

ABSTRACT
Prion diseases are fatal neurodegenerative disorders associated with the conversion of cellular prion protein (PrPC) into its aberrant infectious form (PrPSc). There is no treatment available for these diseases. The bile acids tauroursodeoxycholic acid (TUDCA) and ursodeoxycholic acid (UDCA) have been recently shown to be neuroprotective in other protein misfolding disease models, including Parkinson's, Huntington's and Alzheimer's diseases, and also in humans with amyotrophic lateral sclerosis. Here, we studied the therapeutic efficacy of these compounds in prion disease. We demonstrated that TUDCA and UDCA substantially reduced PrP conversion in cell-free aggregation assays, as well as in chronically and acutely infected cell cultures. This effect was mediated through reduction of PrPSc seeding ability, rather than an effect on PrPC. We also demonstrated the ability of TUDCA and UDCA to reduce neuronal loss in prion-infected cerebellar slice cultures. UDCA treatment reduced astrocytosis and prolonged survival in RML prion-infected mice. Interestingly, these effects were limited to the males, implying a gender-specific difference in drug metabolism. Beyond effects on PrPSc, we found that levels of phosphorylated eIF2α were increased at early time points, with correlated reductions in postsynaptic density protein 95. As demonstrated for other neurodegenerative diseases, we now show that TUDCA and UDCA may have a therapeutic role in prion diseases, with effects on both prion conversion and neuroprotection. Our findings, together with the fact that these natural compounds are orally bioavailable, permeable to the blood-brain barrier, and U.S. Food and Drug Administration-approved for use in humans, make these compounds promising alternatives for the treatment of prion diseases.
IMPORTANCE Prion diseases are fatal neurodegenerative diseases that are transmissible to humans and other mammals. There are no disease-modifying therapies available, despite decades of research. Treatment targets have included inhibition of protein accumulation, clearance of toxic aggregates, and prevention of downstream neurodegeneration. No one target may be sufficient; rather, compounds which have a multimodal mechanism, acting on different targets, would be ideal. TUDCA and UDCA are bile acids that may fulfill this dual role. Previous studies have demonstrated their neuroprotective effects in several neurodegenerative disease models, and we now demonstrate that this effect occurs in prion disease, with an added mechanistic target of upstream prion seeding. Importantly, these are natural compounds which are orally bioavailable, permeable to the blood-brain barrier, and U.S. Food and Drug Administration-approved for use in humans with primary biliary cirrhosis. They have recently been proven efficacious in human amyotrophic lateral sclerosis. Therefore, these compounds are promising options for the treatment of prion diseases.
INTRODUCTION
Prion diseases are fatal neurodegenerative diseases that include Creutzfeldt-Jakob disease in humans, bovine spongiform encephalopathy in cattle, chronic wasting disease in deer, elk, and moose, and scrapie in sheep. These diseases are associated with the misfolding and accumulation of the naturally occurring prion protein (PrPC). The misfolded form (PrPSc) is rich in beta sheet and seeds the ongoing conversion of PrPC with the subsequent development of pathological neuronal loss, astrogliosis, and spongiform change. Human patients usually experience ataxia, dementia, with or without visual disturbances, and late-stage myoclonus. Within months they become akinetic mute, and most die within less than a year from onset. Despite decades of research, there are no disease-modifying therapies available.
Many studies have used PrP as a therapeutic target, either blocking conversion by directly binding PrP or PrPSc, sequestering PrP to a location where it cannot be converted, suppressing PrP expression, interfering with molecules that promote conversion, or enhancing clearance of PrPSc (1). A major reason that these attempts have failed is that by the time symptoms are apparent, pathology is well established and cell death cascades may already be initiated; preventing further PrPSc accumulation at this stage may be insufficient. Instead, compounds that target downstream pathological cascades are required, possibly in combination with PrP conversion inhibitors.
