

Background: Keratinocytes cease proliferation during differentiation, and the mechanism that mediates these events is not well understood.
Results: PRMT5/MEP50 control p21Cip1 promoter methylation to silence gene expression, and this is reversed by PKCδ/p38δ signaling.
Conclusion: PRMT5/MEP50 and PKCδ/p38δ signaling produce opposing actions to control keratinocyte proliferation during differentiation.
Significance: This study describes cross-talk between MAPK signaling and epigenetic mechanisms to control cell proliferation.
Keywords: cancer, cell cycle, cyclin, p38 MAPK, protein arginine N-methyltransferase 5 (PRMT5), protein kinase C (PKC)
Abstract
Protein arginine methyltransferase 5 (PRMT5) is a key epigenetic regulator that symmetrically dimethylates arginine residues on histones H3 and H4 to silence gene expression. PRMT5 is frequently observed in a complex with the cofactor methylosome protein 50 (MEP50), which is required for PRMT5 activity. PKCδ/p38δ signaling, a key controller of keratinocyte proliferation and differentiation, increases p21Cip1 expression to suppress keratinocyte proliferation. We now show that MEP50 enhances keratinocyte proliferation and survival via mechanisms that include silencing of p21Cip1 expression. This is associated with enhanced PRMT5-MEP50 interaction at the p21Cip1 promoter and enhanced arginine dimethylation of the promoter-associated histones H3 and H4. It is also associated with a MEP50-dependent reduction in the level of p53, a key controller of p21Cip1 gene expression. We confirm an important biological role for MEP50 and PRMT5 in regulating keratinocyte proliferation using a stratified epidermal equivalent model that mimics in vivo epidermal keratinocyte differentiation. In this model, PRMT5 or MEP50 knockdown results in reduced keratinocyte proliferation. We further show that PKCδ/p38δ signaling suppresses MEP50 expression, leading to reduced H3/H4 arginine dimethylation at the p21Cip1 promoter, and that this is associated with enhanced p21Cip1 expression and reduced cell proliferation. These findings describe an opposing action between PKCδ/p38δ MAPK signaling and PRMT5/MEP50 epigenetic silencing mechanisms in regulating cell proliferation.
Introduction
PKCδ and p38δ are key components of a cascade that controls keratinocyte differentiation and proliferation (1–7). PKCδ activates a MEKK1/MEK3/p38δ signaling cascade that triggers events that enhance keratinocyte differentiation and suppress proliferation (4, 6, 8, 9). Downstream events include activation of AP1, Sp1, and Kruppel-like transcription factors, leading to activation of differentiation and suppression of proliferation (1, 3, 10–18). This cascade has an important role in controlling differentiation-associated gene expression (15–17), which has been confirmed by transgenic mouse experiments, demonstrating a key role for specific transcription factors and gene response elements in this regulation (15–17, 19).
A recent study indicates that PRMT5, an arginine methyltransferase that symmetrically dimethylates arginine residues on target proteins, acts to antagonize PKCδ/MEK3/p38δ signaling (20). In this context, PRMT5 acts to enhance keratinocyte survival (20). PRMT5 associates with a coactivator called MEP50 (21, 22), an interaction that is required for PRMT5 activation (21, 23). This complex then functions to dimethylate specific arginines on target proteins, leading to altered protein function. Loss of either PRMT5 or MEP50 reduces this activity (21, 22). Histones are important PRMT5 targets. PRMT5 produces changes in histone dimethylation that are part of the epigenetic code that controls gene expression (24, 25). Histones 3 and 4 are important PRMT5 targets. The PRMT5-MEP50 complex symmetrically dimethylates arginine 3 of histone H4 (H4R3me2s),2 and arginine 8 of histone H3 (H3R8me2s), and these modifications are associated with silencing of gene expression (26–28).
Keratinocyte proliferation and differentiation are important processes in the keratinocyte life cycle that must be balanced to produce a stratified tissue that functions as an appropriate barrier (29). Delayed cessation of keratinocyte proliferation, for example, can lead to epidermal disease (19, 30). In this study, we examine the role of MEP50 as a controller of keratinocyte proliferation. We show that MEP50 enhances keratinocyte proliferation. To understand the mechanism, we study the impact of MEP50 expression on p21Cip1 gene expression, a key controller of keratinocyte proliferation (2, 31). We show that MEP50 has a role in stimulating dimethylation of histones associated with key regulator elements in the p21Cip1 gene promoter and that this is associated with silencing of p21Cip1 expression. We further show that PKCδ/p38δ antiproliferation signaling reduces PRMT5 and MEP50 expression and PRMT5-MEP50 association with the p21Cip1 gene. This leads to reduced H3R8me2s and H4R3me2s formation at the p21Cip1 promoter, leading to derepression. We propose that PRMT5-MEP50 silencing of antiproliferation genes is an important mechanism of survival and that PKCδ/p38δ signaling pathways reduce the expression of this PRMT5-MEP50 dimethylation complex as a mechanism to induce p21Cip1 expression and suppress proliferation during keratinocyte differentiation.
Experimental Procedures
Antibodies and Reagents
Rabbit polyclonal antibodies for MEP50 (catalog no. 2823), PRMT5 (catalog no. 2252), and p21Cip1 (catalog no. 2947) were obtained from Cell Signaling Technology (Danvers, MA), and mouse monoclonal anti-β-actin (catalog no. A-5441), anti-FLAG antibody (catalog no. 8592), and anti-FLAG M2-FITC (catalog no. F4049) were purchased from Sigma-Aldrich. Normal rabbit IgG was obtained from Cell Signaling Technology (catalog no. 2729). Rabbit polyclonal antibodies for histone H4 symmetrically dimethylated arginine 3 (H4R3me2s) (catalog no. ab5823) and histone H3 symmetrically dimethylated arginine 8 (H3R8me2s) (catalog no. PA5-27039) were obtained from Abcam (Cambridge, MA) and Thermo Fisher Scientific (Rockford, IL). Mouse anti-MEP50 was purchased from Abcam (catalog no. ab5772). Rabbit anti-PKCδ (catalog no. sc-937), goat anti-PRMT5 (catalog no. sc-22132), and mouse monoclonal anti-p38δ (catalog no. sc-271292) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). The secondary antibodies included peroxidase-conjugated sheep anti-mouse IgG (catalog no. NXA931, GE Healthcare) and peroxidase-conjugated donkey anti-rabbit IgG (catalog no. NA934V, GE Healthcare). Peroxidase-conjugated donkey anti-goat IgG (catalog no. sc-2033) was obtained from Santa Cruz Biotechnology. Phorbol ester, 12-O-tetradecanoylphorbol-13-acetate (TPA), was obtained from Calbiochem (Billerica, MA; catalog no. 524400). We report results using control (catalog no. D-001206-13-05), MEP50 (catalog no. M-006895-01-0005), and PRMT5 (catalog no. M-015817-02-0005) siRNA reagents from Dharmacon Inc (Lafayette, CO). Key findings were confirmed using additional siRNAs that target MEP50 (catalog nos. D-006895-01-0002 and D-006895-02-0002) and PRMT5 (catalog nos. D-015817-01-0002 and D-015817-04-0002).
Cell Culture, Plasmids, and Viruses
Keratinocyte (KERn) serum-free medium (KSFM), 0.25% trypsin, and Hanks' balanced salt solution were purchased from Invitrogen. Foreskin epidermis, obtained from newborn infants, was separated from the dermis by overnight dispase treatment, and KERn were obtained after dispersal with trypsin and maintained in KSFM supplemented with epidermal growth factor and pituitary extract (6, 7). p21-2326, encoding the human p21Cip1 promoter linked to luciferase, was obtained from Dr. Bert Vogelstein (32). The wild-type p21Cip1 promoter and promoter mutants encoding mutant p53 transcription factor binding sites were constructed in pBluescript II KS (+) (2). PG13-Luc was obtained from Dr. Nancy Colburn (33). The human MEP50-encoding plasmid was constructed by primer amplification using plasmid p-OTB7-FLAG-MEP50 (pOTB7-WDR77, MHS1011-202830316) from Open Biosystems (Huntsville, AL) as a template. FLAG-MEP50 was amplified as a BamHI/NotI fragment using 5′-GATC GGA TCC ATG GAC TAC AAG GAC GAC GAC GAC AAG ATG CGG AAG GAA ACC CCA (forward) and 5′-GATC GCG GCC GCC TAC TCA GTA ACA CTT GCA GG (reverse) primers. The ATG start codon is shown in boldface, and the FLAG epitope is underlined. The product was then cloned into pcDNA3 to produce pcDNA3-FLAG-MEP50. Adenoviruses encoding HA-p38δ, PKCδ, and empty control virus (tAd5-HA-p38δ, Ad5-PKCδ, Ad5-FLAG-p38δ, and Ad5-EV) were prepared by propagation in HEK293 cells and followed by cesium chloride gradient centrifugation. For experiments involving adenoviral infection, KERn were treated with adenovirus (multiplicity of infection 15) in KSFM containing 6 μg/ml Polybrene. Tetracycline-inducible viruses (tAd-EV, tAd5-PKCδ, and tAd5-HA-38δ) were coinfected with Ad5-TA-encoding virus to induce PKCδ and p38δ expression (1).
