

Background: HLTF is responsible for template-switching of DNA damage tolerance; HLTF has a novel DNA-binding HIRAN domain, but its function is unknown.
Results: The structure of HIRAN domain bound to DNA reveals that the domain recognizes 3′-end of DNA.
Conclusion: HLTF is recruited to a damaged site via interaction of the HIRAN domain with 3′-end.
Significance: The structure provides a structural basis for the mechanism of template-switching.
Keywords: DNA-binding protein, DNA damage response, DNA replication, protein crystallization, protein structure, protein-DNA interaction, X-ray crystallography, crystal structure, DNA damage tolerance, template-switching, HLTF
Abstract
HLTF (helicase-like transcription factor) is a yeast RAD5 homolog found in mammals. HLTF has E3 ubiquitin ligase and DNA helicase activities, and plays a pivotal role in the template-switching pathway of DNA damage tolerance. HLTF has an N-terminal domain that has been designated the HIRAN (HIP116 and RAD5 N-terminal) domain. The HIRAN domain has been hypothesized to play a role in DNA binding; however, the structural basis of, and functional evidence for, the HIRAN domain in DNA binding has remained unclear. Here we show for the first time the crystal structure of the HIRAN domain of human HLTF in complex with DNA. The HIRAN domain is composed of six β-strands and two α-helices, forming an OB-fold structure frequently found in ssDNA-binding proteins, including in replication factor A (RPA). Interestingly, this study reveals that the HIRAN domain interacts with not only with a single-stranded DNA but also with a duplex DNA. Furthermore, the structure unexpectedly clarifies that the HIRAN domain specifically recognizes the 3′-end of DNA. These results suggest that the HIRAN domain functions as a sensor to the 3′-end of the primer strand at the stalled replication fork and that the domain facilitates fork regression. HLTF is recruited to a damaged site through the HIRAN domain at the stalled replication fork. Furthermore, our results have implications for the mechanism of template switching.
Introduction
DNA damage occurs both naturally and artificially. Damaged template DNA blocks accurate and processive DNA synthesis by replicative DNA polymerase, DNA polymerase δ or DNA polymerase ϵ. Replication arrest leads to collapse of the replication fork and results in genomic instability causing various human diseases, including cancer. To avoid replication arrest and to restart DNA synthesis at the damaged site, cells initiate DNA damage tolerance, also known as post-replication repair (1). DNA damage tolerance can be divided into two pathways that synthesize DNA, translesion and template-switching DNA syntheses. Translesion DNA synthesis (TLS)2 is transient DNA synthesis using a damaged template by error-prone DNA polymerases specialized for DNA damage (TLS polymerases). Template-switching (TS) DNA synthesis, also known as the damage avoidance pathway, in which one newly synthesized strand is utilized as an undamaged template for replication by replicative polymerases, is an error-free process.
DNA damage tolerance is initiated by the ubiquitination of the Lys-164 of PCNA, a scaffold protein that stimulates DNA synthesis by DNA polymerases. Mono-ubiquitination of PCNA at Lys-164 by RAD6-RAD18, an E2-E3 complex, activates the TLS pathway. Poly-ubiquitination of PCNA at Lys-164 promotes the TS pathway. The ubiquitin chain is formed by Lys-63 linkage of ubiquitin. The Lys-63-linked ubiquitin chains are involved in various cellular signaling processes including DNA repair. The Lys-63-linked poly-ubiquitination of PCNA is performed by MMS2-UBC13 (E2) and RAD5 (E3). RAD5 (REV2) was initially identified in yeast as a repair protein with DNA-dependent ATPase activity (2, 3). Yeast RAD5 has been shown to be an E3 ubiquitin ligase responsible for Lys-63-linked poly-ubiquitination of PCNA (4). Mammalian cells contain two RAD5 homologs, HLTF (helicase-like transcription factor) and SHPRH (SNF2 histone-linker PHD finger RING finger helicase), both of which are involved in Lys-63-linked poly-ubiquitination of PCNA at Lys-164 and in the regulation of DNA damage tolerance (5–9). Loss of either HLTF or SHPRH increases the frequency of chromosome abnormalities under DNA damage, indicating that both proteins are important for the maintenance of genomic stability under such conditions (6, 7). Recently, it has been shown in vitro that HLTF forms a thiol-linked ubiquitin chain on UBC13 and then transfers the chain to RAD6, after which the chain is transferred to un-modified Lys-164 of PCNA by RAD18 (10).
HLTF and SHPRH possess a SWI/SNF helicase domain that is divided into N- and C-terminal parts by an insertion of RING domain involved in the poly-ubiquitination of PCNA (see Fig. 1A). For several years, helicase activities of HLTF have been characterized in vitro. Human HLTF shows double-stranded DNA translocase activity with 3′-5′ polarity, thereby facilitating regression of the replication fork (11). Furthermore, a recent study has shown that HLTF facilitates the strand invasion and formation of a D-loop structure (12). Although these biochemical studies have revealed the functional significance of HLTF in the TS pathway, the structural basis of this HLTF function remains unclear. Interestingly, sequence analysis has suggested that HLTF contains a novel domain referred to as the HIRAN (HIP116 and RAD5 N-terminal) domain that is hypothesized to be involved in DNA binding (see Fig. 1A) (13). The HIRAN domain has been identified in various proteins involved in DNA metabolism; however, the structural basis of, and functional evidence for, the HIRAN domain in DNA binding has remained unclear until now.
FIGURE 1.

