

Background: The transcriptional regulation of keratin (KRT) 13 and its underlying mechanism has not been fully elucidated.
Results: Up-regulation of KRT13 by Krüppel-like factor 4 (KLF4) is mediated through GKRE in the KRT13 promoter.
Conclusion: KLF4 induces differentiation of ESCC by promoting KRT13 transcription.
Significance: KLF4 plays a significant and previously unrecognized role in regulation of KRT13 and cell differentiation.
Keywords: cell cycle, differentiation, keratin, Kruppel-like factor 4 (KLF4), transcription regulation
Abstract
Squamous cell differentiation requires the coordinated activation and repression of genes specific to the differentiation process; disruption of this program accompanies malignant transformation of epithelium. The exploration of genes that control epidermal proliferation and terminal differentiation is vital to better understand esophageal carcinogenesis. KLF4 is a member of the KLF family of transcription factors and is involved in both cellular proliferation and differentiation. This study using immunohistochemistry analysis of KLF4 in clinical specimens of esophageal squamous cell carcinoma (ESCC) demonstrated that decreased KLF4 was substantially associated with poor differentiation. Moreover, we determined that both KLF4 and KRT13 levels were undoubtedly augmented upon sodium butyrate-induced ESCC differentiation and G1 phase arrest. Conversely, silencing of KLF4 and KRT13 abrogated the inhibition of G1-S transition induced by sodium butyrate. Molecular investigation demonstrated that KLF4 transcriptionally regulated KRT13 and the expression of the two molecules appreciably correlated in ESCC tissues and cell lines. Collectively, these results suggest that KLF4 transcriptionally regulates KRT13 and is invovled in ESCC cell differentiation.
Introduction
Esophageal cancer is an aggressive disease with a propensity to spread both locoregionally and distally and leads to the sixth leading cause of cancer-related death worldwide (1). Distinct histological, genetic and epigenetic alterations and sites of origin characterize esophageal cancer subtypes and result in a different prognosis to surgical resection, chemotherapy and radiotherapy. In East Asian countries, ESCC3 is the most prevalent distribution (2). Although surgery is considered the best treatment for esophageal cancer in terms of locoregional control and long term survival, recurrence and metastases to the liver have occurred in most cases who undergo complete carcinoma resection, which results in only a 25% 5-year survival rate for patients (3). The absence of effective targeted therapies and poor response to standard chemotherapy often attribute to a rapidly fatal outcome for this disease. Thus, understanding the molecular mechanisms that drive ESCC and translating this knowledge into new encouraging intervention strategies and treatment options are needed.
KLF4 is an epithelium enriched, zinc finger-containing transcription factor that plays an important role in cell differentiation and proliferation. Mice homozygous for a null mutation in KLF4 die by postnatal day 1 and show selective perturbation of late stage differentiation structures in the epidermis (4). Squamous epithelium-specific KLF4 knock-out mice manifest an increased basal cell proliferation and a delayed cellular maturation, and this develops into epithelial hypertrophy and subsequent dysplasia, accompanying down-regulation of critical late stage differentiation factors, which show that KLF4 is essential for squamous epithelial differentiation and homeostasis maintenance (5). KLF4 knock-out mice exhibit a 90% decrease in the number of goblet cells and abnormal expression of the goblet cell-specific marker Muc2, which demonstrates that KLF4 is also required for terminal differentiation of goblet cells in the colon (6). Moreover, KLF4 promotes the context of enterocyte differentiation by transcriptionally activating the differentiation marker gene intestinal alkaline phosphatase (7). Ablation of KLF4 leads to increased proliferation and altered differentiation in the gastric epithelium of mice, and the absent expression of KLF4 in human gastric cancer suggests that failure to activate KLF4 during normal cellular differentiation is a common feature of gastric cancers (8). KLF4 also induces postnatal maturation of corneal epithelium and maintenance of the ocular surface via up-regulating the expression of KRT12 and aquaporin 5 (9). Analysis of microarray data shows that nine KRT genes are up-regulated by KLF4, seven of which are located in a specific region on chromosome 12 (10). These results indicate that KLF4 may be involved in inducing epithelial differentiation through the locus control regions.
KRT is a family of intermediate filament proteins expressed by epithelial cells in a cell type-specific and differentiation-dependent manner, which has been classified into two types and encoded by more than 50 genes in separate clusters on chromosomes 12 and 17 (11). Cumulative evidence showed that the differential expression of individual KRTs in various carcinomas made them useful markers for the histopathological carcinoma subtype and provided relevant information concerning the differentiation state and origin of carcinomas, especially in patients with metastases. The directed transcriptional regulation of distinct KRT genes and the properties of their encoded protein resulted in the specific structural or regulatory functions of KRT filaments to act in a tissue-specific manner (12). KRT13, one of the type I KRT, is often paired with KRT4, which expresses in suprabasal layers of noncornified and stratified squamous epithelium (13). Furthermore, during the urothelial differentiation, a switch from a KRT13 (low)/KRT14 (high) to a KRT13 (high)/KRT14 (low) phenotype is observed when a transitional cell morphology was acquired (14). Alternatively, heterozygous missense mutation of mucosal KRT13 results in white sponge nevus, an autosomal dominant inherited form of mucosal leukokeratosis (15). In squamous cell carcinomas of the head and neck, KRT13 is much lower in neoplastic tissue compared with the matching normal squamous epithelium, and this suggests that KRT13 may be used as a new biomarker for squamous cell carcinomas of the head and neck and to monitor progression in individual patients (16). KRT13 is also thought to be an appropriate candidate for characterizing oral cancer, and it has been considered as a marker for detecting the micrometastases in cervical lymph nodes of oral cancer (17). In tongue squamous cell carcinoma, loss of KRT13 expression presents a high potential recurrence (18). In addition, cumulative evidence documented that the expression of KRT13 could be regulated by variety of factors. For instance, phosphatidylinositol 3-kinase activation promotes the expression of KRT13 in papilloma cells and induces the normal differentiation of human mucosal keratinocytes (19). While in breast cancer, ligand-differential recruitment of the estrogen receptor and coactivators elevates the KRT13 level and substantiates that the estrogen-response elements and Sp1 sites are involved in its ligand-dependent regulation (13). The structure and function of KLF4 are similar to Sp1, so we speculate that it may participate in regulating the expression of KRT13 at a transcriptional level.
Butyrate, a short chain fatty acid derived from dietary fiber, is involved in the regulation of cell growth and differentiation of the colonic epithelia, and it protects against the incidence of colorectal cancer (20). It has been reported that butyrate increased the expression of KLF4 coincident with differentiation and growth arrest along the goblet and absorptive cell lineages by interacting with the promoter and promoting histone acetylation (21). Treatment of oral squamous cell carcinoma with SB inhibited growth and elevated differentiation of the tumor accompanied by significant keratinization and increased expression of KRT13 (22), implicated that SB suppressed cell growth and induced cell differentiation possibly through mechanisms involving KRT13. Moreover, our previous results showed that the expressions of KLF4 and KRT13 are both down-regulated in ESCC compared with their normal counterparts by using a cDNA microarray (23). In this study, we confirmed that the expression of KLF4 was decreased in the clinical ESCC samples compared with their matched normal esophageal epithelium. Further analysis on the relationship between KLF4 expression and the clinicopathologic parameters of ESCC revealed that the KLF4 level is significantly associated with the differentiation of ESCC. Moreover, we verified the induction of morphologic differentiation of ESCC cells in concomitance with an increased expression of KRT13 upon SB in a KLF4-dependent manner and characterized the transcriptional regulation of KRT13 by KLF4.