Promoter Luciferase Assay
p21Cip1 promoter reporter plasmid (0.5 μg) and 1 μg of pcDNA3 or pcDNA3-FLAG-MEP50 were mixed with 4.5 μl of FuGENE6 (Promega) diluted in 95.5 μl of KSFM. After a 20-min incubation, this mixture was added to 2 ml of KSFM in dishes containing 50% confluent KERn cultures. After 24 h, cell lysates were collected and processed for the luciferase activity assay (1).
Keratinocyte Electroporation and Cell Proliferation Assay
The AMAXA electroporator and VPD-1002 nucleofection kit was used for keratinocyte electroporation. KERn were harvested with trypsin and replated 1 day prior to electroporation. The cells were then reharvested with trypsin, and 1 million cells were used per electroporation. The cells were suspended in 100 μl of keratinocyte nucleofection solution containing 3 μg of control, MEP50, or PRMT5 siRNA. The mixture was mixed by gentle pipetting, transferred to the electroporation cuvette, and electroporated using the T-018 setting. KSFM (500 μl) was added, and the mixture was transferred to a 55-cm2 dishes containing 10 ml of KSFM. The cells were maintained for various time points before the extracts were prepared for mRNA or protein analysis. This method achieves electroporation efficiencies of >90% efficiency (1).
Cells used for proliferation experiments were double-electroporated. This involved an initial electroporation with 3 μg of appropriate siRNA, recovery in culture for 72 h, a repeat electroporation with 3 μg of siRNA, and 24 h of recovery in culture. The cells were then harvested and seeded at low density (15,000 cells/well) in 35-mm dishes, and the cells were counted at 48, 72, and 96 h.
Immunoblot
Cell extracts were prepared in cell lysis buffer (Cell Signaling Technology, catalog no. 9803) containing protease inhibitor (Calbiochem, catalog no. 539134). Equivalent amounts of protein were electrophoresed on 12% denaturing polyacrylamide gels and transferred to nitrocellulose membranes. The membranes were blocked with 5% skimmed milk in Tris-buffered saline containing 0.1% Tween 20 and incubated overnight with primary antibody and horseradish peroxidase-conjugated secondary antibody for 2 h. β-actin served as a gel loading control.
Quantitative RT-PCR
Total RNA was isolated using the RNAspin system (GE Healthcare) and reverse-transcribed using Superscript III reverse transcriptase (Invitrogen). RNA (1 μg) was used for cDNA preparation. Light Cycler 480 SYBR Green I master mix (Roche Diagnostics) was used to measure mRNA levels. The signals were normalized to the level of cyclophillin A mRNA. The gene-specific primers used for detection of mRNA levels were as follows: p21Cip1, 5′-AAG ACC ATG TGG ACC TGT CAC TGT (forward) and 5′-AGG GCT TCC TCT TGG AGA AGA TCA (reverse); MEP50, 5′-TTG CTC AGC AGG TGG TAC TGA GTT (forward) and 5′-AAT CTG TGA TGC TGG CTT GGG ACA (reverse); p53, 5′- TAA CAG TTC CTG CAT GGG CGG C (forward) and 5′- AGG ACA GGC ACA AAC ACG CAC C (reverse); and cyclophilin A, 5′-CAT CTG CAC TGC CAA GAC TGA (forward) and 5′-TTC ATG CCT TCT TTC ACT TTGC (reversed).
Immunostaining
Cells maintained on coverslips were fixed for 20 min in phosphate-buffered saline containing 4% paraformaldehyde and permeabilized with methanol chilled at −20 °C. The coverslips were subsequently incubated with the appropriate primary and secondary antibodies for 1 h each. Cells were then incubated with Hoechst 33258 (1:2000) for 5 min, washed, and mounted on glass slides using Fluoromount (Sigma, catalog no. F4680). An Olympus OX81 spinning disc confocal microscope was used to collect fluorescent images. Paraffin-embedded and formalin-fixed foreskin sections were immunostained as described previously using the fluorescence or peroxidase methods. MEP50 antibody (catalog no. ab5772) staining was detected using an appropriate fluorophore-conjugated or biotinylated secondary antibody. Biotinylated secondary antibody was obtained as part of the mouse IgG Vectastain ABC kit (catalog no. PK-6102, Vector Laboratories, Burlingame, CA).
ChIP
ChIP assays were performed using the Diagenode Low Cell ChIP assay kit (C01010073: kch-maglow-G48). 0.5 × 106 cells were infected with empty adenovirus (multiplicity of infection 15) or adenoviruses encoding HA-p38δ or PKCδ, and, after 48 h, 1 million 1 × 106 cells were used for shearing and 0.1 × 105 cells were used for immunoprecipitation per antibody. Enrichment of MEP50-, PRMT5-, H4R3me2s, and H3R8me2s-associated DNA sequences in immunoprecipitated samples were detected by quantitative RT-PCR using sequence-specific primers and LightCycler 480 SYBR Green I master mix. Primers were designed to detect the human p21Cip1 promoter p53 binding site located at nucleotides −1426/−1310 (forward, 5′-CCG AGG TCA GCT GCG TTA GAG G; reverse, 5′-AGA ACC CAG GCT TGG AGC AGC).
Epidermal Equivalent Cultures
Freshly isolated foreskin keratinocytes (1.5 × 106) were electroporated with 3 μg of control, MEP50, or PRMT5 siRNA. After 72 h of recovery, the cells were harvested, and 2 × 106 cells from each group were electroporated again with 3 μg of the same siRNA. The cells were then seeded onto Millicell-PCF chambers (0.4 μm, 12 mm, catalog no. PIHP01250) in KSFM. After 24 h, the cells were shifted to Epilife medium containing 1.4 mm calcium chloride and 5 μg/ml vitamin C and cultured at the air-liquid interface (3, 34). Fresh Epilife medium was added every 2 days. After 5 days, the epidermal equivalents were harvested for preparation of RNA and protein and histology. Total RNA was isolated for quantitative RT-PCR using the Illustra RNAspin mini kit (GE Healthcare). For protein lysates, the inserts were washed twice with phosphate-buffered saline, and the cells were harvested in 0.0625 m Tris-HCl (pH 7.5) containing 10% glycerol, 5% SDS, and 5% β-mercaptoethanol. Then the cells were sonicated and centrifuged at 10,000 rpm for 5 min, and the supernatant was collected for immunoblot analysis (34).
Results
MEP50 Distribution in the Epidermis
MEP50 localizes in the nucleus or cytoplasm in a tissue- and cell type-dependent manner that also depends upon the environment (35–37). To assess MEP50 localization in keratinocytes, we stained paraffin-embedded sections of foreskin epidermis with anti-MEP50. Fig. 1A identifies MEP50 as being present in all epidermal layers. Cytoplasmic localization predominates in the basal and spinous layers, whereas nuclear distribution is prominent in the granular layers. We next compared the distribution of endogenous and FLAG-tagged expressed MEP50 in cultured normal epidermal foreskin keratinocytes. Fig. 1B shows a mixed cytoplasmic and nuclear distribution in these cultures and that expressed FLAG-MEP50 assumes a similar distribution as endogenous MEP50. This is consistent with a previous report showing that MEP50 localizes in the nucleus and cytoplasm in lung cancer cell lines (38). These data also show that vector-expressed FLAG-MEP50 colocalizes with endogenous MEP50.
FIGURE 1.