Structure of the HIRAN domain of human HLTF. A, schematic features of the primary structures of human HLTF. Human HLTF is composed of 1,009 amino acid residues with a molecular mass of 114 kDa. HLTF consists of HIRAN, SWI/SNF helicase, and E3 RING domains. The helicase domain is divided into N-terminal (helicase motifs I, Ia, II, and III) and C-terminal (helicase motifs IV, V, and VI) parts by insertion of the RING domain. B, overall structure of the HIRAN domain in complex with DNA. Structure of the HIRAN domain is shown by ribbon representations colored in red (α-helices), yellow (β-strands), and green. The phosphate backbones of the DNA strands are colored in orange. C, topology diagram of the HIRAN domain. Secondary structures, α-helices and β-strands, are shown by arrows and cylinders, respectively. N and C termini and secondary structural elements are also labeled. Circles indicate residues involved in interaction with DNA. The colors of the structural elements correspond to those of panel B. D, stereo view of structural comparison of the HIRAN domains. The crystal structure of the DNA-free form (cyan) and the NMR structure (yellow) were superimposed to the structure of DNA-bound form (green). Structures are shown by Cα-tracing models. Side chains involved in interaction with DNA are shown by stick models (Tyr-72, Tyr-73, Asn-91, Tyr-93, Asp-94, His-110, and Phe-142).
Here we show the crystal structures of the HIRAN domain of human HLTF and its DNA complex. This study reveals the function of the novel DNA-binding domain as a sensor to the 3′-end of the primer strand and provides a significant clue to clarifying the molecular mechanism behind TS in DNA damage tolerance pathways.
Experimental Procedures
Purification of HLTF HIRAN Domain
The cDNA encoding residues 58–174 of human HLTF HIRAN domain (HLTF58–174) was inserted into the BamHI-SalI site of pGEX-6P1 (GE Healthcare). Escherichia coli strain Rosetta2(DE3) harboring the expression vector encoding GST-fused HLTF58–174 was cultured in LB medium at 37 °C. When optical density reached 0.5–0.6, expression was induced by 0.2 mm isopropyl-1-thio-β-d-galactopyranoside, and the strain was further cultured for 16 h at 18 °C. Harvested cells were suspended in buffer containing 40 mm HEPES-NaOH, pH 7.4, 500 mm NaCl, 200 mm Arg-HCl, 10 mm 2-mercaptoethanol, and 1 mm EDTA, and then disrupted by sonication. Cell lysate was clarified by centrifugation, supernatant was applied onto glutathione-Sepharose 4B resin (GE Healthcare), and unbound proteins were washed with buffer containing 20 mm Tris-HCl, pH 8.0, 1.0 m NaCl, 10 mm 2-mercaptoethanol, 10% glycerol, and 1 mm EDTA. The resin was further washed with buffer containing 20 mm Tris-HCl, pH 8.0, 100 mm NaCl, 10 mm 2-mercaptoethanol, 10% glycerol, and 1 mm EDTA. The bound protein was eluted with buffer containing 20 mm Tris-HCl, pH 8.0, 100 mm NaCl, 10 mm 2-mercaptoethanol, 1 mm EDTA, 10% glycerol, 30 mm reduced glutathione, and 10 mm MgCl2. The GST tag of the fusion protein was cleaved by HRV3C protease. After cleavage by the protease, amino acid residues, GPLGS, were inserted at the N terminus before Val-58 of HLTF. The reaction mixture was applied onto HiTrap SP (GE Healthcare) pre-equilibrated by buffer containing 50 mm HEPES-NaOH, pH 7.4, and 10 mm 2-mercaptoethanol. The bound protein was eluted with a liner gradient to 1.0 m NaCl. Fractions containing HLTF58–174 were collected and applied onto HiLoad 16/600 Superdex 75 pg (GE Healthcare) pre-equilibrated with buffer composed of 10 mm HEPES-NaOH, pH 7.4, and 100 mm NaCl. Fractions containing HLTF58–174 were concentrated up to 10 mg/ml, frozen with liquid N2, and stored at −80 °C until use.
The preparation of His tag-fused HLTF58–174 (His-HLTF58–174) for crystallization of the DNA-free form has been described previously (14). In brief, His-HLTF58–174 was expressed by E. coli strain BL21(DE3). His-HLTF58–174 was purified by nickel-nitrilotriacetic acid-agarose resin (Qiagen), HiTrap SP, and HiLoad Superdex 75. Purified His-HLTF58–174 was concentrated, frozen with liquid N2, and stored at −80 °C until use. The His tag-fused protein contained amino acid residues, MRGSHHHHHHGSENLYFQGGS, at the N terminus before Val-58 of HLTF.
Crystallographic study
An ssDNA mt04-13 (5′-ACCGCCGGGTGCC-3′) for crystallization of the DNA complex was commercially synthesized (Invitrogen) (see Table 2). Prior to crystallization, equal volumes of HLTF58–174 (0.7 mm) and ssDNA (0.8 mm) were mixed on ice. Suitable crystals for x-ray diffraction study were obtained by hanging drop vapor diffusion using a reservoir solution composed of 0.1 m Bis-Tris propane, pH 6.5, 0.2 m sodium acetate, and 20% PEG3350. For phase determination, selenomethionyl protein was overexpressed in methionine auxotrophic E. coli strain B834(DE3)pLysS with a minimum medium including 50 μg/ml ampicillin, 34 μg/ml chloramphenicol, and 25 μg/ml seleno-l-methionine. Selenomethionyl protein was purified and crystallized by a procedure similar to that of non-labeled protein. Crystals were transferred to a cryo-buffer composed of a reservoir solution and ethylene glycol in a ratio of 4:1. Then, the crystals were frozen in N2 gas stream at 100 K. X-ray diffraction data were collected at the Photon Factory beamline NE-3A (Tsukuba, Japan). Diffraction data were processed using the program HKL2000 (15). Phase calculation was performed using the program SOLVE (16), and the initial phase was improved using the program RESOLVE (17). Model building and fitting were performed using the program COOT (18). The structure was refined using the programs CNS (19) and REFMAC (20).
TABLE 2.
Sequences of oligonucleotides
Asterisks indicate TAMRA modification at 5′-OH or 3′-OH.