Experimental Procedures
Reagents, Cell Culture, Transfection, and RNA Interference
Sodium butyrate was purchased from Sigma. The human ESCC cell lines, KYSE series, were generously provided by Dr. Y. Shimada (Kyoto University, Kyoto, Japan (24)). Cells were cultured in RPMI 1640 medium with 10% fetal bovine serum and antibiotics. Transfections were performed as described previously (25). Cells were transfected with siRNAs (25 nm) by HiPerFect (Qiagen) following the manufacturers' protocol. The sequences for siRNAs were added in Table 1.
TABLE 1.
siRNA sequences
| siRNA | Sequences |
|---|---|
| siRNA (control) | AATTCTCCGAACGTGTCACGT |
| siKLF4 (GenePharma) | 1) GGACGGCTGTGGATGGAAA |
| 2) GACCTGGACTTTATTCTCT | |
| 3) CCGAGGAGTTCAACGATCT | |
| 4) TGACCAGGCACTACCGTAA | |
| siRNA (control) | TTCTCCGAACGTGTCACGTTT |
| siKRT13 (Qiagen) | 1) CCGCAGTGAGATGGAGTGCCA |
| 2) CTCTGTTACCACCACCTCTAA | |
| 3) CAAGAGTGCAGAGCTGAACAA | |
| 4) TACGCTTTGGTTTCTCAACTT |
Plasmid Construction
KLF4 and KRT13 cDNA coding regions were cloned into the mammalian expression vectors pcDNA3.1, pcDEF and pFLAG-CMV-2. The DNA fragments of KRT13 promoter regions were cloned into pGL3-basic vector. The resulting construct was verified by direct sequencing. Primers used are listed in Table 3.
TABLE 3.
Sequences used in this study
F is forward, and R is reverse.
| Sequences | |
|---|---|
| For cloning | |
| FLAG-KRT13 (HindIII/BamHI) | F, TTAAGCTTAGCCTCCGCCTGCAGAGC |
| R, TTGGATCCGCAGATTTAAGGCCTACGGACATC | |
| For luciferase and mutant assay | |
| pGL3-KRT13 (512 bp) | F, TCGGGGTACCGGATCCAGGACATCCCAG |
| R, GCCGCTCGAGTGGTGAGAGCAGGATTGAG | |
| GKRE mutant | F, GCCTTTCGAGGGCTACGGTGACCTTGCAA |
| R, CCTTTTTTCTATACCTAAAACTTC | |
| For ChIP | |
| GKRE region | F, CGAACCAAGCAAAGTTTGTCATC |
| R, ACCCAGTATTAGAACGGGACCT | |
| Negative control region | F, TCCATTGTCAGCCCTGGAAT |
| R, TCAGGCTGGGAGAAAAGGAC | |
Tissue Specimens
Tissue microarray from 106 patients with ESCC was obtained at the First Affiliated Hospital of Anhui Medical University (Anhui province, China). Patients received no treatment before surgery and signed informed consent in accordance with the Institutional Review Board of the Chinese Academy of Medical Sciences Cancer Institute and Anhui Medical University standards and guidelines. Representative primary tumor regions and the corresponding histologically normal esophageal mucosa from each patient were snap-frozen in liquid nitrogen and stored at −80 °C (27). Additional sections were collected and embedded in paraffin for histological examination (28, 29).
Immunohistochemistry
The human ESCC tissue microarray was subjected to immunohistochemistry using antibodies against KLF4 (sc-20691; Santa Cruz Biotechnology) and KRT13 (C0791; Sigma), respectively. Briefly, deparaffinized tissue sections were treated by 3% hydrogen peroxide and processed for antigen retrieval (by heating in a microwave oven at 96 °C, in 0.01 m citrate buffer, pH 6, for 15 min), and after cooling at room temperature, they were blocked by normal goat serum for 30 min and incubated with anti-KLF4 or anti-KRT13 antibody at 4 °C overnight. The reaction products were colorized with PV9003 immunohistochemistry kit (Zhong Shan Goldenbridge, Beijing, China) and 3′,3-diaminobenzidine substrate-chromogen kit (Zhong Shan Goldenbridge), resulting in a brown signal. The evaluation of immunohistochemistry staining was described previously (30). Briefly, specimens were reviewed with staining intensity and staining extent. Staining intensity was rated as follows: 0, negative; 1, faint; 2, moderate; 3, strong. The expression extent was graded on a scale from 0 to 4 as follows: 0, no positive staining or only a few scattered positive cells; 1, cluster(s) of positively stained cells that accounted for <25% of the cells within a visual field; 2, cluster(s) of positively stained cells that accounted for 25–50% of the cells within a visual field; 3, cluster(s) of positively stained cells that accounted for 50–75% of the cells within a visual field; 4, cluster(s) of positively stained cells that accounted for >75% of the cells within a visual field. The final score was achieved by multiplication of the extent of positivity and intensity.
RNA Isolation and PCR Analysis
RNA purification and RT-PCR were performed as described previously (25). The primers used are listed in Table 2.
TABLE 2.
RT-PCR primers
F is forward, and R is reverse.
| RT-PCR primers | Sequences |
|---|---|
| KLF4 F | CCACCGGCCGGCTGCACACGACT |
| KLF4 R | TCATCTGAGCGGGCGAATTTCCATCCACA |
| GAPDH F | ACCACAGTCCATGCCATCAC |
| GAPDH R | TCCACCACCCTGTTGCTGTA |
| KRT 13 F | CCAACACTGCCATGATTCAG |
| KRT 13 R | CGTGTCTTGATGTCCAGCAG |
| SPRR2A F | AAAGTGCCCAGAACCATGTC |
| SPRR2A R | GGAACGAGGTGAGCCAAATA |
| Involucrin F | AGAGCAGCAGGTAGGACAGC |
| Involucrin R | GCTGGTGCTCTACAGGAAGC |
| KRT4 F | GCCTCTTGGAGTTTGGTAGC |
| KRT4 R | CCGGGTGTTGGAGAAGTAGT |
| CystatinA F | TCCAGCAAAGAAGCAATCAGCCAAAA |
| CystatinA R | GTTGAAGGAATCTAGAACACTTTGGG |
Electrophoretic Mobility Shift Assays (EMSA)
EMSA was performed as described previously (25). Biotin-labeled and unlabeled wild-type oligonucleotide probes contained the binding motif of KLF4 (5′-ATAGAAAAAAGGAAGGTCGAGGGCTAC-3′) and unlabeled mutant probes contained a substitution of 2 bp (underlined, 5′-ATAGAAAAAAGGAATTTCGAGGGCTAC-3′ (designated mutant 1), 5′-ATAGAAAAAAGGCCGGTCGAGGGCTAC-3′ (designated mutant 2), and 5′-ATAGAAAAAAGGAAGGTTTAGGGCTAC-3′ (designated mutant 3)).
Chromatin Immunoprecipitation Assay (ChIP)
ChIP was performed as described previously (26). The antibodies used were anti-KLF4 and anti-tubulin (Sigma). Primers used are listed in Table 3.
Western Blot Analysis
Western blots were performed as described previously (26). Antibodies used were anti-KLF4 and anti-KRT13.
