Average B-factor, water/ions (Å2) | 30.2/35.4 |
| Wilson B-factor (Å2) | 44.1 |
| r.m.s.d. bond lengths (Å) | 0.005 |
| r.m.s.d. bond angles (degrees) | 1.2 |
| Ramachandran analysis: no. of favored/allowed residues/outliers (%) | 94.2/4.3/1.5 |
| Protein Data Bank code | 4Y8W |
a Numbers in parentheses refer to the outer shell.
b Refinement outer shell 2.71–2.64 Å.
c Belonging to three independent chains: for molecule A, residues 29–266/276–326/333–411/414–485; for molecule B, 29–267/276–324/333–484; for molecule C, 29–266/275–325/333–485.
FIGURE 9.
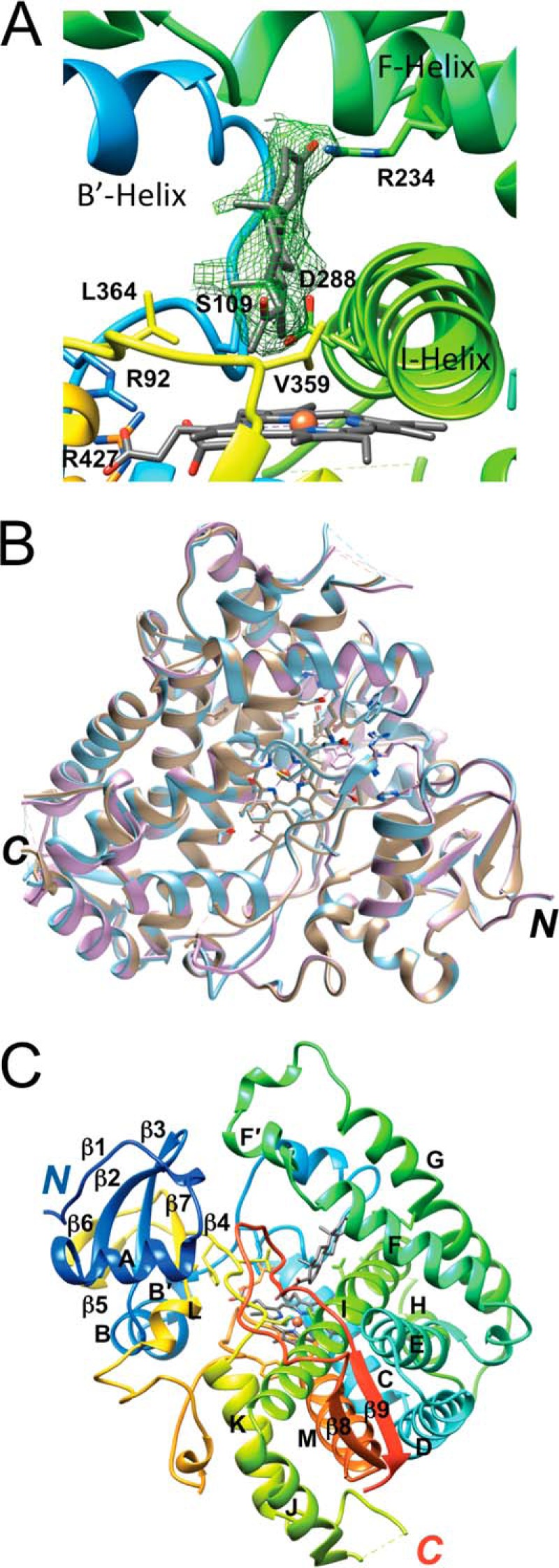
Crystal structure of the human P450 21A2·progesterone complex. A, quality of the final 2Fo − Fc omit electron density (1.2σ level) around the progesterone substrate. Helices and selected amino acids are labeled, including Val-359 (V359), which limits 16α-hydroxylation activity (12). B, superimposition of the three independent complex molecules per asymmetric unit. Complexes A, B, and C are colored in beige, cyan, and lilac, respectively. C, tertiary structural fold of complex A. The residue ranges for α-helices A–M and β-strands 1–9 are as follows: β1, 32–36; A, 45–56; β2, 59–64; β3, 67–72; B, 75–84; B′, 86–89; C, 115–131; D, 136–153; E, 161–178; F, 188–202; F′, 205–212; G, 224–246; H, 257–264; I, 279–310; J, 312–322; K, 344–356; β4, 364–368; β5, 372–374; β6, 377–379; β7, 384–387; L, 389–394; M, 432–447; β8, 450–454; β9, 479–483.
FIGURE 10.

Primary and tertiary structure comparison of human and bovine P450 21A2 proteins. A, sequence alignment for human (h, top line) and bovine (b, bottom line) P450 21A2. The sequence alignment was generated with CLUSTAL Omega (33). For the nucleotide sequence and the protein sequence and numbering of human P450 21A2 (normal 21-hydroxylase B gene), see Ref. 42. B, r.m.s.d. values (in Å) for backbone atoms in the three-dimensional structures of human and bovine P450 21A2 along the entire chain. The r.m.s.d. values from the superimposition were calculated with the Superpose routine (43) in CCP4 (23).
FIGURE 11.

Comparison between the crystal structures of the human P450 21A2·progesterone and bovine P450 21A2·17α-OH-progesterone complexes. A, overall view of the superimposed complexes (human P450 21A2 and bovine P450 21A2 are colored in green and pink, respectively). The overlay was generated using the Matchmaker option in UCSF Chimera (26). B, close-up view of the active sites in the superimposed complexes. Selected amino acid side chains in the two structures are depicted and labeled, with carbon, oxygen, nitrogen, and iron atoms colored red, blue, and beige, respectively (in green for human P450 21A2 and pink for bovine P450 21A2). Selected hydrogen bonds are indicated with dashed lines and distances in Å. C, significant deviations in the conformation of the central region of helix I in the human and bovine P450 21A2 structures. D, differences in the conformation of the loop region connecting helices F′ and G in the human and bovine P450 21A2 structures and evidence for the binding of a second progesterone molecule at a distal site in the structure of the human protein complex. Fourier 2Fo − Fc sum electron density at the 0.7σ level (green meshwork) in the human P450 21A2 structure is consistent with the presence of a partially occupied progesterone molecule at virtually the same location where 17α-OH-progesterone (pink wire diagram) resided in the previously determined structure of the bovine P450 21A2 complex (6).
