

Background: The Vps4 ATPase powers the endosomal sorting complexes required for transport (ESCRT) pathway.
Results: Peptide binding to hexameric Vps4 is promoted by nucleotides that can mimic ADP, ATP, and the transition state.
Conclusion: ESCRT-III substrates bind Vps4 MIT domains and then bind the central pore of an asymmetric, nucleotide-bound Vps4 hexamer.
Significance: Mechanistic understanding of Vps4-substrate interactions is advanced by this work.
Keywords: ATPases associated with diverse cellular activities (AAA), endosomal sorting complexes required for transport (ESCRT), enzyme mechanism, fluorescence anisotropy, protein-protein interaction
Abstract
The endosomal sorting complexes required for transport (ESCRT) pathway drives reverse topology membrane fission events within multiple cellular pathways, including cytokinesis, multivesicular body biogenesis, repair of the plasma membrane, nuclear membrane vesicle formation, and HIV budding. The AAA ATPase Vps4 is recruited to membrane necks shortly before fission, where it catalyzes disassembly of the ESCRT-III lattice. The N-terminal Vps4 microtubule-interacting and trafficking (MIT) domains initially bind the C-terminal MIT-interacting motifs (MIMs) of ESCRT-III subunits, but it is unclear how the enzyme then remodels these substrates in response to ATP hydrolysis. Here, we report quantitative binding studies that demonstrate that residues from helix 5 of the Vps2p subunit of ESCRT-III bind to the central pore of an asymmetric Vps4p hexamer in a manner that is dependent upon the presence of flexible nucleotide analogs that can mimic multiple states in the ATP hydrolysis cycle. We also find that substrate engagement is autoinhibited by the Vps4p MIT domain and that this inhibition is relieved by binding of either Type 1 or Type 2 MIM elements, which bind the Vps4p MIT domain through different interfaces. These observations support the model that Vps4 substrates are initially recruited by an MIM-MIT interaction that activates the Vps4 central pore to engage substrates and generate force, thereby triggering ESCRT-III disassembly.
Introduction
The cellular endosomal sorting complexes required for transport (ESCRT)3 pathway functions in reverse topology membrane fission events in which the membranes are drawn toward the cytoplasm or nucleoplasm (1–3), including the sorting of ubiquitylated cargo proteins into multivesicular bodies (4); the abscission step of cytokinesis (5, 6); repair of the plasma membrane (7); exosome (8–10), shedding vesicle (11, 12), and nuclear vesicle formation (13); and the budding of many retroviruses, including HIV (14–16). This pathway comprises multiple protein complexes and accessory proteins, which converge to create ESCRT-III filaments that constrict the membrane neck (17, 18). The assembled ESCRT-III subunits are resolved by recruitment of the AAA ATPase Vps4 to the membrane neck immediately prior to fission (19, 20), whereupon individual ESCRT-III subunits are released (21, 22). The bodies of ESCRT-III subunits comprise an N-terminal four-helix bundle that can fold against helix 5 and can mediate lattice formation (23–25). The C-terminal tails contain MIT-interacting motifs (MIMs) that recruit Vps4 by binding the enzyme's N-terminal MIT domains (26–29). Three Saccharomyces cerevisiae ESCRT-III proteins (Vps2p, Did2p, and Ist1p) possess Type 1 MIMs that form amphipathic helices that bind in the groove between MIT helices 1 and 3 (26, 27, 30). Other ESCRT-III proteins, including Snf7p, Vps20p, and Ist1p, possess alternative Type 2 MIMs that bind as extended strands in the groove between MIT helices 2 and 3 (29, 31, 32).
Eukaryotic Vps4 enzymes comprise the N-terminal MIT domain, an ∼40-residue linker, a two-domain AAA ATPase cassette, a “β-domain” that is inserted within the small domain of the ATPase cassette, and a C-terminal helix that binds against the large ATPase domain. Vps4 functions as a higher-order oligomer. Although the subunit stoichiometry and structure have been controversial (33–36), we have shown that the active Vps4 enzyme is a hexamer (37), like all other well characterized Type I AAA ATPases (38, 39). Our working structural model for the Vps4 hexamer is based on superposition of the known crystal structure of the Vps4 ATPase cassette (33, 37, 40–43) onto the structure of the p97 D1 hexamer (44) and is supported by mutational analysis of proposed hexameric interface residues (37, 40, 41). This model places the Vps4 β-domains on the periphery, where they can bind the VSL domain of the Vta1p/LIP5 cofactor that promotes Vps4 assembly and stimulates ATPase activity (32, 40, 45–49). The Vps4 hexamer has a central pore that is lined by two conserved loops: pore loop 1 and pore loop 2. Vps4 pore loop 1 displays an aromatic hydrophobic dipeptide that is conserved across AAA ATPases that have polypeptide substrates (40, 41), and pore loop 2 contains a series of charged residues that are conserved and functionally important in the related ATPase spastin (50, 51). Mutations in either Vps4 pore loop inhibit HIV budding (40, 41), although these mutations may also destabilize the Vps4 hexamer to some extent.
Hanson and colleagues (52) have reported that overexpressed human ESCRT-III subunits CHMP2A and CHMP1B that lack their terminal MIM elements nevertheless co-sediment with human VPS4B(E235Q) from cell lysate. This interaction requires ESCRT-III helix 5 and surrounding loops, and this same region also contributes to stimulating VPS4A ATP hydrolysis when it is present within C-terminal fragments of ESCRT-III proteins (53). These observations suggest that the ESCRT-III helix 5 region may bind preferentially to the pore of the Vps4 hexamer. To investigate this idea further, we characterized the Vps4-ESCRT-III interaction using purified recombinant Saccharomyces cerevisiae proteins Vps4p and Vps2p (CHMP2 homolog). We found that the AAA ATPase cassette of a Vps4p construct that lacked the MIT domain formed a stable hexamer with the flexible nucleotide mimics ADP·AlFx or ADP·BeFx, and that this hexameric complex bound a single Vps2p helix 5 peptide in an interaction that was mediated by the Vps4p pore loop residues. Vps2p helix 5 binding was autoinhibited by the MIT domains, and this inhibition was alleviated by association of the MIT domain with an MIM1 or an MIM2 sequence. These observations, together with previous studies (21, 52–54), support a model in which ESCRT-III complexes are disassembled by pulling ESCRT-III helix 5 into the central pore of asymmetric Vps4p hexamers in a manner that is initially primed and activated by binding of MIT domains to ESCRT-III MIM sequences.
Experimental Procedures
Proteins and Peptides
Vps4p and the VSL domain of Vta1p (Vta1pVSL, residues 280–330) were expressed and purified as described (35, 37, 40, 41). Bacterial expression vectors are listed in Table 1. Mutations were generated by QuikChange mutagenesis and verified by DNA sequencing. Peptides were synthesized on a Prelude peptide synthesizer (Protein Technologies, Inc.) using standard Fmoc chemistry (55). Following cleavage from resin, the peptides were precipitated with ice-cold ether, washed thoroughly with ether, dissolved in water/acetonitrile, and lyophilized for long-term storage. 5(6)-Carboxyfluorescein (Acros Organics) was used to fluorescently label the peptides.
TABLE 1.
