

Background: IL-31RA is a novel Type I cytokine receptor that pairs with oncostatin M receptor to mediate IL-31 signaling.
Results: Th2 cytokines up-regulate IL-31RA signaling in macrophages.
Conclusion: We demonstrate that the Th2 cytokines IL-4 and IL-13 were capable of up-regulating IL-31RA expression on macrophages.
Significance: This newly identified counter-regulatory role between Th2 cytokine and the IL-31 signaling cascade may provide valuable insights into the pathobiology of allergic diseases.
Keywords: asthma, cytokine, inflammation, macrophage, signaling, interleukin-31, STAT6, Th2 cytokines, allergic asthma
Abstract
Interleukin 31 receptor α (IL-31RA) is a novel Type I cytokine receptor that pairs with oncostatin M receptor to mediate IL-31 signaling. Binding of IL-31 to its receptor results in the phosphorylation and activation of STATs, MAPK, and JNK signaling pathways. IL-31 plays a pathogenic role in tissue inflammation, particularly in allergic diseases. Recent studies demonstrate IL-31RA expression and signaling in non-hematopoietic cells, but this receptor is poorly studied in immune cells. Macrophages are key immune-effector cells that play a critical role in Th2-cytokine-mediated allergic diseases. Here, we demonstrate that Th2 cytokines IL-4 and IL-13 are capable of up-regulating IL-31RA expression on both peritoneal and bone marrow-derived macrophages from mice. Our data also demonstrate that IL-4Rα-driven IL-31RA expression is STAT6 dependent in macrophages. Notably, the inflammation-associated genes Fizz1 and serum amyloid A (SAA) are significantly up-regulated in M2 macrophages stimulated with IL-31, but not in IL-4 receptor-deficient macrophages. Furthermore, the absence of Type II IL-4 receptor signaling is sufficient to attenuate the expression of IL-31RA in vivo during allergic asthma induced by soluble egg antigen, which may suggest a role for IL-31 signaling in Th2 cytokine-driven inflammation and allergic responses. Our study reveals an important counter-regulatory role between Th2 cytokine and IL-31 signaling involved in allergic diseases.
Introduction
IL-31 is a cytokine with a predicted arrangement consisting of a four-helix bundle structure and based on homology, it is a member of the IL-6 family of cytokines (1). IL-31 signals through a heterodimeric receptor complex of interleukin 31 receptor α (IL-31RA)3 and oncostatin M receptor β (OSMRβ) subunits. IL-31 is the only known ligand for IL-31RA, which belongs to the large family of glycoprotein 130 (gp130) receptors (2). In the mouse, IL-31RA transcripts exist in two different isoforms: IL-31RAv1 and IL-31RAv2 (1). IL-31RAv1 is homologous to human IL-31RAv4 and is expressed as a membrane-bound form, whereas IL-31RAv2 is a soluble form consisting of a cytokine-binding domain and fibronectin type III domains (1, 2). Both the membrane-bound and soluble forms of the receptor bind to IL-31 with comparable affinity and initiate intracellular signal transduction resulting in secretion of several pro-inflammatory cytokines and chemokines, which play a critical role in Th2 cytokine-mediated inflammatory diseases (3, 4). Although the expression and distribution of IL-31RA in different cells or tissues have not been well studied, IL-31RA is constitutively expressed by non-hematopoietic cells, such as lung epithelial cells, and also exhibits limited expression on activated monocytes and eosinophils (1, 5, 6). Recently, it has been suggested that IL-31 is selectively produced by activated CD4+ T cells, skewed toward a Th2-type cytokine profile (5). Human eosinophils and epidermal keratinocytes also have been shown to express IL-31RA and produce pro-inflammatory cytokines in response to IL-31 (6).
The transcripts and protein levels of IL-31 and the number of Th2 cells that secrete IL-31 are elevated in various skin diseases including atopic dermatitis, atopic contact dermatitis, and purigo nodularis (7–9). IL-31 is thought to play an important role in the induction of pruritus, which is a characteristic feature of these skin diseases (10). Notably, IL-31 expression has been shown to correlate with the expression of Th2 cytokines IL-4 and IL-13 in human skin diseases (11). In support of this, overexpression of IL-31 in transgenic mice results in a severe pruritus, alopecia, and skin lesions (1). Moreover, neutralization of IL-31 has been shown to ameliorate scratching behavior in a mouse model of atopic dermatitis (12). Recent studies suggest that IL-31 signaling is required to limit Th2 cytokine-driven inflammation in the lung and that a complete genetic loss of IL-31RA might result in increased activity and signaling by oncostatin M (OSM) via OSMRβ (13). These reports strongly support a need to investigate the molecular mechanisms that regulate IL-31/IL-31RA interactions involved in inflammation and tissue remodeling.
Macrophages are key regulators of the inflammatory response in tissues and exhibit diversity in phenotypes and plasticity based on tissue environment and cytokine stimuli (14). The Th1 cytokine IFN-γ polarizes macrophages toward classically activated macrophages (M1 phenotype) with an induced expression of iNOS. In contrast, Th2 cytokines IL-4 and IL-13 skew macrophages toward alternatively activated macrophages (M2 phenotype) with expression of Ym1, Arg1, and Fizz1 (15, 16). These macrophage phenotypes are responsible for effective immune responses in tissues to target antigens or pathogens. However, downstream mediators of different macrophage phenotypes involved in allergic diseases are largely unknown (17). A recent study showed that IL-31 stimulation of lung epithelial cells along with Th2 cytokines IL-4 and IL-13 induces the expression of epidermal growth factors, vascular endothelial growth factors, and chemokines, all of which are known to contribute to bronchial inflammation (18). However, mechanisms that explain the cross-talk between the Th2 cytokine and IL-31 pathways during inflammatory and allergic responses are poorly defined. In the current study, we explored the immunoregulatory role of Th2 cytokine in altering IL-31/IL-31RA interactions and signaling involved in inflammation and tissue remodeling. Our findings demonstrate that IL-4 and IL-13 increase the expression of IL-31RA and alter the IL-31 signaling involved in allergic responses.
Materials and Methods
Mice
C57BL/6 (Jackson Laboratories, Bar Harbor, ME), Balb/C (Jackson Laboratories), and IL-4Rα knock-out (KO) (C57BL/6 background, inbred) mice at 8–14 weeks of age were used for all of the experiments. IL-13Rα1−/− and IL-13Rα2−/− mice (both on a C57BL/6 background >10 generations) were obtained from the NIAID-Taconic contract. Mice were housed in either the Cincinnati Children's Hospital Medical Center animal facility or at the National Institutes of Health in facilities approved by the American Association for the Accreditation of Laboratory Animal Care. All mice were maintained under aseptic conditions and received sterile food and water. Experiments were performed following IACUC regulations.
Macrophage Isolations and Treatments
A 4% thioglycollate medium (BD Bioscience, Sparks, MD) was prepared in a sterile saline solution and 3 ml was injected intraperitoneally into each mouse. On day 5 after injection, mice were sacrificed and peritoneal macrophages were harvested with 5 ml of phosphate-buffered saline (PBS) wash containing 2% fetal bovine serum (FBS) (Atlanta Biologicals, Flowery Branch, GA). Cells were washed twice with RPMI 1640 medium (GibcoTM, Life TechnologiesTM, ThermoFisher Scientific, Waltham, MA) containing 5% FBS and 1% each of penicillin and streptomycin (Gibco) and cultured overnight in 12-well plates. Non-adherent cells were removed and the medium was replaced with fresh complete RPMI 1640 medium.