The overall structure adopted by human P450 21A2 exhibits the typical P450 fold that consists of at least 12 α-helices and a β-sheet domain near the N terminus; it resembles a triangular prism (34). Human P450 21A2 features 13 α-helices (treating B and B′ as well as F and F′ as single helices; Fig. 9C) and a total of nine β-strands. The latter fold into an N-terminal β-sheet domain (β1–β3 (blue) and β4–β7 (yellow); Fig. 9C) and a C-terminal sheet (β8 (orange) and β9 (red); Fig. 9C). Thus, human and bovine P450 21A2 exhibit very similar secondary structure arrangements (Fig. 11A), and differences in the distribution of α-helices and β-strands are most likely not at the origin of the considerable conformational differences indicated by the above r.m.s.d. values. Rather, the relative orientations of helices and sheets as well as deviating conformations in the extensive loop regions are probably more important in this regard.
Inspection of the progesterone substrates at the active sites of the three human P450 21A2 complexes shows a roughly 45° orientation of the long axis of the substrate relative to the heme plane. The spacing between heme and substrate is quite large in all cases, with the average distance between the progesterone C21 atom and Fe3+ in the heme prosthetic group amounting to 4.15 Å; Fig. 11B). However, if one assumes a perferryl state with a single oxygen wedged between iron and the progesterone carbon atom (Fe4+-O···C21), the Fe···C distance is reduced to ∼3.0 Å, and that between the iron and the H21(C21) atom is reduced to ∼2.3 Å. At the other end of the substrate molecule, the carbonyl group (O3) is engaged in a hydrogen bond with the guanidino moiety of Arg-234 in the G helix (average distance of 2.66 Å between O3 and NH2), which protrudes from the ceiling of the active site (Fig. 11C). The inclination between the plane defined by the core rings of the progesterone substrate and the heme plane is ∼45°. The substrate cavity wall facing the β-face interacts primarily with the I-helix, via the hydrophobic side chain atoms of Val-287 and Ile-291 as well as the Gly-292 residue (Fig. 11, B and C). The α-face does not make any noticeable contacts with nearby loops between helices B′ and C, helix K and β4, and between the β8 and β9 strands. The C19 methyl group of progesterone lies close to Trp-202 from the F-helix and probably engages in an H···π type interaction. Other noteworthy residues located in the vicinity of the substrate are Asp-107, with its carboxylate moiety residing at ∼3.90 Å from the C6 atom of progesterone, Asp-288 from helix I (average distances of 3.34 Å from C6 and 3.36 Å from C7), and Ser-109, with Cγ located 3.37 Å from C7 of progesterone (Fig. 11B).
Comparison between the Structures of the Human P450 21A2·Progesterone and Bovine P450 21A2·17α-OH-Progesterone Complexes
The superimposition of the overall structures of human and bovine P450 21A2 reveals conformational similarities (Fig. 11A), consistent with the high sequence identity (Fig. 10A), but also clear differences. However, there are some notable differences as far as the substrate binding sites are concerned. Thus, in the case of bovine P450 21A2, two distinct 17α-OH-progesterone molecules are visible in all four independent complex molecules, with the proximal one located close to the heme and the distal one residing in the substrate access channel, located between the F′-helix and the β4/β7 sheet. In the human P450 21A2 complexes, only the proximal site is fully occupied by progesterone at first observation. However, inspection of the electron density map at a reduced threshold in the region of the human P450 21A2 structure that corresponds to the distal 17α-OH-progesterone binding site in the crystal structure of the bovine P450 21A2 complex reveals density that is consistent with a progesterone molecule of partial occupancy (Fig. 11D). Further, in the structure of the bovine P450 21A2 complex, accommodation of 17α-OH-progesterone leads to a movement of the I-helix relative to the structure of human P450 21A2·progesterone complexes. This helical shift is stabilized by an OH···π interaction between the Thr-177 OγH and the Phe-288 ring (Fig. 11C). In the 17α-OH-progesterone complexes with bovine P450 21A2, the 17α-OH group makes a hydrogen bond with the carbonyl moiety of Gly-290 (average distance 3.37 Å). However, without a structure of 17α-OH-progesterone in complex with human P450 21A2 available, it is difficult to assess exactly how the human enzyme accommodates 17α-OH-progesterone. An overlay of the active sites in the human P450 21A2·progesterone and bovine P450 21A2·17α-OH-progesterone complex is depicted in Fig. 11B. As can be seen, the majority of residues are conserved, and only a few are different: Gly-468 (bovine) versus Val-470 (human); Ser-97 (bovine) versus Thr-97 (human); and Met-197 (bovine) versus Leu-199 (human) (Fig. 10A). Interestingly, all residues interacting with the porphyrin moiety are identical except for one residue (Ile-108 (bovine) versus Leu-108 (human)). Another residue close to the C11 atom of the substrate C ring is Met-197 (bovine) (average distance 4.04 Å); Leu-199(human) features an average distance of 3.78 Å.
A further difference seen in the structures of the complexes of human and bovine P450 21A2 concerns the acetyl group of progesterone and 17α-OH-progesterone. In the human P450 21A2·progesterone complex, the acetyl carbon is pointing toward the heme moiety, whereas in the bovine P450 21A2·17α-OH-progesterone complex, the carbonyl group is pointing away from the heme (Fig. 11B). Acetyl groups in steroid molecules can rotate more or less freely, and both orientations are possible in principle. Another factor contributing to the different orientations of the acetyl groups could be constituted by the interaction between the progesterone O20 atom and a methyl group from Val-470 (average distance 3.14 Å).