Bacterial expression vectors
All expression plasmids have been deposited at the DNASU Plasmid Repository at Arizona State University (71).
| Construct | Insert | Backbone | Internal ID |
|---|---|---|---|
| Vps4pΔMIT | Vps4p (81–437) | pET151-D-Topo | CPH2473 |
| Vps4pΔMIT E243A | Vps4p (81–437, E243A) | pET151-D-Topo | CPH2675 |
| Vps4pΔMIT E247A | Vps4p (81–437, E247A) | pET151-D-Topo | CPH2678 |
| Vps4pΔMIT E243A, E247A | Vps4p (81–437, E243A,E247A) | pET151-D-Topo | CPH2779 |
| Vps4pΔMIT T240A | Vps4p (81–437, T240A) | pET151-D-Topo | CPH2673 |
| Vps4pΔMIT T240K | Vps4p (81–437, T240K) | pET151-D-Topo | CPH2783 |
| Vps4pΔMIT T240V | Vps4p (81–437, T240V) | pET151-D-Topo | CPH2803 |
| Vps4pΔMIT T240F | Vps4p (81–437, T240F) | pET151-D-Topo | CPH2784 |
| Vps4pΔMIT R241A | Vps4p (81–437, R241A) | pET151-D-Topo | CPH2575 |
| Vps4pΔMIT R251A | Vps4p (81–437, R251A) | pET151-D-Topo | CPH2576 |
| Vps4pΔMIT W206A | Vps4p (81–437, W206A) | pET151-D-Topo | CPH2574 |
| Vta1pVSL | Vta1p (280–330) | pET151-D-Topo | CPH2570 |
| MIM1-Vps4p | Vps2p(218–232)-(GGGGS)3-Vps4p | pET151-D-Topo | CPH2802 |
| MIM1(L225D)-Vps4p | Vps2p(218–232, L225D)-(GGGGS)3-Vps4p | pET151-D-Topo | CPH3093 |
| MIM2-Vps4p | Vfa1p(183–203)-(GGGGS)3-Vps4p | pET151-D-Topo | CPH3077 |
| MIM2(F198D)-Vps4p | Vfa1p(183–203, F198D)-(GGGGS)3-Vps4p | pET151-D-Topo | CPH3078 |
| GST | GST | pET151-D-Topo | CPH3094 |
| GST-peptide A | GST-Vps2p (165–201) | pET151-D-Topo | CPH2914 |
For N-terminally labeled peptides, the fluorescein group was coupled to the N-terminal α-amine by standard coupling conditions. All other amines within the peptide were t-butyloxycarbonyl-protected. For C-terminally labeled peptides, an orthogonal lysine deprotection method was used. At the first position of the peptide (C terminus), Dde-Lys(Fmoc)-OH (AAPPTec) was coupled to the resin. Fmoc removal was performed with 20% piperidine, followed by fluorescein coupling to the ϵ-amine on the lysine side chain. The Dde group (on the lysine α-amine) was then removed with 3% hydrazine in N,N-dimethylformamide, and the remaining peptide was synthesized using standard protocols. Unlabeled N termini were blocked with an acetyl group, and unlabeled C termini were amides. Peptide quality was verified by reversed phase C18 HPLC. For experiments described below, peptide concentrations were determined by infrared spectroscopy using a Direct Detect® spectrometer (EMS Millipore). The identities of all peptides and recombinant proteins were verified by mass spectrometry.
Analytical Size-exclusion Chromatography
A Superdex 200 column was equilibrated in running buffer (10 mm MgCl2, 100 mm NaCl, 20 mm HEPES, pH 7.0, and when appropriate, 3 mm nucleotide). To prepare ADP metal fluoride stock solutions, 1 m NaF was added dropwise to a 10 mm solution of ADP to a final concentration of 62.5 mm with stirring, and then 1 m AlCl3 or BeCl2 was slowly added to a final concentration of 12.5 mm. Vps4p proteins (200 or 50 μm in subunit concentration, corresponding to 33 or 8.3 μm hexamer, respectively) were preincubated with a 2-fold molar (subunit) excess of Vta1pVSL in running buffer at 4 °C for at least 2 h. The column was calibrated using protein standards (Bio-Rad).
Fluorescence Anisotropy Binding Assays
To generate the data shown in Figs. 1B, 3A, 4B, and 6A, a dilution series of Vps4p-Vta1pVSL complex (Vta1pVSL was in 2-fold excess over Vps4p subunits, total volume 60 μl) in binding buffer (10 mm MgCl2, 100 mm NaCl, 20 mm HEPES, pH 7.0, and where indicated, 3 mm nucleotide) was incubated with fluorescein-labeled peptides (1 nm) at 25 °C for at least 3 h before measuring parallel and perpendicular fluorescence intensity using excitation/emission wavelengths of 485/535 nm on a Tecan Infinite 200 microplate reader. Anisotropy values were calculated and plotted against the Vps4p hexamer concentration. Dissociation constants (KD values) were estimated by fitting the data to the equation, FA = [Vps4p hexamer]/(KD + [Vps4p hexamer]), where FA is the normalized fluorescence anisotropy and corresponds to “fraction bound,” using GraphPad Prism 5 (GraphPad Software, Inc.). Error bars show the S.D. from three independent experiments and are shown for every data point in the figures.
FIGURE 1.

Vps2p helix 5 peptides bind the Vps4p ATPase cassette. A, the Vps2p MIM1 and A, B, and C peptides used in this study are indicated. The predicted secondary structure of Vps2p is shown as a schematic. B, binding between peptides and Vps4p was assayed by fluorescence anisotropy. Peptides A, B, and C bind Vps4pΔMIT in the presence of ADP·AlFx or ADP·BeFx, but not in the presence of other nucleotides tested. The control MIM1 peptide also binds, but ∼15-fold more weakly. KD values and S.D. value are shown to the right. C, Vps4pΔMIT forms a stable hexamer in the presence of Vta1pVSL and ADP·AlFx or ADP·BeFx. Vps4pΔMIT at 200 μm subunit concentrations (33 μm hexamer, gray) and 50 μm subunit concentrations (8.3 μm hexamer, black) was run on a gel filtration column in the presence of Vta1pVSL and different nucleotides. Vertical dotted lines indicate calculated elution volumes of the Vps4pΔMIT hexamer and dimer relative to standards. Error bars indicate S.D. from three independent experiments.
FIGURE 3.
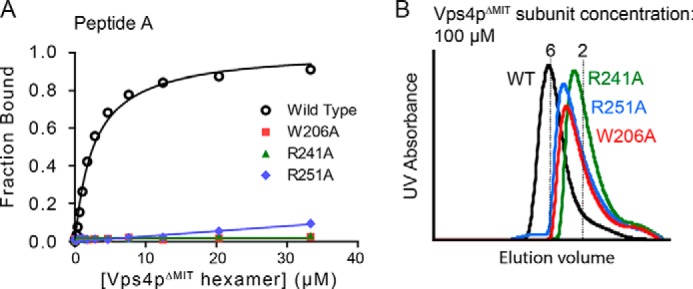
Pore loop mutations equivalent to substitutions reported to block HIV budding abolish peptide binding and reduce Vps4p hexamerization. A, Vps4pΔMIT proteins carrying the mutations W206A, R241A, or R251A are unable to bind peptide A. B, these mutant proteins form less stable hexamers in the presence of Vta1pVSL and ADP·AlFx (100 μm Vps4p subunits, 17 μm hexamers).
FIGURE 4.