Bone marrow (BM)-derived macrophages were generated from mouse BM cells isolated from the tibia and femur by flushing the bones with sterile PBS. Red blood cells (RBC) were lysed with RBC lysis buffer (Gibco). Cells were re-suspended and plated with 15 ng/ml of M-CSF (PeproTech, Rocky Hill, NJ) for 7 days with a one-time medium change on day 3 (19). Macrophages with M1 and M2 phenotype were generated by stimulating cells for 48 h with IFN-γ and IL-13 and/or IL-4 (R&D Systems, Minneapolis, MN), respectively. To study IL-31 signaling, macrophages were stimulated with recombinant IL-31 at 200 ng/ml for 12 h and measured for IL-31-dependent RNA transcripts.
Animal Model of SEA-induced Allergic Asthma
Allergic asthma in animals was induced according to a previously published method (20). In brief, mice were sensitized twice (days 0 and 14) by intraperitoneal injection of 10 μg of soluble egg antigen (SEA) extracted from Schistosoma mansoni parasitic eggs. On days 28 and 31 mice were anesthetized with a mixture of xylazine and ketamine and given an intratracheal airway challenge with 10 μg of SEA. Mice were sacrificed 24 h after the final airway challenge (day 32), and lungs were collected in RNAlater solution (Applied BiosystemsTM, Life TechnologiesTM, ThermoFisher Scientific) and stored at −80 ºC until use.
RNA Isolation, cDNA Synthesis, and Quantitative PCR
RNA was isolated using the RNeasy kit (Qiagen Sciences, Valencia, CA) as described previously (21). A total of 1 μg of RNA was used for cDNA synthesis, and gene expression was measured using the StepOnePlusTM sequence detection system (Applied Biosystems). Relative gene expression was quantified using SYBR® Green PCR Master Mix or TaqMan® assay (Applied Biosystems), and gene expression was normalized to hypoxanthine-guanine phosphoribosyltransferase (HPRT) or 18S RNA. The data were analyzed with StepOnePlusTM software 2.1 (Applied Biosystems) as described by the manufacturer. The mouse probes and primers used in this study are shown in Tables 1 and 2.
TABLE 1.
Mouse probes and primers used for RT-PCR
| Probes | |
|---|---|
| 18S rRNA (TaqMan® probe) | Applied Biosystems® Life Technologies |
| IL-31RA (TaqMan® probe) | Applied Biosystems® Life Technologies (custom designed) |
| OSMR-β (TaqMan® probe) | Applied Biosystems® Life Technologies |
| Fizz1 (TaqMan® probe) | Applied Biosystems® Life Technologies |
TABLE 2.
Primers used for RT-PCR
| Primer | Forward | Reverse |
|---|---|---|
| HPRT | GCCCTTGACTATAATGAGTACTTCAGG | TTCAACTTGCGCTCATCTTAGG |
| OSM | TGCTCCAACTCTTCCTCTCAG | CAGGTTTTGGAGGCGGATA |
| gp130 | TCCCATGGGCAGGAATATAG | CCATTGGCTTCAGAAAGAGG |
| Arg1 | ACAGTCTGGCAGTTGGAAGCATC | GGGAGTCCCCAGGAGAATCCT |
| Ym1 | CATGAGCAAGACTTGCGTGAC | GGTCCAAACTTCCATCCTCCA |
| Fizz1 | CCCTCCACTGTAACGAAGACTC | CACACCCAGTAGCAGTCATCC |
| SAA | CTGCCTGCCAAATACTGAGAGTC | CCACTTCCAAGTTCCTGTTTATTA |
Chromatin Immunoprecipitation (ChIP)
ChIP was performed using the ChIP assay kit (Millipore, Temecula, CA). Briefly, 1 × 107 cells were grown on 100-mm dishes and stimulated with IL-4 (10 ng/ml) for 2 h. Cells were cross-linked for 10 min with 1% formaldehyde at 37 °C. Glycine was then added to a final concentration of 0.125 m to quench unreacted formaldehyde. Cells were washed twice with 1× ice-cold PBS containing protease inhibitors and resuspended in 0.5 ml of SDS lysis buffer. DNA was sheared by cycles of 10 s of sonication followed by 10 s of rest for 20 min using a Bioruptor sonication system (Diagenode, Denville, NJ). The samples were centrifuged at 13,000 rpm at 4 °C, and the cleared supernatants were diluted 10 times with ChIP dilution buffer. 2% of the diluted supernatant was stored at −80 °C as a total input control and eluted in ChIP elution buffer for the reverse cross-linking step. The remaining diluted supernatant was pre-cleared with 75 μl of protein A agarose/salmon sperm DNA for 30 min at 4 °C with agitation collected by a brief centrifugation at 700 rpm for 1 min. For the immunoprecipitation, anti-signal transducer and activator of transcription 6 (STAT6) antibodies (Cell Signaling, Danvers, MA) at 1:50 dilution and an equal concentration of rabbit IgG (Cell Signaling) were used as isotype control. Immunoprecipitation was performed overnight at 4 °C with rotation. After incubation, the samples were added 60 μl of protein A-agarose/salmon sperm DNA for 2 h at 4 °C with rotation and centrifuged at 700 rpm for 1 min. The protein A-agarose-antibody-histone complex was washed consecutively for 3–5 min on a rotating platform with 1 ml of each solution: (a) low salt wash buffer, one wash, (b) high salt wash buffer, two washes, (c) LiCl immune complex wash buffer, one wash, and (d) TE buffer, two washes. The histone complex was eluted by freshly prepared elution buffer (1% SDS and 0.1 m NaHCO3). Following ChIP, reverse cross-linking was performed by addition of 5 m NaCl in eluted DNA and input samples and heated overnight to 65 °C. After incubation, 10 μl of 0.5 m EDTA, 20 μl of 1 m Tris-HCl, pH 6.5, and 2 μl of proteinase K (10 mg/ml) was added to the combined eluates and incubated for 1 h at 45 °C. DNA was purified using a phenol/chloroform method and dissolved in water. PCR was performed in 25 μl of PCR mixture using 1 μl of DNA in a Veriti Thermal Cycler (Applied Biosystems). The primer sequences, annealing temperatures, and PCR product sizes are provided in Table 3. The PCR conditions were 94 °C for 1 min, followed by 37 cycles: denaturation at 94 °C for 30 s, annealing for 20 s, and extension at 72 °C for 1 min. A final extension was performed at 72 °C for 5 min. The PCR product was run electrophoretically on a 2.0% agarose gel with ethidium bromide using 1× TAE buffer (Fisher ScientificTM, ThermoFisher Scientific) and resulting fragments were visualized using a UV transilluminator (FotoDyne, Hartland, WI).
TABLE 3.