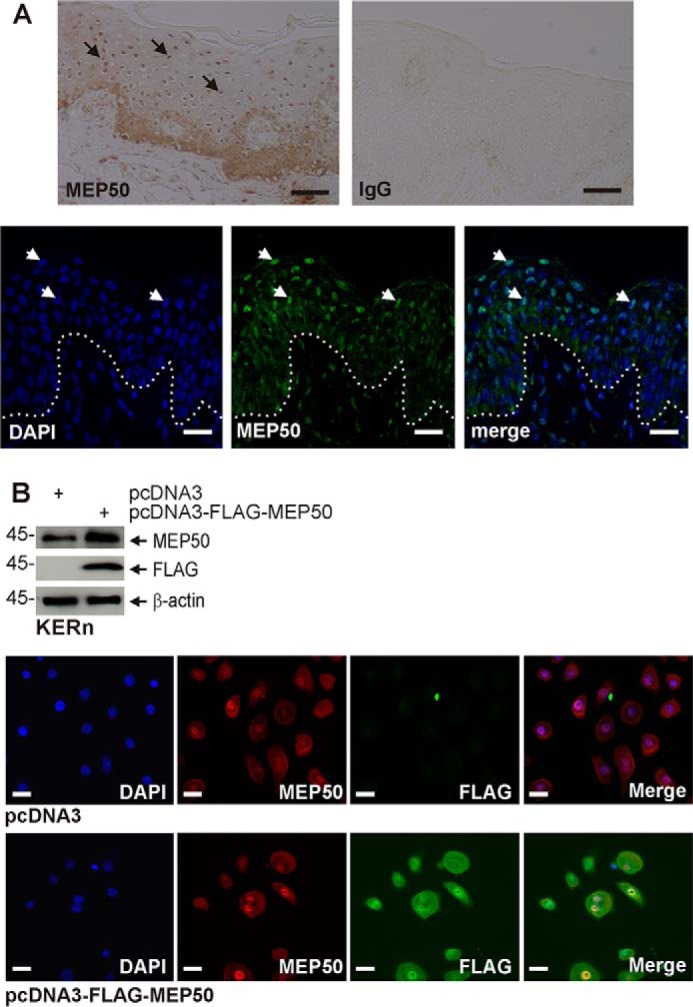
MEP50 is expressed in the human epidermis and in cultured keratinocytes. A, foreskin tissue sections were stained with anti-MEP50, and binding was visualized with peroxidase-conjugated secondary antibody (top panels). The control is IgG. The arrows indicate MEP50 nuclear localization in suprabasal keratinocytes. Foreskin tissue sections (bottom panels) were fixed and stained with anti-MEP50, and antibody binding was visualized using a FITC-conjugated secondary antibody. The arrows indicate nuclear MEP50 accumulation. Scale bars = 10 μm. B, colocalization of endogenous and expressed MEP50. KERn were electroporated with 3 μg of pcDNA3 or pcDNA3-FLAG-MEP50. After 48 h, protein lysates were tested by immunoblot using anti-FLAG and anti-MEP50. β-actin was used as the loading control. After 48 h, the cells were fixed and costained with anti-FLAG (green) and anti-MEP50 (red). Similar results were observed in each of three experiments. The staining indicates MEP50 distribution in the nucleus and cytoplasm. Scale bars = 10 μm.
MEP50 Regulation of Cell Proliferation
A previous study showed that PRMT5 acts to enhance keratinocyte survival and proliferation (20). Because MEP50 is frequently engaged as a PRMT5 cofactor required for PRMT5 activation, we examined whether altering MEP50 expression regulates keratinocyte proliferation. Cells were treated with control, MEP50, or PRMT5 siRNA. Fig. 2A shows that MEP50 or PRMT5 knockdown reduces cell proliferation. Fig. 2B confirms a reduction in MEP50 expression in MEP50 siRNA-treated cells. It also shows that PRMT5 levels are reduced. The reduction in PRMT5 is not surprising considering that these proteins are part of a coregulated complex (2, 21, 23). Simultaneous knockdown of both proteins does not further reduce proliferation, as would be expected if both proteins are a necessary part of the MEP50/PRMT5 complex (data not shown).
FIGURE 2.
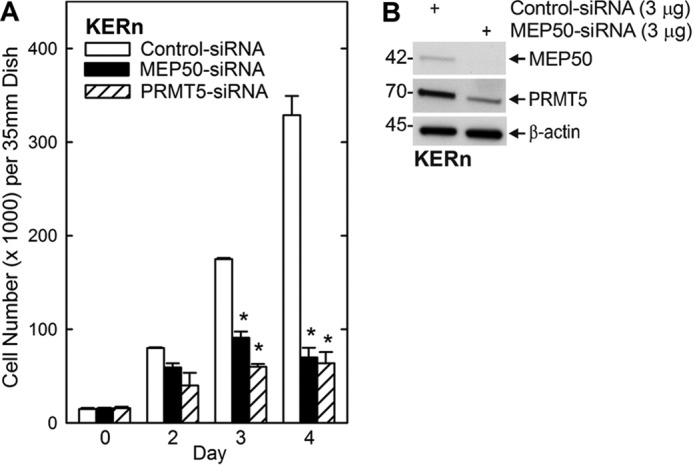
MEP50 and PRMT5 are required for keratinocyte proliferation. A, KERn were electroporated twice with control, MEP50, or PRMT5 siRNA, and 15,000 cells/well were plated. After overnight attachment, the cell number was determined (day 0) and at the indicated times thereafter. The values are mean ± S.E. (n = 3). *, p < 0.005. B, immunoblot detection of MEP50 and PRMT5. The immunoblot confirms a reduction in MEP50 in MEP50 siRNA-treated cultures. PRMT5 is also reduced.
MEP50 Control of p21Cip1 Expression
To gain insights into the mechanism of MEP50 regulation of proliferation, we examined the effect of MEP50 expression on p21Cip1 expression. p21Cip1 is an important cell cycle control regulator in keratinocytes (2, 31). We have shown previously that p21Cip1 is increased when keratinocyte proliferation is inhibited (2) via a mechanism that involves p53 interaction at the p21Cip1 gene promoter (31). Fig. 3A shows that increased MEP50 expression is associated with reduced p21Cip1 mRNA and protein expression. To examine the impact of MEP50 on p21Cip1 gene expression, KERn cells were transfected with constructs encoding various p21Cip1 promoter segments fused to the luciferase reporter gene (2, 31) in the presence or absence of the FLAG-MEP50 expression vector. Fig. 3B shows the structure of the p21Cip1 promoter, which includes a cluster of six Sp1 binding sites in the proximal promoter and two p53 response elements (p53-1 and p53-2) in the distal promoter (2, 31). Fig. 3B shows that MEP50 expression reduces the activity of the full-length p21Cip1 promoter by 70%. As a control, we show that mutation of one or both p53 transcription factor binding sites attenuates the MEP50 impact by reducing the overall promoter activity. These findings indicate that MEP50 and p53 produce opposite effects on p21Cip1 gene expression and that MEP50 expression can antagonize the action of p53.
FIGURE 3.

MEP50 regulation of p21Cip1 promoter activity: a role for p53. A, MEP50 suppresses p21Cip1 expression. KERn were electroporated with the indicated plasmid, and, after 24 h, extracts were prepared for quantitative RT-PCR detection of p21Cip1 mRNA and immunoblot detection of p21Cip1 and MEP50. EV, empty vector. B, opposing action of MEP50 and p53. A schematic of the human p21Cip1 promoter shows the Sp1 and p53 transcription factor response elements. The numbers indicate the distance in nucleotides relative to the transcription start site. KERn were transfected with 0.5 μg of p21-2316, which encodes the full-length wild-type p21Cip1 promoter, or the promoter harboring mutations at the p53-1 or p53-2 sites, linked to luciferase. After 24 h, extracts were prepared for the luciferase activity assay. The values are mean ± S.E. (n = 3). C–E, KERn were transfected with 0.5 μg of PG13-Luc (the p53 gene promoter linked to luciferase) or p21-2326 and the indicated micrograms of pcDNA3 or pcDNA3-FLAG-MEP50. 48 h post-transfection, cell extracts were prepared and assayed for promoter activity. The values are mean ± S.E. (n = 3). *, p < 0.005.
