

Background: To understand the molecular mechanism of cell growth, novel regulators of cell growth should be identified.
Results: LTV1 mutation impairs 40S ribosome subunit synthesis, and it blocks cell growth and ribosome biogenesis induced by dMyc.
Conclusion: LTV1 is essential for cell growth and ribosome biogenesis downstream of dMyc.
Significance: Characterization of LTV1 will be helpful for understanding Myc-induced tumorigenesis.
Keywords: cell growth, Drosophila, Myc (c-Myc), ribosomal RNA processing (rRNA processing), ribosome, LTV1
Abstract
During animal development, various signaling pathways converge to regulate cell growth. In this study, we identified LTV1 as a novel cell growth regulator in Drosophila. LTV1 mutant larvae exhibited developmental delays and lethality at the second larval stage. Using biochemical studies, we discovered that LTV1 interacted with ribosomal protein S3 and co-purified with free 40S ribosome subunits. We further demonstrated that LTV1 is crucial for ribosome biogenesis through 40S ribosome subunit synthesis and preribosomal RNA processing, suggesting that LTV1 is required for cell growth by regulating protein synthesis. We also demonstrated that Drosophila Myc (dMyc) directly regulates LTV1 transcription and requires LTV1 to stimulate ribosome biogenesis. Importantly, the loss of LTV1 blocked the cell growth and endoreplication induced by dMyc. Combined, these results suggest that LTV1 is a key downstream factor of dMyc-induced cell growth by properly maintaining ribosome biogenesis.
Introduction
Cell growth is coordinated with cell differentiation and apoptosis in animal development. To control cell growth precisely, proto-oncogenes and tumor suppressors are tightly regulated by developmental cues and environmental changes. To understand the molecular mechanism of cell growth, novel cell growth regulators should be identified, and their in vivo functions should be demonstrated in animal models. Although multiple cell growth regulators have been discovered and investigated (1, 2), our understanding of cell growth regulation still remains elusive.
During cell growth, de novo synthesis of the ribosome, the machinery required for mRNA translation, is highly induced (3). The ribosome is generated through coordinated multiple processes occurring in the nucleolus, nucleoplasm, and cytosol (4–6). In yeast, the precursor of ribosomal RNA (pre-rRNA)3 is transcribed and simultaneously assembled with the ribosomal proteins imported from the cytoplasm to form the 90S precursor (pre-90S) ribosome particles in the nucleolus (4). The 35S pre-rRNA, the longest precursor, contains 18S, 5.8S, and 25S mature rRNAs that are separated by internal transcribed spacers (ITSs) and flanked by external transcribed spacers. These extra spacer sequences are sequentially removed by endo- and exonucleases to make mature rRNAs (7). In the nucleolus, an internal cleavage of ITS in pre-rRNA separates pre-90S ribosomes into pre-40S and -60S ribosome subunits. Both of these precursor ribosome subunits in the nucleus are exported to the cytosol in a Crm1-Ran GTPase-dependent manner (8, 9). After the export from the nucleus, the precursor ribosomal subunits are further processed to fully mature subunits in the cytosol.
There are ∼200 non-ribosomal proteins that associate and dissociate dynamically with preribosomes during ribosome biogenesis (10). These proteins have indispensable roles in ribosome biogenesis by assisting pre-rRNA processing and modifications, ribosomal protein folding and association, etc. For the synthesis of fully matured 40S ribosome subunits, multiple non-ribosomal proteins, such as Rio2p, Tsr1p, Ltv1p, Enp1p, Nob1p, Hrr25p, Dim1p, and Dim2p, interact with pre-40S ribosome subunits (11). They have various protein domains such as methyltransferase, protein kinase, endoribonuclease, and GTPase, implicating that they are involved in 40S ribosome biogenesis in diverse ways. These non-ribosomal proteins are structurally highly conserved, suggesting that they have similar functions in ribosome biogenesis from yeast to multicellular animals.
Diverse signaling molecules regulate ribosome biogenesis to control cell growth (3, 12). Among these signals, Myc proto-oncogene plays the most important roles at several stages including rRNA transcription (13–15), rRNA processing (16), and the export of ribosome subunits from the nucleus to the cytosol (17, 18). Consistently, Myc transcriptionally induces multiple genes critical for ribosome biogenesis including the genes for ribosomal proteins (19), upstream binding factors (the transcription factors for RNA polymerase I-mediated transcription) (14), and nucleophosmin (a nuclear export chaperone for ribosome) (18, 20).
In this study, we attempted to discover a novel cell growth regulator using a fruit fly system and successfully identified low temperature viability protein 1 (LTV1). LTV1 specifically interacted with ribosomal protein S3 (RpS3) and co-purified with free 40S ribosome subunits. We found that LTV1 is crucial for the biogenesis of 40S ribosome subunits by affecting pre-rRNA processing and the nuclear export of pre-40S ribosome subunits. Furthermore, we showed that LTV1 was transcriptionally regulated by dMyc and was required for dMyc-dependent ribosome biogenesis, cell growth, and endoreplication. Together, our results strongly suggested that dMyc controls ribosome biogenesis and cell growth by directly regulating the gene expression of LTV1 in Drosophila.
Experimental Procedures
Fly Stocks
The following fly lines were used in this study: Cg-GAL4 (Bloomington, 7011), Patched (Ptc)-GAL4 (Bloomington, 2017), UAS-dMyc (Bloomington, 9674), Act>CD2>GAL4 (Bloomington, 4780), UAS-LTV1 RNAi (Vienna Drosophila Resource Center, 33650), UAS-Crm1 RNAi (Vienna Drosophila Resource Center, 3347), UAS-RpI135 RNAi (Vienna Drosophila Resource Center, 37581), UAS-RpS2-3xHA (FlyORF, F000781), and dm4 and the revertant for dm4 (a gift from Dr. Robert Eisenman).
Generation of LTV1E1 Mutant
LTV1E1 was generated in this study through imprecise excision of the P-element from CG7686CB-6202-3 (Kyoto Drosophila Genetic Resource Center, 123972).
Generation of LTV1 Transgenic Flies
LTV1 cDNA was amplified by PCR from expressed sequence tag (Drosophila Genomics Resource Center, LD21529) and subcloned into the EcoRI and XbaI sites followed by HA sequences in pUAST vector. pUAST LTV1-HA plasmid was microinjected into w1118 embryo.
Clone Generation
Homozygous LTV1E1 fat body cells were generated by the FRT/FLP-mediated mitotic recombination (21). Embryos with proper genotypes were collected for 6 h and subsequently incubated at 37 °C for 2 h. To generate transgene-expressing clones, the FRT/FLP-mediated flip-out technique was used (22). Generation of the flip-out clones in the fat body cells did not require heat shock. To induce the flip-out recombination in wing discs, larvae were incubated at 37 °C for 20 min at second instar stage.
RNA Extraction and Quantitative Reverse Transcription PCR Analysis
To induce gene expression of dMyc in the whole body, the third instar larvae with genotypes of Heat shock (Hs)-GAL4, Hs>dMyc (on second chromosome), and Hs>dMyc (on third chromosome) were incubated for 30 min at 37 °C. After recovery for 6 h at 25 °C, the larvae were collected to isolate RNA. For extracting RNA from control and dMyc mutant, first instar larvae of the revertant and dm4 were collected. Total RNA was extracted by TRIzol (Invitrogen) according to the manufacturer's manual. RNA (3 μg) was reverse transcribed by Moloney murine leukemia virus reverse transcriptase (Promega). For quantitative real time PCR (qPCR), the synthesized cDNA was mixed with SYBR Green (Enzynomics, South Korea, RT500) and appropriate primers (300 nm) and then applied to the Bio-Rad iQ5 real time PCR detection system. The following primers were used to amplify cDNA: LTV1 cDNA, 5′-CAGCAGGCACATGTGTTTCTCTGTATTTAC-3′ and 5′-CACGTTTCTTCAGATGCTGCATGTAATCATAG-3′; actin cDNA, 5′-CACACCAAATCTTACAAAATGTGT-3′ and 5′-AATCCGGCCTTGCACATG-3′.
Statistical Analysis
Student's unpaired t test and one-way analysis of variance Dunnett's test (GraphPad Prism 5) were used for statistical comparison.
Immunostaining and Cell Size Measurement
Wandering larvae (for the salivary gland cells) or third instar larvae (for the fat body cells) were cut and turned inside out in phosphate-buffered saline (PBS) and then directly fixed with 4% paraformaldehyde for 20 min at room temperature. After washing for 10 min twice with PBS containing 0.1% Triton X-100 (PBST), the samples were permeabilized in PBS with 0.5% Triton X-100 for 5 min at room temperature and washed once with PBST. The samples were blocked with PBST containing 3% BSA. Primary antibodies and TRITC-labeled phalloidin (Sigma) were applied, and the samples were incubated at 4 °C overnight on a rotator. After three washes for 10 min in PBST, the samples were incubated at room temperature for 2 h with fluorescently labeled secondary antibodies and Hoechst 33258 (Invitrogen). Larvae were washed for 10 min three times with PBST. The salivary glands or the fat body cells were isolated and mounted in 80% glycerol, PBS solution on slide glass. The samples were observed with an LSM710 confocal microscope (Zeiss). Primary antibodies used in this study were as follows: anti-fibrillarin mouse monoclonal antibody (1:200; Abcam, 4566), anti-HA rabbit monoclonal antibody (1:100; Cell Signaling Technology, 3724), and anti-cleaved caspase-3 rabbit polyclonal antibody (1:100; Cell Signaling Technology, 9661). To measure cell size, the fat body cells of third instar larvae were dissected, fixed, and stained by phalloidin and Hoechst to visualize cell boundary and the nucleus, respectively. The samples were mounted with 80% glycerol, PBS solution and then observed by an LSM710 confocal microscope. The cell area was measured by ImageJ.