The sequences of the primer sets used for ChIP Seq
| Location of STAT6 relative to ATG | Sequence of primers | Annealing temperatures | PCR product sizes |
|---|---|---|---|
| Site 1 (−456) | F-ATAATGGGAAGGGGCTCTGG | 62 °C | 216 bp |
| R-CCCACTCACAATGCACACTC | |||
| Site 2 (−5694) | F-TTTGCTTATCTATGAAGTGGAGGT | 58 °C | 200 bp |
| R-CCAGATATTTGCCTTCGGTTTC | |||
| Site 3 (−8991) | F-AAGTGTGCATCGCCTTCA | 58 °C | 225 bp |
| R-TCCCTACACCTAGGATTAGAACA | |||
| Sites 4 and 5 (−9300 and −9334) | F-GGGCTGAAAGACCCATTAGATAG | 58 °C | 212 bp |
| R-GGTCTAGCTGTAATAATGGCAGAA |
Flow Cytometry
Thioglycollate-induced peritoneal macrophages were generated from C57BL/6 and IL-4Rα mice and plated overnight at 37 ºC and 5% CO2 in 12-well tissue culture plates (BD Biosciences). Non-adherent cells were removed, and the medium was replaced with fresh RPMI 1640 medium containing 5% FBS with penicillin and streptomycin and with or without rIL-13 for 24 or 48 h. Cells were dissociated with TripLE Express buffer (Gibco) and stained using a LIVE/DEAD® Fixable Aqua Dead-Cell Stain Kit (Molecular ProbesTM, Life TechnologiesTM, ThermoFisher Scientific). Stained cells were incubated in Fc receptor-blocking medium for 10 min on ice followed by rat anti-mouse IL-31RA antibody for 30 min on ice. Excess antibody was removed by washing the cells with FACS buffer (0.5% BSA in PBS), followed by the addition of a secondary antibody, anti-rat Alexa Fluor® 594 (Molecular Probes), and then cells were washed twice with FACS buffer. Cells were acquired using a BDTM LSRII flow cytometer (BD Biosciences), and only live cell-gated data were analyzed using FlowJo software (Tree Star, Ashland, OR).
Immunofluorescence
Peritoneal macrophages were plated overnight on polylysine-coated coverslips (Sigma). Non-adherent cells were removed and plated with fresh RPMI 1640 medium containing 5% FBS and 1% each of penicillin and streptomycin. Cells were stimulated with IL-4 (10 ng/ml) and IL-13 (20 ng/ml) and incubated at 37 ºC and 5% CO2 for either 12 or 24 h. Cells were fixed with 4% paraformaldehyde for 20 min and washed three times with PBS. Fixed cells were permeabilized with 0.01% of TritonTM X-100 (Sigma) for 5 min and washed with PBS. Cells were then treated with anti-mouse IL-31RA antibody (Bristol-Myers Squibb, Seattle, WA) and a co-localization membrane lipid-raft marker, cholera toxin subunit B conjugated to Alexa Fluor® 594 antibody (Molecular Probes) for 1 h, followed by three washes with PBS. Cells were stained with Alexa Fluor® 594 or Alexa Fluor® 488-conjugated secondary antibody (Molecular Probes) for 30 min, nuclei were stained with DAPI for 5 min, and mounted onto microscope slides with mounting medium. Staining was visualized using a Nikon A1Rsi inverted confocal microscope (Nikon Instruments Inc., Melville, NY), and data were analyzed with Imaris software (Bitplane AG, Zurich, Switzerland).
Statistical Analysis
The data were analyzed using GraphPad Prism (version 6; GraphPad, La Jolla, CA). An unpaired Student's t test was used for comparing between two groups. One-way analysis of variance with Tukey's multiple comparison test was used for comparison of different experimental groups. p values less than 0.05 were considered statistically significant.
Results
IL-4 and IL-13 Up-regulate IL-31RA Expression in Macrophages
To investigate the role of Th2 cytokines in regulating the expression of IL-31RA and OSMRβ, we isolated thioglycollate-induced peritoneal macrophages from C57BL/6 mice stimulated with IL-4 and IL-13. Both of the Th2 cytokines were capable of up-regulating IL-31RA transcripts in a dose-dependent manner, compared with media-treated macrophages (Fig. 1, A and B). However, IL-4-stimulated macrophages had significantly higher expression of IL-31RA transcripts compared with IL-13-stimulated macrophages. We also studied the time kinetics in IL-4-stimulated peritoneal macrophages. IL-31RA expression was significantly up-regulated by IL-4 in time-dependent manner (Fig. 1C). Notably, no significant difference was observed in OSMRβ transcripts when stimulated with either IL-4 or IL-13 (Fig. 1D). These data suggest that Th2 cytokines are involved in IL-31RA expression, but not OSMRβ expression. To investigate whether the expression of IL-31RA was restricted to only thioglycollate-induced macrophages or also occurred in other macrophages, we studied the effect of IL-4 and IL-13 cytokines on IL-31RA expression using bone marrow-derived macrophages (BMDM). Bone marrow cells were isolated from wild-type (WT) mice and differentiated into macrophages with M-CSF according to standard protocols (19). On day 6, BMDM were stimulated with either IL-4 or IL-13. As observed for peritoneal macrophages, Th2 cytokines IL-4 and IL-13 significantly increased IL-31RA expression in BMDM (Fig. 1E).
FIGURE 1.

Th2 cytokines IL-4 and IL-13 signal through IL-4Rα and enhance IL-31RA expression. A, IL-31RA expression was measured by quantitative RT-PCR in peritoneal macrophages of C57BL/6 wild-type mice stimulated with the indicated concentrations of IL-4 for 24 h. B, IL-31RA expression was measured by quantitative RT-PCR in peritoneal macrophages of C57BL/6 wild-type mice stimulated with the indicated concentrations of IL-13 for 24 h. C, IL-31RA expression was measured by quantitative RT-PCR in peritoneal macrophages of wild-type mice treated with IL-4 (10 ng/ml) for the indicated time points. D, OSMRβ expression was measured by quantitative RT-PCR in peritoneal macrophages of C57BL/6 wild-type mice stimulated with IL-4 (20 ng/ml) and IL-13 (20 ng/ml) for 24 h. E, IL-31RA expression was measured by quantitative RT-PCR in bone marrow-derived macrophages of C57BL/6 wild-type mice stimulated with IL-4 (20 ng/ml) and IL-13 (20 ng/ml) for 24 h. F, peritoneal macrophages from wild-type mice were cultured with IL-4 (20 ng/ml) and IL-13 (20 ng/ml) along with anti-4Rα and anti-2RγC for 24 h, and IL-31RA expression was analyzed by quantitative RT-PCR. G, wild-type and IL-13Rα1−/− macrophages were cultured with IL-4 (20 ng/ml) and IL-13 (20 ng/ml), and mRNA level of IL-31RA expression was measured. Data are representative of three independent experiments and are expressed as mean ± S.E. *, p < 0.05; ***, p < 0.001; ****, p < 0.0001.