Bile acids are naturally synthesized molecules derived from cholesterol which have recently been investigated as therapeutic agents for a number of diseases. Although the primary function of bile acids is the solubilization of dietary fats and fat-soluble vitamins in the intestinal lumen, the secondary bile acid ursodeoxycholic acid (UDCA) and its taurine-conjugated form, tauroursodeoxycholic acid (TUDCA), have neuroprotective effects in a number of neurodegenerative disease models (2).
TUDCA and UDCA may partially function as chaperones, reducing the accumulation of toxic aggregates in protein folding disease models (3). However, the actual mechanisms of action are not clear and may be multiple, with evidence for reduction of reactive oxygen species formation (4), prevention of mitochondrial dysfunction (5), and inhibition of apoptosis through intrinsic (6) or extrinsic (7) pathways. They can also alter gene expression via nuclear and G protein-coupled receptors, affecting the regulation of many metabolic pathways (8). Of particular interest to prion disease, these bile acids have been shown to reduce the phosphorylation of eukaryotic initiation factor subunit alpha (eIF2α), which has been implicated in prion pathogenesis in later disease stages (9).
Efficacy has been demonstrated in several animal models of protein folding neurodegenerative disease, including Alzheimer's disease (AD), Huntington's disease (HD), and Parkinson's disease (PD). APP/PS1 transgenic mice (an AD model) fed TUDCA had fewer deposits of pathological protein and fewer memory deficits (10). TUDCA treatment of HD transgenic mice led to reduced neuropathology and an improved clinical phenotype (11). Intraperitoneal injection of TUDCA into a PD mouse model activated prosurvival pathways and prevented dopaminergic cell death (12).
In addition to these promising studies, TUDCA and UDCA have therapeutic advantages because they are orally bioavailable, can cross the blood-brain barrier, and are relatively nontoxic. In addition, TUDCA is a U.S. Food and Drug Administration (FDA)-approved drug used in humans to treat primary biliary cirrhosis. Very recently, TUDCA's efficacy was demonstrated in humans with amyotrophic lateral sclerosis (ALS) (13).
In the present study, we provide evidence for aggregation inhibitory effects and demonstrate neuroprotective roles for TUDCA and UDCA in prion disease in vitro, ex vivo, and in vivo.
MATERIALS AND METHODS
Ethics statement.
This study was carried out in strict accordance with the recommendations in the Canadian Council on Animal Care, as approved by the Animal Care and Use Committee (ACUC) of the University of Alberta (study IDs AUP00000335 and AUP00000914; Animal Welfare Assurance Number A5070-01). All mice were bred in an ACUC approved facility with environmental enrichment. Pups (10 to 12 days old) were used for slice culture; 21- to 28-day-old weaned pups were used for intracerebral inoculation. Any animal showing signs of distress, including the loss of more than 20% body weight, were euthanized by CO2 asphyxiation. Mouse pups were rapidly euthanized by cervical dislocation to provide healthy brain tissue for organotypic cultures.
PrP fibril formation.
Recombinant full-length (residues 23 to 231) and truncated (residues 90 to 231) mouse PrP was expressed and purified as previously described (14). Lyophilized recombinant PrP was dissolved in 6 M guanidine hydrochloride (GdnHCl) with a protein concentration of 5 mg/ml. For aggregation under denaturing conditions, the stock solution was diluted in 50 mM sodium phosphate buffer (pH 7.0) to reach a final concentration of 2 M GdnHCl and a final protein concentration of 0.5 mg/ml. For real-time quaking-induced conversion (RT-QuIC) conditions, the stock solution was diluted in 20 mM sodium phosphate buffer (pH 7.4) and 130 mM NaCl, 10 μM EDTA, 0.002% sodium dodecyl sulfate (SDS; final concentration) for a final protein concentration of 0.2 mg/ml (and residual 0.2 M GdnHCl). The fibril formation reactions were carried out in 96-well plates (white plate, clear bottom; Costar 3610) covered with thermal adhesive sealing film (08-408-240; Fisherbrand) using reaction volumes of 200 μl/well. The samples were incubated in the presence of 10 μM thioflavin T (ThT) at 37°C with continuous shaking at 500 rpm for denaturing conditions or 41°C and 900 rpm for RT-QuIC conditions. Fluorescence measurements were taken at 445/482-nm excitation/emission, with a 475-nm cutoff (Victor 3 or Spectramax M5). For seeding experiments, the reaction was carried out in the presence of 0.00025% (wt/vol) preformed recombinant PrP fibrils or 0.0007% (wt/vol) RML strain prion-infected brain homogenate. The lag phase was calculated as previously reported (14). There were at least three technical replicates per experiment.