We next examined whether MEP50 expression reduces p53 as part of the mechanism that leads to reduced p21Cip1 promoter activity. Fig. 3C shows that MEP50 expression reduces the level of p53-encoding mRNA, and Fig. 3D shows that this is associated with a reduction in p53 (PG13-Luc) promoter activity. Fig. 3E confirms that this is associated with loss of p21Cip1 promoter activity. These findings suggest that MEP50 suppresses p53 gene expression as part of the mechanism by which MEP50 reduces p21Cip1 expression.
MEP50 and PRMT Interact at the p21Cip1 Promoter
A major role of the PRMT5-MEP50 complex is catalyzing the formation of H4R3me2s and H3R8me2s (39). These modifications occur at histones associated with genes that are undergoing silencing (22, 39). We therefore examined the role of MEP50 in modifying H3 and H4 associated with the p21Cip1 promoter. We selected the biologically important p21Cip1 promoter p53-2 binding response element as a target (2, 31, 32, 40). This region is known to bind p53 as a requirement for increased p21Cip1 expression (32, 40). Fig. 4A uses a ChIP analysis to show that MEP50 knockdown reduces MEP50 and PRMT5 association with the p21Cip1 promoter. Further ChIP analysis (Fig. 4B) shows that loss of MEP50 is associated with reduced p21Cip1 promoter-associated H4R3me2s formation but no reduction in H3R8me2s formation. These findings suggest that the increase in p21Cip1 expression observed in MEP50 knockdown cells is, in part, due to loss of PRMT5 and MEP50 and reduced formation of H4R3me2s in chromatin surrounding the p53-2 DNA response element in the p21Cip1 promoter. It is not clear why H3R8me2s levels are not reduced.
FIGURE 4.
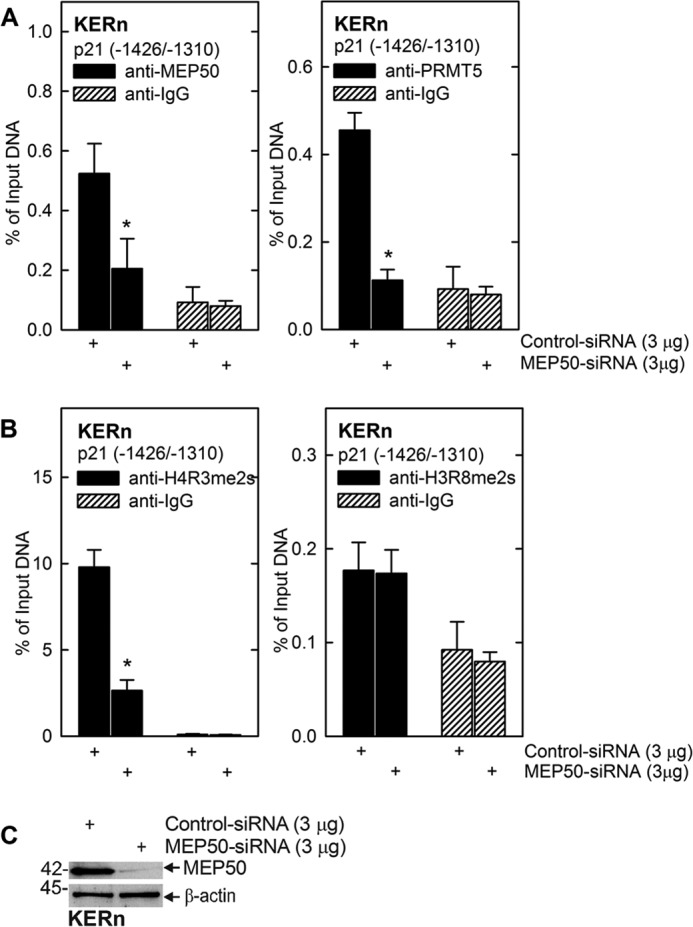
MEP50 knockdown reduces MEP50 and PRMT5 association at the p21Cip1 promoter. A and B, KERn were electroporated with 3 μg of control siRNA or MEP50 siRNA. After 48 h, extracts were prepared for ChIP analysis. DNA from 1 million cells was sheared, and 50,000 cell equivalents of DNA were used for immunoprecipitation. The primers span the p21Cip1 promoter region that includes the p53-2 site. The values are mean ± S.E. (n = 3). *, p < 0.005. C, extracts were prepared from the electroporated cells after 48 h to confirm MEP50 knockdown.
PKCδ and p38δ Regulation of the MEP50 Level
The PKCδ, MEKK1, MEK3, and p38δ signaling cascade is a central controller of keratinocyte proliferation. Activation of this cascade by overexpression of PKCδ or p38δ suppresses proliferation and enhances differentiation (9), which is associated with a specific increase in p21Cip1 gene expression (1, 2, 31). We wanted to assess whether activation of this cascade results in reduced expression of MEP50 or PRMT5. Keratinocytes were grown in the presence of empty virus or PKCδ- or p38δ-encoding adenovirus, and, after 48 h, extracts were prepared to assess the impact on total MEP50, PRMT5, H4R3me2s, and H3R8me2s levels. Fig. 5A shows that PKCδ or p38δ expression reduces the total MEP50 and PRMT5 levels and the total cellular levels of H3R8me2s and H4R3me2s while increasing the p21Cip1 level. We next assessed, by ChIP analysis, whether PKCδ or p38δ expression is associated with reduced MEP50 or PRMT5 interaction at the p21Cip1 gene promoter p53-2 response element. Fig. 5, B and C, shows that PKCδ or p38δ expression results in reduced MEP50 and PRMT5 interaction and reduced H3R8me2s and H4R3me2s histone modification at the p21Cip1 promoter p53-2 site.
FIGURE 5.

PKCδ and p38δ regulate MEP50 and PRMT5 level and activity. A, KERn were infected with tAd5-EV, Ad5-PKCδ, or tAd5-HA-p38δ (multiplicity of infection (MOI) 10), and, at 48 h, extracts were prepared for detection of PKCδ, MEP50, H3R8me2s, and H4R3me2s. Similar results were obtained in three different experiments. EV, empty vector. B and C, KERn were infected as above, and, at 48 h, extracts were prepared for ChIP using primers spanning the p21Cip1 promoter region that includes the p53-2 site. The values are mean ± S.E. (n = 3). *, p < 0.005).
Impact of TPA Treatment on Promoter Activity
We also examined whether a known activator of the PKCδ/p38δ cascade (4) alters MEP50 and PRMT5 expression. Fig. 6A shows that TPA treatment reduces total cellular levels of PRMT5 and MEP50. Moreover, the ChIP analysis in Fig. 6, B and C, shows that this treatment reduces MEP50 and PRMT5 levels at the p21Cip1 promoter p53-2 response element and confirms that this is associated with reduced element-associated H4R3me2s and H3R8me2s. Fig. 6D confirms that reduced modification of methylation of histones at the p21Cip1 promoter is associated with an increased p21Cip1 protein level.
FIGURE 6.