Luciferase Assay
The luciferase assay was performed as described previously (26).
Cell Cycle Analysis
The cell cycle analysis was performed as described previously (25).
MTS Cell Proliferation Assay
MTS cell proliferation assay was performed as described previously (25). Briefly, cells were seeded in 96-well plates at a density of 1 × 103 cells/well and then incubated in the absence or presence of 4 mm SB at 48 h intervals. CellTiter 96 AQueous (MTS) One Solution reagent (Promega, WI) was added, and the absorbance was recorded at 490 nm using a BioTek ELx800 absorbance microplate reader.
Statistical Analysis
We statistically evaluated experimental results using a two-paired sample t test, one-way analysis of variance test, and Pearson correlation analysis. All other data were expressed as the means ± S.D. A p value of <0.05 was considered to be statistically significant.
Results
Down-regulation of KLF4 Is Associated with ESCC Dedifferentiation
Our previous results of gene expression profiles in ESCC showed that genes associated with squamous cell differentiation, including KLF4 and cytokeratins, were coordinately down-regulated in cancer tissues compared with the matched normal counterparts (23), which implicated that KLF4 may be involved in regulating the pathogenesis of ESCC. To determine the alteration of KLF4 expression in ESCC and to analyze the correlation between KLF4 and clinicopathologic features, we conducted immunohistochemistry analysis for the expression of KLF4 in a tissue microarray containing 106 paired esophageal cancer tissues and their normal counterparts, and we assessed the correlation between KLF4 protein level and clinicopathologic parameters in 95 pairs of the samples, which lose 11 adjacent normal tissue specimens. As shown in Fig. 1A, the staining of KLF4 protein was predominantly localized in the nucleus of both normal esophageal epithelia and esophageal cancer tissues. KLF4 only exhibited focal positive staining in certain well differentiated regions and presented negative staining in other less differentiated sections. In contrast, the staining of KLF4 was weak and sporadic or completely undetectable in the moderately or poorly differentiated carcinomas. The immunohistochemistry analysis showed that a significantly reduced expression of KLF4 was observed in ESCC samples versus matched normal epithelial tissues (Fig. 1A, right panel). Down-regulation of KLF4 was substantially correlated with ESCC dedifferentiation (Table 4) but had no relationship with clinical stage, depth of tumor invasion, or lymph node metastasis (data not shown). To further characterize the expression pattern of KLF4 during the course of differentiation, we examined the induction of cumulative differentiation stage-specific genes by KLF4. Transient overexpression of KLF4 in KYSE150 cells promoted the expression of the early differentiation markers KRT4 and KRT13 (Fig. 1B), which verified our previous results from cDNA microarray. These data suggested that KLF4 may participate in esophageal cancer cell terminal differentiation.
FIGURE 1.

Reduced expression of KLF4 is associated with the dedifferentiation of ESCC. A, example case showed the expression of KLF4 in esophageal tumors by immunohistochemisry staining on the tissue microarray. Representative pictures of KLF4 in normal esophageal epithelium ➀, well differentiated ➁, moderately differentiated ➂, and poorly differentiated ➃ carcinoma tissues are shown (bar, 15 μm). Quantitative analysis of the KLF4 staining between ESCC tissues and the matched normal esophageal epithelia are shown on the right. B, KYSE150 cells were transiently transfected with pcDNA3.1 and pcDNA3.1-KLF4 vector. 48 h later, RT-PCR analysis was performed to examine mRNA expression of KLF4 and a selected group of terminal differentiation genes as indicated. GAPDH was used as internal control. The band intensities of mRNA expression level were quantified by densitometry and normalized against GAPDH. The data represent the means ± S.D. *, p < 0.05.
TABLE 4.
Comparison of expression levels of KLF4 with differentiation in ESCC
| Differentiation | No. | Mean ±S.D. | p value |
|---|---|---|---|
| Well | 47 | 5.315 ± 2.848 | <0.05a |
| Moderately | 39 | 3.373 ± 2.796 | <0.05a |
| Poorly | 9 | 0.167 ± 0.408 |
a Statistical significance (p < 0.05) was performed by the one-way analysis of variance test.
Correlated Expression of KRT13 and KLF4 in Esophageal Squamous Carcinoma Cell Lines and Tissues
To gain insight into the proposed regulation of differentiation by KLF4, we conducted immunohistochemistry analysis for the expression of an epithelial specific differentiation marker, KRT13, in the tissue microarray mentioned above. As shown in Fig. 2A, KRT13 exhibited positive staining in the cornified stratified squamous epithelium and keratinized areas of well differentiated esophageal cancer foci. On the contrary, staining for KRT13 was decreased or undetectable in the suprabasal layers of epithelium and less differentiated carcinoma. The immunohistochemistry analysis showed that a significantly reduced expression of KRT13 was observed in ESCC samples versus matched normal epithelial tissues (Fig. 2A, right panel). Down-regulation of KRT13 was substantially correlated with ESCC dedifferentiation (Table 5), and a positive correlation between KLF4 and KRT13 protein expression was observed in ESCC samples (Table 6). To generalize the association of KRT13 with KLF4 in esophageal cancer, we examined KLF4 and KRT13 gene expression data in a large cohort of patients with esophageal cancer and different esophageal cancer cell lines, which are the gene expression profiling of array associated with esophageal cancer available from the public database (GSE23400, GSE33103, GSE9982, and GSE21293). In all data sets, the expression of KRT13 was significantly correlated with that of KLF4 (Fig. 2B). Furthermore, we verified the correlation between KRT13 and KLF4 among a panel of esophageal cancer cell lines. As shown in Fig. 2C, the mRNA and protein level of KRT13 and KLF4 were concordantly exhibited. To further investigate the effect of KLF4 on the regulation potential of KRT13, we overexpressed KLF4 in KYSE150 cells, which showed nearly undetectable KLF4 expression and silenced KLF4 in KYSE450 and KYSE30 cells with moderate endogenous KLF4 expression. The results showed that overexpression of KLF4 significantly increased the expression of KRT13 (Fig. 2D). Meanwhile, knockdown of KLF4 in KYSE30 and KYSE450 cells with a pool of four distinct KLF4-specific siRNAs (siKLF4) showed that ablation of KLF4 potently decreased the KRT13 protein level (Fig. 2E). Taken together, these data showed that KRT13 expressed in concomitance with KLF4 and may be positively regulated by KLF4.
FIGURE 2.

Expression of KRT13 and KLF4 are correlated in esophageal squamous carcinoma cell lines and tissues. A, representative images of KRT13 staining are shown in normal esophageal epithelium ➀, well differentiated ➁, moderately differentiated ➂, and poorly differentiated ➃ carcinoma tissue sections (bar, 15 μm). Quantitative analysis of the KRT13 staining between ESCC tissues and the matched normal esophageal epithelia are shown on the right. B, statistically significant positive correlation between klf4 and KRT13 mRNA was observed by Pearson's method in esophageal squamous cell carcinoma cell lines and tissues in four independent published data sets (➀ GSE9982, ➁ GSE21293, ➂ GSE23400, and ➃ GSE33103). C, expression of KLF4 and KRT13 in a series of esophageal cancer cell lines was determined by RT-PCR (upper panel) and Western blot analysis (lower panel). D, KYSE150 cells were transiently transfected with the pcDNA3.1 and pcDNA3.1-KLF4 vector; cells were harvested, and Western blot was performed to measure the protein expression of KLF4 and KRT13. E, Western blot analysis showed the protein expression of KLF4 and KRT13 in KYSE30 and KYSE450 cells transfected with KLF4 siRNA pools (si-KLF4) or nontargeting control (nc). β-Actin serves as an internal control. The band intensities of KRT13 protein were quantified by densitometry and normalized against β-actin. The data represent the means ± S.D. *, p < 0.05.