Surface plasmon resonance (SPR) measurements.
Interactions between recombinant PrP and bile acids were studied using a Biacore X100 system (Biacore, Uppsala, Sweden). Histidine-tagged PrP(90-231) or PrP(23-231) was immobilized on an NTA sensor chip by injecting a 0.05 μM solution of protein for 150 s at a flow rate of 10 μl/min. On average, 300 relative units (RU) of PrP were immobilized. Various concentrations of analytes were injected in running buffer (10 mM HEPES, 150 mM NaCl, 50 μM EDTA, 0.05% [vol/vol] surfactant P20). Bile acids and Congo red were injected for 60 and 180 s, respectively, at a flow rate of 20 μl/min. The dissociation rate was monitored for 120 s.
CD.
A total of 200 μl of PrP(90-231) at 0.5 mg/ml was incubated in the aggregation assays buffers for 2 h at room temperature in the absence or presence of 1 mM TUDCA and then transferred to a 1-mm cuvette for circular dichroism (CD) measurements. The spectra were recorded on a Chirascan CD spectrometer (Applied Photophysics) instrument between 200 and 260 nm, with sampling points every 1 nm. For each sample, 10 scans were averaged, and baseline spectra were subtracted. Thermal denaturation was monitored by heating the sample from 20 to 80°C at 1°C/min in a 1-mm cuvette. The ellipticity was recorded at 222 nm, and the percentage of unfolded protein was plotted. The data were processed using an Applied Photophysics Chirascan viewer and Microsoft Excel.
Cell culture conditions and treatment.
ScN2a (scrapie-infected mouse neuroblastoma cells) (15) were grown in Dulbecco modified Eagle medium (DMEM; Invitrogen, Carlsbad, CA) containing 5% fetal bovine serum (FBS) and 1% penicillin–streptomycin at 37°C with 5% CO2. For treatment, the cells were seeded in a six-well plate at 40 to 50% confluence. The cells were mechanically passaged every 2 to 3 days in the presence or absence of bile acids for a total of 14 days.
Prion-infected brain homogenate for infection.
For the infection of L929 cells, cerebellar slice cultures, and animals, we used a 10% (wt/vol; in water) brain homogenate prepared from a pool of RML prion-infected C57BL/6 mice. Brains were collected from animals at the end-stage of clinical prion disease, and infection was confirmed by proteinase K (PK) digestion and Western blotting for PrP.
SSCA.
The standard scrapie cell assay (SSCA) was performed as previously described (16) with the following modifications. In brief, L929 cells were exposed for 5 days in 96-well culture plates to 0.1% (wt/vol) RML brain homogenates. The cells were passaged three times (1:4 and 1:7), with 20,000 cells collected at the third passage, and loaded onto MultiscreenHTS IP 96-well, 0.45-μm-pore-size filter plates (Millipore, Billerica, MA). The cells were first subjected to PK digestion (5 μg/ml) for 1 h, followed by denaturation with 3 M guanidine thiocyanate. The enzyme-linked immunospot (ELISPOT) reaction was performed with a mouse anti-PrP antibody (SAF83, 1:1,000) and a goat anti-mouse alkaline phosphatase (AP)-conjugated secondary antibody (1:5,000). The plates were developed using BCIP/NBT. The plates were analyzed using an Autoimmun Diagnostika GmbH ELISPOT plate reader (ELR07).