BrdU Incorporation
Wandering larvae (for the salivary glands) or mid-third instar larvae (for the fat body cells) were dissected on Shields and Sang M3 medium (Sigma, S8398) and then incubated in M3 medium containing 0.1 mg/ml BrdU (Roche Applied Science, 10 280879 001) for 30 min on a rotator at room temperature. After three brief washes with PBS, larvae were fixed in 4% paraformaldehyde for 20 min. The fat body samples were washed three times in PBST and then incubated with 5 units of DNase I (Promega, M6101) in 200 μl of PBS containing DNase I buffer for 1.5 h at 37 °C. The salivary gland samples were washed three times in PBST and then incubated in 2 n hydrochloric acid for 30 min. Larvae were washed three times in PBST followed by incubation with anti-BrdU mouse monoclonal antibody (1:100; BD Biosciences, 555627) in blocking solution.
TUNEL Assay
Wing discs were dissected on PBS and subsequently fixed with 4% paraformaldehyde for 20 min at room temperature. After washing with PBST three times, discs were incubated in 0.1% sodium citrate in PBST for 30 min at 65 °C. The samples were washed three times in PBST followed by incubation in 3% BSA, PBST blocking solution and then incubated with labeling and enzyme solution (In Situ Cell Death Detection kit, Fluorescein, Roche Applied Science, 11 684 795 910) for 2 h at 37 °C. After staining with Hoechst and washing with PBST, wing discs were mounted in 80% glycerol, PBS.
Drosophila S2 Cell Culture
Drosophila S2-DRSC cells were obtained from Drosophila Genomics Resource Center and maintained in Shields and Sang M3 medium with 10% fetal bovine serum (Gibco) and 10% insect medium supplement (Sigma, 17267) at 25 °C. Transfection was performed using Effectene (Qiagen, 301425) according to the manufacturer's instructions on 6-well plates.
Generation of RpS3 Antibody
The RpS3-specific antibody was generated by immunizing rabbits with the peptide MNANLPISKKRKFVS.
Cloning of LTV1 and Ribosomal Protein S3 for Transfection in S2 Cells
To generate the C-terminal V5-tagged LTV1 plasmid, LTV1 cDNA was subcloned into the EcoRI and XbaI sites of pAc5.1-V5/His vector (Invitrogen). For the C-terminal HA-tagged LTV1 plasmid, LTV1-HA containing a stop codon was amplified by PCR from the template pUAST LTV1-HA (see above) and cloned into the EcoRI and NotI sites of pAc5.1-V5/His vector. The RpS3 ORF was amplified by PCR from expressed sequence tag (Drosophila Genomics Resource Center, LD21061). To generate the C-terminal mCherry-tagged RpS3 plasmid, the RpS3 ORF without a stop codon was subcloned into the EcoRI and XhoI sites of pAc5.1-V5/His vector. Subsequently, the mCherry ORF containing a stop codon was subcloned into the XhoI and XbaI sites of pAc5.1-RpS3-V5/His plasmid. The C-terminal V5-tagged RpS3 plasmid was generated by subcloning RpS3 ORF into the EcoRI and XhoI sites of pAc5.1-V5/His vector.
Double-stranded RNA Synthesis and Bathing
For synthesis of double-stranded RNA (dsRNA), double-stranded DNA was amplified by PCR from genomic DNA that was extracted from w1118 with primers containing a T7 polymerase binding site (5′-TAATACGACTCACTATAGGGAGA-3′) at the 5′-end. dsRNA was transcribed using PCR product as a template with the MEGAscript T7 kit (Ambion, AM1334). Synthesized dsRNA was purified using the MEGAclearTM kit (Ambion, AM1908). The following primers were used for dsRNA synthesis: luciferase dsRNA, 5′-TAATACGACTCACTATAGGGAGAGGCCCGGCGCCATTCTATC-3′ and 5′-TAATACGACTCACTATAGGGAGAGATTGGGAGCTTTTTTTGCACG-3′; LTV1 dsRNA, 5′-TAATACGACTCACTATAGGGAGACGGAACTGGGCGACTTGGCTCT-3′ and 5′-TAATACGACTCACTATAGGGAGAACCCCACCACGCAGGACATT-3′; dMyc dsRNA, 5′-TAATACGACTCACTATAGGGAGACAAAGTGACGCATAGCTCCA-3′ and 5′-TAATACGACTCACTATAGGGAGAGGTTATCCTAGCCCTACGCC-3′. For bathing dsRNA, culture medium was replaced by serum-free M3 medium, and S2 cells were diluted to a proper concentration. 1 × 106 cells were applied to a 6-well plate, and 16 μg of dsRNA was added. After incubation for 45 min at 25 °C, serum-containing M3 medium was supplied. S2 cells were lysed after 4 days of incubation at 25 °C.
Generation of LTV1-expressing Stable Cell Line
To generate stably expressing S2 cell lines, each pAc5.1-V5/His or pAc5.1 LTV1-V5/His vector was co-transfected with pCoHygro plasmids (Invitrogen) by Effectene in 6-well plates. Fresh M3 medium containing serum and 300 μg/ml hygromycin (Sigma, H0654) were replaced every 4 days. After 1 month, surviving hygromycin-resistant cells began to proliferate, and they were used for further experiments.
Identification of LTV1-binding Protein
S2 cells stably expressing each empty vector and pAc5.1 LTV1-V5/His were cultured in s T-75 flask, and 1 × 108 cells were collected. After washing twice with 20 ml of cold PBS, S2 cells were lysed with 4 ml of lysis buffer 1 (25 mm Tris, pH 7.9, 150 mm sodium chloride, 10% glycerol, 0.1% Nonidet P-40, 1 mm EDTA, 1 mm dithiothreitol (DTT), 1 μg/ml leupeptin, 1 mm phenylmethylsulfonyl fluoride). The lysates were incubated for 15 min at 4 °C on a rotator. After centrifugation at 13,200 rpm for 15 min at 4 °C, the clarified lysates were transferred. Protein concentration in lysates was measured by Bradford assay. 80 μl of protein G-Sepharose (GE Healthcare, 17-0618-02) was applied to the same concentration of lysates, and then the samples were incubated for 30 min at 4 °C to remove nonspecifically proteins bound to Sepharose. After brief centrifugation, the supernatant was collected. 80 μl of V5-agarose (Sigma, A7345) in a 50% slurry was applied and incubated for 4 h at 4 °C on a rotator. The beads were washed sequentially once with 1 ml of BC150 (20 mm Tris-HCl, pH 7.9, 15% glycerol, 1 mm EDTA, 0.05% Nonidet P-40, 150 mm potassium chloride (KCl)), once with 1 ml of BC300 (20 mm Tris-HCl, pH 7.9, 15% glycerol, 1 mm EDTA, 0.05% Nonidet P-40, 300 mm KCl), twice with 1 ml of BC500 (20 mm Tris-HCl, pH 7.9, 15% glycerol, 1 mm EDTA, 0.05% Nonidet P-40, 500 mm KCl), and once with 1 ml of BC150. The sample buffer (70 mm Tris-HCl, pH 6.8, 2% glycerol, 0.002% bromophenol blue, 3% sodium dodecyl sulfate, 25% β-mercaptoethanol) was directly applied to the washed beads. The samples were boiled and subjected to SDS-PAGE and Coomassie blue staining. Excised gels were sequenced by LC/MS/MS in the Taplin Mass Spectrometry Facility (Harvard Medical School).
Immunoblotting and Immunoprecipitation
S2 cells were grown to a density of 6 × 106 cells/ml, and 1 × 107 cells were collected by pipetting and washed twice with 1 ml of cold PBS. S2 cells were lysed in lysis buffer 1 (described above). The lysates were clarified by centrifugation at 13,200 rpm for 15 min at 4 °C. The whole cell lysates for Western blotting were mixed with the sample buffer and boiled at 95 °C. Anti-HA (MBL International), -V5 (Invitrogen), or -RFP (Abcam) antibody was added to the remaining lysates for immunoprecipitation. The mixture was incubated for 3 h on a rotator at 4 °C. 35 μl of protein G-Sepharose in a 50% slurry was added followed by incubation for 1 h on a rotator at 4 °C. The Sepharose beads were washed five times with 500 μl of washing buffer BC150 (described above). After washing, the immunoprecipitate was boiled with the sample buffer. For immunoblotting of the lysates from flies, 15 adult flies were collected in a tube and homogenized in 150 μl of lysis buffer 1. After centrifugation at 13,200 rpm for 15 min at 4 °C, the cleared lysates were carefully transferred and boiled with the sample buffer. For the treatment with RNase A, the lysates were incubated with 0.1 μg/ml RNase A for 2 h at room temperature, and immunoprecipitation was further conducted. To confirm 18S rRNA degradation, total RNA was extracted by TRIzol-LS (Invitrogen) and reverse transcribed by Moloney murine leukemia virus reverse transcriptase (Promega). Synthesized cDNA was amplified by PCR with the following primers: 5′-CATTCATGTTGGCAGTAAAATGCTTATTGTGTTTG-3′ and 5′-GATCCTTCCGCAGGTTCACCTACG-3′. For immunoblotting, primary antibodies were applied as follow: anti-V5 mouse monoclonal antibody (1:5,000; Invitrogen, 46-0705), anti-HA 3F10 rat monoclonal antibody (1:1,000; Roche Applied Science, 11867423001), anti-β-tubulin mouse monoclonal antibody (E7; 1:500; Developmental Studies Hybridoma Bank), and anti-RFP rabbit polyclonal antibody (1:1,000; Abcam, Ab62341).