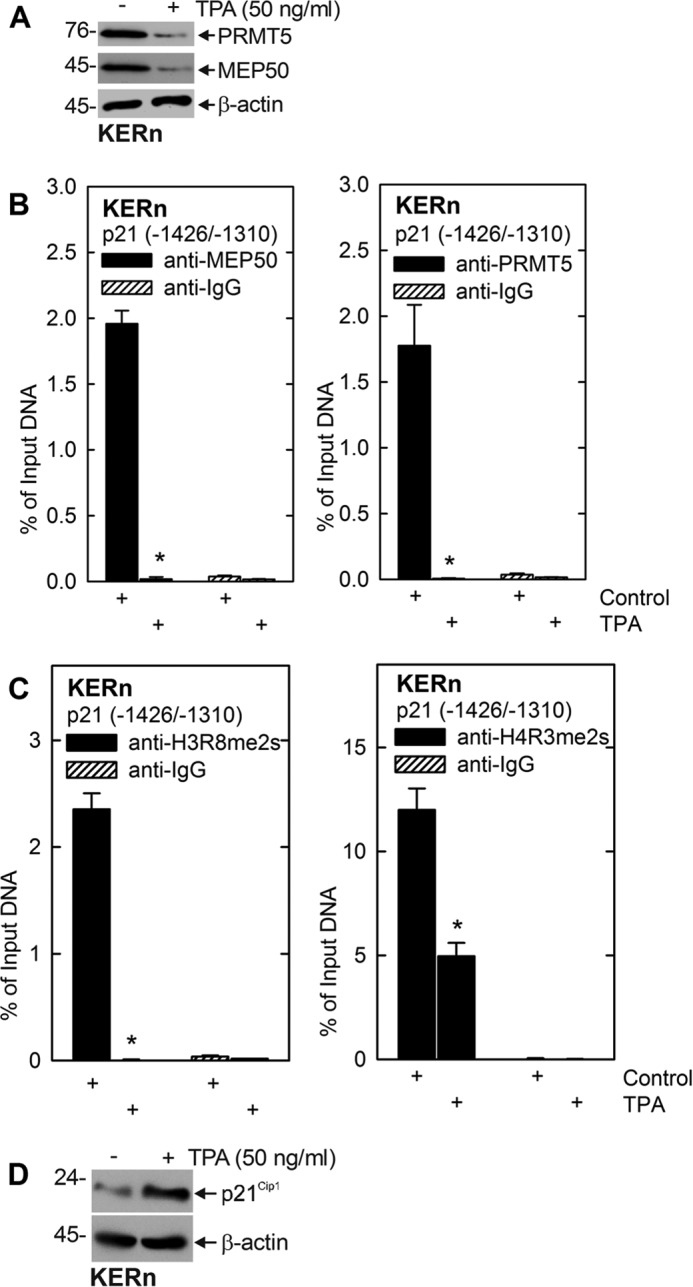
TPA regulates MEP50 and PRMT5 levels and activity. A, KERn were treated with 50 ng/ml TPA for 48 h, and extracts were prepared for detection of MEP50, PRMT5, H3R8me2s, and H4R3me2s. Similar results were obtained in three different experiments. B and C, KERn were treated with 50 ng TPA/ml, and, after 48 h, mRNA extracts were isolated for ChIP analysis and detection of MEP50 and PRMT5 interaction and H3R8-me2s and H4R3me2s formation at the p21Cip1 promoter. Similar results were obtained in three different experiments. D, cells were treated for 48 h with TPA, and p21Cip1 levels were monitored.
Biological Relevance of PRMT5 and MEP50 Regulation
To further assess the biological relevance of this regulation, we examined the impact of altering the MEP50 and PRMT5 levels on proliferation using a keratinocyte epidermal equivalent model. In this model, keratinocytes are grown at the air-liquid interface to produce a stratified, multilayered, and differentiated epidermal equivalent (41). This system is a faithful mimic of in vivo-like stratification (41). Primary foreskin keratinocytes were electroporated with control, MEP50, or PRMT5 siRNA and then transferred to Millicell chambers for growth as epidermal equivalent cultures.
To assess the biological impact of MEP50 or PRMT5 knockdown on differentiation and proliferation, we monitored the effect on morphology of the epidermal equivalent cultures. Fig. 7, A and B, shows that cultures expressing normal endogenous levels of MEP50 and PRMT5 undergo appropriate proliferation and differentiation to produce a normal multilayered tissue and stable cornified layer. In contrast, knockdown of either MEP50 or PRMT5 produces a significantly thinner epidermal equivalent. In particular, the viable middle layers are reduced relative to the cornified (top) dead layers, suggestive of reduced proliferation in the MEP50- and PRMT5-deficient cultures. To test this, we generated epidermal equivalents from cells electroporated with control, MEP50, and PRMT5 siRNA and then stained with anti-Ki67 as a marker of cell proliferation (42). Fig. 7, C and D, shows that MEP50 or PRMT5 knockdown reduces the number of Ki67-positive (proliferating) cells (green stain, arrows).
FIGURE 7.
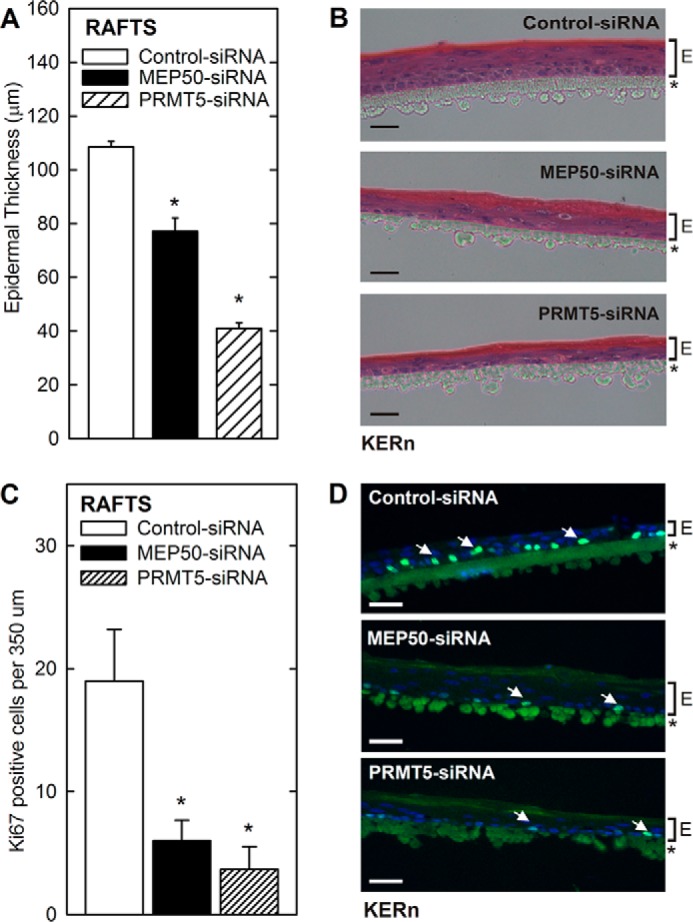
MEP50 and PRMT5 regulate differentiation and proliferation in an epidermal equivalent model. KERn were electroporated twice with control or MEP50 or PRMT5 siRNA and seeded for epidermal equivalent culture. After 4 days of exposure at the air-liquid interface, the equivalents were harvested and sectioned. A and B, MEP50 and PRMT5 are required for appropriate skin equivalent formation. KERn were electroporated twice with control, MEP50, or PRMT5 siRNA and seeded for epidermal equivalent culture. After 4 days of exposure at the air-liquid interface, the equivalents were harvested and stained with hematoxylin and eosin. The nylon support membrane is indicated by an asterisk, and the extent of the epidermis is indicated (E). Similar results were observed in three separate experiments. The graph compares epidermal equivalent thickness among control, MEP50, and PRMT5 siRNA cultures. The values are mean ± S.E. (n = 3). *, p < 0.005. Scale bars = 100 μm. C and D, MEP50 and PRMT5 are required for cell proliferation. KERn were electroporated twice with control, MEP50, or PRMT5 siRNA and seeded for epidermal equivalent culture. After 4 days, the equivalents were stained with Ki67 antibody and Hoechst. The membrane (asterisk) and extent of epidermis (E) are shown. The graph quantitates the number of Ki67-positive cells in the control, MEP50, and PRMT5 siRNA rafts. Similar results were observed in three separate experiments. Significant differences were determined using Student's t test (*, p < 0.005). Note that the blue-green spots are Ki67 staining compared with the larger stained circular structures, which are nonspecific staining to the nylon membrane.
Discussion
Keratinocytes constitute the major cell type of the epidermis (29). These cells begin as proliferative cells in the epidermal basal layer and undergo a highly orchestrated differentiation program leading to cornified envelope formation. The result is the formation of a multilayered epidermis in which differentiated cells are released from the surface (29). Decoding the mechanisms that control epidermal homeostasis is an important goal, as is understanding how deregulation of this process leads to disease. Novel PKC isoforms play pivotal roles in the regulation of epidermal homeostasis (5, 7, 43). They activate MAPK signaling to direct specific transcription factors to increase expression of differentiation-associated genes (4). Simultaneously, they control keratinocyte proliferation by increasing the expression of growth-suppressing genes, including p21Cip1 (2). Previous studies from our group have shown that PKCδ activates a MEKK1/MEK3/p38δ cascade that stimulates KLF4, Sp1, and p53 transcription factor association with the p21Cip1 promoter, leading to increased p21Cip1 expression and reduced cell proliferation (2, 34). However, less is known about mechanisms that antagonize this action to maintain keratinocyte proliferative potential.