Crystals of His-HLTF58–174 in DNA-free form were obtained by sitting drop vapor diffusion method using a reservoir solution containing 0.2 m lithium acetate and 20% PEG3350. Prior to the diffraction study, crystals were transferred in buffer composed of 0.2 m lithium acetate, 20% PEG3350, and 20% ethylene glycol for cryo-protection, and then the crystals were frozen in liquid N2. X-ray diffraction data were collected at the SPring-8 beamline BL-26B2 (Sayo, Japan). Diffraction data were processed using the programs XDS (21) and SCALA (22). Crystal structure of the DNA-free form of His-HLTF58–174 was determined by molecular replacement methods, followed by structure refinement using the programs PHENIX (23) and REFMAC (20). Data collection, phasing, and refinement statistics are given in Table 1. Structural superimposition was performed using the program LSQKAB (24). The evolutionary conservation scores of amino acid positions in the HIRAN domains are calculated by the program CONSURF (25).
TABLE 1.
Data collection and refinement statistics
| HLTF58–174-DNA complex | Selenium-labeled HLTF58–174-DNA complex | DNA-free form (His-HLTF58–174) | |
|---|---|---|---|
| Data collection | |||
| Wavelength (Å) | 1.00000 | 0.97956 | 1.00000 |
| Space group | P21212 | P21212 | P41212 |
| a (Å) | 147.3 | 147.4 | 129.5 |
| b (Å) | 33.4 | 33.4 | 129.5 |
| c (Å) | 36.7 | 36.9 | 144.3 |
| Resolution (Å) | 20.0-1.38 | 20.0-2.00 | 20.0-2.40 |
| Observed reflections | 209,981 | 135,517 | 682,821 |
| Unique reflections | 36,316 | 11,324 | 48,327 |
| R-merge | 0.059 (0.261) | 0.080 (0.165) | 0.099 (0.792) |
| Completeness (%) | 94.1 (91.5) | 86.7 (87.5) | 99.8 (100) |
| 〈I〉/σ〈I〉 | 17.8 (7.8) | 21.2 (16.5) | 19.5 (3.7) |
| Refinement | |||
| Resolution (Å) | 20.0-1.38 | 20.0-2.40 | |
| Refined reflections | 34,441 | 45,744 | |
| Free reflections | 1,807 | 2,430 | |
| R | 0.166 | 0.214 | |
| R-free | 0.206 | 0.231 | |
| Root mean square deviation | |||
| Bond length (Å) | 0.021 | 0.014 | |
| Bond angles (°) | 2.018 | 1.628 | |
| Protein Data Bank code | 4XZF | 4XZG | |
DNA Binding Assay
To perform an EMSA, an alanine mutation was introduced into HLTF58–174 by the QuikChange protocol, a PCR-based site-directed mutagenesis using mismatched primers (Stratagene). The GST-fused HLTF58–174 mutant was overexpressed and purified with glutathione-Sepharose 4B resin. The GST tag of the fusion protein was cleaved by HRV3C protease, followed by gel filtration using HiLoad Superdex 75. Purified mutant proteins were concentrated, frozen with liquid N2, and stored at −80 °C until use.
To investigate the preference of HLTF58–174 for DNA structures, EMSA was performed using ssDNA (mt04-13), blunt-ended (mt03-13/mt04-13), 5′-overhanging (HF1-13/HF3-13), and 3′-overhanging (HF1-13/HF4-13) dsDNAs (Table 2 and Fig. 3A). DNA and HLTF58–174 wild type were mixed and incubated overnight at 4 °C. The concentration of DNA in the mixed solutions was 6.7 μm. The concentration of HLTF58–174 in the mixed solutions was 6.7, 13.3, or 26.7 μm. To identify the residues responsible for DNA binding, mt04-13 was used in EMSA (see Fig. 3B). DNA and HLTF58–174 wild type or mutant were mixed and incubated overnight at 4 °C. The concentration of DNA in the mixed solutions was 6.7 μm. The concentration of HLTF58–174 in the mixed solutions was 0.67, 3.3, or 6.7 μm. These solutions were separated by electrophoresis at 4 °C on 1% agarose gel containing GelRed DNA stain (Biotium). Bands were detected using a ChemiDoc Touch Imaging System (Bio-Rad Laboratories). EMSA was performed at least three times, and band intensities were measured with the program ImageJ (National Institutes of Health). The relative band intensity was calculated with division of the intensity of fully shifted band by the total intensity in the lane and normalized by the values of 6.7 μm wild type (see Fig. 3B, lane 4).
FIGURE 3.

Specific recognition of DNA by the HIRAN domain. A, DNA binding of HLTF58–174. Lane 1 is HLTF58–174 only. Lanes 2, 6, 10, and 14 are DNA only. Lanes 3–5, 7–9, 11–13, and 15–17 are ssDNA, blunt-ended dsDNA, 5′-overhanging dsDNA, and 3′-overhanging dsDNA incubated with HLTF58–174, respectively. Schematic drawings of DNA structures are shown above the blot. B, DNA binding activity of HLTF58–174 mutants. Lanes 1, 2–4, and 5–28 are DNA alone, HLTF58–174 wild type with ssDNA, and HLTF58–174 mutants with ssDNA, respectively. The relative band intensity was shown below the blot. A schematic drawing of the DNA structure is shown above the blot. Error bars indicate mean ± S.D. C, DNA binding specificity of HLTF58–174. Lane 1 is HLTF58–174 only. Lanes 2, 6, 10, 14, 18, and 22 are DNA only. Lanes 3–5, 7–9, 11–13, 15–17, 19–21, and 23–25 are 5′-modified ssDNA, 3′-modified ssDNA, 3′-modified-5′-overhanging dsDNA, 3′-modified-3′-overhanging dsDNA, 5′-modified-5′-overhanging dsDNA, and 5′-modified-3′-overhanging dsDNA incubated with HLTF58–174, respectively. Schematic drawings of DNA structures are shown above the blot. Asterisks indicate TAMRA modification at 5′-OH or 3′-OH.