Sucrose Density Gradient Analysis
After 200 μg/ml cycloheximide treatment for 10 min at 25 °C, 1 × 107 of S2 cells were washed twice with 1 ml of cold PBS and lysed by incubation for 10 min at 4 °C with 900 μl of lysis buffer 2 (10 mm Tris-HCl, pH 7.3, 5 mm MgCl2, 100 mm KCl, 0.5% Triton X-100, 2 mm DTT, 100 μg/ml cycloheximide, protease inhibitor mixture (Calbiochem, 535140), 200 units of Superase-In RNase inhibitor (Ambion, AM2696)). For analysis of ribosome content in LTV1-HA-overexpressing larvae, 30 third instar larvae were collected and washed twice with 1 ml of cold PBS and then homogenized in 900 μl of lysis buffer 2. After centrifugation at 10,000 relative centrifugal force for 15 min at 4 °C, 850 μl of the cleared lysates was transferred. Protein concentration in the lysates was measured by Bradford assay, and the samples were adjusted to the same protein concentration and volume. 10 ml of 10–50% sucrose gradient in 20 mm HEPES-KOH, 5 mm MgCl2, 100 mm KCl, 2 mm DTT, 100 μg/ml cycloheximide was prepared 1 day before cell harvest. 800 μg of the lysates was carefully applied on the top of the sucrose gradient and then separated by centrifugation at 32,000 rpm for 3.5 h at 4 °C using an SW41Ti rotor (Beckman). The fractions were collected by a fraction collector (Bio-Rad), and UV absorbance at 260 nm was measured for each fraction.
Chromatin Immunoprecipitation Assay
The dsRNA for luciferase and dMyc were bathed in 1 × 106 cells in 6-well plates. After 4 days, S2 cells were incubated in 1% formaldehyde for 10 min at 25 °C for cross-linking. The reaction was terminated by incubation in 0.24 m glycine for 5 min at room temperature. S2 cells were washed twice by 1 ml of cold PBS, collected in a 1.5-ml tube, and lysed with 500 μl of freshly prepared SDS lysis buffer (1% SDS, 10 mm EDTA, 50 mm Tris-HCl, pH 8.1, protease inhibitor mixture). The samples were incubated for 5 min at 4 °C and sonicated 10 times with 5-s/30-s pulses in cold ice water slurry to be sheared to 200–1,000-bp genomic fragments. The samples were clarified by centrifugation at 13,200 rpm for 10 min at 4 °C. 10 μl of the lysates was kept for input, and 400 μl of the lysates was mixed with 3.6 ml of chromatin immunoprecipitation (ChIP) dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mm EDTA, 16.7 mm Tris-HCl, pH 8.1, 167 mm NaCl, protease inhibitor mixture). 50 μl of protein A-Sepharose (GE Healthcare, 17-0974-04) was applied to the diluted lysates followed by incubating on a rotator for 30 min at 4 °C to remove proteins nonspecifically bound to Sepharose in the lysates. After brief centrifugation, the supernatant was transferred, and 1.2 μg of dMyc antibody (Santa Cruz Biotechnology, sc-28208) was added. The same amount of rabbit immunoglobin G (Santa Cruz Biotechnology, sc-2027) was added as a control. The samples were incubated on a rotator overnight at 4 °C. 50 μl of protein A-Sepharose was added to the samples and incubated on a rotator for 1 h at 4 °C. The Sepharose beads were sequentially washed using the following washing buffers: once with low salt washing buffer (0.1% SDS, 1% Triton X-100, 2 mm EDTA, 20 mm Tris-HCl, pH 8.1, 150 mm NaCl, protease inhibitor mixture), once with high salt washing buffer (0.1% SDS, 1% Triton X-100, 2 mm EDTA, 20 mm Tris-HCl, pH 8.1, 500 mm NaCl, protease inhibitor mixture), once with lithium chloride (LiCl) washing buffer (0.25 m LiCl, 1% Nonidet P-40, 1% deoxycholate, 1 mm EDTA, 10 mm Tris-HCl, pH 8.1, protease inhibitor mixture), and twice with TE buffer (10 mm Tris-HCl, pH 8.0, 1 mm EDTA, protease inhibitor mixture). The immunoprecipitates were eluted twice by incubating in elution buffer (0.1% SDS, 0.1 m sodium bicarbonate) on a rotator for 15 min at room temperature. The DNA fragments of input and immunoprecipitate were reverse cross-linked in 200 mm NaCl for 4 h at 65 °C. The samples were further incubated in 10 mm EDTA, 40 mm Tris-HCl, pH 6.5, 40 μg/ml proteinase K for 1 h at 65 °C. The DNA fragments were purified by phenol/chloroform extraction. The contents of DNA fragments were amplified by qPCR. The results were quantified by the ΔCt method and normalized to the input control. The following primers were used for qPCR: LTV1 region A, 5′-CAAATTTGTCTTTCCATACTTATTTCAGACTTG-3′ and 5′-CATGAAATTAATAGTTTTTGAGATTTTGTTCTG-3′; LTV1 region B, 5′-GGCGCCTTTTTTCGAATTTGAGTGTGC-3′ and 5′-ATTGGATTGTTTTCGTAAAATCTTTTGTAAATAC-3′; RpI135, 5′-CTTTTATACAACTTGGATACAATTCAAAATTGG-3′ and 5′-CAGATTAATAGAACACACTAAAATAACCTTAAAG-3′.
Northern Blotting
Total RNA was isolated from second instar larvae by TRIzol. An equal amount of denatured RNA (4 μg) was loaded on a 1.5% agarose gel. The separated RNA was transferred by the capillary method to Hybond N membrane (GE Healthcare, RPN303). The cross-linked membranes were blocked by hybridization buffer (5× SSC, 0.5% SDS) and then incubated with hybridization buffer containing probes at 65 °C overnight. The probe for 5′-end of internal transcribed spacer 1 (5ITS1) was synthesized by a random prime labeling system (GE Healthcare, RPN1633) according to the manufacturer's instructions. To synthesize the template for random prime labeling, the following primers were used: 5ITS1, 5′-TTGTATAATATCCTTACCGTTAATAAATATTTGTAATTATAC-3′ and 5′-GAAACGCCGTTGTTGTAAGTACTCGCCAC-3′. Synthesized probes were purified by a Sephadex G-50 column (GE Healthcare, 27-5330-01) and denatured at 95 °C. The probes for 18S and 28S rRNA were prepared by oligonucleotide 5′-end labeling. 10 pmol of oligonucleotides was mixed with T4 polynucleotide kinase (Takara, 2021A), reaction buffer, and [γ-32P]ATP (3,000 Ci/mmol, 10.0 mCi/ml; PerkinElmer Life Sciences, NEG502A). The mixture was incubated for 1 h at 37 °C, and the probes were purified by Sephadex G-50 column. The following primers were used for oligonucleotide 5′-end labeling: 18S rRNA, 5′-CTTCCTCTAAATAATCAAGTTCGGTCAACTTTTGCGAAACAACCGTAACACGC-3′; 28S rRNA, 5′-GTAACTAGCGCGGCATCAGGTGATCGAAGATCCTCCC-3′.
Electron Microscopy
The salivary glands of wandering larvae were fixed with 3% glutaraldehyde in PBS for 3 h. They were washed five times with 0.1 m cacodylate buffer, pH 7.2 containing 0.1% calcium chloride (CaCl2) at 4 °C and then postfixed with 1% osmium tetroxide in 0.1 m cacodylate buffer, pH 7.2 containing 0.1% CaCl2 for 2 h at 4 °C. After rinsing with cold distilled water, the tissue samples were dehydrated slowly with an ethanol series and propylene oxide at 4 °C. The samples were embedded in Embed-812 (Electron Microscopy Sciences). After polymerization of the resin at 60 °C for 36 h, serial sections were cut with a diamond knife on an Ultracut UC7 ultramicrotome (Leica, Austria) and mounted on Formvar-coated slot grids. The sections were stained with 4% uranyl acetate for 10 min and lead citrate for another 10 min and then observed using a Tecnai G2 Spirit Twin transmission electron microscope (FEI Co.).
Results
LTV1 Is a Novel Cell Growth Regulator
We conducted an RNAi knockdown-based screen of novel cell growth regulators in Drosophila. Because many growth-regulating genes were critical for development, the majority of their mutants exhibited lethality at early larval stages (data not shown). Therefore, we randomly generated RNAi transgene-expressing clone cells by flip-out recombination (22). We examined the effects of RNAi transgenes on the size of the fat body cells because they could be easily observed. Fortunately, compared with the neighboring cells expressing no transgenes, CG7686 RNAi-expressing fat body cells (the GFP-positive cell in Fig. 1, A and A′) displayed decreased cell and nucleus size phenotypes when the cell boundary and DNA were stained with phalloidin and Hoechst, respectively (Fig. 1, A and A′). A BLAST search indicated that CG7686 is the Drosophila orthologue of the mammalian and yeast LTV1. To characterize the function of LTV1 in vivo, we generated deletion mutants of LTV1 in Drosophila. For this purpose, we performed imprecise excisions using the CG7686CB-6202-3 line that contained a P-element in the 5′-untranslated region of LTV1. As a result, we isolated one deletion mutant, LTV1E1, in which the first half of the coding region of LTV1 (including the start codon) was removed (Fig. 1B). The mutant did not express LTV1 transcripts as shown by quantitative RT-PCR in the mutant (Fig. 1C), was arrested at the second larval stage, and died ∼6 days after egg hatching (Fig. 1D and data not shown). To confirm that all these phenotypes were caused by the loss of LTV1, we generated LTV1 transgenic flies. We first expressed LTV1 transgene using a ubiquitously expressed GAL4 driver, Daughterless (Da)-GAL4, and detected the LTV1 protein at ∼65 kDa by immunoblotting (Fig. 1E). When LTV1 was ectopically expressed in LTV1E1 mutant by Da-GAL4, the developmental arrest and lethality of LTV1E1 mutant were completely rescued, confirming that these defective phenotypes are due to the LTV1 mutation (Fig. 1D).