Our recent mass spectrometry analysis identified PRMT5, a type II symmetric arginine methyltransferase, as a novel component of the PKCδ/p38δ regulatory complex (20). We further showed that PRMT5 acts to enhance cell survival by catalyzing symmetric dimethyl arginine modification of target proteins in this complex, leading to inhibition of p38δ activity (20). This represents a mechanism whereby PRMT5 acts to suppress activity in a growth suppression signaling cascade.
However, PRMT5 is also known to act at the level of chromatin to increase histone arginine dimethylation as a mechanism to silence gene expression (44). Silencing is mediated by PRMT5-MEP50-dependent histone arginine dimethylation of H4R3 and H3R8 to form H4R3me2s and H3R8me2s (21, 44, 45). In this study, we examine the impact of PRMT5 on histone arginine dimethylation of the p21Cip1 promoter as a mechanism of keratinocyte growth control. We focus on the role of MEP50, a cofactor of PRMT5 required for PRMT5 catalytic activity. We find that MEP50 is abundantly expressed in the foreskin epidermis and monolayer keratinocyte cultures, where it is present in both the nucleus and cytoplasm. This localization is similar to that observed in other cell types (38). We show that MEP50 knockdown leads to a substantial reduction in cell proliferation. Moreover, the reduction is associated with increased expression of p21Cip1, a known inhibitor of keratinocyte proliferation (2, 31). Knockdown studies show that the MEP50 level is inversely correlated with the p21Cip1 mRNA level and promoter activity.
Previous studies show that the p53 transcription factor is a key positive activator of p21Cip1 expression (40) and that it acts by binding to two p53 response elements, p53-1 and p53-2, in the distal region of the p21Cip1 promoter. In an effort to dissect the mechanism of MEP50 action, we manipulated MEP50 expression using siRNA and expression vectors and monitored the impact on p21Cip1 gene expression, promoter activity, and modification of histone surrounding the p53-2 response element in the p21Cip1 promoter. These studies showed an inverse relationship between the MEP50 level and p21Cip1 expression and promoter activity. We further show that MEP50 interaction at the p53-2 response element is associated with the acquisition of the histone silencing marks H4R3me2s and H3R8m2s. This is a particularly important finding because the p53-2 response element (nucleotides −1393/−1374) in the p21Cip1 promoter is a key positive regulatory element (31). This suggests that MEP50, acting with PRMT5, inhibits p21Cip1 by producing silencing histone marks in this region of chromatin. We have not assayed the impact of these manipulations on the arginine dimethylation of histones H4 and H3 at the p53-1 site (nucleotides −2281/−2261), but we assume that the results would be similar.
Few studies have examined the interplay between cell cycle regulators such as p21Cip1 and PRMT5-MEP50. However, one study suggests that MEP50 is phosphorylated by CDK4 and that this leads to increased PRMT5-MEP50 activity, which enhances events leading to cyclin D1-dependent cell proliferation (46). A second study shows that PRMT5 accelerates cell cycle progression through G1 by up-regulation of CDK4, CDK6 and cyclins D1, D2, and E1 and that this was associated with activation of PI3K/AKT activity (47). Therefore, our findings are consistent with cell cycle regulation being a target of PRMT5-MEP50 activity and with PRMT5-MEP50 acting to enhance cell proliferation.
A novel aspect of this work is identifying an opposing/antagonistic relationship between PKCδ/p38δ signaling and MEP50-PRMT5 action. We show that activation of PKCδ/p38δ signaling, by expression of either of these kinases or by treatment with an activator of PKCδ (TPA) results in increased p21Cip1 expression via direct transcriptional activation of p21Cip1 gene expression (2, 31). This study shows that PRMT5-MEP50 opposes this regulation and that this is associated with symmetric dimethyl arginine modification of histones on the p21Cip1 promoter. Therefore, this study establishes a new biochemical link that may help explain the opposing actions of PKCδ/p38δ signaling and MEP50-PRMT5 as regulators of the p21Cip1 locus. This would be consistent with our previous report showing that PRMT5 dimethylates proteins in a p38δ complex, leading to reduced p38δ phosphorylation and activity in this cascade (20). Together, these findings suggest that PRMT5-MEP50 inhibits PKCδ/p38δ signaling in keratinocytes via multiple mechanisms.
An additional significant finding is that PKCδ/p38δ-dependent signaling reduces the MEP50 and PRMT5 protein level and histone arginine dimethylation. Specifically, H3R8me2s and H4R3me2s levels are reduced at key transcriptional elements in the p21Cip1 promoter. This suggests that activity in this prodifferentiation/antiproliferation cascade actively suppresses function of MEP50-PRMT5 pro-proliferation/prosurvival regulation. This, in theory, suggests that this combination of events leads to a more efficient induction of gene (p21Cip1) expression. These findings are also interesting from the perspective that PRMT5 and MEP50 have been shown to be prosurvival proteins in a variety of cancer models (35–38, 46, 48–50), which supports the idea that these proteins are elevated under conditions where cells need to retain proliferative potential.
To better assess the biological relevance of MEP50, we used an epidermal equivalent model that efficiently mimics in vivo keratinocyte differentiation (41). Our study shows that knockdown of MEP50 or PRMT5 results in a significant reduction in thickness of the epidermis. This is in agreement with the observation that MEP50 or PRMT5 knockdown decreases keratinocyte cell number in studies of monolayer cultures. Additionally, staining of sections from the control, MEP50, and PRMT5 knockdown epidermal equivalents show fewer Ki67-positive cells in the basal layer of MEP50 or PRMT5 knockdown cells compared with the control equivalents. These data strongly suggest that MEP50 and PRMT5 are likely to have a physiologically meaningful role in controlling keratinocyte proliferation in vivo.
In summary, we have shown that a PKCδ/p38δ cascade activates p53 interaction at the p21Cip1 promoter p53 response elements to increase transcription of the p21Cip1 gene (Fig. 8). p21Cip1 inhibits cyclin-dependent kinase activity to suppress cell cycle progression and reduce cell proliferation (2, 31). PRMT5 and MEP50 act to antagonize this action by arginine-dimethylating proteins that are part of a p38δ regulatory complex to reduce p38δ activity (20) and also act at the p21Cip1 gene level to arginine-dimethylate histones H3 and H4 to produce closed chromatin and p21Cip1 gene silencing. This leads to enhanced proliferation. We further show that activation of the PKCδ/p38δ cascade by PKCδ/p38δ overexpression or TPA treatment stimulates p21Cip1 expression by three mechanisms (Fig. 8). First, it acts by stimulating p53 binding to the p53 response elements in the p21Cip1 promoter to increase transcription. Second, it suppresses production of PRMT5 and MEP50 mRNA and protein, leading to reduced arginine dimethylation of histones H3 and H4 and derepression of p21Cip1 gene expression (Fig. 8). Third, loss of PRMT5-MEP50 reduces inhibition of p38δ kinase activity, strengthening the stimulus to increase p21Cip1 levels.
FIGURE 8.
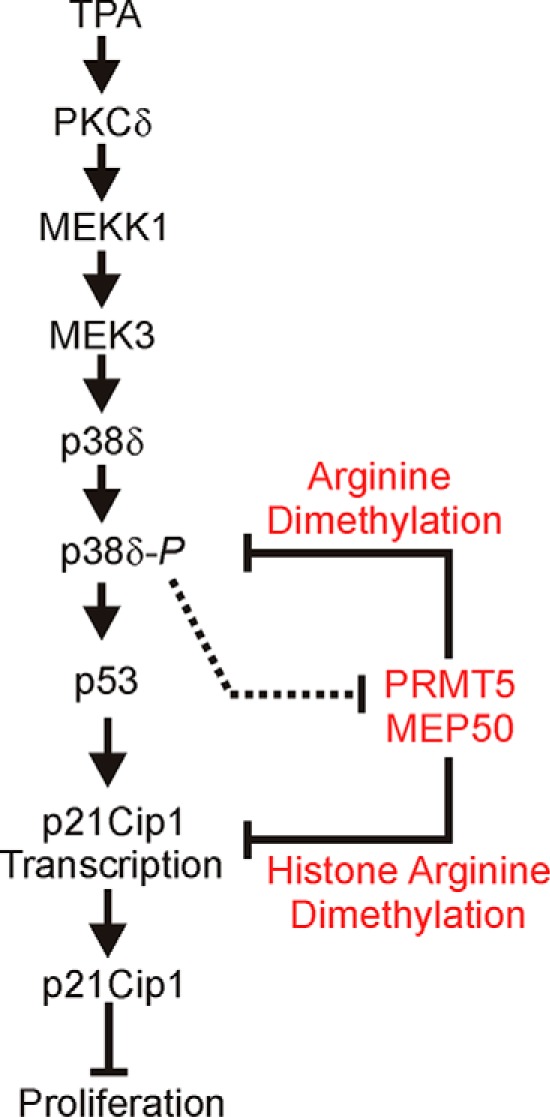
Proposed regulatory model: a balance between transcriptional and epigenetic regulation. Arrows indicate a stimulus, and flat-headed bars inhibition. TPA stimulation or PKCδ overexpression activates MEKK1, MEK3, p38δ, and p53 to increase p21Cip1 expression, which leads to reduced keratinocyte proliferation. PRMT5/MEP50 arginine dimethylates proteins in the p38δ complex to inhibit p38δ (20), and arginine dimethylates histones H3R8me2s and H4R3me2s in the p21Cip1 promoter to reduce p21Cip1 expression, leading to increased cell proliferation. We propose that the PKCδ signaling is dominant in differentiated/non-proliferative cells and that PRMT5-MEP50 is dominant in non-differentiated/proliferative cells. The dashed line indicates that PKCδ/p38δ signaling can reduce PRMT5 and MEP50 expression to suppress pro-proliferation signaling.