To investigate whether HLTF prefers the 3′-end of DNA, oligonucleotides modified by TAMRA in the 5′ or 3′ terminus were commercially synthesized (Operon Biotechnologies Inc. or Fasmac Co. Ltd.) (Table 2). 5′-overhanging and 3′-overhanging dsDNAs with TAMRA modification were prepared as shown in Table 2 and Fig. 3C. DNA and HLTF58–174 were mixed and incubated for 60 min on ice. The concentration of DNA in the mixed solutions was 6.7 μm. The concentration of HLTF58–174 in the mixed solutions was 6.7, 13.3, or 26.7 μm. These solutions were separated by electrophoresis at 4 °C on 1% agarose gel. Bands were detected using an LAS4000 CCD image analyzer with 575DF20 Cy3 filter (GE Healthcare) (see Fig. 3C, lanes 1–17) or GelRed staining and ChemiDoc Touch Imaging System (see Fig. 3C, lanes 18–25).
Figure Preparation
Figures of protein structures were prepared with the program PyMOL (Schrödinger, LLC). All of the figures were modified with the program Illustrator (Adobe Systems).
Results and Discussion
Overall structure of the DNA Complex of Human HLTF HIRAN Domain
The crystal structures of HLTF58–174 in complex with DNA and its DNA-free form were determined at 1.38 and 2.40 Å resolution, respectively (Fig. 1, B and D). The results showed that the HIRAN domain adopts a β-barrel structure composed of six β-strands flanked with two α-helices (Fig. 1, B and C), resulting in an OB-fold structure that contains oligonucleotide-binding proteins such as RPA or telomere-binding protein (26). Typical OB-fold proteins possess a concave surface to accommodate a ligand. In general, ssDNA-binding proteins such as RPA or telomere-binding protein are composed of multiple OB-fold domains to extensively accommodate ssDNA. For instance, RPA70 (residues 181–422) adopts a tandem OB-fold structure, and an ssDNA traverses the successive concave areas of the tandem OB-folds (27). HLTF58–174 also has a concave surface that interacts with the two bases of ssDNA (Fig. 2B), and the concave surface is evolutionally conserved (Fig. 2C). In contrast to RPA and other ssDNA-binding proteins, the 3′-end of DNA strand is specifically bound to a pocket composed of β3, β4, and α1 of HLTF58–174 (Figs. 1B and 2, B and D). Interestingly, ssDNA used in crystallization forms a duplex DNA due to the 2-fold crystallographic symmetry, despite some pairing of mismatched bases (Fig. 2A). Gua4 and Thy10 formed typical TG-mismatched structure. In contrast, electron densities of Gua7 and Gua7 were ambiguous, and thus these could form a Hoogsteen base pair with dual conformations. Details of the interaction of HLTF58–174 with DNA are described and discussed in later sections.
FIGURE 2.
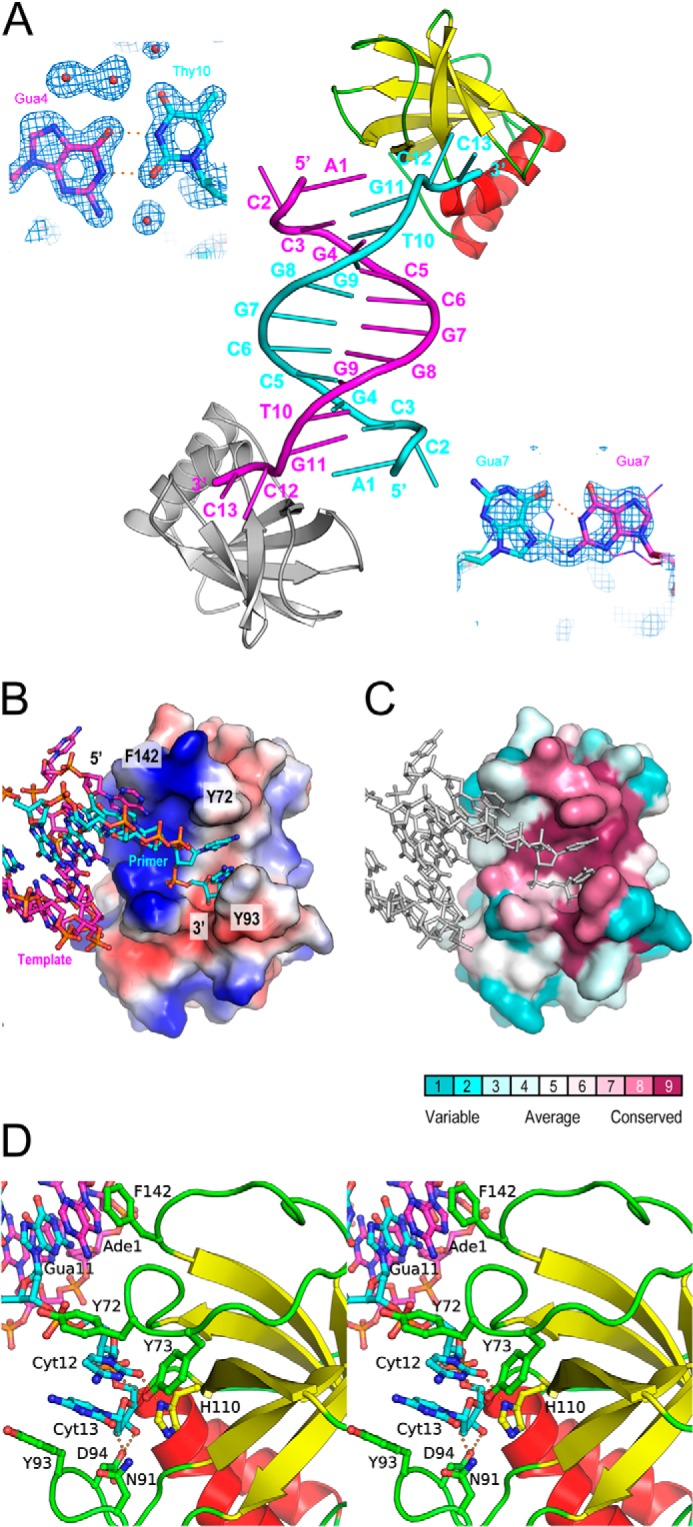
Interaction of the HIRAN domain with DNA. A, the crystal structure of DNA complex. The asymmetric unit contains one HLTF58–174 (yellow, red, and green) and one ssDNA (cyan). The DNA duplex was formed by the crystallographic 2-fold symmetry. The symmetry-related HLTF58–174 and ssDNA are shown in gray and magenta, respectively. Structures of mismatched base pairs are also shown by stick models with the weighted 2Fo − Fc map contoured at 1σ. The Hoogsteen base pair formed by the Gua7-Gua7 pair with dual conformations was shown stick or line models. B, the electrostatic potential of the HIRAN domain on the molecular surface. Blue and red surfaces indicate positive and negative potential, respectively. DNA bound to the HIRAN domain is shown by stick representation. C, the evolutionary conservation of amino acid on the molecular surface. Conservation scores and color scheme are shown at the bottom. DNA bound to the HIRAN domain is shown by stick representation colored in gray. D, stereo view of the detailed interaction between the HIRAN domain and DNA. Side chains of residues involved in the interaction are shown as stick models. Colors correspond to those of Fig. 1B. Electrostatic interactions are shown by orange dots.