Negatively charged residues on pore loop 2 of the Vps4p hexamer modulate binding to Vps2p helix5. A, Vps4pΔMIT carrying pore loop 2 mutations T240A, T240K, T240F, E243A, or E247A can form hexamers as assayed by gel filtration chromatography (50 μm Vps4p subunits, 8.3 μm hexamers) in the presence of Vta1pVSL and ADP·AlFx. B, Vps4pΔMIT proteins carrying mutations on Thr-240 (upper panel) or mutations of acidic pore loop 2 residues (lower panel) were tested for binding to peptide C by fluorescence anisotropy. When compared with Vps4pΔMIT, Vps4pΔMIT(T240K) and Vps4pΔMIT(E243A,E247A) displayed increased binding. C, mutations of the acidic residues on pore loop 2 of Vps4p impaired the ATPase activity of Vps4p. A double mutation (E243A,E247A) on the negatively charged collar reduced the ATPase activity by ∼55% when compared with wild-type Vps4p. The ATPase inactive mutant E233Q was used as a negative control. Error bars indicate S.D. from three independent experiments.
FIGURE 6.

MIT-MIM interactions regulate binding of Vps2p-helix 5 peptide C to the Vps4p AAA ATPase cassette. A, full-length Vps4p, MIM1(L225D)-Vps4p, and MIM2(F198D)-Vps4p bind peptide C with lower affinity than Vps4pΔMIT, MIM1-Vps4p, and MIM2-Vps4p. Error bars indicate S.D. from three independent experiments. B, Vps4p constructs used in the binding experiments. C, dissociation constants (KD) between peptides and different Vps4p constructs. D, MIM1-Vps4p and MIM1(L225D)-Vps4p oligomerize similarly as assayed by gel filtration chromatography.
To generate the data shown in Fig. 2, fluorescence anisotropy was measured during titration of Vps4pΔMIT-Vta1pVSL hexamer into 40 μm peptide C in the binding buffer (above). Fraction bound was plotted versus the [Vps4pΔMIT hexamer]/[peptide] ratio. At these high peptide concentrations (17-fold over KD), the stoichiometry of peptide binding to the Vps4pΔMIT hexamer can be estimated by the ratio at which saturation binding was reached. Data are shown for three independent experiments performed with protein from independent preparations.
FIGURE 2.
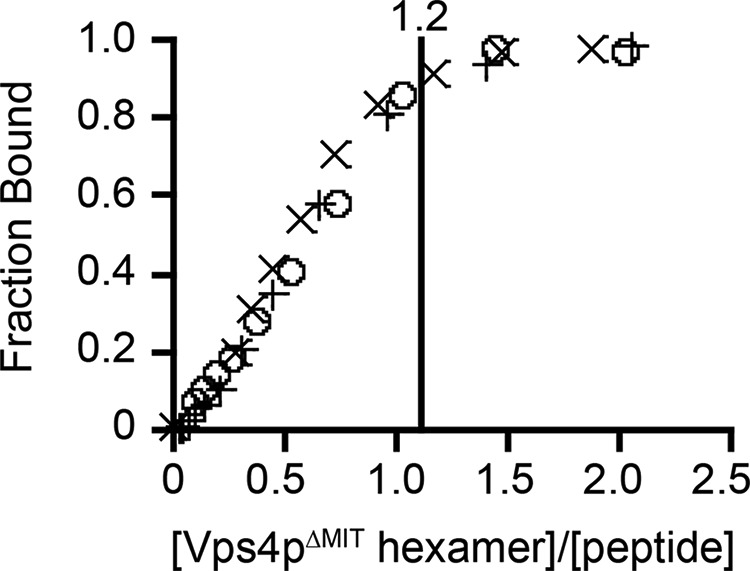
One Vps4pΔMIT hexamer binds one Vps2p helix 5 peptide. Fluorescence anisotropy was measured during titration of 40 μm (∼20-fold over KD) peptide C with the Vps4pΔMIT-Vta1pVSL hexamer. Saturation of the signal occurred when the stoichiometry of the complex was close to 1:1. Data from three independent experiments are shown here, and the indicated stoichiometry represents the ratio of Vps4pΔMIT-Vta1pVSL hexamer to peptide C at saturation when all data sets were fit globally.
GST Pulldown Assays
An N-terminal GST fusion of Vps2p residues 165–201 was used for pulldown assays. The fusion protein was expressed from a pET151-D-Topo vector with an N-terminal tobacco etch virus protease-cleavable His6 tag and purified using the procedure described above for Vps4p proteins. Purified Vps4p at the indicated subunit concentration, Vta1pVSL (2-fold excess over Vps4p), and GST fusion proteins (1 μm) were mixed in wash buffer (10 mm MgCl2, 100 mm NaCl, 20 mm HEPES, pH 7.0, with or without 3 mm ADP·AlFx) and incubated for 3 h at 4 °C. 20 μl of glutathione agarose (Amersham Biosciences) was added to 1 ml of protein samples and incubated for 1 h at 4 °C. Unbound proteins were removed by washing with wash buffer (four washes with 1 ml of buffer). The glutathione resin was subsequently resuspended in SDS-PAGE loading buffer and incubated for 5 min at 95 °C, and bound proteins were visualized by SDS-PAGE.
ATPase Assay
To generate the data shown in Fig. 5A, Vps4pΔMIT (final concentration 0.3 μm subunits, corresponding to 50 nm hexamer) was mixed with Vta1pVSL (2-fold excess over Vps4p subunits) and 2 mm ATP in reaction buffer (100 mm NaCl, 10 mm MgCl2, 1 mm DTT, and 20 mm HEPES, pH 7.5), together with the indicated concentrations of Vps2p peptide C. After 10 min, 2 volumes of malachite green solution (14 mm ammonium molybdate, 1.3 m HCl, 1.5 mm malachite green) were added, followed by 21% citric acid (same volume as the malachite green solution). Absorbance at 650 nm was measured using a POLARstar OPTIMA (BMG Labtech) plate reader, and phosphate concentrations were calculated using a sodium phosphate standard curve. To generate the data shown in Fig. 4C, Vps4pΔMIT and its pore loop 2 mutants (final concentration 0.5 μm subunits) were mixed with Vta1pVSL (2-fold excess over Vps4p subunits), and their ATPase activity was measured as described above.
FIGURE 5.
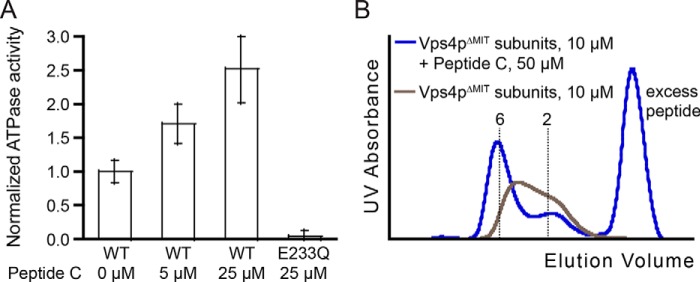
Vps2p helix 5 peptide C stimulates Vps4p ATPase activity and stabilizes the hexamer. A, ATPase activity of Vps4p was measured in the presence of different concentrations of peptide C. Normalization of ATPase activity was carried out with respect to wild-type Vps4p. B, oligomerization of Vps4pΔMIT-Vta1pVSL (10 μm Vps4p subunits, 1.7 μm hexamers) was measured by gel filtration in the presence of ADP·AlFx and in the presence (blue) or absence (gray) of 50 μm peptide C. Error bars indicate S.D. from three independent experiments.