TABLE 5.
Comparison of expression levels of KRT13 with differentiation in ESCC
| Differentiation | No. | Mean ±S.D. | p value |
|---|---|---|---|
| Well | 47 | 4.535 ± 2.154 | <0.01a |
| Moderately | 39 | 1.641 ± 2.072 | <0.01a |
| Poorly | 9 | 0.000 ± 0.000 |
a Statistical significance (p < 0.05) was performed by the one-way analysis of variance test.
TABLE 6.
The correlation between KLF4 and KRT13 expression in ESCCs
| Group | No. | Pearson correlation | p value |
|---|---|---|---|
| ESCC | 95 | 0.263 | <0.01a |
a Statistical significance (p < 0.05) was by the Pearson correlation analysis.
Up-regulation of KRT13 upon KLF4 Is Mediated through the GKRE in KRT13 Promoter
To explore the potential role of KLF4 on regulation of KRT13 expression, we searched for putative KLF4-responsive element (GKRE) upstream of the KRT13 5′-flanking region and identified one potential KLF4-binding site residing at −411 to −399 bp upstream of the KRT13 ATG codon. Homology search showed that the predicted GKRE site was highly conserved among different species (Fig. 3A). To determine whether KRT13 is regulated by KLF4 through direct binding to its promoter, we subcloned the −1.8 kb area of the 5′-flanking region of KRT13 into the pGL3 reporter vector (pGL3-KRT13-P1 for the full-length WT plasmid) and generated several truncated mutants of the promoter-luciferase constructs, including P2 (−1.1 kb), P3 (−0.5 kb), P4 (−0.4 kb), and P5 (−0.2 kb). These constructs were cotransfected with or without pcDNA3.1-KLF4, and luciferase activity assay showed that KLF4 significantly induced P1, P2, and P3 reporter activity. However, deletion of the −450 to −350 bp led to a significant impairment of KLF4-mediated activation of the KRT13 promoter (Fig. 3B), suggesting that this segment was required for KLF4-induced KRT13 transcriptional activation. Moreover, transfected HEK293T cells with pcDNA3.1-KLF4 and the −0.5 kb deletion mutant showed that KLF4 effectively increased the transcriptional activation of KRT13 in a dose-dependent manner, whereas the activated effect was evidently suppressed on the matching construct harboring a mutation in the GKRE, demonstrating that the GKRE element was at least one of the main elements on the KLF4-mediated transcriptional activation (Fig. 3C). To further explore whether KLF4 could directly bind the putative GKRE of KRT13, we performed EMSA using biotinylated probes and the pcDNA3.1-KLF4 expression plasmid. The results showed that KLF4 bound a double strand probe containing the putative GKRE of KRT13 (Fig. 3D, lane 2), and the excess specific competitor KRT13 GKRE (with the putative GKRE) effectively attenuated formation of the probe-KLF4 complex in a dose-dependent manner (Fig. 3D, lanes 3 and 4). Mutant competitors M1 and M2 (with the mutant GKRE) abolished the inhibition of biotinylated probe binding to KLF4 (Fig. 3D, lanes 5 and 6), whereas competitor M3, which contained a mutation near the GKRE, suppressed the biotinylated probe binding to KLF4 (Fig. 3D, lane 7). Although the application of anti-KLF4 antibody to the probe/protein mixture, further shifted binding was not observed, the probe-KLF4 complex was predominantly mitigated (Fig. 3D, lane 10). These data substantiated that the putative GKRE of KRT13 represented a bona fide site for KLF4 binding. ChIP analysis revealed that KLF4 specifically associated with the promoter region containing the conserved GKRE but not the control distal region of the KRT13 promoter in KYSE150 cells. Conversely, tubulin did not precipitate detectable DNA (Fig. 3E), thus providing additional evidence to support the active role of KLF4 in KRT13 gene transcription in vivo. To further confirm the putative GKRE involved in transcriptional activation of KRT13, we constructed the GKRE mutant promoter-luciferase plasmid. As expected, mutation of GKRE completely abolished KLF4-mediated activation of the KRT13 promoter luciferase activity (Fig. 3F). Taken together, these findings demonstrated unequivocally that KLF4 transactivated KRT13 by binding the putative GKRE.
FIGURE 3.

Up-regulation of KRT13 upon KLF4 is mediated through the GKRE in KRT13 promoter. A, GKRE resides at −411 to −399 bp upstream of the KRT13 ATG codon and is highly conserved in different species. B, luciferase reporter assay was performed using different vectors containing predicted KLF4-binding sites upstream of the KRT13. GKRE is indicated by arrowhead. Bars represent the means ± S.D. C, luciferase reporter assay was performed using increased amounts of pcDNA3.1-KLF4 as indicated. Bars represent the means ± S.D. D, EMSA were performed using 3 fmol of labeled oligonucleotides and 5 μg of nuclear extracts (NE) obtained from HEK293T cells transfected with pcDNA3.1-KLF4. Competitor oligonucleotides were added at 50- and 200-fold molar excess of the labeled probe. In experiments involving antibody, 2 μg of anti-KLF4 polyclonal antibody was added to the reaction. The positions of free probe (F) and shifted bands (S) are indicated. M1, M2, and M3 are the mutant oligonucleotides used as competitor DNA. E, KYSE150 cells were transfected with pcDNA3.1-KLF4, and 48 h later, ChIP assays were performed with antibodies against KLF4 or tubulin, IgG control, respectively. The presence of the KRT13 5′-flanking region containing GKRE or not was assayed by quantitative RT-PCR. Bars represent the means ± S.D. F, luciferase reporter assay was performed using different vectors containing wild-type (WT) or mutant-type (MT) KLF4-binding sites upstream of the KRT13. Putative KLF4-responsive sequences (AAGG) are underlined. Mutant of the KLF4-binding site is represented by shading. Bars represent the means ± S.D. *, p < 0.05.
KLF4 and KRT13 Are Induced during Esophageal Cancer Cell Differentiation
SB was an inhibitor of histone deacetylases and an inducer of differentiation. It has been demonstrated that SB inhibited cell growth and induced accumulation of the G1 phase (31). Previous study also showed that SB directly transactivated KLF4 expression in an Sp1-dependent manner in colon cancer cells and promoted the differentiation of oral squamous cancer cells concomitant with increased expression of KRT13 (21, 22). Therefore, we speculated that SB may induce cell differentiation and growth inhibition by increasing the expression of KLF4 and KRT13, both of which are substantially associated with the development of ESCC as our results had shown. To clarify the alteration of KLF4 and KRT13 levels during ESCC differentiation, we first analyzed the expression of KLF4 and KRT13 in KYSE150 cells treated with SB. Western blot results clearly showed that SB induced the expression of KLF4 and KRT13 (Fig. 4A). We subsequently examined the expression of KLF4 and KRT13 by different dosages of SB from 0.5 to 5 mm, and a dose-dependent stimulation of KLF4 and KRT13 was observed (Fig. 4B). Calcium is another commonly used differentiation inducer, and our previous study showed that calcium induced the expression of a series of differentiation-associated genes in esophageal cancer cells, inhibited cell growth, and induced G1/S phase arrest and cell apoptosis. Moreover, calcium treatment induced an apparent cell-stratified and dramatic change in cell-cell contact (28), the effect of which coincides with SB. To further confirm the increased expression of KLF4 and KRT13 during the differentiation induction, we treated esophageal cancer cell lines KYSE150 and KYSE450 with 2.4 mm calcium that had been corroborated effectively by our previous results. Western blot analysis revealed that 2.4 mm CaCl2 apparently elevated KLF4 and KRT13 protein levels in KYSE150 and KYSE450 cells (Fig. 4C). Therefore, we selected KYSE150 and KYSE450 cells to perform phenotypic characterization in the following experiments.