An NMR structure of the HIRAN domain of human HLTF (residues 50–171) had previously been deposited in the Protein Data Bank (PDB ID: 2L1I). That structure was distinctly different from our structure in complex with DNA, as it was composed of one β-sheet flanked with four α-helices. Root mean square deviation values for structural superimposition between the crystal structure and the NMR structures (20 ensembles) are in the range of 7.248–8.509 Å for 114 comparable Cα atoms in residues 58–171 (Fig. 1D). To investigate whether the DNA binding imposes structural change on the HIRAN domain, we also determined the crystal structure of a DNA-free form of His-HLTF58–174 by molecular replacement using the structure of the DNA-bound form. Nine molecules of the DNA-free form were contained in the asymmetric unit. Our DNA-free structures were virtually identical to the DNA-bound structure with root mean square deviation values in the range of 0.469–0.646 Å for superimposable 116 Cα atoms in residues 58–173 (Fig. 1D), indicating that the HIRAN domain does not undergo a large structural change upon DNA binding. Actually, residues responsible for DNA binding in DNA-free and DNA-bound forms are located on similar positions (Fig. 1D). It is possible that the structural differences between the structure previously deposited in the Protein Data Bank and our own, if these differences do exist, may be due to the structural plasticity of the HIRAN domain.
Interaction between HIRAN Domain and DNA
As described above, the crystal structure showed that HLTF58–174 interacts with dsDNA (Fig. 2A). Thus, to investigate that the HIRAN domain interacts with duplex DNA, we performed EMSA using blunt-ended and overhanging dsDNAs (Fig. 3A). The result clearly shows that the HIRAN domain actually interacts not only with the ssDNA (Fig. 3A, lanes 3–5) but also with dsDNAs (Fig. 3A, lanes 7–9, 11–13, and 15–17). Interestingly, the HIRAN domain appears to prefer 3′-overhanging dsDNA rather than blunt-ended dsDNA (Fig. 3A, lanes 7–9 and 15–17), suggesting that the HIRAN domain interacts with DNA including the exposed single-stranded structure. In fact, the crystal structure reveals that the single-stranded part of DNA is bound to HLTF58–174 along with the positive concave (Fig. 2B), and the 3′-end of the DNA is specifically bound to the conserved pocket of the HIRAN domain (Fig. 2, B–D). Asn-91 interacts with the Cyt13 of the 3′ terminus by hydrogen bond. His-110 stacks with the ribose ring of Cyt13. Furthermore, Asp-94 interacts with the 3′-OH of Cyt13 at the terminus of DNA. Tyr-73 forms hydrogen bonds with the Cyt12 adjacent to the 3′-terminal base and with the His-110 that recognizes the ribose ring at the 3′ terminus. Two aromatic residues, Tyr-72 and Tyr-93, stack with these two bases, Cyt12 and Cyt13, respectively. Those stacking interactions pinch the two bases of the single-strand moiety (Fig. 2, B and C). In addition, Phe-142 stacks with the Ade1 of the symmetry-related DNA strand that forms dsDNA in the crystal. More interestingly, the HIRAN domain has affinity for the 5′-overhanging dsDNA at a level similar to 3′-overhanging dsDNA (Fig. 3A, lanes 11–13 and 15–17). This implies that the HIRAN domain may facilitate unwinding of 3′-overhanging dsDNA, a simple model of the primer-template DNA, thereby interacts with the single-stranded 3′-end. That issue is discussed in the later section.
To clarify the significant residues that interacted with DNA, we performed an EMSA (Fig. 3B). Mutations in residues involved in binding to the terminal nucleotide (N91A and H110A) drastically decreased DNA binding (Fig. 3B, lanes 11–13 and 20–22), and mutation in Asp-94 interacting with 3′-OH caused little defect in DNA binding (Fig. 3B, lanes 17–19) when compared with wild type (Fig. 3B, lanes 2–4, WT). Y73A mutation also remarkably impaired DNA binding (Fig. 3B, lanes 8–10). As described above, Tyr-73 interacts not only with Cyt12 but also with the His-110 that stacks with the terminal ribose moiety. Thus, the Y73A mutation may have affected the interaction of His-110 with the ribose, thereby indicating a significant reduction in DNA binding. As expected from the crystal structure, the mutation in Tyr-72 involved in pinching Cyt12-Cyt13 of the ssDNA strongly reduced DNA binding (Fig. 3B, lanes 5–7). In contrast, the mutation in Tyr-93 that was also involved in the base pinching did not have a large effect on DNA binding (Fig. 3B, lanes 14–16), indicating that role of Tyr-72 is more significant than that of Tyr-93 in the base pinching. The Y72A/Y93A double mutant protein lost DNA binding activity (Fig. 3B, lanes 26–28). Mutation in Phe-142 stacking with the symmetry-related Ade1 in the crystal structure slightly decreased the DNA binding of HLTF58–174 (Fig. 3B, lanes 23–25). This EMSA has revealed that Tyr-72, Tyr-73, Asn-91, and His-110 are crucial for DNA binding.