Results
Vps2p Peptides Bind the ATPase Cassette of an Asymmetric Vps4p Hexamer
Quantitative fluorescence anisotropy assays were performed to test whether peptides that spanned the C-terminal end of Vps2p helix 5 (Fig. 1A) bound a pure recombinant protein complex comprising a Vps4p construct that lacked the N-terminal MIT domain (Vps4pΔMIT; residues 81–437) and the Vta1p VSL domain (Vta1pVSL, residues 280–330). Tight peptide binding (KD = 2–10 μm) was observed in the presence of ADP·AlFx or ADP·BeFx, but not in the presence of ATP, ADP, AMPPNP, or ATPγS (Fig. 1B). Binding specificity was validated by the observation that a control peptide corresponding to the downstream Vps2p MIM1 motif (residues 218–232) bound 15-fold less tightly than Vps2p helix 5 peptides A and C, and by the demonstration that different versions of peptide A that were fluorescently labeled at the N or the C terminus bound equivalently (within 2-fold, data not shown).
Peptide binding affinities to the Vps4pΔMIT-Vta1pVSL complex in the presence of ADP·AlFx were calculated assuming that Vps4pΔMIT is present as a hexamer, and that one Vps4p hexamer binds one peptide (see below). Peptides A (residues 165–201; KD = 2.5 ± 0.5 μm) and C (residues 165–184; KD = 2.4 ± 0.5 μm) bound modestly more tightly than peptide B (residues 144–184; KD = 6.1 ± 2.4 μm) (Fig. 1B), indicating that the minimal peptide C construct contains the primary binding site, and suggesting that the patch of highly acidic residues within the N-terminal extension of peptide B may weaken the binding slightly.
The unique ability of ADP·AlFx and ADP·BeFx to support binding likely reflects the ability of these nucleotide analogs to mimic multiple states, including ATP, ADP, and transition state-bound nucleotide complexes (56–59), and suggests that different subunits within each Vps4p hexamer may need to bind different nucleotide states and adopt different conformations to generate a stable, asymmetric substrate-binding site. Consistent with this idea, hexameric Vps4pΔMIT-Vta1pVSL complexes were more stable in the presence of ADP·AlFx or ADP·BeFx than in the presence of ATP, ADP, or ATPγS, or in the absence of nucleotide, as shown by size-exclusion chromatography (note the relative mobilities of the different complexes at 50 μm Vps4pΔMIT subunit concentrations, corresponding to 8.3 μm hexamer concentrations, Fig. 1C). Thus, Vps4pΔMIT is hexameric in the concentration range used for the fluorescence anisotropy binding assay in the presence of ADP·AlFx or ADP·BeFx, and the fluorescence anisotropy signal can be used as a readout to quantify peptide binding, especially since substrate binding further stabilizes hexamer formation (see below).
The Vps4p Hexamer Binds a Single Peptide
To verify the stoichiometry of binding, we performed a fluorescence anisotropy binding experiment in which Vps4pΔMIT-Vta1pVSL hexamers in the presence of ADP·AlFx were titrated into a solution of fluorescently labeled peptide C at high concentration (∼20-fold above the KD). Peptide binding saturated when the stoichiometry was close to one peptide per Vps4p hexamer (Fig. 2), supporting the idea that each asymmetric Vps4p hexamer contains a single binding site for ESCRT-III helix 5.
Pore Loop Residues Bind Directly to Vps2p-derived Peptides
To determine whether residues of the Vps4p hexamer pore contribute to binding, we tested binding to pore loop mutants that have been previously shown to inhibit HIV budding when equivalent mutations are present in human Vps4 homologs (40, 41). These correspond to W206A (pore loop 1), R241A (pore loop 2), and R251A (adjacent to pore loop 2) of S. cerevisiae Vps4p (40, 41). None of these mutants bound peptides with appreciable affinity (Fig. 3A). However, gel filtration experiments, performed at 100 μm Vps4pΔMIT subunit concentration (17 μm hexamer) for a more sensitive readout of hexamer dissociation, indicated that the hexamer stability of these mutants was reduced (Fig. 3B).
To identify pore loop mutations that do not destabilize the Vps4p hexamer, we substituted 10 different pore loop residues individually with alanines and assayed oligomerization by analytical size-exclusion chromatography at a Vps4p subunit concentration of 50 μm (8.3 μm hexamer). At this concentration, the wild-type Vps4pΔMIT protein elutes as a stable hexamer in the presence of ADP·AlFx (Figs. 1C and 4A). Three of the Vps4p substitution mutants (T240A, E243A, and E247A) displayed essentially unchanged oligomerization, even at low protein concentrations (Fig. 4A). These pore loop alanine substitution mutants were therefore assayed for peptide binding. Remarkably, introducing E243A or E247A point mutations increased the affinity of peptide C binding (by 3- and 4-fold, respectively, Fig. 4B). Moreover, the double mutant, Vps4pΔMIT(E243A,E247A), bound peptides 4-fold (peptide C) or 10-fold (peptide B, not shown) more tightly than wild-type Vps4p (Fig. 4B, lower panel). Because different alanine pore loop substitutions can either increase or decrease peptide binding affinity (Fig. 4B) without substantially changing Vps4p hexamerization, we conclude that substrates bind the pore loops directly. This conclusion is reinforced by the observation that the double mutation, which gives the tightest binding, actually reduces hexamerization slightly (Fig. 4A, lower panel), thereby emphasizing that the mutations uncouple reduction of binding from reduction of hexamerization. Similar to the effect of removing acidic pore loop residues, we also find that introducing a positive charge (T240K) enhances peptide binding (Fig. 4B, top panel) without affecting oligomerization (Fig. 4A, top panel), whereas the T240F mutation has no effect.
We also tested the effects of the pore loop 2 glutamate mutations on the basal ATPase activity of Vps4pΔMIT. As shown in Fig. 4C, both the E243A and the E247A mutations reduced ATPase activity, but the double mutation did not further reduce ATPase activity (p value between E243A and the double mutant is 0.17). Although hexamerization was not impaired when assayed by gel filtration, a possible explanation is that reduced ATPase activity reflects reduced hexamerization at the concentration used to assay ATP hydrolysis (0.3 μm versus 50 μm subunit concentrations), whereas the increased peptide binding reflects improved interactions between the pore loop residues and the substrate. Alternatively, the reduced activity of these mutants may reflect allosteric coupling of the enzymatic active site to the central peptide-binding site (and altered coupling in the mutants).
Peptide Binding Stimulates ATPase Activity and Stabilizes Hexamerization
A previous study showed that VPS4A ATPase activity is stimulated by C-terminal ESCRT-III constructs that span the MIM motif and helix 5 (53). Having mapped relatively high affinity binding to the helix 5 region of Vps2p, we tested whether this pore-binding element could also stimulate Vps4pΔMIT ATPase activity. As shown in Fig. 5A, the addition of peptide C stimulated the Vps4pΔMIT ATPase activity by 2.5-fold at the highest concentration tested. To test the possibility that this effect resulted from increased stabilization of the Vps4p hexamer in the presence of peptide C, we characterized the oligomerization of Vps4pΔMIT in the presence of Vta1pVSL and ADP·AlFx and in the presence or absence of peptide C. Analytical gel filtration was performed at (low) Vps4p concentrations where the hexameric complex was appreciably dissociated (10 μm Vps4pΔMIT subunits, 1.7 μm hexamer). Under these conditions, Vps4pΔMIT eluted as a hexamer in the presence but not the absence of peptide C (Fig. 5B). Thus, peptide binding stabilizes the Vps4p hexamer. We note that ATPase activity was assayed at a concentration where inactive monomers and dimers of Vps4pΔMIT likely predominated over active hexamers, and the observed peptide stimulation of ATPase activity is therefore likely due, at least in part, to substrate-mediated stabilization of the Vps4pΔMIT hexamer.