Macrophages express both Type I and Type II Th2 cytokine receptors involved in signaling for IL-4 and IL-13 (15). To determine the role of different Th2 cytokine receptors in IL-31RA expression, macrophages were treated with IL-4 or IL-13 in the presence or absence of neutralizing antibodies to IL-4Rα or IL-2RγC that can selectively block signaling receptors for IL-4 and IL-13 (21). IL-4, but not IL-13 signals via the Type I IL-4 receptor, which is a heterodimeric complex comprising IL-4Rα and IL-2RγC. Blockade of Type I IL-4 receptor with anti-IL-2RγC attenuated IL-4-driven IL-31RA expression (Fig. 1F). Both IL-4 and IL-13 bind and signal through Type II IL-4 receptor, which is a heterodimeric receptor complex formed by IL-4Rα and IL-13Rα1. Notably, neutralizing antibodies to IL-4Rα reduced both IL-4- and IL-13-induced expression of IL-31RA (Fig. 1F). These data suggest that IL-4Rα signaling is indispensable for IL-4/IL-13-induced IL-31RA expression. To further identify IL-13Rα1 receptor signaling in IL-4/IL-13-induced IL-31RA expression, we measured IL-31RA transcripts in peritoneal macrophages of IL-13Rα1 KO mice treated with IL-4 and IL-13. Consistent with our other findings, the absence of IL-13Rα1 attenuated IL-13-driven IL-31RA expression (Fig. 1G). Conversely, the absence of IL-13Rα1 augmented the expression of IL-31RA in IL-13Rα1-deficient macrophages compared with WT cells treated with IL-4. These findings suggest that the presence of IL-13Rα1 receptor (Type II IL-4 receptor) antagonizes Type I IL-4 receptor-driven IL-31RA expression in macrophages (Fig. 1G).
IL-4Rα-driven IL-31RA Expression is STAT6-dependent
STAT6 is a major transcription factor involved in the expression of IL-4/IL-13-driven genes (22, 23). To confirm whether IL-4Rα-mediated STAT6 signaling is necessary for IL-31RA expression, peritoneal macrophages of WT and IL-4Rα or STAT6 KO mice were treated with IL-13. Notably, WT macrophages had a severalfold increase in IL-31RA expression; however, no significant changes were observed in the IL-13-induced IL-31RA expression in either IL-4Rα KO or STAT6 KO macrophages compared with media-treated controls (Fig. 2, A and B). We also investigated whether IL-31 regulates its own receptor in an autocrine fashion by measuring IL-31RA expression in IL-13- or IL-13/IL-31-stimulated macrophages, and found that IL-13 and IL-31 in combination had no further effect on IL-31RA expression compared with IL-13 alone (supplemental Fig. S1). These results suggest that IL-4Rα-mediated STAT6 signaling is crucial for IL-31RA expression.
FIGURE 2.
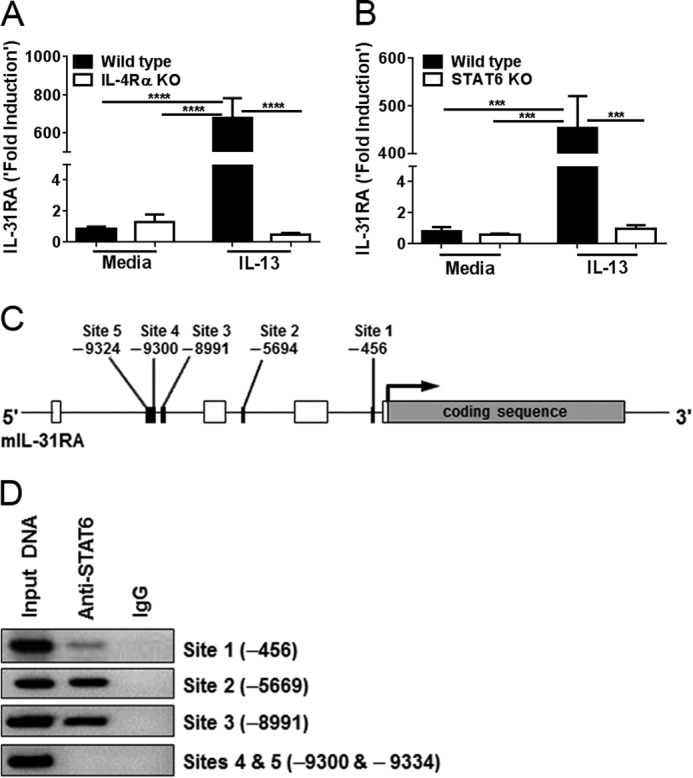
IL-4Rα-mediated STAT6 signaling is critical for IL-13-induced IL-31RA expression. A, peritoneal macrophages from wild-type C57BL/6 and IL-4Rα KO mice were stimulated with IL-13 (10 ng/ml) for 24 h, and IL-31RA expression was measured by quantitative RT-PCR. Data are representative of two independent experiments and expressed as mean ± S.E. ***, p < 0.001. B, peritoneal macrophages from wild-type and STAT6 KO mice were stimulated with IL-13, and IL-31RA transcripts were measured by quantitative RT-PCR. Data are representative of two independent experiments and expressed as mean ± S.E. ***, p < 0.001. C, schematic representation of the mouse IL-31RA gene showing the location of the putative STAT6 binding sites. Open boxes indicate untranslated regions followed by coding sequence (exon). Sequences of STAT6 are represented by black boxes and their respective binding sites are indicated. D, peritoneal macrophages from wild-type mice were cultured with IL-4 (10 ng/ml) for 2 h. Cell lysates were prepared, and the ChIP assay was performed with PCR using anti-STAT6 antibody and normal rabbit IgG as a negative control. Non-immunoprecipitated DNA is represented as Input DNA. Data are representative of two independent experiments.
To determine the location of Th2 cytokine-induced STAT6 in the mouse IL-31RA gene, we performed a computational analysis of the sequence, which revealed the presence of several STAT6 binding sites in the promoter region of the gene (Fig. 2C). To further validate the binding capacity of the STAT6 motifs identified in the mouse IL-31RA promoter, peritoneal macrophages from WT mice were stimulated with IL-4 (10 ng/ml) for 2 h, and ChIP assays were performed. ChIP analysis using STAT6-specific antibodies revealed that the transcription factor bound more efficiently to Site 2 (−5669) and Site 3 (−8991) as compared with Site 1 (−456) of the IL-31RA promoter (Fig. 2D), suggesting that Sites 2 and 3 are more relevant for IL-4-mediated IL-31RA expression. In contrast, no signal was observed with primers designed for Sites 4 and 5 (−9300 and −9334) of the IL-31RA promoter, suggesting that these sites may play a limited role in IL-4-mediated IL-31RA expression. Together, these data confirm that IL-4-induced signaling for IL-31RA expression was mediated by binding of the STAT6 transcription factor to Sites 2 and 3 of the IL-31RA promoter.
Surface Expression of IL-31RA on Macrophages
IL-31RA is expressed as two different variants (IL-31RAv1 and IL-31RAv2) and IL-31RAv2 is a soluble form of IL-31RA that can decoy the signaling of IL-31 (2). To assess whether the membrane-bound form of IL-31RA protein (IL-31RAv1) was being expressed in our experiments, we developed a flow cytometry method to detect the membrane-bound form of IL-31RA using a mouse anti-IL-31RA antibody. Peritoneal macrophages isolated from WT and IL-4Rα KO mice were stimulated with IL-13 for 12 and 24 h and stained for IL-31RA. Significantly increased expression of the membrane-bound form of IL-31RA was observed in IL-13-induced WT macrophages; however, no difference was observed in IL-4Rα KO macrophages (Fig. 3, A and B). These results are consistent with the level of IL-31RA transcripts measured using RT-PCR. Furthermore, macrophages stained for IL-31RA also were used to obtain immunofluorescence images by fluorescence microscopy. IL-13-stimulated macrophages had increased expression of IL-31RA protein at 12 and 24 h compared with unstimulated macrophages (Fig. 3C), suggesting that Th2-cytokines up-regulate the membrane form of IL-31RA, which was absent in IL-4Rα KO macrophages. Notably, a similar expression of IL-31RA along with co-localization with a membrane lipid-raft marker, cholera toxin, was observed in IL-4-stimulated peritoneal macrophages (Fig. 3D). To further determine IL-31RA expression on macrophages with M1 or M2 phenotypes, we stimulated macrophages with IL-13 and IFN-γ individually and also in combination. Not surprisingly, macrophages transformed into M1 or M2 phenotypes, expressing iNOS or Arg1/Ym1, respectively (supplemental Fig. S2). IFN-γ stimulation that induced an M1 phenotype had no effect on IL-31RA expression; however, the presence of IFN-γ attenuated IL-13-driven IL-31RA expression on both peritoneal macrophages and BMDM (Fig. 4, A and B).