FIGURE 4.
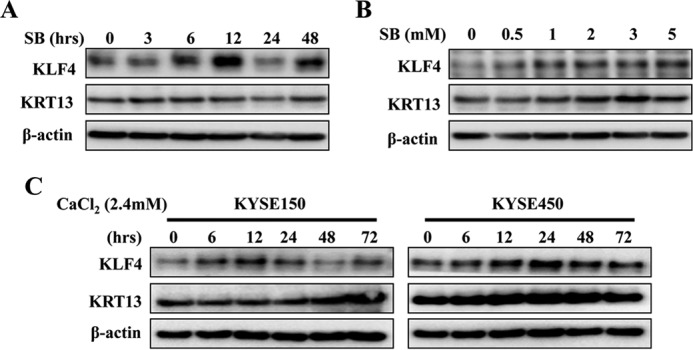
KLF4 and KRT13 expression are regulated during SB and calcium-induced esophageal cancer cell differentiation. A, KYSE150 cells were incubated with 4 mm SB for the indicated time, and the expression of KLF4 and KRT13 was determined by Western blot. B, KYSE150 cells were incubated with increasing concentrations (0–5 mm) of SB for 48 h. The expression of KLF4 and KRT13 was determined by Western blot. C, KYSE150 and KYSE450 cells were incubated with 2.4 mm CaCl2 for the indicated times, and the expression of KLF4 and KRT13 was determined by Western blot.
KRT13 Promotes the Cell Cycle Arrest and Growth Inhibition of Esophageal Cancer Cells Induced by SB
To investigate the contribution of KRT13 on SB-mediated esophageal cancer cell differentiation, we used KYSE150 cells that exhibited low expression of KRT13 and could be dramatically induced by SB. Western blot analysis showed that KRT13 was effectively overexpressed and further induced by SB (Fig. 5A, upper panel). Quantification of the DNA content by flow cytometry (FACS) after propidium iodide staining confirmed that overexpression of KRT13 accumulated esophageal cancer cells at the G1 phase, which indicated that KRT13 participated in the G1/S transition. Similarly, treatment with SB also impeded the cell cycle progression by arresting cells in G1 phase. Concurrent treatment with SB in KRT13 overexpressed KYS150 cells resulted in a significant decline in the G2/M phase and a strong G1/S arrest in ESCC, relative to either treatment alone (Fig. 5A, lower panel), suggesting that KRT13 facilitated SB-mediated cell cycle arrest in esophageal cancer cells. Furthermore, we tested the effect of KRT13 overexpression on cell growth in the absence or presence of SB by a direct viable cell count assay. The results showed that overexpression of KRT13 decreased proliferation with or without SB treatment after 48 h, and the difference was still statistically significant after 4 days (Fig. 5B). These data indicated that overexpression of KRT13 conferred cell growth inhibition induced by SB. To further test this, knockdown of KRT13 in KYSE450 cells with a pool of four distinct KRT13-specific siRNAs (siKRT 13) potently decreased the proportion of G1 phase cells induced by SB (Fig. 5C). These results indicate a suppressive role of KRT13 in esophageal cancer cell growth and cell cycle progression.
FIGURE 5.

KRT13 promotes SB-induced cell growth inhibition. A, empty vector or KRT13-transfected KYSE150 cells were seeded at 1 × 105 cells per well in conventional medium treated or untreated (control) with 4 mm SB on 6-well plates; cells were stained with propidium iodide and analyzed by flow cytometry at 48 h (lower panel), and Western blotting was performed to measure the protein expression of KRT13 (upper panel). B, cell viability of KRT13-transfected KYSE150 cells compared with empty vector-transfected KYSE150 cells treated or untreated (con) with 4 mm SB at selected time points. Bars represent the means ± S.D. C, KYSE450 cells were transfected with KRT13 siRNA (siKRT13) or nontargeting control (nc) and treated or untreated (control) with 4 mm SB for 48 h. Cell cycle distribution was assessed by flow cytometry. Bars represent the means ± S.D. (left panel). Western blot analysis shows the knockdown of KRT13 in KYSE450 cells (right panel). β-Actin served as an internal control. *, p < 0.05.
SB Regulates Cell Cycle through KLF4 and KRT13
Because KLF4 was a putative target activated by SB, we assessed whether the targeting was involved in SB-induced cell cycle arrest. Western blot analysis showed that KLF4 expression was rigorously mitigated in KLF4 siRNA-transfected cells (Fig. 6A). Cell cycle analysis showed that individual silencing of KLF4 in the basal condition marginally promoted the G2/M phase transition. However, when cells were treated with SB, the percentage of cells in the G1 phase were substantially reduced in KLF4-silenced cells compared with the control cells (Fig. 6B). The result implicated that KLF4 activity was required for SB-induced transition from G1 to S phase. To examine whether KRT13 was involved in KLF4-mediated cell cycle arrest induced by SB, we cotransfected KYSE450 cells with KRT13 expression vector and KLF4-specific siRNAs, and the results of flow cytometry indicated that re-expression of KRT13 marginally restored the G1/S arrest induced by SB when endogenous KLF4 was knocked down (data not shown). We then examined the function of KLF4 mediated by KRT13 through proliferation. Results showed that deletion of KLF4 repressed the proliferation inhibition induced by SB, whereas ectopic expression of KRT13 partially reversed the promotional effect on cell proliferation (Fig. 6D), which indicated that SB-induced cell differentiation was dependent on KLF4, and KRT13 was one of its downstream effectors. The expressions of KLF4 and KRT13 were confirmed by Western blotting (Fig. 6C). Taken together, our results showed the important role of the KLF4-KRT13 signaling pathway in mediating the effect of SB on esophageal cancer cell differentiation.
FIGURE 6.

KLF4 and KRT13 are required for SB-induced cell cycle arrest. A, KYSE450 cells transfected with KLF4 siRNA pools (si-KLF4) or nontargeting control (nc) treated or untreated with 4 mm SB for 48 h. Western blotting was performed to determine the expression of KLF4. β-Actin served as an internal control. B, KYSE450 cells transfected with KLF4 siRNA pools (siKLF4) or nontargeting control (nc) were seeded at 1.5 × 105 cells per well in conventional medium and treated or untreated (control) with 4 mm SB on 6-well plates; cells were stained with propidium iodide and analyzed by flow cytometry at 48 h. Bars represent the means ± S.D. C, KYSE450 cells were cotransfected FLAG-KRT13 or vector with KLF4 siRNA (siKLF4) or nontargeting control (nc) and treated with 4 mm SB for 48 h. Western blot analysis showed the expression of KLF4 and KRT13 in KYSE450 cells. β-Actin served as an internal control. D, cell viability at selected time points of each indicated treatment was analyzed by MTS cell proliferation array. Bars represent the means ± S.D. *, p < 0.05.