Discussion
Congenital adrenal hyperplasia is one of the most common heritable metabolic diseases in humans, and >95% of cases are attributed to mutations in P450 21A2 (1, 2). Much of the literature involving P450 21A2 biochemistry used enzymes from other species, and this appears to be the first reported purification of human P450 21A2. We previously published a crystal structure of bovine P450 21A2 with its substrate 17α-OH-progesterone (6), but this is apparently the first structure of a P450 21A2 enzyme with the substrate progesterone. The human P450 21A2-progesterone structure has single ligand occupancy and differs from the bovine structure, which had two molecules of the substrate 17α-OH-progesterone bound (6). We also report kinetic characterization of the human P450 21A2 enzyme and conclude that the rate-limiting step(s) differs from that reported for bovine P450 21A2 by another group (17).
The catalytic activities of our purified human P450 21A2 enzyme were higher than reported for the bovine enzyme, which had been used in some previous kinetic studies (14, 15, 17). Some of the differences may be due to the enzyme reconstitution conditions, in that previous studies included relatively high concentrations of detergents and glycerol, and our own studies with the bovine enzyme yielded higher rates (see above; Table 2), but not as high as for the human enzyme. The kcat values for progesterone and 17α-OH-progesterone 21-hydroxylation were similar (Table 1), but due to a lower Km, the reaction was more efficient with progesterone. The catalytic efficiency (for progesterone 21-hydroxylation) appears to be the highest reported for any mammalian P450 enzyme (∼1.3 × 107 m−1 s−1 at 37 °C), which compares favorably with the apparent kon rate estimated for the reaction (at 23 °C) (Fig. 6). Of the other mammalian P450s examined in detail, only human P450 7A1 (2 × 106 m−1 s−1) (35) and human P450 27C1 (8 × 105 m−1 s−1)5 have been found to be close to being this efficient (3).
Because of the high catalytic efficiency of human P450 21A2, we were interested in what the rate-limiting step(s) in catalysis is. Substrate binding was relatively fast, calculated to be 2.4 × 107 m−1 s−1 (at 23 °C) (Fig. 5, C and D). If we consider the Kd values of the substrates and products (Fig. 5, A and B) and assume that the kon rates are similar for the products, the koff values for the products 11-deoxycorticosterone and 11-deoxycortisol are 40–60 s−1 (at 23 °C), which is not very rate-limiting for steady-state reactions with rates of 3 s−1 (Fig. 4).
The KIE studies provide strong evidence that C–H bond breaking (step 7 in Fig. 2) is rate-limiting. A non-competitive intermolecular KIE provides the most direct evaluation of the extent of rate limitation of the C–H bond-breaking step in an enzyme reaction (32, 36). The DV and D(V/K) values were high for the P450 21A2 reactions (5–11) (Fig. 8 and Table 1), strongly indicating that C–H bond breaking is at least partially rate-limiting. Yoshimoto et al. (13) also reported significant KIE values for human P450 21A2 using other KIE experimental designs, and the results are consistent with ours.
The high KIEs and rapid product koff rates are not consonant with the conclusions of Kominami et al. (17) with bovine P450 21A2, who concluded that product dissociation was rate-limiting in the case of progesterone 21-hydroxylation but not 17α-OH-progesterone. In contrast, we found high KIEs for both steroid hydroxylations with human P450 21A2. The conclusion that product (11-deoxycorticosterone) dissociation is rate-limiting in the progesterone 21-hydroxylation reaction was based largely on the appearance of a burst in the pre-steady-state reaction (17) (the reaction with 17α-OH-progesterone did not show a burst). We also observed a partial burst in the progesterone reaction (Fig. 7A). The partial (20%) burst (cf. 40% for bovine P450 21A2 in the report of Kominami et al. (17)) has several possible explanations. One is that only part of the P450 21A2 is catalytically active, although this scenario would not explain the complete absence of a burst with the 17α-OH-progesterone reaction (17) (Fig. 7B). The possibility can be considered that the product release is only partially rate-limiting, although modeling with the program DynaFit (37) did not yield partial bursts when koff was systematically lowered (results not presented). Another possibility is one that we established for DNA polymerases (38). The appearance of a quantitative burst is evidence that the rate-limiting step occurs after product formation, but a reversible equilibrium of an enzyme-substrate complex between an active and an alternate form can explain a partial burst (38, 39), and this proved to be the case in DynaFit kinetic modeling (results not presented). Such an explanation is possible here, in that it is known that the enzyme also forms an alternate product from progesterone, 16α-OH-progesterone (this reaction (i.e. 16α-hydroxylation) does not occur with the substrate 17α-OH-progesterone (13)). Thus, one can explain the partial burst with an alternate conformation of the enzyme-substrate complex (the “16α-OH” form) in equilibrium. The equilibrium could be estimated by kinetic modeling (38) but the equilibrium may not be reflected in the comparison of the ratio of the 21- and 16α-hydroxylation ratios.