For bile acid treatments, 100 μM TUDCA or UDCA was either used as a pretreatment before infection, added to cells immediately after infection, or added after the first passage. For pretreatment, 100 μM bile acid was incubated with 1% (wt/vol) brain homogenate at 4°C for 24 h before infection. Treatment concentrations were maintained by adding 100 μM bile acids to the media at each cell passage.
Prion-infected organotypic slice culture assay (POSCA).
Prion-infected cerebellar slice cultures were established as previously described (17, 18), using 10- to 12-day-old tga20/+ mice (19). Sagittal slices (350 μm thick) of cerebellum were prepared and treated with 1.0 μg of RML prion-infected or uninfected brain homogenates/ml as free-floating sections for 1 h on ice before washing and plating. The slices were cultured in 50% (vol/vol) MEM, 25% (vol/vol) basal medium Eagle medium, and 25% (vol/vol) horse serum supplemented with 0.65% (wt/vol) glucose, penicillin-streptomycin, and GlutaMAX (Invitrogen), with medium changes three times per week.
Immunofluorescence and microscopy.
POSCA inserts were fixed with fresh 4% paraformaldehyde (PFA; Invitrogen) for 15 min, washed three times, permeabilized with 0.25% Triton X-100 for 15 min, and washed three more times. Slices were blocked with 1% goat serum–1% bovine serum albumin (BSA) for 1 h, treated with 1:4,000 anti-mouse Calbindin (Abcam) in blocking buffer for 1 h, washed three times, treated with 1:4,000 anti-goat Alexa Fluor 488 (Invitrogen) and 1 μg of DAPI (4′,6′-diamidino-2-phenylindole; Roche)/ml for 30 min, and washed three more times.
For the TUNEL (terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling) assay, a DeadEnd fluorometric TUNEL assay (Promega) was used. Slices on inserts were fixed in 4% PFA in phosphate-buffered saline (PBS) for 25 min at 4°C, washed three times, permeabilized with 0.25% Triton X-100 at room temperature for 10 min, washed three more times, equilibrated in equilibration buffer for 10 min, and then treated with rTdT incubation buffer at 37°C for 1 h in a humidified chamber to allow the tailing reaction to occur. The reaction was stopped using 2× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate) at room temperature for 15 min, and the slices were washed three times. The immunofluorescence labeling then proceeded from the blocking step as described above.
All washes were performed apically and basolaterally with PBS (pH 7.4). All stains were prepared in PBS (pH 7.4). After labeling, the membranes were removed from the insert support and placed on a slide with the apical surface of the tissue up. Three drops of Prolong gold (Invitrogen) were placed on the membrane insert, and a coverslip was affixed to the slide. Slides were cured a minimum of 24 h. Twelve bit 2× images were acquired using an InCell Analyser 2000 (GE Healthcare) and analyzed using Imaris 7.1.1 software.
N2a and ScN2a neuroblastoma cells were grown on glass coverslips to 80 to 90% confluence in DMEM (high glucose; Gibco) supplemented with 10% heat-inactivated FBS (Thermo Fisher) with Pen-Strep (Gibco) and GlutaMAX (Gibco). The cells were fixed with fresh 4% PFA for 15 min, washed three times, permeabilized with 0.25% Triton X-100 for 10 min, washed three more times, immersed in 3 M guanidine thiocyanate (Thermo Fisher) for 2 h at 4°C, and blocked with 1% BSA in PBS containing 0.25% Triton X-100 (PBST) for 30 min at room temperature. The primary antibodies were diluted 1:1,000 in PBST, and the samples were incubated in primary solution at 4°C overnight, washed three times, treated with 1:2,000 anti-goat Alexa Fluor 488 (Invitrogen) and DAPI (Roche) at 1 μg/ml for 30 min, and washed three more times. Two drops of Prolong gold (Invitrogen) were added. After labeling, the coverslips were removed from the well and placed on a slide. The slides were cured a minimum of 24 h before imaging. Sixteen bit ×20 images were acquired with a Zeiss LSM 700, ×20 objective lens, and analyzed using Imaris 7.1.1 software.