As the crystal structure had indicated that HLTF58–174 might prefer the 3′-end of DNA, we investigated whether it actually did by performing EMSA using DNAs modified by TAMRA at the 3′- or 5′-end (Fig. 3C). HLTF58–174 bound to the ssDNA or overhanging dsDNAs modified at the 5′-end at a level comparable with the unmodified ssDNA (Fig. 3A, lanes 3–5, and 3C, lanes 3–5, 19–21, and 23–25). In contrast, TAMRA modifications at the 3′-end in the ssDNA (Fig. 3C, lanes 7–9), in the 5′-overhanging (Fig. 3C, lanes 11–13), or in the 3′-overhanging dsDNA (Fig. 3C, lanes 15–17) significantly impaired the interaction. These results clearly indicate that HLTF58–174 specifically recognizes the 3′-end of DNA.
Recruitment of HLTF to the Stalled Replication Fork and the Mechanism of the TS Pathway
As described above, DNA bound to HLTF58–174 forms DNA duplex by crystallographic 2-fold symmetry (Fig. 2A). Furthermore, EMSA reveals that HLTF58–174 actually interacts with dsDNA (Fig. 3A). Thus, it is possible that the duplex mimics a primer-template DNA and that the crystal structure presents a snapshot of how HLTF interacts with the stalled replication fork. Moreover, specific recognition of the 3′-end and the base-pinching activity of the HIRAN domain imply a significant role for the domain in the process of fork regression. The crystal structure reveals specific interaction of the HIRAN domain with the 3′-end, indicating that the domain acts as a sensor of the 3′-end. Furthermore, the structure suggests that the HIRAN domain may be capable of unwinding the primer-template DNA by the base pinching with 3′-5′ polarity. Consistent with this hypothesis, HLTF actually exhibits 3′-5′ translocase activity (11). Structural study of UvrD DNA helicase with 3′-5′ polarity has revealed that an aromatic residue, Tyr-621, acts as the separation pin that buttresses the end of DNA duplex (28). The Tyr-72 and Phe-142 of the HIRAN domain are located in the end of DNA duplex (Fig. 2D). Those aromatic residues may play a role similar to the separation pin of UvrD to drive unwinding of the primer-template DNA. In fact, Tyr-72 is significantly involved in interaction with the primer strand (Fig. 3B, lanes 5–7). Therefore, Tyr-72 could be crucial for both separation and DNA binding.
The process of DNA damage tolerance involves either the TLS or the TS pathway, but how one pathway is chosen over the other remains a mystery. TLS appears to be a rapid process because it is performed by several TLS polymerases and does not require fork regression (1). In contrast, TS is a much more complicated process including regression of the stalled replication fork, DNA synthesis, and reversal of the regressed fork (1). It is plausible that the TS pathway is actuated when TLS fails due to DNA damage that is too severe for TLS polymerases to use it as a template (Fig. 4). In such a situation, the HIRAN domain of HLTF may sense the exposed 3′-end of the primer stand in the stalled replication fork, thereby recruiting MMS2-UBC13-HLTF to the stalled replication fork. If MMS2-UBC13-HLTF and RAD6-RAD18 had already been recruited to the damaged site to initiate the TLS pathway by mono-ubiquitination of PCNA, they could also perform poly-ubiquitination of PCNA. Additional RAD6-RAD18 might accompany MMS2-UBC13-HLTF, as it has been shown that RAD18 interacts with RAD5 (4). Recently, several groups have reported that the Lys-63-linked poly-ubiquitin chain of PCNA at Lys-164 is recognized by ZRANB3 (zinc finger, RAN-binding domain containing 3), alternatively termed AH2 (annealing helicase 2), that plays an important role in restarting the stalled replication fork (29–31). ZRANB3 contains not only PCNA-binding motifs but also the NZF (NPL4 zinc finger) domain that is a specialized ubiquitin-binding domain for the Lys-63-linked poly-ubiquitin chain. ZRANB3 is also a SWI/SNF DNA helicase that facilitates fork regression but disrupts the D-loop structure that is a product of strand invasion, suggesting that ZRANB3 suppresses unfavorable sister-chromatid exchange, also known as recombination-mediated TS (29). Interestingly, HLTF is capable of strand invasion and D-loop formation in a RAD51-independent manner (12). Considering this, these two SWI/SNF DNA helicases, HLTF and ZRANB3, might be involved in the early and late stages of the TS pathway, respectively. More complicatedly, mammalian cells have homologs that are functionally related to these DNA helicases. As described above, SHPRH is another RAD5 homolog, whereas interestingly, SHPRH lacks the HIRAN domain, suggesting that HLTF and SHPRH are not functionally redundant. In fact, HLTF and SHPRH have been shown to suppress mutagenesis in a damage-specific manner (32). Furthermore, mammalian cells have a ZRANB3 homolog, SMARCAL1 (SWI/SNF-related, matrix-associated, actin-dependent regulator of chromatin, subfamily A-like 1), that is responsible for Schimke immuno-osseous dysplasia (33). SMARCAL1 catalyzes fork regression and Holliday junction migration (34). It is a puzzle how cells differently utilize these SWI/SNF DNA helicases to maintain genome stability during DNA replication. Structural studies of these DNA helicases in complex with DNA are urgently required to unravel the mystery of the functional diversity of the helicases and the molecular mechanism behind TS. We are continuing to work on these problems.
FIGURE 4.