The MIT Domain Inhibits Substrate Binding in the Absence of an MIM Interaction
To determine whether the MIT domains influence substrate binding to the Vps4p pore, we compared binding of the A, B, and C peptides to various Vps4p constructs (Fig. 6B) and found that full-length Vps4p binds each of these peptides with ∼10-fold lower affinity than does Vps4pΔMIT (Fig. 6, A and C). To determine whether this inhibitory effect of the MIT domain could be alleviated by MIM binding, we designed a Vps4p variant (MIM1-Vps4p) that included an N-terminal extension of the Vps2p MIM1 motif (residues 218–232) covalently linked to the MIT domain through a (GGGGS)3 linker that was long enough to allow an intramolecular MIM1-MIT interaction (Fig. 6B). The equivalent construct bearing the MIM1 L225D mutation (MIM1(L225D)-Vps4p) was used as a negative control because this mutation disrupts the interaction between MIM1 and MIT (26). The MIM1-Vps4p fusion and Vps4pΔMIT proteins bound peptides with similar affinities, whereas the binding affinity of MIM1(L225D)-Vps4p was similar to that of full-length Vps4p (Fig. 6, A and C). Additional validation was provided by a GST affinity co-purification pulldown assay using Vps2p helix 5 residues fused to GST. This construct bound both Vps4pΔMIT and MIM1-Vps4p, whereas binding to both Vps4p and MIM1(L225D)-Vps4p was greatly diminished (Fig. 7). These data indicate that MIM1 binding alleviates inhibition of substrate pore binding by the MIT domains. This effect does not appear to reflect MIT domain inhibition of Vps4p oligomerization because MIM1-Vps4p and MIM1(L225D)-Vps4p displayed equivalent migration on gel filtration chromatography in the presence of ADP·AlFx (Fig. 6D). Essentially identical results were found for binding of peptide C to MIM2-Vps4p constructs (Vfa1p residues 183–203) (Fig. 6C) that were designed in light of the recently reported Vfa1p MIM2-MIT complex structure (60).
FIGURE 7.
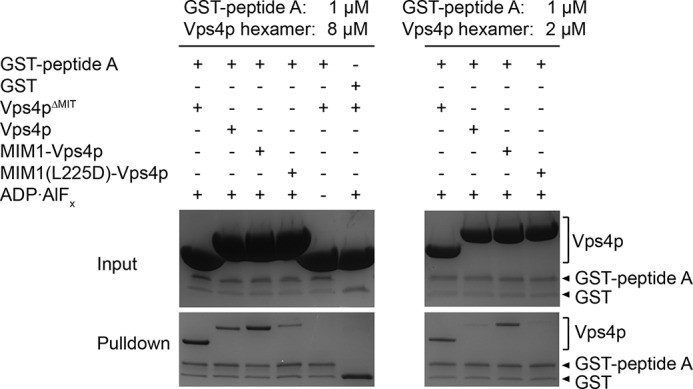
Pulldown assays demonstrating that GST-peptide A can bind Vps4p in the presence of ADP·AlFx and that GST-peptide A pulls down Vps4pΔMIT or MIM1-Vps4p better than Vps4p or MIM1(L225D)-Vps4p.
Discussion
ESCRT-mediated membrane fission requires remodeling and depolymerization of ESCRT-III filaments by the Vps4 AAA ATPase, which is recruited, at least in part, by binding of its N-terminal MIT domain to C-terminal ESCRT-III MIM elements. Earlier studies indicated that an additional interaction occurs for human VPS4A between the ATPase cassette and sequences from the helix 5 region of ESCRT-III subunits, that deletion of the MIT domain and linker sequence enhances ATPase activity in human VPS4A, and that mutation of pore loop residues could either increase or reduce ESCRT-III-induced ATPase activity (52, 53). We have verified and extended these findings by performing quantitative binding studies using purified reagents, delineating effects due to peptide binding versus those due to stabilization or destabilization of the active hexamer, and demonstrating effects of MIM1 and MIM2 interactions on substrate engagement. The earlier studies on VPS4A were prompted in part by the relatively low ATPase activity of human VPS4 proteins (53), which suggested that fundamental differences in Vps4 regulation might exist between species but might be explained by the different assay conditions such as the presence/absence of Vta1/LIP5. In addition to providing new insights, our work using the S. cerevisiae Vps2p and Vps4p proteins therefore indicates that key principles of Vps4 regulation are likely to be conserved across species.
Our fluorescence anisotropy studies demonstrated that a 20-residue peptide from the helix 5 region of Vps2p/ESCRT-III binds the hexameric Vps4p AAA ATPase cassette with an affinity similar to that of the primary contact between the isolated Vps4p MIT domain and the Vps2p/ESCRT-III MIM (albeit without enhancing avidity effects). This interaction seems unlikely to have absolute sequence specificity, and we anticipate that many protein sequences could similarly engage the Vps4 ATPase cassette, consistent with the reports that overlapping fragments of multiple ESCRT-III family members are able to co-sediment with VPS4B(E235Q) (52) and stimulate ATPase activity of human VPS4A (53) or yeast Vps4p (61, 62). We also demonstrated that binding occurs with a stoichiometry of one peptide per hexamer. This further validates the conclusion that Vps4 is active as a hexamer (37) and supports the model that substrates bind in the central Vps4 pore. We also found that peptide binding stimulates ATPase activity, at least in part by stabilizing the hexamer.
Although residues of the pore loops have previously been implicated in Vps4 function (40, 41), it was formally possible that these mutations inhibited activity by reducing the stability of the active Vps4 oligomer. We have now identified point mutants in Vps4p pore loop 2 that increased binding without decreasing hexamerization, thereby solidifying the idea that substrates bind in the central pore of the hexamer. We were unable to perform equivalent experiments with pore loop 1 mutants, however, because although we identified many pore loop 1 mutations that diminished binding, they also diminished hexamerization, at least to some degree.
An important conclusion from our work is that Vps4p functions as an asymmetric hexamer that translocates substrate into the central pore. This follows from our observations that a single peptide binds per hexamer, which necessarily requires an asymmetric interface, and is dependent upon the presence of ADP·AlFx or ADP·BeFx, which allow the ATPase to adopt multiple nucleotide-bound states. This is consistent with models of other ring-like ATPases, including F1 ATPase (63), E1 helicase (64), Rho helicase (59), the 26S proteasome (65, 66), the bacterial enhancer-binding protein NtrC1 (58), and N-ethylmaleimide-sensitive factor (NSF) (67), which all display asymmetry of the subunits that results in a spiral staircase arrangement of the pore loops. An attractive model is that Vps4 translocates substrate, at least partially, through the central pore, driven by changes in pore loop conformations that propagate around the hexamer ring in concert with the ATP hydrolysis cycle.
Peptide binding was not observed with ADP or ATP, which presumably gave a mixture of ATP and ADP because the binding assay was performed at a temperature that allows for hydrolysis, and nor was binding observed in the presence of non-hydrolyzable analogs of ATP. Presumably, ADP metal fluorides enable the formation of asymmetric, peptide-bound complexes but do not drive translocation, whereas binding under conditions of active ATP turnover is transient and therefore not captured by our assay. The transcriptional activator PspF, a related AAA+ ATPase, also requires ADP·AlFx to form a complex with σ54 that can be detected in vitro (68).