Cytotoxicity assay.
Cytotoxicity of bile acids was measured according to mitochondrial activity by the MTS [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt] assay. The cells were passaged into a 96-well plate at 40 to 50% confluence and grown in the presence of the compounds for 48 h. Then, 20 μl of MTS solution was added to the wells containing 100 μl of cell media, and the plate was incubated at 37°C for 2 h in a humidified, 5% CO2 atmosphere. The absorbance was measured at 490 nm.
ToxiLight assay.
Inserts with two to three cerebellar slices were exposed to plain media or media containing increasing concentrations of TUDCA or UDCA. After 48 h, the medium was collected, and the toxicity of the compounds was measured using a ToxiLight assay kit according to the manufacturer's protocol (Cambrex Bioscience Rockland). This assay determines the number of dying cells based on the release of adenylate kinase into the media.
Immunoblots.
Cells or cerebellar slices were rinsed twice with PBS buffer and then homogenized in 25 μl of ice-cold radioimmunoprecipitation assay buffer. The total protein concentration was determined by a bicinchoninic acid (BCA) assay. To detect PrPres, 8 to 12 μg of protein (in 10 μl) was treated with PK (20 μg/ml; Invitrogen) at 37°C for 1 h. Then, 2× loading buffer (187.5 mM Tris-HCl [pH 6.8], 15% glycerol, 15% SDS, 9 mM EDTA, 8 M urea, 8% β-mercaptoethanol) was added, the samples were boiled for 10 min, and 15 μl was loaded onto a Novex 4 to 12% Bis-Tris polyacrylamide gel (NuPAGE; Invitrogen). The gels were blotted onto a polyvinylidene difluoride membrane and blocked with 5% milk in TBS supplemented with Tween (TBS-T; 150 mM NaCl, 10 mM Tris-HCl, 0.1% [vol/vol] Tween 20). To determine total PrP, 5 μg of protein was loaded onto the gel. PrP was detected using SAF83 antibody (1:10,000; Cayman Chemical) in 5% BSA–TBS-T. Anti-GRP78/BiP and anti-β-actin (Sigma-Aldrich) were used at a 1:5,000 dilution. Anti-PSD95 (Abcam) was used at a 1:10,000 dilution. Anti-eIF2α (phospho S51; Abcam) was used at a 1:3,000 dilution. Secondary antibodies used were AP-tagged anti-mouse IgG (Promega). Blots were developed using AttoPhos substrate (Promega) and detected on an ImageQuant LAS 4000 (GE Healthcare).
Source of mice and husbandry.
Tg(FVB/N)-Tg(Gfap-luc)-Xen mice or Tg(Gfap-luc) mice are hemizygous and are maintained on site by breeding with FVB/N mice (20). The presence of the Gfap-luc transgene in offspring was determined by PCR. Homozygous tga20 mice (C57BL/6 background) (19) and C57BL/6 mice are also bred on site.
Mouse inoculation and treatment.
Portions (30 μl) of a 1% or 0.1% RML prion-infected brain homogenate were injected into the anterior fontanelles of Gfap-luc or C57BL/6 mice, respectively. UDCA was orally administered by preparing a 0.03 and 0.05% (wt/vol) water solution for Gfap and C57BL/6 mice, respectively. Based on the average water consumption of 5 ml per mouse per day, this translates to daily dosages of 0.006 and 0.01% (wt/wt). In comparison, the recent human ALS pilot study used doses of 1 g twice a day, which, in a 70-kg human, was 0.003% (wt/wt) daily. The treatment was initiated at 14 and 35 days postinfection (dpi) for Gfap mice and 35 dpi for C57BL/6 mice and continued until the mice were euthanized. Timing of euthanasia was based on the loss of coordinated motor function, specifically when mice could no longer escape capture and lost their righting reflex.
Bioluminescence imaging.