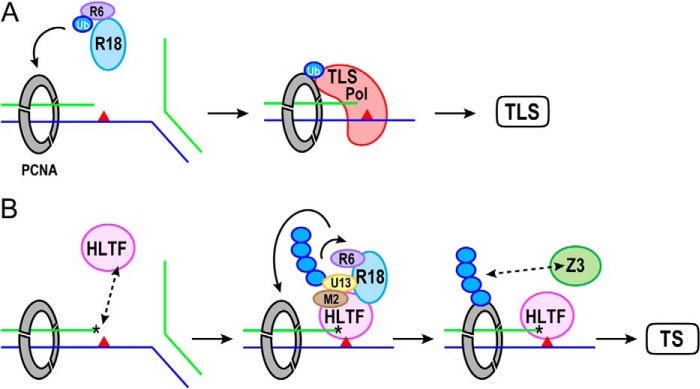
Model of recruitment of HLTF to the stalled fork in TS pathway. Colors represent the following proteins: ubiquitin (Ub), blue; RAD6 (R6), purple; RAD18 (R18), light blue; PCNA, gray; TLS polymerase (TLS Pol), red; UBC13 (U13), yellow; MMS2 (M2), brown; HLTF, pink; and ZRANB3 (Z3), green. Primer and template strands are shown by green and blue lines, respectively. DNA damage in the template strand is shown by the red triangle. The asterisk indicates the 3′-end of the primer strand recognized by the HIRAN domain of HLTF. A, RAD6-RAD18 mono-ubiquitinates PCNA to activate TLS. TLS polymerase, which possesses a ubiquitin-binding domain and a PCNA-interacting motif, is recruited to the damaged site by interaction with mono-ubiquitinated PCNA and performs DNA synthesis. B, the exposed 3′-end may be sensed by the HIRAN domain of HLTF when TLS fails due to severe DNA damage. The Lys-164 of PCNA is poly-ubiquitinated by MMS2-UBC13-HLTF and RAD6-RAD18 complexes. The Lys-63-linked poly-ubiquitin chain of PCNA recruits ZRANB3, another SWI/SNF helicase, to facilitate the fork regression. The HIRAN domain may also be involved in the unwinding of the primer-template DNA.
The HIRAN domain has been identified in proteins involved in DNA transaction events, including DNA repair and chromatin remodeling (13). Here we have shown the first structure of the HIRAN domain in complex with DNA. This is the first evidence showing that the HIRAN domain is a DNA-binding domain. The structure reveals the function of the novel DNA-binding domain as a sensor of the 3′-end and implies a significant role in recruiting HLTF to the damaged site, thereby facilitating poly-ubiquitination of PCNA and fork regression. Although the detailed molecular mechanism behind the TS pathway is still an open question and structural study of full-length HLTF is required, our results are the first to shed light on the mechanisms underlying the DNA transaction events in which the HIRAN domains are involved.
Acknowledgments
We acknowledge the kind support of the beamline staff of PF and SPring-8 for data collection. We thank Philip Hawke of the University of Shizuoka Scientific English Program for comments on the English in the manuscript.
This work was supported by KAKENHI (Grants 23770125, 26440034, 25121728, and 25291017), the National Project on Protein Structural and Functional Analyses (Protein 3000 Project), and the Targeted Proteins Research Program (TPRP) from Ministry of Education, Culture, Sports, Science and Technology (MEXT) (to A. H., T. S., M. S., and H. H.). This work was also supported by grants from the Takeda Science Foundation and the Naito Foundation (to H. H.).
The atomic coordinates and structure factors (codes 4XZF and 4XZG) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- TLS
- translesion DNA synthesis
- TS
- template-switching
- HLTF
- helicase-like transcription factor
- SHPRH
- SNF2 histone-linker PHD finger RING finger helicase
- HIRAN
- HIP116 and RAD5 N-terminal
- PCNA
- proliferating cell nuclear antigen
- Bis-Tris
- 2-(bis(2-hydroxyethyl)amino)-2-(hydroxymethyl)propane-1,3-diol
- TAMRA
- 6-carboxytetramethylrhodamine
- RPA
- replication factor A.
References
- 1. Friedberg E. C., Elledge S. J., Lehmann A. R., Lindahl T., Muzi-Falconi M., eds (2014) DNA repair, Mutagenesis, and Other Responses to DNA Damage, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 2. Johnson R. E., Henderson S. T., Petes T. D., Prakash S., Bankmann M., Prakash L. (1992) Saccharomyces cerevisiae RAD5-encoded DNA repair protein contains DNA helicase and zinc-binding sequence motifs and affects the stability of simple repetitive sequences in the genome. Mol. Cell Biol. 12, 3807–3818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Johnson R. E., Prakash S., Prakash L. (1994) Yeast DNA repair protein RAD5 that promotes instability of simple repetitive sequences is a DNA-dependent ATPase. J. Biol. Chem. 269, 28259–28262 [PubMed] [Google Scholar]
- 4. Hoege C., Pfander B., Moldovan G.-L., Pyrowolakis G., Jentsch S. (2002) RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 419, 135–141 [DOI] [PubMed] [Google Scholar]
- 5. MacKay C., Toth R., Rouse J. (2009) Biochemical characterisation of the SWI/SNF family member HLTF. Biochem. Biophys. Res. Commun. 390, 187–191 [DOI] [PubMed] [Google Scholar]
- 6. Motegi A., Sood R., Moinova H., Markowitz S. D., Liu P. P., Myung K. (2006) Human SHPRH suppresses genomic instability through proliferating cell nuclear antigen polyubiquitination. J. Cell Biol. 175, 703–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Motegi A., Liaw H.-J., Lee K.-Y., Roest H. P., Maas A., Wu X., Moinova H., Markowitz S. D., Ding H., Hoeijmarkers J. H. J., Myung K. (2008) Polyubiquitination of proliferating cell nuclear antigen by HLTF and SHPRH prevents genomic instability from stalled replication forks. Proc. Natl. Acad. Sci. U.S.A. 105, 12411–12416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Unk I., Hajdú I., Fátyol K., Szakál B., Blastyák A., Bermudez V., Hurwitz J., Prakash L., Prakash S., Haracska L. (2006) Human SHPRH is a ubiquitin ligase for Mms2-Ubc13-dependent polyubiquitylation of proliferating cell nuclear antigen. Proc. Natl. Acad. Sci. U.S.A. 103, 18107–18112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Unk I., Hajdú I., Fátyol K., Hurwitz J., Yoon J.