FIGURE 1.

LTV1 is required for cell growth and normal development. A and A′, LTV1 RNAi-expressing clones (GFP-positive) indicated by the arrowhead were generated, and the nuclei and cell size were examined by staining with Hoechst (blue) and phalloidin (red), respectively. B, a schematic representation of LTV1 genomic locus and the genomic deletion region of LTV1E1. The exons of LTV1 are marked by boxes, and the coding regions of LTV1 are by colored black. The P-element, LTV1CB-6202-3, is inserted in the 5′-untranslated region of LTV1. C, qPCR analysis of LTV1 transcripts in w1118 and LTV1E1 flies. Actin expression was used as a loading control. D, larvae of w1118, LTV1 mutants, and LTV1 mutants expressing exogenous LTV1 in the whole body were photographed at 24, 48, and 120 h after egg laying (AEL). E, LTV1 was overexpressed by Da-GAL4. The whole cell lysates were immunoblotted by anti-HA and -β-tubulin antibodies. F and F', FRT/FLP-mediated recombination was used to generate homozygous LTV1E1 mutant clones (arrowhead) that are GFP-negative. G and G', in the fat body expressing exogenous LTV1, LTV1E1 mutant clones (arrowhead) that are GFP-negative were generated by FRT/FLP-mediated recombination. Ectopic expression of LTV1 rescued the decreased cell size of the mutant clones back to that of control cells. F′ and G′, Hoechst (blue) and phalloidin (red) staining was performed to observe the nuclei and cell boundary, respectively. Genotypes are as follows: y w hsflp; UAS-LTV1 RNAi; Act>CD2>GAL4 UAS-GFP (A); y w hsflp; Cg-GAL4 FRT42D UAS-GFP/FRT42D LTV1E1 (F); and y w hsflp; Cg-GAL4 FRT42D UAS-GFP/FRT42D LTV1E1; UAS-LTV1-HA (G).
We also generated mosaic clones of the LTV1 mutant in the fat body using FLP/FRT-mediated mitotic recombination (21). Similar to the cell growth defects of LTV1 RNAi-expressing cells (Fig. 1, A and A′), homozygous LTV1 deletion mutant cells (GFP-negative cells) were much smaller than neighboring control cells (GFP-positive cells) (Fig. 1, F and F′). Moreover, the decreased cell size of LTV1-deleted cells was completely rescued when we overexpressed exogenous LTV1 in the fat body using Cg-GAL4, a fat body-specific driver (Fig. 1, G and G′). These results indicated that LTV1 is required for normal cell growth.
LTV1 Interacts with RpS3 and Co-purifies with Free 40S Ribosome Subunits
To investigate the mechanisms by which LTV1 regulates cell growth, we decided to identify novel proteins that interact with LTV1. We generated an S2 cell line stably expressing the C-terminal V5-tagged LTV1 and immunoprecipitated LTV1 using anti-V5 antibody. Notably, we detected a 32-kDa protein specifically bound to the LTV1 protein (Fig. 2A) and identified the protein as RpS3 in the subsequent mass spectrometry analysis (data not shown). To validate the result, LTV1-V5 and the C-terminal mCherry-tagged RpS3 were transfected in S2 cells. We observed that LTV1 was co-precipitated with RpS3 (Fig. 2B). Additionally, when we immunoprecipitated LTV1, exogenous RpS3 was also co-precipitated (Fig. 2C). Moreover, we generated an antibody against RpS3 to determine whether endogenous RpS3 interacts with LTV1. As expected, endogenous RpS3 was co-precipitated with overexpressed LTV1, further confirming the specific interaction between these two proteins (Fig. 2D). Because ribosomal proteins are assembled with rRNA to form ribosome subunits, we examined whether rRNA is required for the interaction between LTV1 and RpS3. Notably, when we degraded rRNA by treatment with RNase A, LTV1 still interacted with RpS3 (Fig. 2E). This result indicated that the interaction between LTV1 and RpS3 is not dependent on the integrity of 40S ribosome subunits.
FIGURE 2.

LTV1 interacts with RpS3 and co-purifies with 40S ribosome subunits. A, LTV1-V5 and RpS3-mCherry were co-transfected in S2 cells. The immunoprecipitates of LTV1-V5 were separated by SDS-PAGE and subsequently stained with Coomassie Blue. RpS3 was identified by mass spectrometry analysis. Immunoglobin heavy and light chains are indicated by asterisks. B, LTV1-V5 and RpS3-mCherry were expressed in S2 cells by transfection. The whole cell lysates were immunoblotted by anti-RFP and -V5 antibodies, and RpS3-mCherry was immunoprecipitated (IP) by anti-RFP antibody followed by immunoblotting (IB) with anti-RFP and -V5 antibodies. The asterisk indicates immunoglobin heavy chain. C, LTV1-V5 and RpS3-mCherry were transfected in S2 cells. The whole cell lysates were immunoblotted by anti-V5 and -RFP antibodies. LTV1-V5 was immunoprecipitated by anti-V5 antibody, and the immunoprecipitates were blotted by anti-V5 and -RFP antibodies. The asterisk indicates nonspecifically bound proteins. D, transfected LTV1-HA was immunoprecipitated by anti-HA antibody, and LTV1-HA and endogenous RpS3 were detected by anti-HA and -RpS3 antibodies, respectively. The whole cell lysates were blotted by anti-HA and -RpS3 antibodies. E, LTV1-HA and RpS3-V5 were transfected in S2 cells. The lysates were incubated without or with 0.1 μg/ml RNase A for 2 h at room temperature, and RpS3-V5 was immunoprecipitated. The whole cell lysates and immunoprecipitates were immunoblotted by anti-HA and -V5 antibodies. Degradation of 18S rRNA was confirmed by RT-PCR. F, LTV1-HA was transfected in S2 cells. The clarified lysates were applied to a 10–50% sucrose gradient. After centrifugation, the absorbance at 260 nm was measured, and each fraction was collected and analyzed by immunoblotting with anti-HA antibody. The peaks of 40S and 60S ribosome subunits, 80S monosomes, and polysomes are indicated.
Moreover, previous studies in yeast demonstrated that LTV1 associates with pre-40S ribosome subunits, and LTV1-RpS3 subcomplex could be released from pre-40S ribosome subunits in a high salt buffer (11, 23). Therefore, we questioned whether Drosophila LTV1 also associates with 40S ribosome subunits. We expressed LTV1-HA in S2 cells and analyzed its co-localization with 40S ribosome subunits using sucrose gradient experiments. Notably, our immunoblot analyses revealed that LTV1 was co-purified with the fractions containing 40S ribosome subunits but not with 60S ribosome subunits or 80S monosomes (Fig. 2F), suggesting that LTV1 can specifically associate with free 40S ribosome subunits.
LTV1 Is Required for the Biogenesis of 40S Ribosome Subunit
Next, we determined whether LTV1 is required for ribosome biogenesis. We examined the ribosomes isolated from LTV1 knockdown cells on sucrose density gradient. The LTV1 transcripts were efficiently depleted by dsRNA-based knockdown in S2 cells (data not shown). In LTV1 knockdown cells, the amounts of free 40S ribosome subunits and 80S monosomes were severely reduced, whereas free 60S ribosome subunits accumulated compared with the luciferase knockdown controls (Fig. 3A). This imbalanced ratio of 40S/60S ribosome subunits indicated that 40S ribosome biogenesis was defective without LTV1. In addition, we analyzed the amount of 40S and 60S ribosome subunits in LTV1 mutant larvae by hybridizing 18S and 28S mature rRNA, respectively. Consistent with the results from the sucrose gradient analysis, the ratio of 18S to 28S rRNA was significantly reduced by ∼25% in the LTV1 mutant (Fig. 3, B and C). This defect was also rescued by the ectopic expression of LTV1 in the entire body, indicating that it was caused by LTV1 mutation (Fig. 3, B and C).
FIGURE 3.

LTV1 is crucial for 40S ribosome biogenesis. A, the lysates were extracted from S2 cells treated with dsRNA for luciferase (red) and LTV1 (blue). Ribosomes were separated on a 10–50% sucrose gradient. The absorbance at 260 nm was measured. Luciferase dsRNA-treated cell lysates were used for control. B, total RNA was extracted from the second instar larvae of w1118, LTV1 mutant, and LTV1 mutant expressing exogenous LTV1. RNA was subjected to formaldehyde-agarose gel electrophoresis and assessed by Northern blotting. The amounts of 18S and 28S rRNA were analyzed by Northern blotting. C, the relative ratio of 18S and 28S rRNA amounts was quantified. The intensity of each band was measured by MultiGauge. The means of three independent experiments are presented. Error bars show S.E. **, p < 0.005, n = 3. D, LTV1 was ectopically expressed by Da-GAL4. Control (red) and LTV1-HA-expressing (green) third instar larvae were collected and lysed. Ribosomes were separated by a 10–50% sucrose gradient from the lysates, and the absorbance at 260 nm was measured. E–G″, RpS2-HA was immunostained by anti-HA antibody (red) in the fat body cells expressing RpS2-HA (E), RpS2-HA and Crm1 RNAi (F), and RpS2-HA and LTV1 RNAi (G). Transgene-expressing cells are marked by the presence of GFP. Hoechst (blue) was used to stain the nucleus. Genotypes are as follows: y w hsflp; Act>CD2>GAL4 UAS-GFP/UAS-RpS2-3xHA (E); y w hsflp; UAS-Crm1 RNAi; Act>CD2>GAL4 UAS-GFP/UAS-RpS2-3xHA (F), and y w hsflp; UAS-LTV1 RNAi; Act>CD2>GAL4 UAS-GFP/UAS-RpS2-3xHA (G).