Mice were imaged weekly, as previously described (21). In brief, mice were injected intraperitoneally with a solution of d-luciferin potassium salt in PBS (pH 7.4), receiving 150 mg of d-luciferin per kg of body weight. The mice were then anesthetized using an isoflurane-oxygen gas mix and imaged after 15 min for 30 s under anesthesia. Bioluminescence was quantified from images using Living Image 3.0 software (Caliper).
RESULTS
Antiaggregation effect of TUDCA in Gdn-IF with or without seeding by recPrP fibrils.
The inhibitory effect of TUDCA on thermal aggregation of other proteins has been reported (22, 23), so we first assessed TUDCA's ability to prevent the aggregation of prion protein in a range of concentrations from 10 μM to 1 mM. Guanidinium-induced fibrillization (Gdn-IF) assays were performed at pH 7.0 in the presence of 2 M GdnHCl to facilitate amyloid formation (14) and the kinetics of amyloid formation were followed by changes in thioflavin T (ThT) fluorescence intensity. The formation of amyloids is a nucleated polymerization where the rate-limiting step is nucleus formation, seen as a lag phase in our ThT fluorescence assay, followed by exponential growth of the fibrils. The expected sigmoidal curve of amyloid fibril formation was obtained (Fig. 1A) with a lag phase of 23 ± 1.5 h (mean ± the standard error [SE]) in untreated samples and in samples treated with 10 or 100 μM TUDCA. Only at 1 mM TUDCA was a significant increase in the lag phase seen (47 ± 1.5 h) with an ∼20% reduction in ThT intensity by the end of the reaction. Although this could indicate an interaction between TUDCA and the recombinant PrP monomer or other early oligomer (prior to seed formation), no change in growth rate (slope of the curve) was seen, indicating that, once formed, the seed was able to outcompete TUDCA for the monomer/oligomer substrate.
FIG 1.

Effect of TUDCA on PrP(90-231) fibril formation, as monitored by ThT fluorescence. (A and B) PrP(90-231) at 22 μM was incubated under Gdn-IF conditions (50 mM phosphate buffer [pH 7.0] and 2 M GdnHCl at 37°C and 500 rpm) in the absence (A) or presence (B) of seeds. (A) Unseeded PrP(90-231) was aggregated alone (○) or with the addition of 0.01 mM (□), 0.1 mM (△), and 1 mM (♢) TUDCA. B, PrP(90-231) seeded with 0.00025% (wt/vol) preformed fibrils was aggregated with (◆) or without (●) the addition of 1 mM TUDCA. (C) Immunoblot of total aggregated PrP(90-231) formed from under seeded conditions (corresponding to the kinetics curves from panel B) with (+) or without (−) 1 mM TUDCA. The total volume of the aggregation reaction was centrifuged at 10,000 × g for 30 min, and the pellet was resuspended in sample buffer and analyzed by immunoblotting with SAF83. (D and E) PrP(90-231) at 10 μM was incubated under RT-QuIC conditions (10 mM phosphate buffer [pH 7.4], 130 mM NaCl, 10 μM EDTA, and 0.002% SDS at 41°C and 900 rpm) in the presence of normal mouse brain homogenate (NBH) or prion-infected brain homogenate. (D) PrP(90-231) was aggregated in the presence of 0.0007% (wt/vol) NBH (○) or RML brain homogenate with (◆) or without (●) 1 mM TUDCA. (E) PrP(90-231) was unseeded (asterisk) or seeded with 0.0007% (wt/vol) RML (circles), ME7 (squares) and 22L (triangle) strains with (empty symbols) or without (filled symbols) 1 mM TUDCA. Each kinetic was performed in triplicate, and the means ± the standard errors of the mean (SEM) are shown.
To better test whether 1 mM TUDCA could alter aggregation kinetics in the presence of seeds, we seeded the reactions with 0.00025% (wt/vol) preformed fibrils of PrP(90-231). As expected, the addition of seeds reduced the lag phase of control reactions from 23 ± 1.5 h to 11 ± 0.8 h (Fig. 1B). A 1 mM concentration of TUDCA did not prolong the lag phase or alter the growth rate in these seeded assays, confirming that TUDCA did not significantly affect monomer incorporation into the fibrils. However, it did lead to a final reduction in ThT intensity by >50% (Fig. 1B).