FIGURE 3.

IL-13 up-regulates a membrane-bound form of IL-31RA. Macrophages from wild-type and IL-4Rα KO mice were treated with media or IL-13 for 24 h, stained using anti-mouse IL-31RA antibody, and analyzed by flow cytometry. A, a shaded histogram plot indicates the unstained isotype control, and an open histogram indicates wild-type cells stained with anti-mouse IL-31RA antibody. B, mean fluorescence intensity (MFI) of IL-31RA expression in wild-type and IL-4Rα KO macrophages. Data are representative of three independent experiments and are expressed as mean ± S.E. *, p < 0.05. C, immunofluorescence of peritoneal macrophages for IL-31RA expression. Macrophages were plated on polylysine-coated coverslips overnight and stimulated with IL-13 for 12 and 24 h. Cells were fixed in 4% paraformaldehyde and stained with mouse anti-IL-31RA antibody. Nucleus, blue; IL-31RA, red. Scale bar: 100 μm. D, immunofluorescence of peritoneal macrophages for IL-31RA expression stimulated with IL-4 for 24 h, using anti-mouse IL-31RA antibody along with a co-localization membrane lipid-raft marker, cholera toxin subunit B. Nucleus, blue; IL-31RA, green; cholera toxin, red. Scale bar: 50 μm.
FIGURE 4.
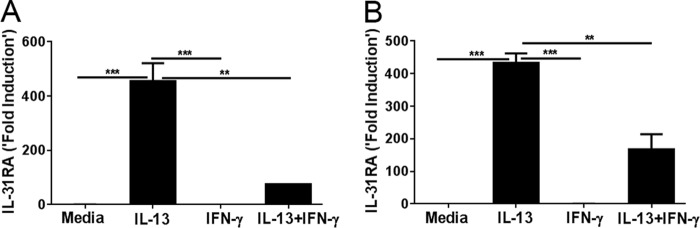
The role of IFN-γ on IL-13-induced IL-31RA expression. Macrophages were stimulated with IL-13 and IFN-γ, independently or in combination, and IL-31RA gene expression was measured in peritoneal macrophages (A) and bone-marrow-derived macrophages (B). Data are representative of three independent experiments and expressed as mean ± S.E. **, p < 0.01; ***, p < 0.001.
Th2 Cytokines Augment IL-31RA-driven Signaling
To study the effects of Th2-cytokine-induced IL-31RA expression on IL-31 signaling, we pre-treated peritoneal macrophages from WT and IL-4Rα KO mice with both IL-4 and IL-13 for 48 h and stimulated cells with media or IL-31 alone for 12 h. Macrophages treated with IL-31 expressed more inflammatory-response proteins, in particular Fizz1 (found in inflammatory zone) and SAA, compared with cells stimulated by media; however, no difference was observed in macrophages isolated from IL-4Rα KO mice (Fig. 5, A and B). Furthermore, no increase in SAA expression was observed in IL-31-treated macrophages deficient for IL-31RA (supplemental Fig. S3). These results suggest that IL-4Rα signaling is critical for IL-31-induced Fizz1 and SAA expression in macrophages.
FIGURE 5.
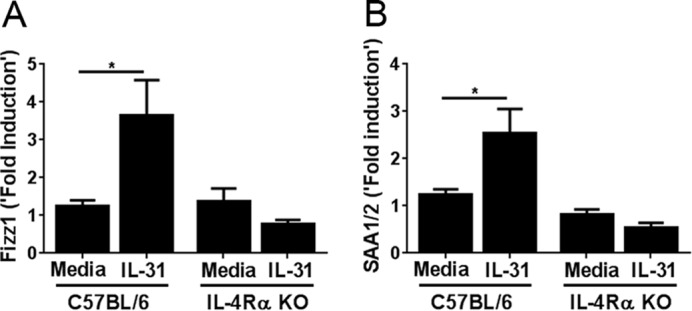
IL-31 induces Fizz1 and SAA expression. Peritoneal macrophages from wild-type and IL-4Rα mice were cultured with IL-4 (20 ng/ml) and IL-13 (20 ng/ml) for 48 h. Cells were induced with IL-31 (200 ng/ml) for 12 h, and expression of Fizz1 (A) and SAA (B) were measured. Data are representative of four independent experiments and expressed as mean ± S.E. *, p < 0.05.
Altered IL-31RA Expression in Vivo during SEA-induced Allergic Disease
The SEA of S. mansoni has been shown to induce strong Th2 responses. Previously, we have demonstrated that SEA acts as a potent allergen and induces Th2-cytokine-driven allergic inflammation and airway hyper-responsiveness in a mouse model of allergic asthma (20, 24). To identify the effect of Type II Th2 cytokine receptors on IL-31RA expression in vivo during allergic lung disease, we studied IL-31RA expression in WT and IL-13Rα2−/− mice exposed to SEA. As shown in Fig. 6A, mice were sensitized and challenged twice with either SEA or PBS as a control. As described previously, SEA-induced allergic disease requires no adjuvant and is sufficient to induce inflammation and hyper-responsiveness in airways (20, 24). PBS- or SEA-challenged lungs were collected at 24 h after the final challenge. Mice exposed to SEA showed a significant increase in the expression of IL-31RA compared with mice exposed to PBS (Fig. 6B). Notably, the increase in IL-31RA transcripts during SEA-induced allergic disease was attenuated in mice deficient for Type II IL-4 receptors. Conversely, an increase in IL-31RA expression was observed in IL-13Rα2−/− mice during SEA-induced allergic disease (Fig. 6B). OSM is another member of the IL-6 group of cytokines that is also known to play an important role in Th2-cytokine-mediated diseases (13). OSM signals through a heterodimeric receptor, consisting of OSMRβ and gp130. To determine whether IL-13Rα1 signaling alters other members of the IL-6 group of cytokines and their receptor subunits, we quantified OSM, OSMRβ, and gp130 expression during SEA-induced allergic disease in IL-13Rα1−/− and IL-13Rα2−/− mice. No significant differences were observed in OSM, OSMRβ, or gp130 expression between the different groups of mice (Fig. 6, C–E), suggesting that IL-13Rα1 signaling was involved in altering IL-31/IL-31RA interactions, but had no effect on OSM and OSMRβ expression in the lungs during SEA-induced allergic disease in mice.
FIGURE 6.

IL-13Rα1 is required for IL-31RA expression in vivo during SEA-induced allergic disease. A, timeline of the experiments showing the days of sensitization and challenges using SEA. B–E, wild-type, IL-13Rα1−/−, and IL-13Rα2−/− mice were sensitized and challenged with either SEA or PBS as a control. Lung transcripts for IL-31RA, OSM, OSMRβ, and gp130 were measured by normalizing to PBS-treated wild-type mice. The data are shown as mean ± S.E. (n = 4/group). Statistical significance is indicated above the bars as determined using one-way analysis of variance followed by Tukey's test, and the groups include SEA-challenged wild-type, IL-13Rα1−/−, and IL-13Rα2−/− mice. *, p < 0.05; ***, p < 0.001.