This work was supported, in whole or in part, by National Institutes of Health Grants R21AR065266 and RO1CA131064 (to R. L. E.). This work was also supported by a pilot award from the Greenebaum Cancer Center.
- H4R3me2s
- symmetrical dimethylation of arginine 3 of histone H4
- H3R8me2s
- symmetrical dimethylation of arginine 8 of histone H
- TPA
- 12-O-tetradecanoylphorbol-13-acetate
- KERn
- keratinocyte(s)
- KSFM
- keratinocyte serum-free medium.
References
- 1. Adhikary G., Chew Y. C., Reece E. A., Eckert R. L. (2010) PKC-δ and -η, MEKK-1, MEK-6, MEK-3, and p38-δ are essential mediators of the response of normal human epidermal keratinocytes to differentiating agents. J. Invest. Dermatol. 130, 2017–2030 [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 2. Chew Y. C., Adhikary G., Wilson G. M., Reece E. A., Eckert R. L. (2011) PKCδ suppresses keratinocyte proliferation by increasing p21CIP1 level by a KLF4-dependent mechanism. J. Biol. Chem. 286, 28771–28782 [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 3. Chew Y. C., Adhikary G., Xu W., Wilson G. M., Eckert R. L. (2013) Protein kinase C δ increases Kruppel-like factor 4 Protein, which drives involucrin gene transcription in differentiating keratinocytes. J. Biol. Chem. 288, 17759–17768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Efimova T., LaCelle P., Welter J. F., Eckert R. L. (1998) Regulation of human involucrin promoter activity by a protein kinase C, Ras, MEKK1, MEK3, p38/RK, AP1 signal transduction pathway. J. Biol. Chem. 273, 24387–24395 [DOI] [PubMed] [Google Scholar]
- 5. Efimova T., Deucher A., Kuroki T., Ohba M., Eckert R. L. (2002) Novel protein kinase C isoforms regulate human keratinocyte differentiation by activating a p38 δ mitogen-activated protein kinase cascade that targets CCAAT/enhancer-binding protein α. J. Biol. Chem. 277, 31753–31760 [DOI] [PubMed] [Google Scholar]
- 6. Efimova T., Broome A. M., Eckert R. L. (2003) A regulatory role for p38 δ MAPK in keratinocyte differentiation: evidence for p38 δ-ERK1/2 complex formation. J. Biol. Chem. 278, 34277–34285 [DOI] [PubMed] [Google Scholar]
- 7. Efimova T., Broome A. M., Eckert R. L. (2004) Protein kinase Cδ regulates keratinocyte death and survival by regulating activity and subcellular localization of a p38δ-extracellular signal-regulated kinase 1/2 complex. Mol. Cell Biol. 24, 8167–8183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Eckert R. L., Efimova T., Balasubramanian S., Crish J. F., Bone F., Dashti S. (2003) p38 mitogen-activated protein kinases on the body surface: a function for p38δ. J. Invest. Dermatol. 120, 823–828 [DOI] [PubMed] [Google Scholar]
- 9. Eckert R. L., Crish J. F., Efimova T., Dashti S. R., Deucher A., Bone F., Adhikary G., Huang G., Gopalakrishnan R., Balasubramanian S. (2004) Regulation of involucrin gene expression. J. Invest. Dermatol. 123, 13–22 [DOI] [PubMed] [Google Scholar]
- 10. Welter J. F., Eckert R. L. (1995) Differential expression of fos and jun family members c-fos, fosB, Fra-1, Fra-2, c-jun, junB and junD during human epidermal keratinocyte differentiation. Oncogene 11, 2681–2687 [PubMed] [Google Scholar]
- 11. Welter J. F., Crish J. F., Agarwal C., Eckert R. L. (1995) Fos-related antigen (Fra-1), junB, and junD activate human involucrin promoter transcription by binding to proximal and distal AP1 sites to mediate phorbol ester effects on promoter activity. J. Biol. Chem. 270, 12614–12622 [DOI] [PubMed] [Google Scholar]
- 12. Han B., Rorke E. A., Adhikary G., Chew Y. C., Xu W., Eckert R. L. (2012) Suppression of AP1 transcription factor function in keratinocyte suppresses differentiation. PLoS ONE 7, e36941. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 13. Banks E. B., Crish J. F., Eckert R. L. (1999) Transcription factor Sp1 activates involucrin promoter activity in non-epithelial cell types. Biochem. J. 337, 507–512 [PMC free article] [PubMed] [Google Scholar]
- 14. Banks E. B., Crish J. F., Welter J. F., Eckert R. L. (1998) Characterization of human involucrin promoter distal regulatory region transcriptional activator elements: a role for Sp1 and AP1 binding sites. Biochem. J. 331, 61–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Crish J. F., Zaim T. M., Eckert R. L. (1998) The distal regulatory region of the human involucrin promoter is required for expression in epidermis. J. Biol. Chem. 273, 30460–30465 [DOI] [PubMed] [Google Scholar]
- 16. Crish J. F., Bone F., Banks E. B., Eckert R. L. (2002) The human involucrin gene contains spatially distinct regulatory elements that regulate expression during early versus late epidermal differentiation. Oncogene 21, 738–747 [DOI] [PubMed] [Google Scholar]
- 17. Crish J. F., Gopalakrishnan R., Bone F., Gilliam A. C., Eckert R. L. (2006) The distal and proximal regulatory regions of the involucrin gene promoter have distinct functions and are required for in vivo involucrin expression. J. Invest. Dermatol. 126, 305–314 [DOI] [PubMed] [Google Scholar]
- 18. Crish J. F., Eckert R. L. (2008) Synergistic activation of human involucrin gene expression by Fra-1 and p300: evidence for the presence of a multiprotein complex. J. Invest. Dermatol. 128, 530–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rorke E. A., Adhikary G., Jans R., Crish J. F., Eckert R. L. (2010) AP1 factor inactivation in the suprabasal epidermis causes increased epidermal hyperproliferation and hyperkeratosis but reduced carcinogen-dependent tumor formation. Oncogene 29, 5873–5882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kanade S. R., Eckert R. L. (2012) Protein arginine methyltransferase 5 (PRMT5) signaling suppresses protein kinase Cδ- and p38δ-dependent signaling and keratinocyte differentiation. J. Biol. Chem. 287, 7313–7323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ho M. C., Wilczek C., Bonanno J. B., Xing L., Seznec J., Matsui T., Carter L. G., Onikubo T., Kumar P. R., Chan M. K., Brenowitz M., Cheng R. H., Reimer U., Almo S. C., Shechter D. (2013) Structure of the arginine methyltransferase PRMT5-MEP50 reveals a mechanism for substrate specificity. PLoS ONE 8, e57008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Karkhanis V., Hu Y. J., Baiocchi R. A., Imbalzano A. N., Sif S. (2011) Versatility of PRMT5-induced methylation in growth control and development. Trends Biochem. Sci. 36, 633–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Antonysamy S., Bonday Z., Campbell R. M., Doyle B., Druzina Z., Gheyi T., Han B., Jungheim L. N., Qian Y., Rauch C., Russell M., Sauder J. M., Wasserman S. R., Weichert K., Willard F. S., Zhang A., Emtage S. (2012) Crystal structure of the human PRMT5:MEP50 complex. Proc. Natl. Acad. Sci. U.S.A. 109, 17960–17965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bedford M. T., Richard S. (2005) Arginine methylation an emerging regulator of protein function. Mol. Cell 18, 263–272 [DOI] [PubMed] [Google Scholar]