Next, we examined whether LTV1 overexpression increases the biogenesis of 40S ribosome subunits and 80S monosomes. Exogenous LTV1 was expressed in the entire body using the Da-GAL4 driver, and the ribosome content of the LTV1-expressing larvae was analyzed using sucrose density gradient experiments. As a result, we did not observe significant differences in the amounts of 40S ribosome subunits and 80S monosomes between LTV1-expressing larvae and controls (Fig. 3D). In addition, LTV1 overexpression did not increase the size of the fat body cells (data not shown). These results indicated that LTV1 overexpression is not sufficient for increasing ribosome contents and cell size.
Previous studies in yeast demonstrate that LTV1 is required for the nuclear export of pre-40S ribosome subunits to the cytosol (24). Therefore, we examined the role of LTV1 in the nuclear export of pre-40S ribosome subunits in Drosophila. In yeast and human cells, the localization of pre-40S ribosome subunits can be monitored by immunostaining of RpS2 (25–28). Because no available antibody detects endogenous RpS2, we overexpressed HA-tagged RpS2 and immunostained with an HA antibody. When RpS2 was expressed alone in the fat body cells, it dominantly localized in the cytosol (Fig. 3, E–E″). To verify whether RpS2 could serve as a reporter for the nuclear export of pre-40S ribosome subunits in Drosophila cells, we co-overexpressed an RNAi transgene for Crm1, the receptor for the nuclear export of pre-40S and pre-60S ribosome subunits (29–31). Interestingly, Crm1 knockdown highly induced the accumulation of RpS2 in the nucleus, consistent with yeast and human cells (Fig. 3, F–F″). Notably, similar to the Crm1 knockdown result, LTV1 RNAi expression also increased the concentration of the nuclear RpS2 (Fig. 3, G–G″), suggesting that LTV1 is important for the nuclear export of pre-40S ribosome subunits.
LTV1 Is Required for Pre-rRNA Processing
During ribosome synthesis, pre-rRNA is processed over multiple steps by several nucleases to yield mature rRNA (4, 6). Because LTV1 is required for 40S ribosome biogenesis, we examined whether LTV1 also functions in pre-rRNA processing. In Drosophila, pre-rRNA is processed by two different pathways (Fig. 4A) (32). To observe the pre-rRNA processing for 18S rRNA synthesis, we examined pre-rRNA intermediates using 32P-radiolabeled 5ITS1 as a hybridization probe. In Northern blotting using the 5ITS1 probe, we detected pre-rRNA and intermediate d in w1118 larvae (Fig. 4B, first lane). We did not observe intermediate a in w1118 likely because of its rapid turnover rate (Fig. 4B, first lane). However, the LTV1 mutant larvae accumulated intermediate a and the unreported 2.5-kb intermediate * (Fig. 4B). The size of intermediate * suggested that this intermediate corresponds to mammalian 21S rRNA, which contains 18S rRNA and 5ITS1 (32). This altered pre-rRNA processing pattern suggested that the cleavage of pre-rRNA intermediates was defective in LTV1 mutant larvae. In addition, ectopic expression of LTV1 fully rescued the pre-rRNA processing defects in LTV1 mutant larvae, confirming that the pre-rRNA processing defects were caused by the loss of LTV1.
FIGURE 4.

LTV1 is required for pre-rRNA processing. A, a schematic representation of Drosophila pre-rRNA processing. Pre-rRNA and its intermediates are designated pre, a, and d according to a previous report (32). External transcribed spacer (ETS), ITS1, and ITS2 are indicated. The 5ITS1 probe, which was used in hybridizing the 5′-region of ITS1, is indicated by black bar. B, total RNA was extracted from the second instar larvae of w1118, LTV1 mutant, and LTV1 mutant-expressing exogenous LTV1. Total RNA was separated in a formaldehyde-agarose gel. Pre-rRNA and its intermediates were examined by Northern blotting with the probe for 5ITS1. Each intermediate is indicated. An asterisk marks the 2.5-kb intermediate.
Loss of LTV1 Induces Cell Death
To find the in vivo functions of LTV1 other than cell growth regulation, we expressed LTV1 RNAi in developing tissues. When LTV1 RNAi-expressing GFP-positive clones were generated in wing imaginal discs using flip-out recombination (22), their clone size (Fig. 5A, lower panel) was much smaller than that of the control clones expressing GFP alone (Fig. 5A, upper panel). Moreover, LTV1 RNAi-expressing clones were accumulated in the basal side of the wing epithelia layers (Fig. 5A, lower panel). Because dead cells have been shown to be extruded basally (33, 34), we examined whether LTV1 knockdown induces cell death. Surprisingly, LTV1 RNAi-expressing wing disc cells showed increased TUNEL signals (Fig. 5, C and C′), whereas control cells did not (Fig. 5, B and B′). In addition, we immunostained control and LTV1 knockdown wing imaginal disc cells with the antibody against cleaved caspase-3. In control cells, we observed weak caspase-3 signals (Fig. 5, D and D′). However, we detected increased cleaved and active caspase-3 signals in LTV1 RNAi-expressing cells (Fig. 5, E and E′), suggesting that LTV1 loss of function induces a caspase-dependent cell death. Combined, these data demonstrated that LTV1 is critical for cell survival.
FIGURE 5.

LTV1 is critical for cell survival in wing imaginal disc. A, confocal z-stack analyses of control clones (top) and LTV1 RNAi-expressing clones (bottom) in wing discs. The clonal cells were marked by GFP. DNA (blue) was stained by Hoechst. B–E′, control clones (B and D) and LTV1 RNAi-expressing clones (C and E) (GFP-positive) were immunostained for TUNEL (B and C) and cleaved caspase-3 (D and E) in wing discs. Hoechst (blue) staining marks the nuclei. Genotypes are as follows: y w hsflp; Act>CD2>GAL4 UAS-GFP (top) and y w hsflp; UAS-LTV1 RNAi; Act>CD2>GAL4 UAS-GFP (bottom) (A); y w hsflp; Act>CD2>GAL4 UAS-GFP (B and D); and y w hsflp; UAS-LTV1 RNAi; Act>CD2>GAL4 UAS-GFP (C and E).
LTV1 Is a Direct Transcriptional Target of dMyc
To understand the upstream regulator of LTV1, we examined dMyc (also known as diminutive (dm)), the oncogenic transcription factor that regulates multiple genes involved in ribosome biogenesis. We determined whether dMyc overexpression increases the level of LTV1 mRNA. For this experiment, we used two transgenic flies of dMyc in which dMyc transgene was inserted in two different chromosome locations. Notably, the ectopic expression of dMyc in the entire body of two different transgenic flies using Hs-GAL4 increased the level of LTV1 mRNA by ∼3.5-fold (Fig. 6A). Because dMyc regulates cell growth and ribosome biogenesis in the salivary glands (35, 36), we also examined LTV1 transcripts in the salivary gland tissues expressing ectopic dMyc. Similar to the results for expression in the entire body, the amount of LTV1 mRNA increased by ∼2-fold when dMyc transgene was expressed in the salivary glands using Ptc-GAL4 (Fig. 6B). By contrast, the transcript level of LTV1 decreased by ∼75% in dm4 (dMyc-null mutant) compared with that of control flies (a revertant of dm4) (Fig. 6C). These results indicated that the level of LTV1 mRNA is positively regulated by dMyc.
FIGURE 6.

dMyc directly regulates LTV1 transcription. A, LTV1 transcripts were measured upon induction of dMyc by heat shock with Hs-GAL4 in the third instar larvae. Two transgenic flies for dMyc were used. One has dMyc transgene in the second chromosome (ch2) and the other in third chromosome (ch3). dMyc was induced by heat shock at 37 °C for 30 min and the larvae were subsequently incubated at 25 °C for 6 h. B, the transcript level of LTV1 in the dMyc-expressing salivary glands was determined. C, the mRNA levels for LTV1 were examined in the first instar larvae of the revertant (RV) and dMyc-null mutant, dm4. A–C, after total RNA extraction and reverse transcription, qPCR was used to quantify LTV1 mRNA. Actin was used as a loading control. Error bars show S.E. A, ***, p < 0.0005, n = 3; B, *, p < 0.05 and **, p < 0.01, n = 3; C, ***, p < 0.0001, n = 4. D, a schematic representation of putative LTV1 promoter region. The transcription start site and the start codon of LTV1 are indicated by arrows. The genomic regions A and B used for ChIP-qPCR are indicated by black bars. E, S2 cells were treated with dsRNA for luciferase and dMyc. After 4 days, protein and DNA were cross-linked by formaldehyde. S2 cells were lysed and immunoprecipitated (IP) by anti-dMyc antibody. Immunoglobin G was used in controls. The genomic sequence (regions A and B) of the LTV1 promoter region was amplified by qPCR. The promoter sequence of RpI135 was used as a positive control. Relative -fold changes over IgG controls were measured. Error bars show S.E. r Region A, *, p < 0.05, n = 3; Region B, **, p < 0.01, n = 3; RpI135, *, p < 0.05, n = 3; ns, non-significant. Genotypes are as follows: Hs-GAL4, UAS-dMyc (ch2); Hs-GAL4, UAS-dMyc (ch3)/Hs-GAL4 (A); Ptc-GAL4, Ptc-GAL4/UAS-dMyc (ch2), Ptc-GAL4; UAS-dMyc (ch3) (B).