Our observations that the pore loop mutants E243A and E247A bind more tightly (especially for peptide B) indicate that the negatively charged pore residues Glu-243 and Glu-247 diminish binding of peptides rich in acidic residues (such as peptide B). Thus, the pore is not optimized to bind all peptides equally or with maximal binding affinity. Our findings are somewhat discordant with conclusions from pioneering work from the Hanson laboratory, which provided motivation for this study but also concluded that acidic residues preferentially bind (52) and stimulate ATPase activity (53) of human VPS4A. Regardless, the overall impression is that there is a pore-selective-binding element located within helix 5 of Vps2p, but also that different amino acid sequences can bind the Vps4p pore with appreciable affinity, which is consistent with the idea that once an ESCRT-III subunit is engaged, it can be processively translocated through the hexamer pore. In principle, this action could translocate the ESCRT-III substrates completely, although the extent of translocation remains to be determined experimentally.
In contrast to human VPS4A (53), deletion of the MIT domain does not seem to increase the basal rate of ATP hydrolysis for yeast Vps4p (36, 61, 62). However, our observation that the MIT domain inhibits peptide binding to the hexamer pore in the absence of an MIT-MIM interaction suggests a two-step model of Vps4-mediated ESCRT-III lattice disassembly (Fig. 8). We propose that the MIM-MIT interaction serves to recruit Vps4 to ESCRT-III polymers and to unmask or activate the binding site at the hexamer pore, which possesses an inherent ability to bind and translocate a wide variety of sequences starting at an internal loop, such as has been described for the proteasome (69, 70). An attractive feature of this model is that it provides a mechanism for avoiding Vps4 pore engagement of inappropriate substrates. The mechanism of MIT inhibition is unclear at this time, and might include direct occlusion or indirect stabilization of an inhibited pore conformation. It is also unclear whether all of the MIT domains in a Vps4 hexamer need to be engaged by MIMs to allow binding at the pore and stimulate ATPase activity, although it is attractive to speculate that multiple MIT-MIM interactions are required because that would provide an effective mechanism for restricting activity to ESCRT-III polymers. This model provides a foundation for future structural and mechanistic studies to understand in more detail how Vps4 functions in membrane fission and the dissociation of ESCRT-III filaments.
FIGURE 8.
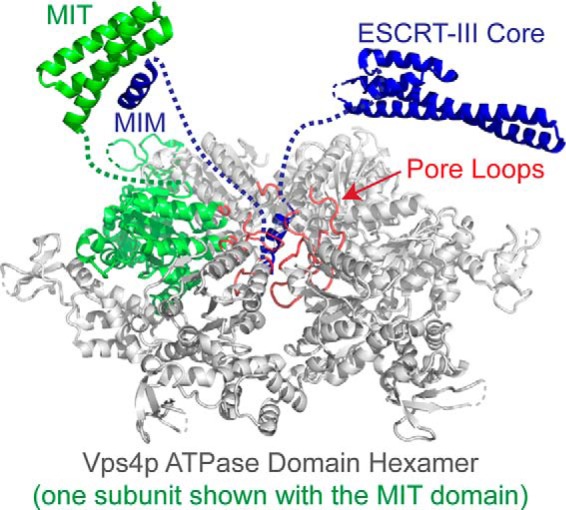
Two-step model for disassembly of the ESCRT-III complex by Vps4. The active Vps4 hexamer is shown with one subunit colored green. ESCRT-III substrates are recognized by binding of their MIM elements to Vps4 MIT domains, which release MIT/linker-mediated autoinhibition and allow the Vps4 pore loops (red) to engage a high affinity binding site on helix 5 of the ESCRT-III subunits and initiate translocation.
Acknowledgments
We thank Matthew Weinstock, Michael Jacobsen, and Michael Kay for peptide synthesis and Frank Whitby for helpful discussions and comments on the manuscript. Portions of this work (mass spectrometry and DNA sequencing) were performed in Core Facilities at the University of Utah, which were supported by National Institutes of Health Grant NCI P30CA042014.
This work was supported by National Institutes of Health Grants P50 GM082545 (to C. P. H.), RO1 AI051174 (to W. I. S.), and T32 AI055434 (to N. M.). This work was also supported by Grants PBZHP3-135952 and PBZHP3-141465 from the Swiss National Science Foundation (to N. M.) and Deutsche Forschungsgemeinschaft (DFG) Fellowship VO 1836/1-1 (to J. V.).
- ESCRT
- endosomal sorting complexes required for transport
- Dde
- 1-(4,4-dimethyl-2,6-dioxocyclohex-1-ylidene)ethyl
- MIT
- microtubule-interacting and trafficking
- MIM
- MIT-interacting motif
- Fmoc
- N-(9-fluorenyl)methoxycarbonyl
- AMPPNP
- 5′-adenylyl-β,γ-imidodiphosphate
- ATPγS
- adenosine 5′-(3-thiotriphosphate).
References
- 1. McCullough J., Colf L. A., Sundquist W. I. (2013) Membrane fission reactions of the mammalian ESCRT pathway. Annu. Rev. Biochem. 82, 663–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hurley J. H., Hanson P. I. (2010) Membrane budding and scission by the ESCRT machinery: it's all in the neck. Nat. Rev. Mol. Cell Biol. 11, 556–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Henne W. M., Buchkovich N. J., Emr S. D. (2011) The ESCRT pathway. Dev. Cell 21, 77–91 [DOI] [PubMed] [Google Scholar]
- 4. Hanson P. I., Cashikar A. (2012) Multivesicular body morphogenesis. Annu. Rev. Cell Dev. Biol. 28, 337–362 [DOI] [PubMed] [Google Scholar]
- 5. Carlton J. G., Martin-Serrano J. (2007) Parallels between cytokinesis and retroviral budding: a role for the ESCRT machinery. Science 316, 1908–1912 [DOI] [PubMed] [Google Scholar]
- 6. Morita E., Sandrin V., Chung H. Y., Morham S. G., Gygi S. P., Rodesch C. K., Sundquist W. I. (2007) Human ESCRT and ALIX proteins interact with proteins of the midbody and function in cytokinesis. EMBO J. 26, 4215–4227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jimenez A. J., Maiuri P., Lafaurie-Janvore J., Divoux S., Piel M., Perez F. (2014) ESCRT machinery is required for plasma membrane repair. Science 343, 1247136. [DOI] [PubMed] [Google Scholar]
- 8. Mathivanan S., Simpson R. J. (2009) ExoCarta: A compendium of exosomal proteins and RNA. Proteomics 9, 4997–5000 [DOI] [PubMed] [Google Scholar]
- 9. Théry C., Boussac M., Véron P., Ricciardi-Castagnoli P., Raposo G., Garin J., Amigorena S. (2001) Proteomic analysis of dendritic cell-derived exosomes: a secreted subcellular compartment distinct from apoptotic vesicles. J. Immunol. 166, 7309–7318 [DOI] [PubMed] [Google Scholar]
- 10. Baietti M. F., Zhang Z., Mortier E., Melchior A., Degeest G., Geeraerts A., Ivarsson Y., Depoortere F., Coomans C., Vermeiren E., Zimmermann P., David G. (2012) Syndecan-syntenin-ALIX regulates the biogenesis of exosomes. Nat. Cell Biol. 14, 677–685 [DOI] [PubMed] [Google Scholar]