The three-dimensional overlay of the human and bovine P450 21A2 complex structures revealed that the two proteins exhibit significant deviations even in their main chain conformations despite the high sequence identity (Figs. 10 and 11). Some of these are accrued within secondary structure elements, as established for a section of helix I (Fig. 11C). It should not come as a surprise, however, that the most noticeable conformational differences occur in loop regions. A good case in point is offered by the loop connecting the F′ and G helices that maps to the surface of the protein (Fig. 11D). The F′ helix lies adjacent to the distal binding site occupied by 17α-OH-progesterone in the structure of bovine P450 21A2, and together with the β4/β7 sheet, the loop connecting β2 and β3 as well as a portion of the loop between helices B′ and C, forms the outer section of the substrate access channel. The loop leading from helix F′ to helix G features a short curl and makes a tighter turn between residues Val-212 and Arg-217 in the structure of human P450 21A2 compared with the bovine protein (Fig. 12A). The tighter turn is caused in part by a sulfate ion that stitches together the side chains of Arg-217, Arg-103, and Lys-99. The orientations of side chains from the corresponding residues Arg-215, Gln-103, and Lys-99, respectively, in the structure of the bovine protein are substantially different, and it is unlikely that these could shape the loop conformation in bovine P450 21A2 to resemble the one in human P450 21A2, even if a sulfate ion were present and could be trapped between Arg-215 and Lys-99. Although the r.m.s.d. values for main chain atoms discussed above offer a measure for the distinct geometries adopted by the human and bovine P450 21A2 proteins, the differences are even more obvious at the level of amino acid side chains, as demonstrated by the loop region between the F′ and G helices. Differences between the conformations of the loop region connecting helices F′ and G in the two structures (Fig. 12A) are not the result of deviating crystal packing interactions. Both space groups and packing interactions differ between the human and bovine P450 21A2 crystal structures. Bovine P450 21A2 crystals are of space group P21 with four molecules per asymmetric unit. Helices F, G, and J are involved in crystal packing interactions. In the case of human P450 21A2 crystals, the space group is C2 with three molecules per asymmetric unit. Crystal packing interactions are primarily mediated by helices B, B′, C, J, and M. The loop region between helices F′ and G is not involved in packing in either the crystal of human P450 21A2 or that of the bovine protein. Instead, residues from loops participating in lattice interactions in the former include 35–36, 66–67, 213–223, 247–252, 380–382, 412–414, and terminal residues of helices F (position 189) and H (position 264).
FIGURE 12.
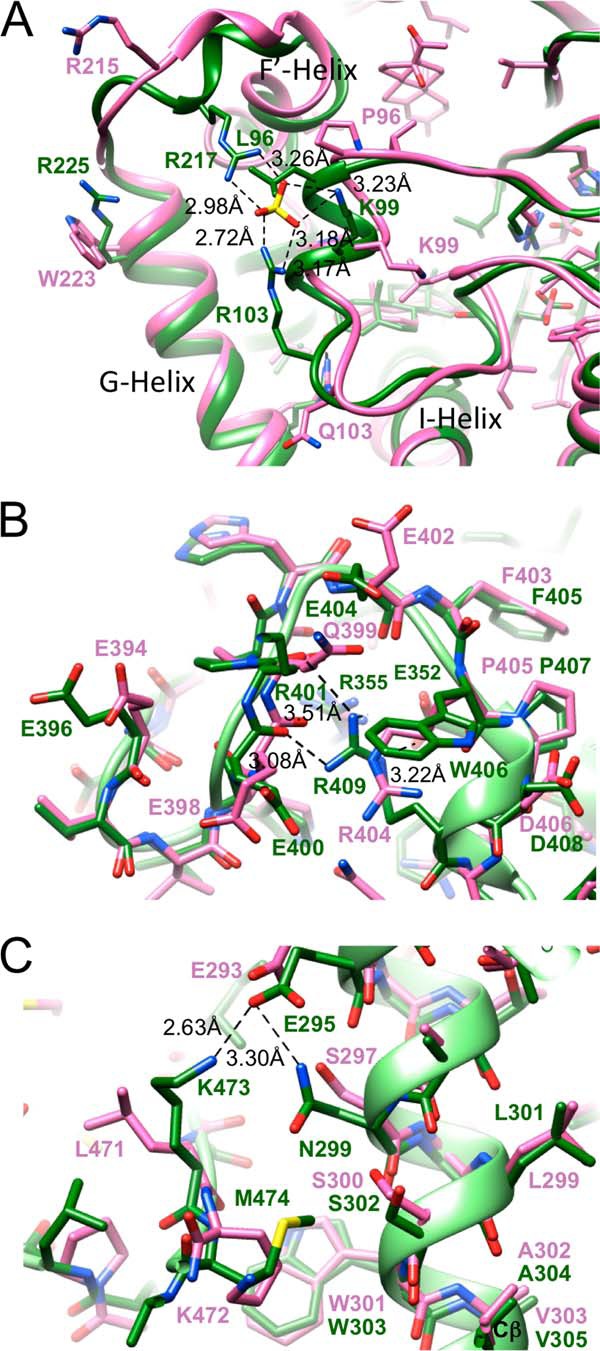
Distinct side chain interactions in the structures of human and bovine P450 21A2 complexes and insights into the structural consequences of mutations. A, a sulfate ion is bound by amino acids from loop regions connecting helices F and G (top left, Arg-217) and helices B and C (center, Lys-99 and Arg-103), respectively, in the structure of human P450 21A2. The corresponding residues in bovine P450 21A2 are Arg-215, Lys-99, and Gln-103, respectively, which exhibit completely different relative orientations compared with the situation in the structure of the human protein. B, view of a portion of an extended loop (meander) region connecting helices L and M, with Arg-409 in human P450 21A2 engaging in interactions with main chain oxygen atoms of amino acids Glu-400 and Arg-401 as well as the carboxylate moiety of the Glu-352 side chain from the K helix. A further amino acid in close vicinity of Arg-409 is Arg-355. The R409H and R352K mutations (7) as well as the R409C and R409L mutations (6, 44–47) could disrupt the ERR triad. C, view of interactions between I helix residue Glu-295 and the side chains of the adjacent Asn-299 and Lys-473 located in the β8-β9 loop. The V305D mutation was predicted to result in a salt bridge between Asp-305 and Lys-473 (7), but the structure of the human P450 21A2 complex reveals that the two residues are spaced too far apart.