To further determine whether dMyc directly regulates LTV1 transcription, we conducted a ChIP assay on S2 cells. We used the identical dMyc antibody used in a previous genome-wide ChIP-sequencing analysis of Drosophila cells (37). As a positive control, we observed that dMyc associated with the promoter region of RpI135, a known target gene of dMyc (Fig. 6E) (35). To confirm the specificity of the association, we depleted dMyc by treatment with dsRNA, which blocked the association of dMyc with the RpI135 promoter region (Fig. 6E). We then examined two different regions (regions A and B) in the putative LTV1 promoter region. Region A is located away from the LTV1 transcription start site, and region B contains the LTV1 transcription start site (Fig. 6D). Notably, dMyc bound to both regions A and B. However, when dMyc was down-regulated, region A was still enriched in dMyc immunoprecipitates, indicating that region A was not specifically associated with dMyc. By contrast, region B was barely detected in dMyc immunoprecipitates from dMyc knockdown cells (Fig. 6E). These results suggested that dMyc directly associates with the promoter region nearby the transcription start site of LTV1 and regulates LTV1 transcription.
LTV1 Is Critical for the Ribosome Biogenesis Induced by dMyc
Because dMyc is a well established regulator of ribosome biogenesis, we investigated whether LTV1 is required for ribosome biogenesis downstream of dMyc. To assess this possibility, we examined the nucleolus size, an index of ribosome biogenesis (35). When the nucleolus was stained in dMyc-overexpressing salivary glands using an antibody for fibrillarin (a nucleolar marker), the size of the nucleolus dramatically increased, indicating that ribosome biogenesis was stimulated (Fig. 7, A and B). By contrast, LTV1 knockdown suppressed the enlargement of the nucleolus of the dMyc-expressing salivary glands (Fig. 7, B and C). A transmission electron microscopy analysis also showed that LTV1 depletion reduced the enlarged nucleolus of the dMyc-overexpressing salivary glands (Fig. 7, D–F). Moreover, the amount of ribosomes and rough endoplasmic reticulum network increased after dMyc overexpression as described previously (Fig. 7, D′ and E′) (35). These increases were also suppressed by LTV1 RNAi expression to levels even lower than those in control (Fig. 7, D′–F′). Collectively, these results showed that LTV1 is critical for the ribosome biogenesis stimulated by dMyc.
FIGURE 7.
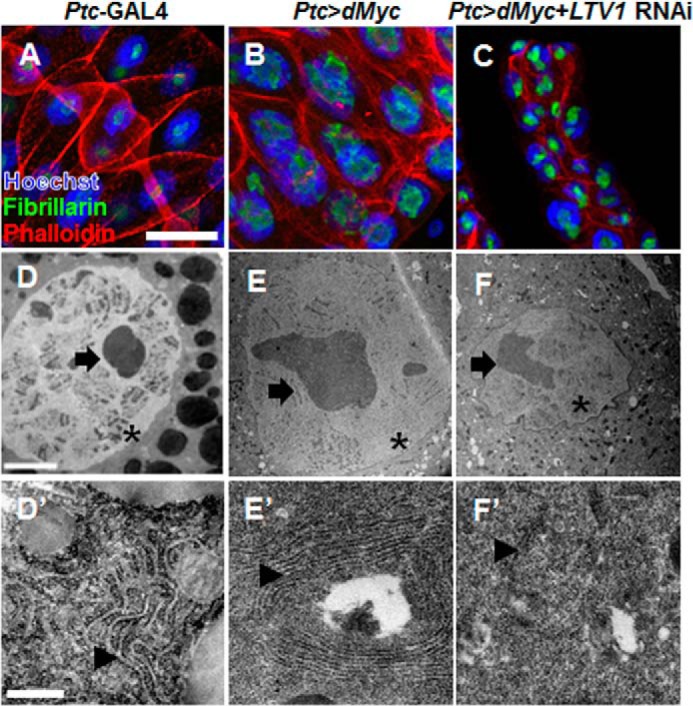
LTV1 knockdown suppresses dMyc-induced ribosome biogenesis. A–C, the nucleus (blue), nucleolus (green), and cell boundary (red) were stained by Hoechst, anti-fibrillarin antibody, and phalloidin, respectively, in the salivary glands of the wandering larvae with the indicated genotypes. D–F′, the nucleus, nucleolus, and rough endoplasmic reticulum of the salivary glands of the wandering larvae with the indicated genotypes were examined by transmission electron microscope. The nuclei are marked by asterisks. The nucleoli are indicated by arrows. The rough endoplasmic reticulum and ribosomes are marked by arrowheads. Scale bars: 500 μm (A–C), 5 μm (D–F), and 500 nm (D′–F′). Genotypes are as follows: Ptc-GAL4 (A and D), Ptc-GAL4/UAS-dMyc (ch2) (B and E), Ptc-GAL4/UAS-dMyc (ch2), UAS-LTV1 RNAi (C and F).
dMyc Requires LTV1 to Promote Cell Growth and Endoreplication
Because ribosome biogenesis is critical for cell growth, we hypothesized that LTV1 is also important for dMyc-induced cell growth. To test this possibility, we determined whether dMyc overexpression can increase the size of LTV1 RNAi-expressing cells. To examine cell size, we expressed transgenes clonally in the fat body cells and measured the cell area of the transgene-expressing cells relative to that of the neighboring cells expressing no transgenes. Control cells expressing only GFP exhibited a cell size similar to that of neighboring cells (Fig. 8, A and G). However, as shown in the previous results (Fig. 1, A and A′), LTV1 RNAi expression dramatically decreased cell size up to ∼90% (Fig. 8, B and G). We also observed highly increased cell sizes of ∼50% (Fig. 8, D and G) when we overexpressed dMyc as shown by others (36). However, when dMyc was simultaneously overexpressed in LTV1 RNAi-expressing cells, dMyc failed to increase cell size (Fig. 8, E and G), indicating that LTV1 is highly important for dMyc-induced cell growth. We speculated that other target genes of dMyc involved in ribosome biogenesis are also important for dMyc-induced cell growth. To investigate this possibility, we expressed RNAi for RpI135 (a subunit of the RNA polymerase I complex) with or without dMyc transgene. RpI135 RNAi expression also decreased cell size by up to ∼90% (Fig. 8, C and G). Moreover, dMyc overexpression did not increase the cell size of RpI135 knockdown cells, similar to the results obtained from LTV1 knockdown cells (Fig. 8, F and G). These results suggested that ribosome biogenesis is critical for dMyc-induced cell growth, and LTV1 is one of the critical dMyc target genes involved in ribosome biogenesis.
FIGURE 8.

dMyc requires LTV1 to promote cell growth and endoreplication. A–F, the fat body clone cells expressing only GFP (control) (A), LTV1 RNAi (B), RpI135 RNAi (C), dMyc (D), both dMyc and LTV1 RNAi (E), and both dMyc and RpI135 RNAi (F) were generated by the flip-out technique. The clone cells were marked by the presence of GFP. The cell boundary and nuclei were stained by phalloidin and Hoechst, respectively, at the mid-third instar larvae (96 h after egg laying). G, the area of the transgene-expressing cells relative to that of the neighboring cells was measured. Error bars show S.E. ***, p < 0.0001; ns, non-significant; n = 23 (control), n = 21 (dMyc), n = 22 (LTV1 RNAi), n = 41 (dMyc + LTV1 RNAi), n = 21 (RpI135 RNAi), and n = 22 (dMyc + RpI135 RNAi). H, the fat body clone cells expressing only GFP (control), dMyc, LTV1 RNAi, and both dMyc and LTV1 RNAi were induced by flip-out recombination. DNA replication of the fat body cells in the mid-third instar larvae (96 h after egg laying) was examined by immunostaining with incorporated BrdU, and the BrdU-positive clone cells were counted. BrdU-positive clone cells/total clone cells were as follows: 10/109 (control), 0/71 (LTV1 RNAi), 35/66 (dMyc), and 6/83 (dMyc + LTV1 RNAi). I–K′, BrdU (green) was incorporated in the salivary glands of the wandering larvae with the indicated genotypes. The nuclei were stained by Hoechst (blue). FB and the dotted line indicate the fat body cells that were contaminated in the preparation of the salivary glands. Genotypes are as follows: y w hsflp; Act>CD2>GAL4 UAS-GFP (A); y w hsflp; UAS-LTV1 RNAi, Act>CD2>GAL4 UAS-GFP (B); y w hsflp; Act>CD2>GAL4 UAS-GFP/UAS-RpI135 RNAi (C); y w hsflp; UAS-dMyc (ch2); Act>CD2>GAL4 UAS-GFP (D); y w hsflp; UAS-dMyc (ch2)/UAS-LTV1 RNAi; Act>CD2>GAL4 UAS-GFP (E); y w hsflp; UAS-dMyc (ch2); Act>CD2>GAL4 UAS-GFP/UAS-RpI135 RNAi (F); y w hsflp; Act>CD2>GAL4 UAS-GFP (control); y w hsflp; UAS-LTV1 RNAi, Act>CD2>GAL4 UAS-GFP (LTV1 RNAi); y w hsflp; UAS-dMyc (ch2); Act>CD2>GAL4 UAS-GFP (dMyc); y w hsflp; UAS-dMyc (ch2)/UAS-LTV1 RNAi; Act>CD2>GAL4 UAS-GFP (dMyc+LTV1 RNAi) (H); Ptc-GAL4 (I); Ptc-GAL4/UAS-dMyc (ch2) (J); Ptc-GAL4/UAS-dMyc (ch2), UAS-LTV1 RNAi (K).