- 11. Nabhan J. F., Hu R., Oh R. S., Cohen S. N., Lu Q. (2012) Formation and release of arrestin domain-containing protein 1-mediated microvesicles (ARMMs) at plasma membrane by recruitment of TSG101 protein. Proc. Natl. Acad. Sci. U.S.A. 109, 4146–4151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wehman A. M., Poggioli C., Schweinsberg P., Grant B. D., Nance J. (2011) The P4-ATPase TAT-5 inhibits the budding of extracellular vesicles in C. elegans embryos. Curr. Biol. 21, 1951–1959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Webster B. M., Colombi P., Jäger J., Lusk C. P. (2014) Surveillance of nuclear pore complex assembly by ESCRT-III/Vps4. Cell 159, 388–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bieniasz P. D. (2009) The cell biology of HIV-1 virion genesis. Cell Host Microbe 5, 550–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Weiss E. R., Göttlinger H. (2011) The role of cellular factors in promoting HIV budding. J. Mol. Biol. 410, 525–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Votteler J., Sundquist W. I. (2013) Virus budding and the ESCRT pathway. Cell Host Microbe 14, 232–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Guizetti J., Schermelleh L., Mäntler J., Maar S., Poser I., Leonhardt H., Müller-Reichert T., Gerlich D. W. (2011) Cortical constriction during abscission involves helices of ESCRT-III-dependent filaments. Science 331, 1616–1620 [DOI] [PubMed] [Google Scholar]
- 18. Hanson P. I., Roth R., Lin Y., Heuser J. E. (2008) Plasma membrane deformation by circular arrays of ESCRT-III protein filaments. J. Cell Biol. 180, 389–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Baumgärtel V., Ivanchenko S., Dupont A., Sergeev M., Wiseman P. W., Kräusslich H. G., Bräuchle C., Müller B., Lamb D. C. (2011) Live-cell visualization of dynamics of HIV budding site interactions with an ESCRT component. Nat. Cell Biol. 13, 469–474 [DOI] [PubMed] [Google Scholar]
- 20. Elia N., Sougrat R., Spurlin T. A., Hurley J. H., Lippincott-Schwartz J. (2011) Dynamics of endosomal sorting complex required for transport (ESCRT) machinery during cytokinesis and its role in abscission. Proc. Natl. Acad. Sci. U.S.A. 108, 4846–4851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hill C. P., Babst M. (2012) Structure and function of the membrane deformation AAA ATPase Vps4. Biochim. Biophys. Acta 1823, 172–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lata S., Schoehn G., Jain A., Pires R., Piehler J., Gottlinger H. G., Weissenhorn W. (2008) Helical structures of ESCRT-III are disassembled by VPS4. Science 321, 1354–1357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xiao J., Chen X. W., Davies B. A., Saltiel A. R., Katzmann D. J., Xu Z. (2009) Structural basis of Ist1 function and Ist1-Did2 interaction in the multivesicular body pathway and cytokinesis. Mol. Biol. Cell 20, 3514–3524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Muzioł T., Pineda-Molina E., Ravelli R. B., Zamborlini A., Usami Y., Göttlinger H., Weissenhorn W. (2006) Structural basis for budding by the ESCRT-III factor CHMP3. Dev. Cell 10, 821–830 [DOI] [PubMed] [Google Scholar]
- 25. Bajorek M., Schubert H. L., McCullough J., Langelier C., Eckert D. M., Stubblefield W. M., Uter N. T., Myszka D. G., Hill C. P., Sundquist W. I. (2009) Structural basis for ESCRT-III protein autoinhibition. Nat. Struct. Mol. Biol. 16, 754–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Obita T., Saksena S., Ghazi-Tabatabai S., Gill D. J., Perisic O., Emr S. D., Williams R. L. (2007) Structural basis for selective recognition of ESCRT-III by the AAA ATPase Vps4. Nature 449, 735–739 [DOI] [PubMed] [Google Scholar]
- 27. Stuchell-Brereton M. D., Skalicky J. J., Kieffer C., Karren M. A., Ghaffarian S., Sundquist W. I. (2007) ESCRT-III recognition by VPS4 ATPases. Nature 449, 740–744 [DOI] [PubMed] [Google Scholar]
- 28. Scott A., Gaspar J., Stuchell-Brereton M. D., Alam S. L., Skalicky J. J., Sundquist W. I. (2005) Structure and ESCRT-III protein interactions of the MIT domain of human VPS4A. Proc. Natl. Acad. Sci. U.S.A. 102, 13813–13818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kieffer C., Skalicky J. J., Morita E., De Domenico I., Ward D. M., Kaplan J., Sundquist W. I. (2008) Two distinct modes of ESCRT-III recognition are required for VPS4 functions in lysosomal protein targeting and HIV-1 budding. Dev. Cell 15, 62–73 [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 30. Dimaano C., Jones C. B., Hanono A., Curtiss M., Babst M. (2008) Ist1 regulates Vps4 localization and assembly. Mol. Biol. Cell 19, 465–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Samson R. Y., Obita T., Freund S. M., Williams R. L., Bell S. D. (2008) A role for the ESCRT system in cell division in archaea. Science 322, 1710–1713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shestakova A., Hanono A., Drosner S., Curtiss M., Davies B. A., Katzmann D. J., Babst M. (2010) Assembly of the AAA ATPase Vps4 on ESCRT-III. Mol. Biol. Cell 21, 1059–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hartmann C., Chami M., Zachariae U., de Groot B. L., Engel A., Grütter M. G. (2008) Vacuolar protein sorting: two different functional states of the AAA-ATPase Vps4p. J. Mol. Biol. 377, 352–363 [DOI] [PubMed] [Google Scholar]
- 34. Landsberg M. J., Vajjhala P. R., Rothnagel R., Munn A. L., Hankamer B. (2009) Three-dimensional structure of AAA ATPase Vps4: advancing structural insights into the mechanisms of endosomal sorting and enveloped virus budding. Structure 17, 427–437 [DOI] [PubMed] [Google Scholar]
- 35. Yu Z., Gonciarz M. D., Sundquist W. I., Hill C. P., Jensen G. J. (2008) Cryo-EM structure of dodecameric Vps4p and its 2:1 complex with Vta1p. J. Mol. Biol. 377, 364–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Babst M., Wendland B., Estepa E. J., Emr S. D. (1998) The Vps4p AAA ATPase regulates membrane association of a Vps protein complex required for normal endosome function. EMBO J. 17, 2982–2993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Monroe N., Han H., Gonciarz M. D., Eckert D. M., Karren M. A., Whitby F. G., Sundquist W. I., Hill C. P. (2014) The oligomeric state of the active Vps4 AAA ATPase. J. Mol. Biol. 426, 510–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Erzberger J. P., Berger J. M. (2006) Evolutionary relationships and structural mechanisms of AAA+ proteins. Annu. Rev. Biophys. Biomol. Struct. 35, 93–114 [DOI] [PubMed] [Google Scholar]
- 39. Hanson P. I., Whiteheart S. W. (2005) AAA+ proteins: have engine, will work. Nat. Rev. Mol. Cell Biol. 6, 519–529 [DOI] [PubMed] [Google Scholar]
- 40. Scott A., Chung H. Y., Gonciarz-Swiatek M., Hill G. C., Whitby F. G., Gaspar J., Holton J. M., Viswanathan R., Ghaffarian S., Hill C. P., Sundquist W. I. (2005) Structural and mechanistic studies of VPS4 proteins. EMBO J. 24, 3658–3669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gonciarz M. D., Whitby F. G., Eckert D. M., Kieffer C., Heroux A., Sundquist W. I., Hill C. P. (2008) Biochemical and structural studies of yeast Vps4 oligomerization. J. Mol. Biol. 384, 878–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Inoue M., Kamikubo H., Kataoka M., Kato R., Yoshimori T., Wakatsuki S., Kawasaki M. (2008) Nucleotide-dependent conformational changes and assembly of the AAA ATPase SKD1/VPS4B. Traffic 9, 2180–2189 [DOI] [PubMed] [Google Scholar]