Discussion
Our findings revealed that KLF4 and KRT13 expressions were significantly correlated with differentiation of ESCC. KLF4 up-regulated the expression of KRT13 by binding directly to the putative GKRE motif within the KRT13 promoter. Enhancement of KLF4 and KRT13 could be evoked by SB, and it facilitated SB-induced G1/S phase arrest and growth inhibition of esophageal cancer cells.
Epithelial differentiation is a complex process and requires the coordinated stepwise change in expression of multiple genes. Disruption of normal differentiation is an important characteristic of tumorigenesis. The progression of esophageal cancer is associated with loss of gene function governing differentiation. Disturbance of transcription factors, especially those that confer the differentiation of stem cells in the normal esophageal epithelium, can block normal differentiation of squamous epithelial cells and lead to cancer. A better understanding of the underlying molecular events regulating differentiation would offer possible targets for tumor therapeutic intervention or potential biomarkers. KLF4 is such a transcription factor participating in regulating stem cell transformation, cell cycle and differentiation (32). Cumulative evidence illustrated that hemizygous deletion, hypermethylated 5′-untranslated region and point mutation in the open reading frame are attributed with the loss of function of KLF4 in colorectal cancer (33). Here, our immunohistochemistry analysis using clinical ESCC samples revealed that the expression of KLF4 predominantly decreased in ESCC tissues compared with their normal counterparts and was remarkably correlated with the differentiation of ESCC (Fig. 1A and Table 4). KLF4 epithelial cells in transgenic mice manifested an increased expression of KLF4 in maturing PCNA-negative cells (34), which provide additional evidence for the role of KLF4 in squamous cell differentiation. Molecular investigation further supported the morphologic findings as KLF4 regulated the expression level of epithelial differentiation markers such as KRT4 and KRT13 (Fig. 1B). In the epidermis, KLF4 had been reported to be activated by protein kinase Cδ and to drive the expression of involucrin, another major components of cornified envelope and appropriate markers for terminal differentiation (35). However, in our study, we did not observe any altered expression of involucrin by KLF4. We speculated that the context-specific function of KLF4 and the drastic alterations in gene expression programming accompanied by the dynamic transition between normal epithelium and cancer were considerable possibilities.
Recent studies have revealed that a number of transcription factors are involved in regulating transcription of differentiation-specific genes. For example, STAT3 activated transcription of the KRT13 promoter (36). Sp1 and AP2 were important for regulating the expression of KRT during epithelial cell differentiation (37, 38). KLF5 played an essential role in the differentiation and homeostasis of epithelial cells, smooth muscle cells and adipocytes by regulating the expression of KRT5, SM22α, peroxisome proliferator-activated receptor γ and others (39, 40). Our previous results showed that KLF4 could bind directly to the promoter of S100A14 and activate its transcription, which modulated the terminal differentiation in esophageal cancer (26, 28). cDNA microarray analysis also revealed a group of KRT genes, especially those located in a specific region on chromosome 12 could be induced by KLF4 (10). Here, we further delineated that transfection of KLF4 into KYSE150 cells increased the expression of KRT13 (Fig. 2D), although knockdown of KLF4 in KYSE30 and KYSE450 cells inhibited its expression (Fig. 2E). KRT13 together with KRT4 composed a KRT network in the suprabasal cells of stratified squamous epithelia and the reduction of KRT13 often resulted in an abnormal differentiation (13). In this study, we corroborated that the expression of KRT13 was pervasive in accordance with KLF4 both in esophageal cancer cell lines and the patient tissue samples (Table 6 and Fig. 2C). These results were also supported by gene expression profiling data from published studies (Fig. 2B). The effect of KLF4-mediated promotion of KRT13 expression could be direct or indirect or both. Our studies identified that the potential KLF4-binding sites were located on −411 to −399 bp of the KRT13 promoter region, and these sequences conferred KLF4 binding directly with the KRT13 promoter (Fig. 3, D and E). In addition, deletion or mutation of the GKRE within KRT13 promoter abolished the interaction between the KRT13 promoter and KLF4 (Fig. 3D). Furthermore, luciferase assay demonstrated substantially that KLF4 transactivated KRT13 by binding the putative GKRE (Fig. 3, B and C), and impairment of the interaction between KLF4 and KRT13 promoter by the GKRE mutant blunted the transcriptional capacity of KRT13 induced by KLF4 (Fig. 3F). In summary, these data evidently elucidated that KLF4-mediated transactivation of KRT13 at least partially accounted for the relevant expression of KRT13 in ESCC.
To clarify the biological significance of KLF4-mediated transcriptional regulation of KRT13, we used SB, a putative agonist of both KLF4 and KRT13, to incubate with esophageal cancer cells. SB is a short chain fatty acid released from dietary carbohydrates by symbiotic bacteria in gastrointestinal tracts. It has been reported that SB leads to alteration of chromosomal structure and gene expression by suppressing histone deacetylation (41), and it was known to induce differentiation and apoptosis and to inhibit proliferation in many cancer cell lines (42, 43). The chemopreventive benefits of SB remain debatable because apparent inconsistencies have been documented (44). The discrepant effects of SB depend in part on amount, time of exposure with respect to the tumorigenic process, and the type of fat in the diet (20). In this context, we showed that SB substantially invigorated KLF4 and KRT13 protein expression in a time-dependent (Fig. 4A) and dose-dependent (Fig. 4B) manner. The protein level of KLF4 and KRT13 could also be up-regulated by calcium treatment (Fig. 4C), another differentiation inducer. Alteration of KRT protein expression and cross-linked envelopes generally reflected the extrinsic growth regulation in primary esophageal carcinomas (45). Here, we illustrated that overexpression of KRT13 promoted SB-mediated cell cycle arrest and growth inhibition in KYSE150 cells (Fig. 5, A and B), whereas the depletion of KRT13 impaired the inhibition of G1/S transition induced by SB in the KYSE450 cells (Fig. 5C). Deletion of KLF4 almost completely antagonized the accumulation of G1 phase and proliferation inhibition induced by SB (Fig. 6, B and D), and reconstituting KRT13 evidently restored the cell proliferation inhibition mediated by SB while knocking down endogenous KLF4 (Fig. 6D), but it had moderate effects on cell cycle arrest (data not shown). It could not exclude that other functional downstream targets of KLF4 participated in its mediated cell cycle arrest, such as p21 and cyclin D1, which antagonized its function. However, we observed that ectopic expression of KRT13 at least partially restored the proliferation inhibition (Fig. 6D) induced by SB when endogenous KLF4 was knocked down, suggesting that KLF4 and KRT13 were involved in SB-mediated cell differentiation. We speculated that the effect of KRT13 overexpression was negligible on KYSE450 cell proliferation because the endogenous KRT13 was saturated.
These results indicated that KLF4 and KRT13 were required for the cell cycle arrest and growth inhibition in response of the esophageal cancer cells to SB. Therefore, SB-induced differentiation was conferred at least partially by the KLF4-KRT13 signaling pathway. KLF4 was well documented to function as a molecular switch that converts the cellular program from stem maintenance to differentiation in many cell types (6, 46, 47). In this regard, it was conceivable that KLF4 acted as a program activator during esophageal cancer cell differentiation.