With the human P450 21A2 crystal structure now in hand, it is interesting to evaluate the structural consequences of some selected missense mutations in human P450 21A2 that cause complete loss of function in some cases and congenital adrenal hyperplasia, resulting in salt-wasting disease, or steep reductions in enzyme activity and simple virilizing disease. An example of the former is constituted by the E352K mutation located in helix K (7). Glu-352 is part of the so-called ERR triad together with Arg-355 and Arg-409, the latter mapping to the meander region leading up to helix M (Fig. 12B). Using a model of human P450 21A2 based on the crystal structure of the bovine protein, it was predicted that the E352K mutation would disrupt the triad as a result of repulsions between the three basic side chains (7). The crystal structure of human P450 21A2 indeed confirms this view in that the ERR triad seen in the bovine protein is maintained and that replacement of the Glu-352 by a lysine would disrupt the salt bridge to Arg-409 (Fig. 12B). Another salt-wasting disease-causing mutation, R409H (7), can also be expected to disrupt the ERR triad, because the shorter histidine side chain might not be able to reach the carboxylate moiety of Glu-352 (Fig. 12B).
Although humanized models of P450 21A2 (6, 7) based on the crystal structure of the bovine protein (6) may be helpful in some cases for interpreting changes at the structural level as a result of disease-causing mutations in patients (e.g. those affecting the above ERR triad), clear differences between the human and bovine 21A2 crystal structures set limitations for the usefulness of a computational model. Thus, the V305D mutation in helix I, which is implicated in S-channel gating (simple virilizing phenotype), was interpreted to result in a salt bridge to Lys-473 based on the modeled structure of human 21A2 (7). However, Lys-473 in the crystal structure of human P450 21A2 and the corresponding Lys-472 in the structure of bovine P450 21A2 adopt very different orientations relative to Val-305 (Glu-305 in the mutant) and Val-303, respectively (Fig. 12C). Lys-473 in human P450 21A2 engages in a salt bridge to Glu-295 located further up in helix I. The same glutamate also forms a hydrogen bond to Asn-299 (Fig. 12C). The amino nitrogen of Lys-473 lies at a distance of 13.9 Å from the Cβ atom of the side chain of Val-305, a gap that obviously cannot be bridged even if valine is mutated to glutamate. Conversely, in the structure of bovine P450 21A2, the corresponding distance between Lys-472 and Val-303 is 8.6 Å, a gap that could conceivably be bridged if valine were replaced by glutamate and the lysine side chain unraveled to assume a more extended conformation. In the crystal structure of bovine P450 21A2, methylene groups of the lysine side chain stack onto Trp-303 (Fig. 12C). In the structure of human P450 21A2, it is the side chain of Met-474 that snakes along the plane of Trp-303 (Fig. 12C).
Two additional salt-wasting mutations in the N-terminal region, G65E (40) and G91V (41), that reduce P450 21A2 activity to <1%,6 can now be readily interpreted in light of the human P450 21A2 crystal structure. Gly-65 maps to a hydrophobic patch, and mutation to a glutamate would disrupt hydrophobic interactions and result in a direct clash with the side chain of Val-212 (Fig. 13A). The mutation can therefore be expected to destabilize the protein and alter the local conformation considerably. The opposite scenario, namely disturbing a largely negatively polarized region by introducing a bulky hydrophobic moiety, would play out in the case of the G91V mutation (Fig. 13B). Thus, in the mutant, the valine side chain would be surrounded by main chain keto groups as well as hydrophilic or negatively charged side chains from threonine or aspartate, respectively. Moreover, the carboxylate moiety of the latter residue, Asp-112, would clash with the Val-91 side chain.
FIGURE 13.
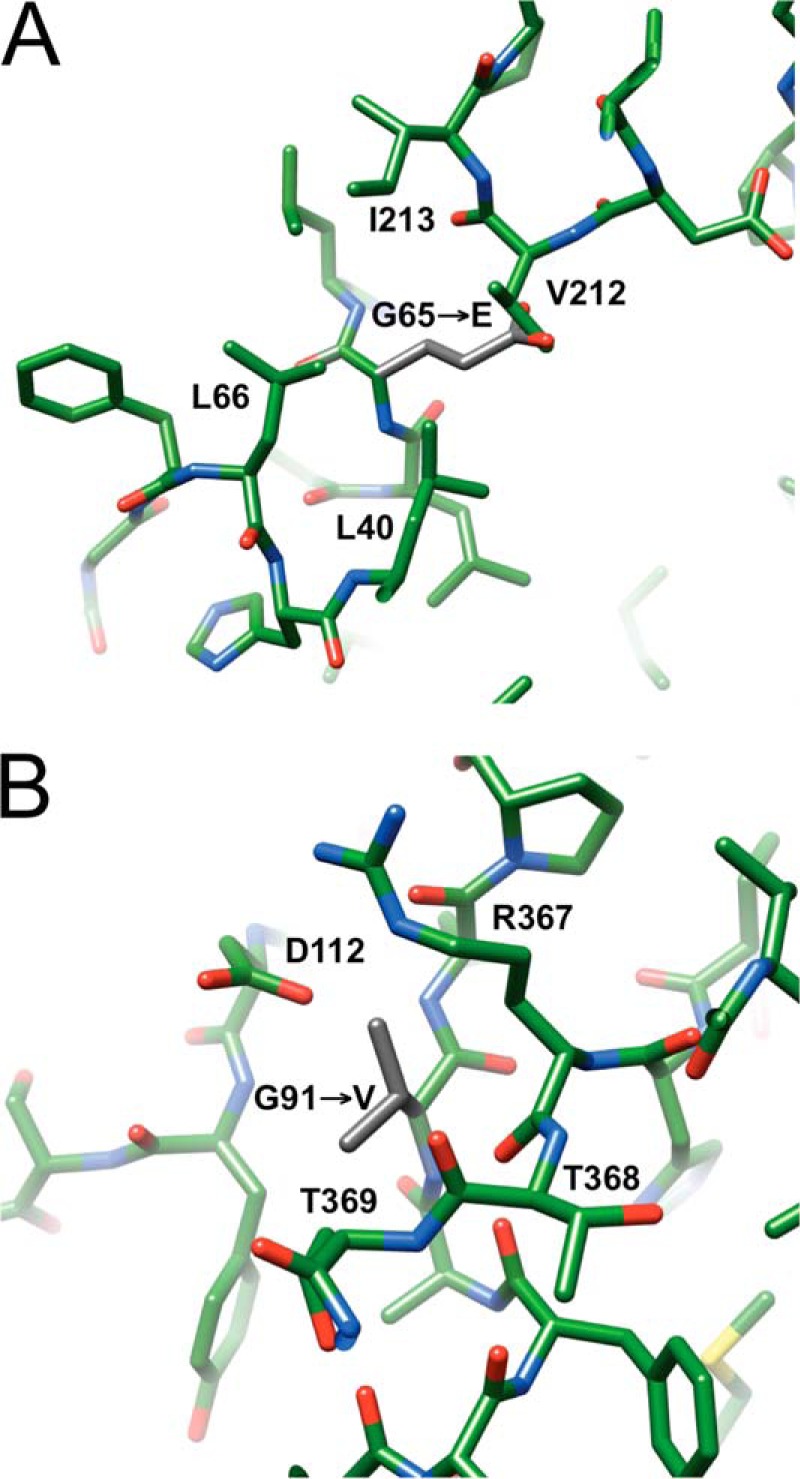
Structural consequences of two salt-wasting mutations in the N-terminal region of human P450 21A2. A, mutation of Gly-65 located in the β2-β3 loop to glutamate (40) (side chain carbon atoms highlighted in gray) introduces a charged residue into a hydrophobic patch and is expected to result in a clash with Val-212, located in the loop region linking helices F and G. B, mutation of Gly-91 located in the loop between the B and B′ helices to valine (41) (side chain carbon atoms highlighted in gray) introduces a hydrophobic residue into a highly polar region near the surface of the protein and is expected to result in a clash with Asp-112, located in the loop region linking helices B′ and C.