- 25. Yang Y., Bedford M. T. (2013) Protein arginine methyltransferases and cancer. Nat. Rev. Cancer 13, 37–50 [DOI] [PubMed] [Google Scholar]
- 26. Fabbrizio E., El Messaoudi S., Polanowska J., Paul C., Cook J. R., Lee J. H., Negre V., Rousset M., Pestka S., Le Cam A., Sardet C. (2002) Negative regulation of transcription by the type II arginine methyltransferase PRMT5. EMBO Rep. 3, 641–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tae S., Karkhanis V., Velasco K., Yaneva M., Erdjument-Bromage H., Tempst P., Sif S. (2011) Bromodomain protein 7 interacts with PRMT5 and PRC2, and is involved in transcriptional repression of their target genes. Nucleic Acids Res. 39, 5424–5438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bedford M. T., Clarke S. G. (2009) Protein arginine methylation in mammals: who, what, and why. Mol. Cell 33, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Eckert R. L., Crish J. F., Robinson N. A. (1997) The epidermal keratinocyte as a model for the study of gene regulation and cell differentiation. Physiol. Rev. 77, 397–424 [DOI] [PubMed] [Google Scholar]
- 30. Rorke E. A., Adhikary G., Young C. A., Roop D. R., Eckert R. L. (2015) Suppressing AP1 factor signaling in suprabasal epidermis produces a keratoderma phenotype. J. Invest. Dermatol. 135, 170–180 [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 31. Chew Y. C., Adhikary G., Wilson G. M., Xu W., Eckert R. L. (2012) Sulforaphane induction of p21(Cip1) cyclin-dependent kinase inhibitor expression requires p53 and Sp1 transcription factors and is p53-dependent. J. Biol. Chem. 287, 16168–16178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. el-Deiry W. S., Tokino T., Velculescu V. E., Levy D. B., Parsons R., Trent J. M., Lin D., Mercer W. E., Kinzler K. W., Vogelstein B. (1993) WAF1, a potential mediator of p53 tumor suppression. Cell 75, 817–825 [DOI] [PubMed] [Google Scholar]
- 33. Li J. J., Cao Y., Young M. R., Colburn N. H. (2000) Induced expression of dominant-negative c-jun downregulates NFκB and AP-1 target genes and suppresses tumor phenotype in human keratinocytes. Mol. Carcinog. 29, 159–169 [DOI] [PubMed] [Google Scholar]
- 34. Saha K., Adhikary G., Kanade S. R., Rorke E. A., Eckert R. L. (2014) p38δ regulates p53 to control p21Cip1 expression in human epidermal keratinocytes. J. Biol. Chem. 289, 11443–11453 [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 35. Peng Y., Chen F., Melamed J., Chiriboga L., Wei J., Kong X., McLeod M., Li Y., Li C. X., Feng A., Garabedian M. J., Wang Z., Roeder R. G., Lee P. (2008) Distinct nuclear and cytoplasmic functions of androgen receptor cofactor p44 and association with androgen-independent prostate cancer. Proc. Natl. Acad. Sci. U.S.A. 105, 5236–5241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ligr M., Patwa R. R., Daniels G., Pan L., Wu X., Li Y., Tian L., Wang Z., Xu R., Wu J., Chen F., Liu J., Wei J. J., Lee P. (2011) Expression and function of androgen receptor coactivator p44/Mep50/WDR77 in ovarian cancer. PLoS ONE 6, e26250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Peng Y., Li Y., Gellert L. L., Zou X., Wang J., Singh B., Xu R., Chiriboga L., Daniels G., Pan R., Zhang D. Y., Garabedian M. J., Schneider R. J., Wang Z., Lee P. (2010) Androgen receptor coactivator p44/Mep50 in breast cancer growth and invasion. J. Cell Mol. Med. 14, 2780–2789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wei T. Y., Hsia J. Y., Chiu S. C., Su L. J., Juan C. C., Lee Y. C., Chen J. M., Chou H. Y., Huang J. Y., Huang H. M., Yu C. T. (2014) Methylosome protein 50 promotes androgen- and estrogen-independent tumorigenesis. Cell. Signal. 26, 2940–2950 [DOI] [PubMed] [Google Scholar]
- 39. Molina-Serrano D., Schiza V., Kirmizis A. (2013) Cross-talk among epigenetic modifications: lessons from histone arginine methylation. Biochem. Soc. Trans. 41, 751–759 [DOI] [PubMed] [Google Scholar]
- 40. Macleod K. F., Sherry N., Hannon G., Beach D., Tokino T., Kinzler K., Vogelstein B., Jacks T. (1995) p53-dependent and independent expression of p21 during cell growth, differentiation, and DNA damage. Genes Dev. 9, 935–944 [DOI] [PubMed] [Google Scholar]
- 41. Poumay Y., Dupont F., Marcoux S., Leclercq-Smekens M., Hérin M., Coquette A. (2004) A simple reconstructed human epidermis: preparation of the culture model and utilization in in vitro studies. Arch. Dermatol. Res. 296, 203–211 [DOI] [PubMed] [Google Scholar]
- 42. Gerdes J., Lemke H., Baisch H., Wacker H. H., Schwab U., Stein H. (1984) Cell cycle analysis of a cell proliferation-associated human nuclear antigen defined by the monoclonal antibody Ki-67. J. Immunol. 133, 1710–1715 [PubMed] [Google Scholar]
- 43. Efimova T., Eckert R. L. (2000) Regulation of human involucrin promoter activity by novel protein kinase C isoforms. J. Biol. Chem. 275, 1601–1607 [DOI] [PubMed] [Google Scholar]
- 44. Pal S., Baiocchi R. A., Byrd J. C., Grever M. R., Jacob S. T., Sif S. (2007) Low levels of miR-92b/96 induce PRMT5 translation and H3R8/H4R3 methylation in mantle cell lymphoma. EMBO J. 26, 3558–3569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Krause C. D., Yang Z. H., Kim Y. S., Lee J. H., Cook J. R., Pestka S. (2007) Protein arginine methyltransferases: evolution and assessment of their pharmacological and therapeutic potential. Pharmacol. Ther. 113, 50–87 [DOI] [PubMed] [Google Scholar]
- 46. Aggarwal P., Vaites L. P., Kim J. K., Mellert H., Gurung B., Nakagawa H., Herlyn M., Hua X., Rustgi A. K., McMahon S. B., Diehl J. A. (2010) Nuclear cyclin D1/CDK4 kinase regulates CUL4 expression and triggers neoplastic growth via activation of the PRMT5 methyltransferase. Cancer Cell 18, 329–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wei T. Y., Juan C. C., Hisa J. Y., Su L. J., Lee Y. C., Chou H. Y., Chen J. M., Wu Y. C., Chiu S. C., Hsu C. P., Liu K. L., Yu C. T. (2012) Protein arginine methyltransferase 5 is a potential oncoprotein that upregulates G1 cyclins/cyclin-dependent kinases and the phosphoinositide 3-kinase/AKT signaling cascade. Cancer Sci. 103, 1640–1650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nicholas C., Yang J., Peters S. B., Bill M. A., Baiocchi R. A., Yan F., Sïf S., Tae S., Gaudio E., Wu X., Grever M. R., Young G. S., Lesinski G. B. (2013) PRMT5 is upregulated in malignant and metastatic melanoma and regulates expression of MITF and p27(Kip1.). PLoS ONE 8, e74710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gu Z., Gao S., Zhang F., Wang Z., Ma W., Davis R. E., Wang Z. (2012) Protein arginine methyltransferase 5 is essential for growth of lung cancer cells. Biochem. J. 446, 235–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gu Z., Zhang F., Wang Z. Q., Ma W., Davis R. E., Wang Z. (2013) The p44/wdr77-dependent cellular proliferation process during lung development is reactivated in lung cancer. Oncogene 32, 1888–1900 [DOI] [PMC free article] [PubMed] [Google Scholar]