In Drosophila polyploidy cells, such as the salivary glands and the fat body, dMyc promotes endoreplication and cell growth (36). We decided to examine whether LTV1 is required for dMyc-promoted endoreplication in the fat body cells. Drosophila polyploid cells replicate DNA by repeating cycles of G and S phases (38). Therefore, we thought that dMyc-overexpressing fat body cells may enter S phase more frequently than controls. To observe S phase cells, we incorporated BrdU into replicating DNA. We observed that ∼9% of control clone cells replicated DNA in the mid-third instar larvae (96 h after egg laying) (Fig. 8H). However, LTV1 RNAi-expressing clone cells did not replicate during the identical time period (Fig. 8H). Consistent with these results, the nuclear size of LTV1 mutant cells was smaller than that of control cells (Fig. 1A, A′, E, and E′). When ectopic dMyc was clonally expressed, the number of cells replicating DNA was increased by ∼6-fold more than control clone cells (Fig. 8H). However, when both dMyc and LTV1 RNAi were clonally expressed, the number of replicating cells drastically decreased to the level of control cells (Fig. 8H). In addition, a previous study reported that dMyc expression also extends the endoreplicating period (36). Similar to the claims of this previous report, when we expressed dMyc in the salivary glands of wandering larvae, we observed the cells replicating DNA (Fig. 8, J and J′), whereas control cells halted replication (Fig. 8, I and I′). Notably, LTV1 knockdown almost completely blocked the endoreplication induced by dMyc expression (Fig. 8, K and K′). Based on these results, we concluded that LTV1 is critical for the cell growth and endoreplication driven by dMyc.
Discussion
In this study, we identified LTV1 as a novel cell growth regulator using a fly system. We generated an LTV1-null animal to investigate the in vivo functions of LTV1 and discovered that LTV1 is required for normal cell growth (Fig. 1, F and G). Subsequently, we found that LTV1 interacted with RpS3 (Fig. 2, A–D) and co-sedimented with 40S ribosome subunits in a sucrose density gradient (Fig. 2F). We also observed that the amount of 40S ribosome subunits was diminished in LTV1 mutants (Fig. 3, A–C) and further demonstrated that the nuclear export of pre-40S ribosome subunits (Fig. 3, E–G) and the processing of pre-rRNA (Fig. 4) were impaired in the mutants. These results suggested that LTV1 is required for cell growth through regulating ribosome biogenesis. We further characterized LTV1 mutant phenotypes and found that LTV1 functions in cell survival and endoreplication (Figs. 5 and 8H). In addition, we discovered that dMyc transcription factor positively regulates the level of LTV1 mRNA and directly binds to the LTV1 promoter (Fig. 6). Notably, LTV1 RNAi expression suppressed dMyc-induced ribosome biogenesis, endoreplication, and cell growth (Figs. 7 and 8), suggesting that LTV1 is an important downstream factor of dMyc in cell growth regulation.
In yeast, Ltv1p is reported to bind with RpS3 (11, 39), a result highly consistent with this study (Fig. 2, A–D). Furthermore, Ltv1p associates with pre-40S ribosome subunits (11, 23). However, in our anti-LTV1 immunoprecipitation assay (Fig. 2A), we could not identify proteins associated in pre-40S ribosomal subunits other than RpS3. We postulated that they were washed out from LTV1 immunoprecipitates during the washing steps as our washing buffers contained higher salt concentrations than the buffers used in a previous study (Fig. 2A) (11). Consistent with this postulation, we observed that LTV1 was co-purified with 40S ribosome subunits in sucrose gradient experiments (Fig. 2F). Moreover, we examined whether rRNA is required for the interaction between LTV1 and RpS3. Notably, when we degraded rRNA by treatment with RNase A, the interaction between the two proteins was not affected (Fig. 2E). These results indicated that the interaction between LTV1 and RpS3 is highly specific and not dependent on the integrity of 40S ribosome subunits in Drosophila.
Mature 40S ribosome subunits are generated through sequential processing steps from the nucleolus to the cytosol. Here, we observed increased nuclearly localized RpS2, a marker for pre-40S ribosome subunits, in LTV1 mutant cells, similar to a previous study in yeast (Fig. 3, E–G) (24). In yeast, 20S rRNA assembled in pre-40S ribosome subunits is exported from the nucleus and subsequently processed in the cytosol. Notably, we found that 21S rRNA, which corresponds to yeast 20S rRNA, accumulated in Drosophila LTV1 mutant larvae, supporting that pre-40S ribosome subunits were stalled in the nucleus of LTV1 mutant cells (Fig. 4B). These data consistently support that LTV1 is required for the nuclear export of pre-40S ribosome subunits both in yeast and Drosophila. In addition, LTV1 may also be involved in other steps of ribosome biogenesis, although we could not further address this issue in this study. Interestingly, the yeast ltv1 mutant shows a defective maturation of pre-40S ribosome subunits in the cytosol (40).
In this study, we showed that LTV1 transcription was directly regulated by dMyc transcription factor (Fig. 6). However, LTV1 could also be regulated at the translation level. The target of rapamycin complex 1 (TORC1) regulates translation of the mRNAs displaying a structural characteristic of the 5′-terminal oligopyrimidine tract (41). Notably, Drosophila LTV1 mRNA has “CCCTTTCT” at the 5′-terminal end (NCBI accession number NM_136776), suggesting that mRNA translation of LTV1 can be a target of TORC1. Consistent with this suggestion, we observed that LTV1 knockdown suppressed the cell growth induced by a constitutively active insulin receptor (an upstream activator of TORC1) in the developing Drosophila eye (data not shown).
Myc regulates transcription of the diverse genes involved in ribosome biogenesis, cell cycle progression, metabolism, etc. to promote cell growth (42). In this study, we identified LTV1 as another direct transcription target of dMyc (Fig. 6) and found that LTV1 is crucial for dMyc-induced ribosome biogenesis (Fig. 7). Notably, dMyc overexpression could not induce cell growth in LTV1-depleted cells (Fig. 8, A, B, D, E, and G), indicating that LTV1 is a critical target of dMyc in cell growth regulation. Moreover, dMyc also regulates transcription of other ribosome biogenesis-related genes including RpI135, a subunit of the RNA polymerase complex I that synthesizes pre-rRNA (35). Interestingly, the knockdown of RpI135 blocked the cell growth stimulated by dMyc overexpression, similar to LTV1. Furthermore, the heterozygotic loss-of-function mutation of the ribosomal protein L24 suppressed the cell size increase and lymphoma formation induced by Myc overexpression in B cells (43). All of these results consistently support that ribosome biogenesis is crucial for Myc-dependent cell size regulation and tumorigenesis.
In summary, we demonstrated that LTV1 is required for Myc-induced ribosome biogenesis and cell growth in Drosophila. We expect that our data may assist in understanding the molecular mechanism of the tumorigenesis induced by Myc in cancers.
Acknowledgments
We thank Dr. Robert Eisenman for providing Drosophila stocks. We are grateful to Bloomington Stock Center, Kyoto Drosophila Genetic Resource Center, FlyORF, and Vienna Drosophila Resource Center. We thank Drs. Narry Kim and Jun Cho for kind support for sucrose density gradient analysis. We appreciate Dr. Sang-Hee Lee for electron microscopic analysis. We thank Taplin Mass Spectrometry Facility for mass spectrometry analysis. We thank Drs. Eunjoo Cho, Minjeong Ryu, Jinwook Choi, Shin Jeon, and Ji Min Lee for helpful discussion.
This work was supported by National Creative Research Initiatives Program Grant 2010-0018291, Korean Ministry of Science, ICT and Future Planning Grant 2012R1A2A1A01009027, and BK21 Plus Program.
- pre-rRNA
- precursor of ribosomal RNA
- LTV1
- low temperature viability protein 1
- dMyc
- Drosophila Myc
- Rp
- ribosomal protein
- rRNA
- ribosomal RNA
- dsRNA
- double-stranded RNA
- ITS
- internal transcribed spacer
- 5ITS1
- 5′-end of internal transcribed spacer 1
- TORC1
- target of rapamycin complex 1
- Da
- Daughterless
- Ptc
- Patched
- Hs
- Heat shock
- FLP
- flippase
- FRT
- flippase recognition target
- qPCR
- quantitative real time PCR
- TRITC
- tetramethylrhodamine isothiocyanate
- RFP
- red fluorescent protein
- dm
- diminutive
- BrdU
- 5-bromo-2′-deoxyuridine
- ch
- chromosome.