Our study provides a complete kinetic framework for human P450 21A2 and the first crystal structure of the human enzyme with bound substrate. The kinetic data demonstrate that human P450 21A2 catalyzes 21-hydroxylation of both progesterone and 17α-OH-progesterone with higher rates than the bovine enzyme and that human P450 21A2 is among the fastest mammalian P450 enzymes if not the fastest (3). The crystal structures of human P450 21A2 in complex with progesterone and the bovine enzyme in complex with 17α-OH-progesterone exhibit significant differences. Importantly, these are not limited to loop regions and areas closer to the surface but are also found within secondary structure elements conserved in P450 enzymes. Could some of these conformational differences be the result of two 17α-OH-progesterone molecules being bound to bovine P450 21A2 but only one progesterone to human P450 21A2? The electron density computed at a lower threshold for the latter structure revealed a second progesterone of lower occupancy that is located at the distal site in the substrate access tunnel (Fig. 11D), where the second 17α-OH-progesterone molecule is lodged in the structure of the bovine P450 21A2 complex. Thus, the differences in the binding modes of progesterone and 17α-OH-progesterone in the structures of the human and bovine P450 21A2 protein complexes are more likely reflective of deviating binding affinities for the substrates and different crystallization conditions. Although the human and bovine P450 21A2 enzymes share 80% sequence identity, the conformational differences in the main chains and orientations of side chains in the two structures are most likely a reflection of intrinsic differences between the two proteins rather than a consequence of the crystallizations with different substrates. This conclusion is also supported by very similar binding modes of progesterone and 17α-OH-progesterone at the proximal sites in the human and bovine complexes (Fig. 11, B and C).
The crystal structure of human P450 21A2 now enables insight into the structural underpinnings of the phenotypes exhibited by the numerous mutations identified for the P450 21A2 enzyme and associated in many cases with congenital adrenal hyperplasia. By analyzing just a few of these mutants in the context of the structure of human P450 21A2, we have demonstrated that a computational model of the three-dimensional structure of human P450 21A2 derived by others (7) on the basis of our previously determined structure of the bovine enzyme (6) does not provide a meaningful understanding of the consequences for structure and stability by mutation in some cases. A more extensive correlative analysis of the structure and phenotypes of the many P450 21A2 mutants identified to date goes beyond the focus of this paper and will be presented elsewhere.
In summary, the human P450 21A2 structure, complexed with progesterone, provides a better template for a biochemical understanding of the disease variants. The juxtaposition of the substrate is considered to be very important in explaining the very efficient catalysis and the rate-limiting nature of the C–H bond cleavage in the reaction. Both the C–H bond-breaking and substrate-binding steps appear to contribute in the kinetic analysis of the overall hydroxylation reaction.
Acknowledgments
We thank K. Trisler for assistance in the preparation of the manuscript. Vanderbilt University is a member institution of the Life Sciences Collaborative Access Team at Sector 21 of the Advanced Photon Source (Argonne, IL). Use of the Advanced Photon Source at Argonne National Laboratory was supported by the United States Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract DE-AC02-06CH11357.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 GM103937 (to M. E. and F. P. G.), T32 ES007028 (to F. K. Y.), and R01 GM086596 (to R. J. A.).
The atomic coordinates and structure factors (code 4Y8W) have been deposited in the Protein Data Bank (http://wwpdb.org/).
L. D. Nagy and F. P. Guengerich, unpublished results.
C. Wang and F. P. Guengerich, unpublished data.
- P450
- cytochrome P450
- KIE
- kinetic isotope effect
- r.m.s.d.
- root mean square deviation
- UPLC
- ultraperformance liquid chromatography.