- 43. Xiao J., Xia H., Yoshino-Koh K., Zhou J., Xu Z. (2007) Structural characterization of the ATPase reaction cycle of endosomal AAA protein Vps4. J. Mol. Biol. 374, 655–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Dreveny I., Kondo H., Uchiyama K., Shaw A., Zhang X., Freemont P. S. (2004) Structural basis of the interaction between the AAA ATPase p97/VCP and its adaptor protein p47. EMBO J. 23, 1030–1039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lottridge J. M., Flannery A. R., Vincelli J. L., Stevens T. H. (2006) Vta1p and Vps46p regulate the membrane association and ATPase activity of Vps4p at the yeast multivesicular body. Proc. Natl. Acad. Sci. U.S.A. 103, 6202–6207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ward D. M., Vaughn M. B., Shiflett S. L., White P. L., Pollock A. L., Hill J., Schnegelberger R., Sundquist W. I., Kaplan J. (2005) The role of LIP5 and CHMP5 in multivesicular body formation and HIV-1 budding in mammalian cells. J. Biol. Chem. 280, 10548–10555 [DOI] [PubMed] [Google Scholar]
- 47. Yeo S. C., Xu L., Ren J., Boulton V. J., Wagle M. D., Liu C., Ren G., Wong P., Zahn R., Sasajala P., Yang H., Piper R. C., Munn A. L. (2003) Vps20p and Vta1p interact with Vps4p and function in multivesicular body sorting and endosomal transport in Saccharomyces cerevisiae. J. Cell Sci. 116, 3957–3970 [DOI] [PubMed] [Google Scholar]
- 48. Yang D., Hurley J. H. (2010) Structural role of the Vps4-Vta1 interface in ESCRT-III recycling. Structure 18, 976–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Azmi I., Davies B., Dimaano C., Payne J., Eckert D., Babst M., Katzmann D. J. (2006) Recycling of ESCRTs by the AAA-ATPase Vps4 is regulated by a conserved VSL region in Vta1. J. Cell Biol. 172, 705–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Roll-Mecak A., Vale R. D. (2008) Structural basis of microtubule severing by the hereditary spastic paraplegia protein spastin. Nature 451, 363–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. White S. R., Evans K. J., Lary J., Cole J. L., Lauring B. (2007) Recognition of C-terminal amino acids in tubulin by pore loops in Spastin is important for microtubule severing. J. Cell Biol. 176, 995–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Shim S., Merrill S. A., Hanson P. I. (2008) Novel interactions of ESCRT-III with LIP5 and VPS4 and their implications for ESCRT-III disassembly. Mol. Biol. Cell 19, 2661–2672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Merrill S. A., Hanson P. I. (2010) Activation of human VPS4A by ESCRT-III proteins reveals ability of substrates to relieve enzyme autoinhibition. J. Biol. Chem. 285, 35428–35438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Martin A., Baker T. A., Sauer R. T. (2008) Pore loops of the AAA+ ClpX machine grip substrates to drive translocation and unfolding. Nat. Struct. Mol. Biol. 15, 1147–1151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chan W. C., White P. D. (eds) (2000) Fmoc Solid Phase Peptide Synthesis: A Practical Approach, Oxford University Press, New York [Google Scholar]
- 56. Braig K., Menz R. I., Montgomery M. G., Leslie A. G., Walker J. E. (2000) Structure of bovine mitochondrial F1-ATPase inhibited by Mg2+ ADP and aluminium fluoride. Structure 8, 567–573 [DOI] [PubMed] [Google Scholar]
- 57. Kagawa R., Montgomery M. G., Braig K., Leslie A. G., Walker J. E. (2004) The structure of bovine F1-ATPase inhibited by ADP and beryllium fluoride. EMBO J. 23, 2734–2744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sysoeva T. A., Chowdhury S., Guo L., Nixon B. T. (2013) Nucleotide-induced asymmetry within ATPase activator ring drives σ54-RNAP interaction and ATP hydrolysis. Genes Dev. 27, 2500–2511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Thomsen N. D., Berger J. M. (2009) Running in reverse: the structural basis for translocation polarity in hexameric helicases. Cell 139, 523–534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Vild C. J., Xu Z. (2014) Vfa1 binds to the N-terminal microtubule-interacting and trafficking (MIT) domain of Vps4 and stimulates its ATPase activity. J. Biol. Chem. 289, 10378–10386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Azmi I. F., Davies B. A., Xiao J., Babst M., Xu Z., Katzmann D. J. (2008) ESCRT-III family members stimulate Vps4 ATPase activity directly or via Vta1. Dev. Cell 14, 50–61 [DOI] [PubMed] [Google Scholar]
- 62. Shestakova A., Curtiss M., Davies B. A., Katzmann D. J., Babst M. (2013) The linker region plays a regulatory role in assembly and activity of the Vps4 AAA ATPase. J. Biol. Chem. 288, 26810–26819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Abrahams J. P., Leslie A. G., Lutter R., Walker J. E. (1994) Structure at 2.8 Ä resolution of F1-ATPase from bovine heart mitochondria. Nature 370, 621–628 [DOI] [PubMed] [Google Scholar]
- 64. Enemark E. J., Joshua-Tor L. (2006) Mechanism of DNA translocation in a replicative hexameric helicase. Nature 442, 270–275 [DOI] [PubMed] [Google Scholar]
- 65. Matyskiela M. E., Martin A. (2013) Design principles of a universal protein degradation machine. J. Mol. Biol. 425, 199–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Förster F., Unverdorben P., Sledź P., Baumeister W. (2013) Unveiling the long-held secrets of the 26S proteasome. Structure 21, 1551–1562 [DOI] [PubMed] [Google Scholar]
- 67. Zhao M., Wu S., Zhou Q., Vivona S., Cipriano D. J., Cheng Y., Brunger A. T. (2015) Mechanistic insights into the recycling machine of the SNARE complex. Nature 518, 61–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Chaney M., Grande R., Wigneshweraraj S. R., Cannon W., Casaz P., Gallegos M. T., Schumacher J., Jones S., Elderkin S., Dago A. E., Morett E., Buck M. (2001) Binding of transcriptional activators to σ54 in the presence of the transition state analog ADP-aluminum fluoride: insights into activator mechanochemical action. Genes Dev. 15, 2282–2294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Rape M., Jentsch S. (2002) Taking a bite: proteasomal protein processing. Nat. Cell Biol. 4, E113–116 [DOI] [PubMed] [Google Scholar]
- 70. Lee C., Prakash S., Matouschek A. (2002) Concurrent translocation of multiple polypeptide chains through the proteasomal degradation channel. J. Biol. Chem. 277, 34760–34765 [DOI] [PubMed] [Google Scholar]
- 71. Cormier C. Y., Park J. G., Fiacco M., Steel J., Hunter P., Kramer J., Singla R., LaBaer J. (2011) PSI:Biology-materials repository: a biologist's resource for protein expression plasmids. J. Struct. Funct. Genomics 12, 55–62 [DOI] [PMC free article] [PubMed] [Google Scholar]