-H., Prakash L., Prakash S., Haracska L. (2008) Human HLTF functions as a ubiquitin ligase for proliferating cell nuclear antigen polyubiquitination. Proc. Natl. Acad. Sci. U.S.A. 105, 3768–3773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Masuda Y., Suzuki M., Kawai H., Hishiki A., Hashimoto H., Masutani C., Hishida T., Suzuki F., Kamiya K. (2012) En bloc transfer of polyubiquitin chains to PCNA in vitro is mediated by two different human E2-E3 pairs. Nucleic Acids Res. 40, 10394–10407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Blastyák A., Hajdú I., Unk I., Haracska L. (2010) Role of double-stranded DNA translocase activity of human HLTF in replication of damaged DNA. Mol. Cell Biol. 30, 684–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Burkovics P., Sebesta M., Balogh D., Haracska L., Krejci L. (2014) Strand invasion by HLTF as a mechanism for template switch in fork rescue. Nucleic Acids Res. 42, 1711–1720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Iyer L. M., Babu M. M., Aravind L. (2006) The HIRAN domain and recruitment of chromatin remodeling and repair activities to damaged DNA. Cell Cycle 5, 775–782 [DOI] [PubMed] [Google Scholar]
- 14. Ikegaya Y., Hara K., Hishiki A., Yokoyama H., Hashimoto H. (2015) Crystallographic study of a novel DNA-binding domain of human HLTF involved in the template-switching pathway to avoid the replication arrest caused by DNA damage. Acta Crystallogr. F, in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 16. Terwilliger T. C., Berendzen J. (1999) Automated MAD and MIR structure solution. Acta Crystallogr. D 55, 849–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Terwilliger T. C. (2000) Maximum-likelihood density modification. Acta Crystallogr. D Biol Crystallogr. 56, 965–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Emsley P., Cowtan K. (2004) COOT: model-building tools for molecular graphics. Acta Crystallogr. D 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 19. Brünger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., Read R. J., Rice L. M., Simonson T., Warren G. L. (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D 54, 905–921 [DOI] [PubMed] [Google Scholar]
- 20. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 21. Kabsch W. (2010) Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. D 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Evans P. (2006) Scaling and assessment of data quality. Acta Crystallogr. D 62, 72–82 [DOI] [PubMed] [Google Scholar]
- 23. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kabsch W. (1976) A solution for the best rotation to relate two sets of vectors. Acta Crystallogr. A 32, 922–923 [Google Scholar]
- 25. Glaser F., Pupko T., Paz I., Bell R. E., Bechor-Shental D., Martz E., Ben-Tal N. (2003) Consurf: Identification of functional regions in proteins by surface-mapping of phylogenetic information. Bioinformatics 19, 163-164 [DOI] [PubMed] [Google Scholar]
- 26. Arcus V. (2002) OB-fold domains: a snapshot of the evolution of sequence, structure and function. Curr. Opin. Struct. Biol. 12, 794–801 [DOI] [PubMed] [Google Scholar]
- 27. Bochkarev A., Pfuetzner R. A., Edwards A. M., Frappier L. (1997) Structure of the single-stranded-DNA-binding domain of replication protein A bound to DNA. Nature 385, 176–181 [DOI] [PubMed] [Google Scholar]
- 28. Lee J. Y., Yang W. (2006) UvrD helicase unwinds DNA one base pair at a time by a two-part power stroke. Cell 127, 1349–1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ciccia A., Nimonkar A. V., Hu Y., Hajdu I., Achar Y. J., Izhar L., Petit S. A., Adamson B., Yoon J. C., Kowalczykowski S. C., Livingston D. M., Haracska L., Elledge S. J. (2012) Polyubiquitinated PCNA recruits the ZRANB3 translocase to maintain genomic integrity after replication stress. Mol. Cell 47, 396–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yuan J., Ghosal G., Chen J. (2012) The HARP-like domain-containing protein AH2/ZRANB3 binds to PCNA and participates in cellular response to replication stress. Mol. Cell 47, 410–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Weston R., Peeters H., Ahel D. (2012) ZRANB3 is a structure-specific ATP-dependent endonuclease involved in replication stress response. Genes Dev. 26, 1558–1572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lin J.-R., Zeman M. K., Chen J.-Y., Yee M.-C., Cimprich K. A. (2011) SHPRH and HLTF act in a damage-specific manner to coordinate different forms of postreplication repair and prevent mutagenesis. Mol. Cell 42, 237–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Boerkoel C. F., Takashima H., John J., Yan J., Stankiewicz P., Rosenbarker L., André J. L., Bogdanovic R., Burguet A., Cockfield S., Cordeiro I., Fründ S., Illies F., Joseph M., Kaitila I., Lama G., Loirat C., McLeod D. R., Milford D. V., Petty E. M., Rodrigo F., Saraiva J. M., Schmidt B., Smith G. C., Spranger J., Stein A., Thiele H., Tizard J., Weksberg R., Lupski J. R., Stockton D. W. (2002) Mutant chromatin remodeling protein SMARCAL1 causes Schimke immuno-osseous dysplasia. Nat. Genet. 30, 215–220 [DOI] [PubMed] [Google Scholar]
- 34. Bétous R., Mason A. C., Rambo R. P., Bansbach C. E., Badu-Nkansah A., Sirbu B. M., Eichman B. F., Cortez D. (2012) SMARCAL1 catalyzes fork regression and Holliday junction migration to maintain genome stability during DNA replication. Genes Dev. 26, 151–162 [DOI] [PMC free article] [PubMed] [Google Scholar]