Discussion
IL-31RA is a gp130-like Type I cytokine receptor that pairs with OSMRβ and forms a signaling receptor for IL-31 (1). Binding of IL-31RA to the IL-31 ligand activates downstream signaling and induces the expression of proinflammatory cytokines and chemokines (2). Therefore, the study of the IL-31/IL-31RA interaction may provide clinical implications for treatment of IL-31-associated diseases. In the present study, we investigated the role of the Th2 cytokines IL-4 and IL-13 in regulating IL-31RA expression and downstream signaling in mouse macrophages. Macrophages stimulated with IL-4 and IL-13 had significantly increased IL-31RA expression, suggesting that Th2 cytokine signaling plays an important role in IL-31RA expression. IL-4 and IL-13 are the key inducers in development of M2 macrophages (15) and signal through binding to a common IL-4Rα chain. IL-2RγC and IL-13Rα1 are the other active constituents to form Type I (IL-4Rα/IL-2RγC) and Type II (IL-4Rα/IL-13Rα1) IL-4 receptors, respectively (25). These receptor complexes are phosphorylated upon ligation with IL-4 and IL-13 and mediate distinct signaling pathways. Studies of mice have shown that IL-4Rα signaling plays a crucial role in the development of M2 macrophages and is associated with Type II immune responses (26). To determine the role of IL-4Rα in Th2 cytokine-mediated IL-31RA expression, we blocked the signaling pathway mediated by Type I IL-4 receptor (IL-4Rα/IL-2Rγ) and measured IL-31RA transcripts in the Th2 cytokine-induced macrophages. IL-31RA expression was attenuated in the presence of antibodies against anti-IL-4Rα, which may suggest that IL-4Rα plays a critical role in Th2 cytokine-mediated IL-31RA expression. However, macrophages deficient for IL-13Rα1−/− displayed heightened IL-31RA expression with IL-4 stimulation, whereas no differences were observed with IL-13. The increased expression of IL-31RA in the absence of the IL-13Rα1 receptor may suggest that Type II IL-4 receptors compete for IL-4 and function as a negative regulator of Type I IL-4 receptor-driven signaling (27). However, mice deficient for IL-13Rα1 showed no significant increase in IL-31RA expression compared with wild-type mice during SEA-induced allergic asthma. One possible explanation for no increase in IL-31RA transcripts in IL-13Rα−/− mice could be in vivo dependence of IL-13 to induce the pathophysiological features of asthma (21, 25, 27, 28). In support, the absence of IL-13Rα2, a decoy receptor of IL-13 has resulted in a further increase in IL-31RA expression compared with wild-type mice during SEA-induced allergic asthma. Furthermore, macrophages deficient for either IL-4Rα or STAT6 showed no increases in IL-31RA expression. In summary, our findings demonstrate that Th2 cytokine-induced IL-31RA expression is regulated by IL-4Rα and STAT6 signaling in murine macrophages.
Previous studies have demonstrated that the IL-31RA protein is expressed as multiple isoforms that are either membrane bound or secreted as a soluble decoy receptor (1, 2). Our flow cytometry and immunofluorescence data demonstrated that IL-4Rα-mediated signaling increased the expression of the membrane-bound form of IL-31RA. Future studies are needed to determine whether any soluble form of IL-31RA protein also is expressed and secreted by macrophages. Previous in vitro studies have shown that the Th1 cytokine IFN-γ up-regulates the expression of IL-31RA in monocytes/macrophages and keratinocytes (1, 29). Therefore, we attempted to determine the effect of IFN-γ alone or in combination with the Th2 cytokine IL-13 on IL-31RA expression. In contrast to observations made using human peripheral blood mononuclear cells (16), we found that mouse macrophages stimulated with IFN-γ did not have altered IL-31RA expression, but did have inhibited IL-13-induced IL-31RA expression. In addition, previous studies also suggest opposing effects of IFN-γ on IL-4/IL-13 signaling and their target gene expression in human primary airway epithelial cells and also airway smooth muscle cells via inhibition of STAT6 activation (30, 31). However, future studies are needed to identify mechanisms that might be involved in IFN-γ inhibition of Th2 cytokine-driven IL-31RA gene expression. Macrophages with an M2 phenotype selectively express Fizz1 and are involved in recruitment of immune cells and tissue remodeling during Th2 cytokine-driven allergic diseases (32). Our new findings demonstrate that enhanced IL-31/IL-31RA interactions also contribute to Fizz1 expression and possibly to elevated inflammatory responses. Furthermore, we observed a significant increase in the expression of SAA in macrophages stimulated with IL-31, but not in macrophages deficient for Th2 cytokine signaling. Together, our findings suggest that IL-4Rα signaling is critical for IL-31-driven Fizz1 and SAA expression involved in inflammatory and allergic responses.
Lung allergic responses induce Th2 cells to secrete IL-4 and IL-13, which increases bronchial smooth muscle contractility, mucus production by goblet cells in airway epithelium, and production of excess collagen by fibroblasts (33–35). The absence of IL-13Rα1 or an excess of soluble IL-13Rα2 has been shown to attenuate these pathologic responses during an allergic asthma (27, 34). To substantiate our findings that Th2 cytokines alter IL-31RA expression, we studied a murine model of SEA-induced allergic disease, as well as an in vivo model for Th2-driven inflammation and remodeling of the lung (20). SEA-challenged IL-13Rα1−/− mice exhibited a significant decrease in expression of IL-31RA compared with WT mice. In contrast, significantly increased expression of IL-31RA was observed in SEA-challenged IL-13Rα2−/− mice. Previous studies have shown that IL-13Rα2 functions as a decoy receptor of IL-13 and inhibits Type II IL-4 receptor complex-dependent STAT6 signaling (36). Therefore, lack of the IL-13Rα2 gene may have augmented IL-13-driven IL-31RA expression during SEA-induced allergic disease. Several previous studies (1, 37, 38) have shown that human IL-31RA is expressed on monocytes/macrophages, keratinocytes, epithelial cells, and dendritic cells. In addition, increased IL-31 and IL-31RA levels have been found in Th2-related allergic diseases, such as atopic dermatitis (39), which is characterized by the polarization of antigen-presenting cells and activation of a Th2 response (40). In a mouse model of atopic dermatitis, increased IL-31 transcripts were associated with scratching behavior (41), whereas a recent study showed that anti-IL-31 neutralizing antibody reduces scratching behavior and dermatitis in mice (12). Based on an accumulating understanding of IL-31 signaling and atopic dermatitis, our findings suggest that signaling induced by Th2 cytokines is critical in regulating IL-31/IL-31RA interactions involved in inflammatory and remodeling responses. Thus, our finding of Th2-dependent IL-31RA expression may provide new insights into the regulation of IL-31 and subsequent immunopathogenesis associated with elevated Th2 responses.