References
- 1. White P. C., Speiser P. W. (2000) Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocr. Rev. 21, 245–291 [DOI] [PubMed] [Google Scholar]
- 2. Miller W. L., Auchus R. J. (2011) The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr. Rev. 32, 81–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Guengerich F. P. (2015) Human cytochrome P450 enzymes. in Cytochrome P450: Structure, Mechanism, and Biochemistry, 4th Ed (Ortiz de Montellano P. R., ed) pp. 523–785, Springer, New York [Google Scholar]
- 4. Dieter H. H., Muller-Eberhard U., Johnson E. F. (1982) Identification of rabbit microsomal cytochrome P-450 isozyme, form 1, as a hepatic progesterone 21-hydroxylase. Biochem. Biophys. Res. Commun. 105, 515–520 [DOI] [PubMed] [Google Scholar]
- 5. Kishimoto W., Hiroi T., Shiraishi M., Osada M., Imaoka S., Kominami S., Igarashi T., Funae Y. (2004) Cytochrome P450 2D catalyzes steroid 21-hydroxylation in the brain. Endocrinology 145, 699–705 [DOI] [PubMed] [Google Scholar]
- 6. Zhao B., Lei L., Kagawa N., Sundaramoorthy M., Banerjee S., Nagy L. D., Guengerich F. P., Waterman M. R. (2012) Three-dimensional structure of steroid 21-hydroxylase (cytochrome P450 21A2) with two substrates reveals locations of disease-associated variants. J. Biol. Chem. 287, 10613–10622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Haider S., Islam B., D'Atri V., Sgobba M., Poojari C., Sun L., Yuen T., Zaidi M., New M. I. (2013) Structure-phenotype correlations of human CYP21A2 mutations in congenital adrenal hyperplasia. Proc. Natl. Acad. Sci. U.S.A. 110, 2605–2610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. White P. C., Grossberger D., Onufer B. J., Chaplin D. D., New M. I., Dupont B., Strominger J. L. (1985) Two genes encoding steroid 21-hydroxylase are located near the genes encoding the fourth component of complement in man. Proc. Natl. Acad. Sci. U.S.A. 82, 1089–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Higashi Y., Yoshioka H., Yamane M., Gotoh O., Fujii-Kuriyama Y. (1986) Complete nucleotide sequence of two steroid 21-hydroxylase genes tandemly arranged in human chromosome: a pseudogene and a genuine gene. Proc. Natl. Acad. Sci. U.S.A. 83, 2841–2845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Therrell B. L., Jr., Berenbaum S. A., Manter-Kapanke V., Simmank J., Korman K., Prentice L., Gonzalez J., Gunn S. (1998) Results of screening 1.9 million Texas newborns for 21-hydroxylase-deficient congenital adrenal hyperplasia. Pediatrics 101, 583–590 [DOI] [PubMed] [Google Scholar]
- 11. Bratland E., Bredholt G., Mellgren G., Knappskog P. M., Mozes E., Husebye E. S. (2009) The purification and application of biologically active recombinant steroid cytochrome P450 21-hydroxylase: the major autoantigen in autoimmune Addison's disease. J. Autoimmun. 33, 58–67 [DOI] [PubMed] [Google Scholar]
- 12. Mizrachi D., Wang Z., Sharma K. K., Gupta M. K., Xu K., Dwyer C. R., Auchus R. J. (2011) Why human cytochrome P450c21 is a progesterone 21-hydroxylase. Biochemistry 50, 3968–3974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yoshimoto F. K., Zhou Y., Peng H. M., Stidd D., Yoshimoto J. A., Sharma K. K., Matthew S., Auchus R. J. (2012) Minor activities and transition state properties of the human steroid hydroxylases cytochromes P450c17 and P450c21, from reactions observed with deuterium-labeled substrates. Biochemistry 51, 7064–7077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kominami S., Ochi H., Kobayashi Y., Takemori S. (1980) Studies on the steroid hydroxylation system in adrenal cortex microsomes. Purification and characterization of cytochrome P-450 specific for steroid C-21 hydroxylation. J. Biol. Chem. 255, 3386–3394 [PubMed] [Google Scholar]
- 15. Arase M., Waterman M. R., Kagawa N. (2006) Purification and characterization of bovine steroid 21-hydroxylase (P450c21) efficiently expressed in Escherichia coli. Biochem. Biophys. Res. Commun. 344, 400–405 [DOI] [PubMed] [Google Scholar]
- 16. Yoshimoto F. K., Peng H. M., Zhang H., Anderson S. M., Auchus R. J. (2014) Epoxidation activities of human cytochromes p450c17 and p450c21. Biochemistry 53, 7531–7540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kominami S., Owaki A., Iwanaga T., Tagashira-Ikushiro H., Yamazaki T. (2001) The rate-determining step in P450 C21-catalyzing reactions in a membrane-reconstituted system. J. Biol. Chem. 276, 10753–10758 [DOI] [PubMed] [Google Scholar]
- 18. Daniels F., Alberty R. A. (1966) Physical Chemistry, 3rd Ed., pp. 330–331, Wiley, New York [Google Scholar]
- 19. Hanna I. H., Teiber J. F., Kokones K. L., Hollenberg P. F. (1998) Role of the alanine at position 363 of cytochrome P450 2B2 in influencing the NADPH- and hydroperoxide-supported activities. Arch. Biochem. Biophys. 350, 324–332 [DOI] [PubMed] [Google Scholar]
- 20. Guengerich F. P. (2014) in Hayes' Principles and Methods of Toxicology, 6th Ed (Hayes A. W., Kruger C. L., eds) pp. 1905–1964, CRC Press, Inc., Boca Raton, FL [Google Scholar]
- 21. Otwinowski Z., Borek D., Majewski W., Minor W. (2003) Multiparametric scaling of diffraction intensities. Acta Crystallogr. A 59, 228–234 [DOI] [PubMed] [Google Scholar]
- 22. Vagin A., Teplyakov A. (2010) Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 66, 22–25 [DOI] [PubMed] [Google Scholar]