In summary, our results characterized a pivotal association between KLF4-KRT13 and the differentiation of ESCC and provided insight into the mechanisms responsible for KRT13 regulated by KLF4. We also demonstrated the importance of the KLF4-KRT13 signaling pathway in SB-mediated differentiation of esophageal cancer cells. Our findings therefore opened a unique avenue for identification of novel biomarkers to predict malignant progression of ESCC via specific evaluation of KLF4 and KRT13 expression alterations.
This work was supported by National Natural Science Foundation of China Grants 81130043, 81302329, and 81420108025 and National Basic Research Program of China Grants 2011CB504205 and 2013CB911004.
- ESCC
- esophageal squamous cell carcinoma
- SB
- sodium butyrate
- MTS
- 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt.
References
- 1. Siegel R., Naishadham D., Jemal A. (2012) Cancer statistics, 2012. CA-Cancer J. Clin. 62, 10–29 [DOI] [PubMed] [Google Scholar]
- 2. Ferlay J., Shin H. R., Bray F., Forman D., Mathers C., Parkin D. M. (2010) Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int. J. Cancer 127, 2893–2917 [DOI] [PubMed] [Google Scholar]
- 3. Mariette C., Piessen G., Triboulet J. P. (2007) Therapeutic strategies in oesophageal carcinoma: role of surgery and other modalities. Lancet Oncol. 8, 545–553 [DOI] [PubMed] [Google Scholar]
- 4. Segre J. A., Bauer C., Fuchs E. (1999) Klf4 is a transcription factor required for establishing the barrier function of the skin. Nat. Genet. 22, 356–360 [DOI] [PubMed] [Google Scholar]
- 5. Tetreault M. P., Yang Y., Travis J., Yu Q. C., Klein-Szanto A., Tobias J. W., Katz J. P. (2010) Esophageal squamous cell dysplasia and delayed differentiation with deletion of kruppel-like factor 4 in murine esophagus. Gastroenterology 139, 171–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Katz J. P., Perreault N., Goldstein B. G., Lee C. S., Labosky P. A., Yang V. W., Kaestner K. H. (2002) The zinc-finger transcription factor Klf4 is required for terminal differentiation of goblet cells in the colon. Development 129, 2619–2628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hinnebusch B. F., Siddique A., Henderson J. W., Malo M. S., Zhang W., Athaide C. P., Abedrapo M. A., Chen X., Yang V. W., Hodin R. A. (2004) Enterocyte differentiation marker intestinal alkaline phosphatase is a target gene of the gut-enriched Kruppel-like factor. Am. J. Physiol. Gastrointest Liver Physiol. 286, G23–G30 [DOI] [PubMed] [Google Scholar]
- 8. Katz J. P., Perreault N., Goldstein B. G., Actman L., McNally S. R., Silberg D. G., Furth E. E., Kaestner K. H. (2005) Loss of Klf4 in mice causes altered proliferation and differentiation and precancerous changes in the adult stomach. Gastroenterology 128, 935–945 [DOI] [PubMed] [Google Scholar]
- 9. Swamynathan S. K., Katz J. P., Kaestner K. H., Ashery-Padan R., Crawford M. A., Piatigorsky J. (2007) Conditional deletion of the mouse Klf4 gene results in corneal epithelial fragility, stromal edema, and loss of conjunctival goblet cells. Mol. Cell. Biol. 27, 182–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen X., Whitney E. M., Gao S. Y., Yang V. W. (2003) Transcriptional profiling of Kruppel-like factor 4 reveals a function in cell cycle regulation and epithelial differentiation. J. Mol. Biol. 326, 665–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hesse M., Magin T. M., Weber K. (2001) Genes for intermediate filament proteins and the draft sequence of the human genome: novel keratin genes and a surprisingly high number of pseudogenes related to keratin genes 8 and 18. J. Cell Sci. 114, 2569–2575 [DOI] [PubMed] [Google Scholar]
- 12. Magin T. M., Vijayaraj P., Leube R. E. (2007) Structural and regulatory functions of keratins. Exp. Cell Res. 313, 2021–2032 [DOI] [PubMed] [Google Scholar]
- 13. Sheng S., Barnett D. H., Katzenellenbogen B. S. (2008) Differential estradiol and selective estrogen receptor modulator (SERM) regulation of keratin 13 gene expression and its underlying mechanism in breast cancer cells. Mol. Cell. Endocrinol. 296, 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Southgate J., Varley C. L., Garthwaite M. A., Hinley J., Marsh F., Stahlschmidt J., Trejdosiewicz L. K., Eardley I. (2007) Differentiation potential of urothelium from patients with benign bladder dysfunction. BJU Int. 99, 1506–1516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nishizawa A., Nakajima R., Nakano H., Sawamura D., Takayama K., Satoh T., Yokozeki H. (2008) A de novo missense mutation in the keratin 13 gene in oral white sponge naevus. Br. J. Dermatol. 159, 974–975 [DOI] [PubMed] [Google Scholar]
- 16. Baker H., Patel V., Molinolo A. A., Shillitoe E. J., Ensley J. F., Yoo G. H., Meneses-García A., Myers J. N., El-Naggar A. K., Gutkind J. S., Hancock W. S. (2005) Proteome-wide analysis of head and neck squamous cell carcinomas using laser-capture microdissection and tandem mass spectrometry. Oral. Oncol. 41, 183–199 [DOI] [PubMed] [Google Scholar]
- 17. Hamakawa H., Fukuzumi M., Bao Y., Sumida T., Kayahara H., Onishi A., Sogawa K. (2000) Keratin mRNA for detecting micrometastasis in cervical lymph nodes of oral cancer. Cancer Lett. 160, 115–123 [DOI] [PubMed] [Google Scholar]
- 18. Yanagawa T., Yoshida H., Yamagata K., Onizawa K., Tabuchi K., Koyama Y., Iwasa S., Shimoyamada H., Harada H., Omura K. (2007) Loss of cytokeratin 13 expression in squamous cell carcinoma of the tongue is a possible sign for local recurrence. J. Exp. Clin. Cancer Res. 26, 215–220 [PubMed] [Google Scholar]
- 19. Dackour R., Carter T., Steinberg B. M. (2005) Phosphatidylinositol 3-kinase regulates early differentiation in human laryngeal keratinocytes. In Vitro Cell. Dev. Biol. Anim. 41, 111–117 [DOI] [PubMed] [Google Scholar]
- 20. Lupton J. R. (2004) Microbial degradation products influence colon cancer risk: the butyrate controversy. J. Nutr. 134, 479–482 [DOI] [PubMed] [Google Scholar]
- 21. Flandez M., Guilmeau S., Blache P., Augenlicht L. H. (2008) KLF4 regulation in intestinal epithelial cell maturation. Exp. Cell Res. 314, 3712–3723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kuroda J., Urade M., Kishimoto H., Noguchi K., Hashitani S., Sakurai K., Nishimura N., Hashimoto-Tamaoki T. (2005) Promotion of cell differentiation, and suppression of cell growth and cyclooxygenase-2 expression by differentiation-inducing agents in human oral squamous carcinoma SCC25 cells. Int. J. Oncol. 26, 361–367 [PubMed] [Google Scholar]