In conclusion, the current study demonstrates that Th2 cytokines are the key inducers for IL-31RA expression and therefore play a critical role in Th2-mediated IL-31/IL-31RA interactions (Fig. 7). We also showed that both Type I and Type II IL-4 receptor mediated STAT6 signaling is crucial for IL-31RA expression. Moreover, our in vivo SEA-induced allergic disease model reveals that IL-13Rα2, a decoy receptor for IL-13, acts as a potent negative regulator of IL-31RA expression. Although future studies warrant delineation of the role of Th2-mediated IL-31/IL-31RA signaling in the manifestation of Th2 cytokine-related inflammatory disease, our current data highlight the counter-regulatory roles between Th2 cytokine signaling and IL-31/IL-31RA interactions in allergic and inflammatory diseases.
FIGURE 7.

Proposed model for STAT6-dependent IL-31RA expression in macrophages. IL-4/IL-13 bind with Type I and Type II IL-4 receptors and activate STAT6-mediated IL-31RA expression. Interaction of IL-31RA with IL-31 induces the expression of Fizz1 and SAA, which are involved in inflammation and tissue remodeling.
Supplementary Material
Acknowledgments
We thank the veterinary services at Cincinnati Children's Hospital Medical Center and the National Institutes of Health for the care of mice used in this study. We also are grateful to Drs. Thomas A. Wynn and William D. Hardie for their valuable suggestions and insights and J. Denise Wetzel, CCHMC Medical Writer, for editing of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grant 1R03AR062832 (to S. K. M.), a Parker B. Francis Fellowship (to S. K. M.), and American Heart Association Grant 12SDG9130040 (to S. K. M.). S. R. Dillon is an employee and stockholder of Bristol-Myers Squibb. None of the remaining authors have a financial relationship with a commercial entity that has an interest in the subject of this manuscript.

This article contains supplemental Figs. S1–S3.
- IL-31RA
- interleukin 31 receptor α
- OSMRβ
- oncostatin M receptor β
- BM
- bone marrow
- SEA
- soluble egg antigen
- BMDM
- bone marrow-derived macrophages
- OSM
- oncostatin M
- STAT6
- signal transducer and activator of transcription 6
- SAA
- serum amyloid A.
References
- 1. Dillon S. R., Sprecher C., Hammond A., Bilsborough J., Rosenfeld-Franklin M., Presnell S. R., Haugen H. S., Maurer M., Harder B., Johnston J., Bort S., Mudri S., Kuijper J. L., Bukowski T., Shea P., Dong D. L., Dasovich M., Grant F. J., Lockwood L., Levin S. D., LeCiel C., Waggie K., Day H., Topouzis S., Kramer J., Kuestner R., Chen Z., Foster D., Parrish-Novak J., Gross J. A. (2004) Interleukin 31, a cytokine produced by activated T cells, induces dermatitis in mice. Nat. Immunol. 5, 752–760 [DOI] [PubMed] [Google Scholar]
- 2. Zhang Q., Putheti P., Zhou Q., Liu Q., Gao W. (2008) Structures and biological functions of IL-31 and IL-31 receptors. Cytokine Growth Factor Rev. 19, 347–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chattopadhyay S., Tracy E., Liang P., Robledo O., Rose-John S., Baumann H. (2007) Interleukin-31 and oncostatin-M mediate distinct signaling reactions and response patterns in lung epithelial cells. J. Biol. Chem. 282, 3014–3026 [DOI] [PubMed] [Google Scholar]
- 4. Cornelissen C., Lüscher-Firzlaff J., Baron J. M., Lüscher B. (2012) Signaling by IL-31 and functional consequences. Eur. J. Cell Biol. 91, 552–566 [DOI] [PubMed] [Google Scholar]
- 5. Maier E., Werner D., Duschl A., Bohle B., Horejs-Hoeck J. (2014) Human Th2 but not Th9 cells release IL-31 in a STAT6/NF-κB-dependent way. J. Immunol. 193, 645–654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cheung P. F., Wong C. K., Ho A. W., Hu S., Chen D. P., Lam C. W. (2010) Activation of human eosinophils and epidermal keratinocytes by Th2 cytokine IL-31: implication for the immunopathogenesis of atopic dermatitis. Int. Immunol. 22, 453–467 [DOI] [PubMed] [Google Scholar]
- 7. Bilsborough J., Leung D. Y., Maurer M., Howell M., Boguniewicz M., Boguniewcz M., Yao L., Storey H., LeCiel C., Harder B., Gross J. A. (2006) IL-31 is associated with cutaneous lymphocyte antigen-positive skin homing T cells in patients with atopic dermatitis. J. Allergy Clin. Immunol. 117, 418–425 [DOI] [PubMed] [Google Scholar]
- 8. Neis M. M., Peters B., Dreuw A., Wenzel J., Bieber T., Mauch C., Krieg T., Stanzel S., Heinrich P. C., Merk H. F., Bosio A., Baron J. M., Hermanns H. M. (2006) Enhanced expression levels of IL-31 correlate with IL-4 and IL-13 in atopic and allergic contact dermatitis. J. Allergy Clin. Immunol. 118, 930–937 [DOI] [PubMed] [Google Scholar]
- 9. Sonkoly E., Muller A., Lauerma A. I., Pivarcsi A., Soto H., Kemeny L., Alenius H., Dieu-Nosjean M. C., Meller S., Rieker J., Steinhoff M., Hoffmann T. K., Ruzicka T., Zlotnik A., Homey B. (2006) IL-31: a new link between T cells and pruritus in atopic skin inflammation. J. Allergy Clin. Immunol. 117, 411–417 [DOI] [PubMed] [Google Scholar]
- 10. Gonzales A. J., Humphrey W. R., Messamore J. E., Fleck T. J., Fici G. J., Shelly J. A., Teel J. F., Bammert G. F., Dunham S. A., Fuller T. E., McCall R. B. (2013) Interleukin-31: its role in canine pruritus and naturally occurring canine atopic dermatitis. Vet. Dermatol. 24, 48–53 [DOI] [PubMed] [Google Scholar]
- 11. Rabenhorst A., Hartmann K. (2014) Interleukin-31: a novel diagnostic marker of allergic diseases. Curr. Allergy Asthma Rep. 14, 423. [DOI] [PubMed] [Google Scholar]
- 12. Grimstad O., Sawanobori Y., Vestergaard C., Bilsborough J., Olsen U. B., Grønhøj-Larsen C., Matsushima K. (2009) Anti-interleukin-31-antibodies ameliorate scratching behaviour in NC/Nga mice: a model of atopic dermatitis. Exp. Dermatol. 18, 35–43 [DOI] [PubMed] [Google Scholar]
- 13. Bilsborough J., Mudri S., Chadwick E., Harder B., Dillon S. R. (2010) IL-31 receptor (IL-31RA) knockout mice exhibit elevated responsiveness to oncostatin M. J. Immunol. 185, 6023–6030 [DOI] [PubMed] [Google Scholar]
- 14. Mosser D. M. (2003) The many faces of macrophage activation. J. Leukoc. Biol. 73, 209–212 [DOI] [PubMed] [Google Scholar]