References
- 1. Hariharan I. K., Bilder D. (2006) Regulation of imaginal disc growth by tumor-suppressor genes in Drosophila. Annu. Rev. Genet. 40, 335–361 [DOI] [PubMed] [Google Scholar]
- 2. Neto-Silva R. M., Wells B. S., Johnston L. A. (2009) Mechanisms of growth and homeostasis in the Drosophila wing. Annu. Rev. Cell Dev. Biol. 25, 197–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ruggero D., Pandolfi P. P. (2003) Does the ribosome translate cancer? Nat. Rev. Cancer 3, 179–192 [DOI] [PubMed] [Google Scholar]
- 4. Tschochner H., Hurt E. (2003) Pre-ribosomes on the road from the nucleolus to the cytoplasm. Trends Cell Biol. 13, 255–263 [DOI] [PubMed] [Google Scholar]
- 5. Venema J., Tollervey D. (1999) Ribosome synthesis in Saccharomyces cerevisiae. Annu. Rev. Genet. 33, 261–311 [DOI] [PubMed] [Google Scholar]
- 6. Fromont-Racine M., Senger B., Saveanu C., Fasiolo F. (2003) Ribosome assembly in eukaryotes. Gene 313, 17–42 [DOI] [PubMed] [Google Scholar]
- 7. Henras A. K., Soudet J., Gérus M., Lebaron S., Caizergues-Ferrer M., Mougin A., Henry Y. (2008) The post-transcriptional steps of eukaryotic ribosome biogenesis. Cell. Mol. Life Sci. 65, 2334–2359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zemp I., Kutay U. (2007) Nuclear export and cytoplasmic maturation of ribosomal subunits. FEBS Lett. 581, 2783–2793 [DOI] [PubMed] [Google Scholar]
- 9. Johnson A. W., Lund E., Dahlberg J. (2002) Nuclear export of ribosomal subunits. Trends Biochem. Sci. 27, 580–585 [DOI] [PubMed] [Google Scholar]
- 10. Karbstein K. (2011) Inside the 40S ribosome assembly machinery. Curr. Opin. Chem. Biol. 15, 657–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schäfer T., Maco B., Petfalski E., Tollervey D., Böttcher B., Aebi U., Hurt E. (2006) Hrr25-dependent phosphorylation state regulates organization of the pre-40S subunit. Nature 441, 651–655 [DOI] [PubMed] [Google Scholar]
- 12. Lempiäinen H., Shore D. (2009) Growth control and ribosome biogenesis. Curr. Opin. Cell Biol. 21, 855–863 [DOI] [PubMed] [Google Scholar]
- 13. Arabi A., Wu S., Ridderstråle K., Bierhoff H., Shiue C., Fatyol K., Fahlén S., Hydbring P., Söderberg O., Grummt I., Larsson L. G., Wright A. P. (2005) c-Myc associates with ribosomal DNA and activates RNA polymerase I transcription. Nat. Cell Biol. 7, 303–310 [DOI] [PubMed] [Google Scholar]
- 14. Poortinga G., Hannan K. M., Snelling H., Walkley C. R., Jenkins A., Sharkey K., Wall M., Brandenburger Y., Palatsides M., Pearson R. B., McArthur G. A., Hannan R. D. (2004) MAD1 and c-MYC regulate UBF and rDNA transcription during granulocyte differentiation. EMBO J. 23, 3325–3335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shiue C. N., Berkson R. G., Wright A. P. (2009) c-Myc induces changes in higher order rDNA structure on stimulation of quiescent cells. Oncogene 28, 1833–1842 [DOI] [PubMed] [Google Scholar]
- 16. Schlosser I., Hölzel M., Mürnseer M., Burtscher H., Weidle U. H., Eick D. (2003) A role for c-Myc in the regulation of ribosomal RNA processing. Nucleic Acids Res. 31, 6148–6156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wu C. H., Sahoo D., Arvanitis C., Bradon N., Dill D. L., Felsher D. W. (2008) Combined analysis of murine and human microarrays and ChIP analysis reveals genes associated with the ability of MYC to maintain tumorigenesis. PLoS Genet. 4, e1000090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Maggi L. B., Jr., Kuchenruether M., Dadey D. Y., Schwope R. M., Grisendi S., Townsend R. R., Pandolfi P. P., Weber J. D. (2008) Nucleophosmin serves as a rate-limiting nuclear export chaperone for the mammalian ribosome. Mol. Cell. Biol. 28, 7050–7065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Boon K., Caron H. N., van Asperen R., Valentijn L., Hermus M. C., van Sluis P., Roobeek I., Weis I., Voûte P. A., Schwab M., Versteeg R. (2001) N-myc enhances the expression of a large set of genes functioning in ribosome biogenesis and protein synthesis. EMBO J. 20, 1383–1393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zeller K. I., Haggerty T. J., Barrett J. F., Guo Q., Wonsey D. R., Dang C. V. (2001) Characterization of nucleophosmin (B23) as a Myc target by scanning chromatin immunoprecipitation. J. Biol. Chem. 276, 48285–48291 [DOI] [PubMed] [Google Scholar]
- 21. Xu T., Rubin G. M. (1993) Analysis of genetic mosaics in developing and adult Drosophila tissues. Development 117, 1223–1237 [DOI] [PubMed] [Google Scholar]
- 22. Pignoni F., Zipursky S. L. (1997) Induction of Drosophila eye development by decapentaplegic. Development 124, 271–278 [DOI] [PubMed] [Google Scholar]
- 23. Schäfer T., Strauss D., Petfalski E., Tollervey D., Hurt E. (2003) The path from nucleolar 90S to cytoplasmic 40S pre-ribosomes. EMBO J. 22, 1370–1380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Seiser R. M., Sundberg A. E., Wollam B. J., Zobel-Thropp P., Baldwin K., Spector M. D., Lycan D. E. (2006) Ltv1 is required for efficient nuclear export of the ribosomal small subunit in Saccharomyces cerevisiae. Genetics 174, 679–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Merwin J. R., Bogar L. B., Poggi S. B., Fitch R. M., Johnson A. W., Lycan D. E. (2014) Genetic analysis of the ribosome biogenesis factor Ltv1 of Saccharomyces cerevisiae. Genetics 198, 1071–1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zemp I., Wild T., O'Donohue M. F., Wandrey F., Widmann B., Gleizes P. E., Kutay U. (2009) Distinct cytoplasmic maturation steps of 40S ribosomal subunit precursors require hRio2. J. Cell Biol. 185, 1167–1180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Grandi P., Rybin V., Bassler J., Petfalski E., Strauss D., Marzioch M., Schäfer T., Kuster B., Tschochner H., Tollervey D., Gavin A. C., Hurt E. (2002) 90S pre-ribosomes include the 35S pre-rRNA, the U3 snoRNP, and 40S subunit processing factors but predominantly lack 60S synthesis factors. Mol. Cell 10, 105–115 [DOI] [PubMed] [Google Scholar]
- 28. Milkereit P., Strauss D., Bassler J., Gadal O., Kühn H., Schütz S., Gas N., Lechner J., Hurt E., Tschochner H. (2003) A Noc complex specifically involved in the formation and nuclear export of ribosomal 40 S subunits. J. Biol. Chem. 278, 4072–4081 [DOI] [PubMed] [Google Scholar]
- 29. Moy T. I., Silver P. A. (2002) Requirements for the nuclear export of the small ribosomal subunit. J. Cell Sci. 115, 2985–2995 [DOI] [PubMed] [Google Scholar]
- 30. Moy T. I., Silver P. A. (1999) Nuclear export of the small ribosomal subunit requires the ran-GTPase cycle and certain nucleoporins. Genes Dev. 13, 2118–2133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ho J. H., Kallstrom G., Johnson A. W. (2000) Nmd3p is a Crm1p-dependent adapter protein for nuclear export of the large ribosomal subunit. J. Cell Biol. 151, 1057–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Long E. O., Dawid I. B. (1980) Alternative pathways in the processing of ribosomal RNA precursor in Drosophila melanogaster. J. Mol. Biol. 138, 873–878 [DOI] [PubMed] [Google Scholar]
- 33. Sedlak B. J., Manzo R., Stevens M. (1984) Localized cell death in Drosophila imaginal wing disc epithelium caused by the mutation apterous-blot. Dev. Biol. 104, 489–496 [DOI] [PubMed] [Google Scholar]
- 34. Mitchell N. C., Johanson T. M., Cranna N. J., Er A. L., Richardson H. E., Hannan R. D., Quinn L. M. (2010) Hfp inhibits Drosophila myc transcription and cell growth in a TFIIH/Hay-dependent manner. Development 137, 2875–2884 [DOI] [PubMed] [Google Scholar]
- 35. Grewal S. S., Li L., Orian A., Eisenman R. N., Edgar B. A. (2005) Myc-dependent regulation of ribosomal RNA synthesis during Drosophila development. Nat. Cell Biol. 7, 295–302 [DOI] [PubMed] [Google Scholar]
- 36. Pierce S. B., Yost C., Britton J. S., Loo L. W., Flynn E. M., Edgar B. A., Eisenman R. N. (2004) dMyc is required for larval growth and endoreplication in Drosophila. Development 131, 2317–2327 [DOI] [PubMed] [Google Scholar]
- 37. Yang J., Sung E., Donlin-Asp P. G., Corces V. G. (2013) A subset of Drosophila Myc sites remain associated with mitotic chromosomes colocalized with insulator proteins. Nat. Commun. 4, 1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Budirahardja Y., Gönczy P. (2009) Coupling the cell cycle to development. Development 136, 2861–2872 [DOI] [PubMed] [Google Scholar]
- 39. Loar J. W., Seiser R. M., Sundberg A. E., Sagerson H. J., Ilias N., Zobel-Thropp P., Craig E. A., Lycan D. E. (2004) Genetic and biochemical interactions among Yar1, Ltv1 and Rps3 define novel links between environmental stress and ribosome biogenesis in Saccharomyces cerevisiae. Genetics 168, 1877–1889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Fassio C. A., Schofield B. J., Seiser R. M., Johnson A. W., Lycan D. E. (2010) Dominant mutations in the late 40S biogenesis factor Ltv1 affect cytoplasmic maturation of the small ribosomal subunit in Saccharomyces cerevisiae. Genetics 185, 199–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Meyuhas O. (2000) Synthesis of the translational apparatus is regulated at the translational level. Eur. J. Biochem. 267, 6321–6330 [DOI] [PubMed] [Google Scholar]
- 42. Dang C. V. (2012) MYC on the path to cancer. Cell 149, 22–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Barna M., Pusic A., Zollo O., Costa M., Kondrashov N., Rego E., Rao P. H., Ruggero D. (2008) Suppression of Myc oncogenic activity by ribosomal protein haploinsufficiency. Nature 456, 971–975 [DOI] [PMC free article] [PubMed] [Google Scholar]