- 23. Luo A., Kong J., Hu G., Liew C. C., Xiong M., Wang X., Ji J., Wang T., Zhi H., Wu M., Liu Z. (2004) Discovery of Ca2+-relevant and differentiation-associated genes downregulated in esophageal squamous cell carcinoma using cDNA microarray. Oncogene 23, 1291–1299 [DOI] [PubMed] [Google Scholar]
- 24. Shimada Y., Imamura M., Wagata T., Yamaguchi N., Tobe T. (1992) Characterization of 21 newly established esophageal cancer cell lines. Cancer 69, 277–284 [DOI] [PubMed] [Google Scholar]
- 25. Li S., Zhou Q., He H., Zhao Y., Liu Z. (2013) Peroxisome proliferator-activated receptor γ agonists induce cell cycle arrest through transcriptional regulation of Kruppel-like factor 4 (KLF4). J. Biol. Chem. 288, 4076–4084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. He H., Li S., Chen H., Li L., Xu C., Ding F., Zhan Y., Ma J., Zhang S., Shi Y., Qu C., Liu Z. (2014) 12-O-Tetradecanoylphorbol-13-acetate promotes breast cancer cell motility by increasing S100A14 level in a Kruppel-like transcription factor 4 (KLF4)-dependent manner. J. Biol. Chem. 289, 9089–9099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang L., Ding F., Cao W., Liu Z., Liu W., Yu Z., Wu Y., Li W., Li Y. (2006) Stomatin-like protein 2 is overexpressed in cancer and involved in regulating cell growth and cell adhesion in human esophageal squamous cell carcinoma. Clin. Cancer Res. 12, 1639–1646 [DOI] [PubMed] [Google Scholar]
- 28. Chen H., Ma J., Sunkel B., Luo A., Ding F., Li Y., He H., Zhang S., Xu C., Jin Q., Wang Q., Liu Z. (2013) S100A14: novel modulator of terminal differentiation in esophageal cancer. Mol. Cancer Res. 11, 1542–1553 [DOI] [PubMed] [Google Scholar]
- 29. He H., Ding F., Li Y., Luo A., Chen H., Wu C., Liu Z. (2012) Migfilin regulates esophageal cancer cell motility through promoting GSK-3β-mediated degradation of β-catenin. Mol. Cancer Res. 10, 273–281 [DOI] [PubMed] [Google Scholar]
- 30. Tong T., Zhong Y., Kong J., Dong L., Song Y., Fu M., Liu Z., Wang M., Guo L., Lu S., Wu M., Zhan Q. (2004) Overexpression of Aurora-A contributes to malignant development of human esophageal squamous cell carcinoma. Clin. Cancer Res. 10, 7304–7310 [DOI] [PubMed] [Google Scholar]
- 31. Abramova M. V., Pospelova T. V., Nikulenkov F. P., Hollander C. M., Fornace A. J., Jr., Pospelov V. A. (2006) G1/S arrest induced by histone deacetylase inhibitor sodium butyrate in E1A + Ras-transformed cells is mediated through down-regulation of E2F activity and stabilization of β-catenin. J. Biol. Chem. 281, 21040–21051 [DOI] [PubMed] [Google Scholar]
- 32. Cui J., Shi M., Quan M., Xie K. (2013) Regulation of EMT by KLF4 in gastrointestinal cancer. Curr. Cancer Drug Targets 13, 986–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhao W., Hisamuddin I. M., Nandan M. O., Babbin B. A., Lamb N. E., Yang V. W. (2004) Identification of Kruppel-like factor 4 as a potential tumor suppressor gene in colorectal cancer. Oncogene 23, 395–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Huang C. C., Liu Z., Li X., Bailey S. K., Nail C. D., Foster K. W., Frost A. R., Ruppert J. M., Lobo-Ruppert S. M. (2005) KLF4 and PCNA identify stages of tumor initiation in a conditional model of cutaneous squamous epithelial neoplasia. Cancer Biol. Ther. 4, 1401–1408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chew Y. C., Adhikary G., Xu W., Wilson G. M., Eckert R. L. (2013) Protein kinase Cδ increases Kruppel-like factor 4 protein, which drives involucrin gene transcription in differentiating keratinocytes. J. Biol. Chem. 288, 17759–17768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wu R., Sun S., Steinberg B. M. (2003) Requirement of STAT3 activation for differentiation of mucosal stratified squamous epithelium. Mol. Med. 9, 77–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wanner R., Zhang J., Henz B. M., Rosenbach T. (1996) AP-2 gene expression and modulation by retinoic acid during keratinocyte differentiation. Biochem. Biophys. Res. Commun. 223, 666–669 [DOI] [PubMed] [Google Scholar]
- 38. Chen T. T., Wu R. L., Castro-Munozledo F., Sun T. T. (1997) Regulation of K3 keratin gene transcription by Sp1 and AP-2 in differentiating rabbit corneal epithelial cells. Mol. Cell. Biol. 17, 3056–3064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dong J. T., Chen C. (2009) Essential role of KLF5 transcription factor in cell proliferation and differentiation and its implications for human diseases. Cell. Mol. Life Sci. 66, 2691–2706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liu R., Zhou Z., Zhao D., Chen C. (2011) The induction of KLF5 transcription factor by progesterone contributes to progesterone-induced breast cancer cell proliferation and dedifferentiation. Mol. Endocrinol. 25, 1137–1144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mali P., Chou B. K., Yen J., Ye Z., Zou J., Dowey S., Brodsky R. A., Ohm J. E., Yu W., Baylin S. B., Yusa K., Bradley A., Meyers D. J., Mukherjee C., Cole P. A., Cheng L. (2010) Butyrate greatly enhances derivation of human induced pluripotent stem cells by promoting epigenetic remodeling and the expression of pluripotency-associated genes. Stem Cells 28, 713–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Heerdt B. G., Houston M. A., Augenlicht L. H. (1994) Potentiation by specific short-chain fatty acids of differentiation and apoptosis in human colonic carcinoma cell lines. Cancer Res. 54, 3288–3293 [PubMed] [Google Scholar]
- 43. Tsutsumi T., Ido A., Nakao K., Hamasaki K., Kato Y., Ohtsuru A., Nakata K., Tamaoki T., Nagataki S. (1994) Reciprocal regulation of α-fetoprotein and albumin gene expression by butyrate in human hepatoma cells. Gastroenterology 107, 499–504 [DOI] [PubMed] [Google Scholar]
- 44. Lupton J. R. (2000) Is fiber protective against colon cancer? Where the research is leading us. Nutrition 16, 558–561 [DOI] [PubMed] [Google Scholar]
- 45. Banks-Schlegel S. P., Quintero J. (1986) Growth and differentiation of human esophageal carcinoma cell lines. Cancer Res. 46, 250–258 [PubMed] [Google Scholar]
- 46. Yu F., Li J., Chen H., Fu J., Ray S., Huang S., Zheng H., Ai W. (2011) Kruppel-like factor 4 (KLF4) is required for maintenance of breast cancer stem cells and for cell migration and invasion. Oncogene 30, 2161–2172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cittelly D. M., Finlay-Schultz J., Howe E. N., Spoelstra N. S., Axlund S. D., Hendricks P., Jacobsen B. M., Sartorius C. A., Richer J. K. (2013) Progestin suppression of miR-29 potentiates dedifferentiation of breast cancer cells via KLF4. Oncogene 32, 2555–2564 [DOI] [PMC free article] [PubMed] [Google Scholar]