- 15. Van Dyken S. J., Locksley R. M. (2013) Interleukin-4- and interleukin-13-mediated alternatively activated macrophages: roles in homeostasis and disease. Annu. Rev. Immunol. 31, 317–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Martinez F. O., Gordon S. (2014) The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 6, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sica A., Mantovani A. (2012) Macrophage plasticity and polarization: in vivo veritas. J. Clin. Invest. 122, 787–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ip W. K., Wong C. K., Li M. L., Li P. W., Cheung P. F., Lam C. W. (2007) Interleukin-31 induces cytokine and chemokine production from human bronchial epithelial cells through activation of mitogen-activated protein kinase signalling pathways: implications for the allergic response. Immunology 122, 532–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Thompson R. W., Pesce J. T., Ramalingam T., Wilson M. S., White S., Cheever A. W., Ricklefs S. M., Porcella S. F., Li L., Ellies L. G., Wynn T. A. (2008) Cationic amino acid transporter-2 regulates immunity by modulating arginase activity. PLoS Pathog. 4, e1000023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wilson M. S., Elnekave E., Mentink-Kane M. M., Hodges M. G., Pesce J. T., Ramalingam T. R., Thompson R. W., Kamanaka M., Flavell R. A., Keane-Myers A., Cheever A. W., Wynn T. A. (2007) IL-13Rα2 and IL-10 coordinately suppress airway inflammation, airway-hyperreactivity, and fibrosis in mice. J. Clin. Invest. 117, 2941–2951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Madala S. K., Pesce J. T., Ramalingam T. R., Wilson M. S., Minnicozzi S., Cheever A. W., Thompson R. W., Mentink-Kane M. M., Wynn T. A. (2010) Matrix metalloproteinase 12-deficiency augments extracellular matrix degrading metalloproteinases and attenuates IL-13-dependent fibrosis. J. Immunol. 184, 3955–3963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Takeda K., Tanaka T., Shi W., Matsumoto M., Minami M., Kashiwamura S., Nakanishi K., Yoshida N., Kishimoto T., Akira S. (1996) Essential role of Stat6 in IL-4 signalling. Nature 380, 627–630 [DOI] [PubMed] [Google Scholar]
- 23. Kaplan M. H., Schindler U., Smiley S. T., Grusby M. J. (1996) Stat6 is required for mediating responses to IL-4 and for development of Th2 cells. Immunity 4, 313–319 [DOI] [PubMed] [Google Scholar]
- 24. Wilson M. S., Pesce J. T., Ramalingam T. R., Thompson R. W., Cheever A., Wynn T. A. (2008) Suppression of murine allergic airway disease by IL-2:anti-IL-2 monoclonal antibody-induced regulatory T cells. J. Immunol. 181, 6942–6954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Munitz A., Brandt E. B., Mingler M., Finkelman F. D., Rothenberg M. E. (2008) Distinct roles for IL-13 and IL-4 via IL-13 receptor α1 and the type II IL-4 receptor in asthma pathogenesis. Proc. Natl. Acad. Sci. U.S.A. 105, 7240–7245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gordon S., Martinez F. O. (2010) Alternative activation of macrophages: mechanism and functions. Immunity 32, 593–604 [DOI] [PubMed] [Google Scholar]
- 27. Ramalingam T. R., Pesce J. T., Sheikh F., Cheever A. W., Mentink-Kane M. M., Wilson M. S., Stevens S., Valenzuela D. M., Murphy A. J., Yancopoulos G. D., Urban J. F., Jr., Donnelly R. P., Wynn T. A. (2008) Unique functions of the type II interleukin 4 receptor identified in mice lacking the interleukin 13 receptor α1 chain. Nat. Immunol. 9, 25–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wills-Karp M., Luyimbazi J., Xu X., Schofield B., Neben T. Y., Karp C. L., Donaldson D. D. (1998) Interleukin-13: central mediator of allergic asthma. Science 282, 2258–2261 [DOI] [PubMed] [Google Scholar]
- 29. Heise R., Neis M. M., Marquardt Y., Joussen S., Heinrich P. C., Merk H. F., Hermanns H. M., Baron J. M. (2009) IL-31 receptor α expression in epidermal keratinocytes is modulated by cell differentiation and interferon γ. J. Invest. Dermatol. 129, 240–243 [DOI] [PubMed] [Google Scholar]
- 30. Moynihan B. J., Tolloczko B., El Bassam S., Ferraro P., Michoud M. C., Martin J. G., Laberge S. (2008) IFN-gamma, IL-4 and IL-13 modulate responsiveness of human airway smooth muscle cells to IL-13. Respir. Res. 9, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Heller N. M., Matsukura S., Georas S. N., Boothby M. R., Rothman P. B., Stellato C., Schleimer R. P. (2004) Interferon-γ inhibits STAT6 signal transduction and gene expression in human airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 31, 573–582 [DOI] [PubMed] [Google Scholar]
- 32. Pesce J. T., Ramalingam T. R., Wilson M. S., Mentink-Kane M. M., Thompson R. W., Cheever A. W., Urban J. F., Jr., Wynn T. A. (2009) Retnla (relmα/fizz1) suppresses helminth-induced Th2-type immunity. PLoS Pathog. 5, e1000393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kuperman D. A., Huang X., Nguyenvu L., Hölscher C., Brombacher F., Erle D. J. (2005) IL-4 receptor signaling in Clara cells is required for allergen-induced mucus production. J. Immunol. 175, 3746–3752 [DOI] [PubMed] [Google Scholar]
- 34. Ohta Y., Hayashi M., Kanemaru T., Abe K., Ito Y., Oike M. (2008) Dual modulation of airway smooth muscle contraction by Th2 cytokines via matrix metalloproteinase-1 production. J. Immunol. 180, 4191–4199 [DOI] [PubMed] [Google Scholar]
- 35. Perkins C., Yanase N., Smulian G., Gildea L., Orekov T., Potter C., Brombacher F., Aronow B., Wills-Karp M., Finkelman F. D. (2011) Selective stimulation of IL-4 receptor on smooth muscle induces airway hyperresponsiveness in mice. J. Exp. Med. 208, 853–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kawakami K., Taguchi J., Murata T., Puri R. K. (2001) The interleukin-13 receptor α2 chain: an essential component for binding and internalization but not for interleukin-13-induced signal transduction through the STAT6 pathway. Blood 97, 2673–2679 [DOI] [PubMed] [Google Scholar]
- 37. Kasraie S., Niebuhr M., Werfel T. (2010) Interleukin (IL)-31 induces pro-inflammatory cytokines in human monocytes and macrophages following stimulation with staphylococcal exotoxins. Allergy 65, 712–721 [DOI] [PubMed] [Google Scholar]
- 38. Horejs-Hoeck J., Schwarz H., Lamprecht S., Maier E., Hainzl S., Schmittner M., Posselt G., Stoecklinger A., Hawranek T., Duschl A. (2012) Dendritic cells activated by IFN-γ/STAT1 express IL-31 receptor and release proinflammatory mediators upon IL-31 treatment. J. Immunol. 188, 5319–5326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nobbe S., Dziunycz P., Mühleisen B., Bilsborough J., Dillon S. R., French L. E., Hofbauer G. F. (2012) IL-31 expression by inflammatory cells is preferentially elevated in atopic dermatitis. Acta Derm. Venereol. 92, 24–28 [DOI] [PubMed] [Google Scholar]
- 40. Brandt E. B., Sivaprasad U. (2011) Th2 cytokines and atopic dermatitis. J. Clin. Cell. Immunol. 2, 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Takaoka A., Arai I., Sugimoto M., Honma Y., Futaki N., Nakamura A., Nakaike S. (2006) Involvement of IL-31 on scratching behavior in NC/Nga mice with atopic-like dermatitis. Exp. Dermatol. 15, 161–167 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.