- 23. Collaborative Computational Project, Number 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 24. Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 26. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E. (2004) UCSF Chimera: a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 27. Richardson T. H., Jung F., Griffin K. J., Wester M., Raucy J. L., Kemper B., Bornheim L. M., Hassett C., Omiecinski C. J., Johnson E. F. (1995) A universal approach to the expression of human and rabbit cytochrome P450s of the 2C subfamily in Escherichia coli. Arch. Biochem. Biophys. 323, 87–96 [DOI] [PubMed] [Google Scholar]
- 28. Omura T., Sato R. (1964) The carbon monoxide-binding pigment of liver microsomes: I. Evidence for its hemoprotein nature. J. Biol. Chem. 239, 2370–2378 [PubMed] [Google Scholar]
- 29. Sandhu P., Guo Z., Baba T., Martin M. V., Tukey R. H., Guengerich F. P. (1994) Expression of modified human cytochrome P450 1A2 in Escherichia coli: stabilization, purification, spectral characterization, and catalytic activities of the enzyme. Arch. Biochem. Biophys. 309, 168–177 [DOI] [PubMed] [Google Scholar]
- 30. Auchus R. J., Sampath Kumar A., Andrew Boswell C., Gupta M. K., Bruce K., Rath N. P., Covey D. F. (2003) The enantiomer of progesterone (ent-progesterone) is a competitive inhibitor of human cytochromes P450c17 and P450c21. Arch. Biochem. Biophys. 409, 134–144 [DOI] [PubMed] [Google Scholar]
- 31. Fersht A. (1999) Structure and Mechanism in Protein Science, pp. 164–166, W.H. Freeman and Co., New York [Google Scholar]
- 32. Northrop D. B. (1982) Deuterium and tritium kinetic isotope effects on initial rates. Methods Enzymol. 87, 607–625 [DOI] [PubMed] [Google Scholar]
- 33. Sievers F., Wilm A., Dineen D., Gibson T. J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Söding J., Thompson J. D., Higgins D. G. (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Johnson E. F., Stout C. D. (2013) Structural diversity of eukaryotic membrane cytochromes P450. J. Biol. Chem. 288, 17082–17090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shinkyo R., Guengerich F. P. (2011) Cytochrome P450 7A1 cholesterol 7α-hydroxylation: individual reaction steps in the catalytic cycle and rate-limiting ferric iron reduction. J. Biol. Chem. 286, 4632–4643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Guengerich F. P., Krauser J. A., Johnson W. W. (2004) Rate-limiting steps in oxidations catalyzed by rabbit cytochrome P450 1A2. Biochemistry 43, 10775–10788 [DOI] [PubMed] [Google Scholar]
- 37. Kuzmic P. (1996) Program DYNAFIT for the analysis of enzyme kinetic data: application to HIV protease. Anal. Biochem. 237, 260–273 [DOI] [PubMed] [Google Scholar]
- 38. Furge L. L., Guengerich F. P. (1999) Explanation of pre-steady-state kinetics and decreased burst amplitude of HIV-1 reverse transcriptase at sites of modified DNA bases with an additional, nonproductive enzyme-DNA-nucleotide complex. Biochemistry 38, 4818–4825 [DOI] [PubMed] [Google Scholar]
- 39. Zhang H., Eoff R. L., Kozekov I. D., Rizzo C. J., Egli M., Guengerich F. P. (2009) Versatility of Y-family Sulfolobus solfataricus DNA polymerase Dpo4 in translesion synthesis past bulky N2-alkylguanine adducts. J. Biol. Chem. 284, 3563–3576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ohlsson G., Müller J., Skakkebaek N. E., Schwartz M. (1999) Steroid 21-hydroxylase deficiency: mutational spectrum in Denmark, three novel mutations, and in vitro expression analysis. Hum. Mutat. 13, 482–486 [DOI] [PubMed] [Google Scholar]
- 41. Nunez B. S., Lobato M. N., White P. C., Meseguer A. (1999) Functional analysis of four CYP21 mutations from Spanish patients with congenital adrenal hyperplasia. Biochem. Biophys. Res. Commun. 262, 635–637 [DOI] [PubMed] [Google Scholar]
- 42. Rodrigues N. R., Dunham I., Yu C. Y., Carroll M. C., Porter R. R., Campbell R. D. (1987) Molecular characterization of the HLA-linked steroid 21-hydroxylase B gene from an individual with congenital adrenal hyperplasia. EMBO J. 6, 1653–1661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Krissinel E. B., Winn M. D., Ballard C. C., Ashton A. W., Patel P., Potterton E. A., McNicholas S. J., Cowtan K. D., Emsley P. (2004) The new CCP4 Coordinate Library as a toolkit for the design of coordinate-related applications in protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 60, 2250–2255 [DOI] [PubMed] [Google Scholar]
- 44. Soardi F. C., Barbaro M., Lau I. F., Lemos-Marini S. H., Baptista M. T., Guerra-Junior G., Wedell A., Lajic S., de Mello M. P. (2008) Inhibition of CYP21A2 enzyme activity caused by novel missense mutations identified in Brazilian and Scandinavian patients. J. Clin. Endocrinol. Metab. 93, 2416–2420 [DOI] [PubMed] [Google Scholar]
- 45. Tardy V., Menassa R., Sulmont V., Lienhardt-Roussie A., Lecointre C., Brauner R., David M., Morel Y. (2010) Phenotype-genotype correlations of 13 rare CYP21A2 mutations detected in 46 patients affected with 21-hydroxylase deficiency and in one carrier. J. Clin. Endocrinol. Metab. 95, 1288–1300 [DOI] [PubMed] [Google Scholar]
- 46. Billerbeck A. E., Bachega T. A., Frazatto E. T., Nishi M. Y., Goldberg A. C., Marin M. L., Madureira G., Monte O., Arnhold I. J., Mendonca B. B. (1999) A novel missense mutation, GLY424SER, in Brazilian patients with 21-hydroxylase deficiency. J. Clin. Endocrinol. Metab. 84, 2870–2872 [DOI] [PubMed] [Google Scholar]
- 47. Yu Y., Wang J., Huang X., Wang Y., Yang P., Li J., Tsuei S. H., Shen Y., Fu Q. (2011) Molecular characterization of 25 Chinese pedigrees with 21-hydroxylase deficiency. Genet. Test. Mol. Biomarkers 15, 137–142 [DOI] [PubMed] [Google Scholar]