To determine whether this reduction in ThT was due to a quenching effect of TUDCA, the ThT fluorescence of preformed fibrils was measured before and after addition of 1 mM TUDCA. No significant changes in fluorescence were observed (1,324 ± 7.320 arbitrary units (a.u.) before, 1,309 ± 30.23 arbitrary units after; mean ± the SE; n = 5). We also ultracentrifuged the final product of the samples from Fig. 1B and performed an immunoblot on the pellet. Banding was less intense for the fibrils formed in the presence of TUDCA (Fig. 1C), indicating that the lower ThT fluorescence did in fact reflect fewer fibrils.
Antiaggregation effect of 1 mM TUDCA in RT-QuIC seeded with three prion strains.
Because TUDCA was able to reduce the total amount of ThT-positive aggregates formed, without a demonstrable effect on the kinetics, we wanted to test TUDCA's effect in more relevant systems. Recombinant PrP fibrils are not infectious under the conditions formed in our study because they are structurally distinct from the infectious form of prion protein (PrPSc) (24). Because aggregation reactions are based on structural conversion, it follows that the structure of the seed might influence the kinetics of aggregation and the efficacy of a conversion inhibitor. We therefore decided to test whether the TUDCA would have any effect on the kinetics induced by infectious seeds, while still using recombinant PrP monomer substrate.
The RT-QuIC assay was developed as a highly sensitive amplification reaction for detection of the infectious form of prion protein (PrPSc) in different prion-infected samples. Although the end products of the reaction do not share all of the morphological and infectious properties of the input PrPSc (25), the assay has been used to evaluate the efficacy of antiprion compounds (26). Using a modified version of RT-QuIC, we tested TUDCA's antiaggregation effect in the presence of infectious prions (Fig. 1D and E). As expected, control reactions incubated with normal brain homogenate (NBH) had a lag phase of at least 30 h (spontaneous seed formation), whereas in the presence of RML-infected brain homogenate (seeded reaction) the lag phase was reduced to 2 h (Fig. 1D). Surprisingly, the presence of 1 mM TUDCA abolished the seeding effect of RML, extending the lag phase to 30 h, which equaled that of spontaneous seed formation in the control. In addition, as seen in the Gdn-IF reactions, the final ThT fluorescence was reduced to ∼30% that of the RML-seeded reaction. Note, this final ThT value was also reduced (by ∼50%) compared to the control spontaneous seed reaction (Fig. 1D).
Since antiprion compounds can have strain-specific efficacy (27), we compared the antiaggregation properties of TUDCA when RT-QuIC was seeded with three different mouse prion strains: RML, 22L, and ME7 (Fig. 1E). Whereas the spontaneous reaction did not aggregate at all within the first 20 h, reaction mixtures seeded with RML, 22L, and ME7 all had lag phases of 2 to 4 h. When 1 mM TUDCA was added, the same extension of the lag phase was observed for all three strains, with no exponential phase observed over the 20 h (Fig. 1E). Of note, when using this modified form of RT-QuIC, we found that there is inherent kinetic variability between different preparations of recombinant PrP, as demonstrated by the kinetic profile differences between Fig. 1D and E, where a different preparation was used for each. Importantly, the kinetics were reproducible within a given preparation, and the inhibitory effects of TUDCA were preserved across the different preparations.
Assessments of TUDCA: PrP binding and structural stability.
The fact that TUDCA could efficiently block aggregation reactions seeded with infectious prions, but not with recombinant PrP fibrils, suggested that TUDCA has a seed-specific effect. Another explanation is that TUDCA was better able to bind monomer under RT-QuIC buffer conditions, which contain less guanidinium. Using surface plasmon resonance (SPR)-based technology, CD, and thermal denaturation, we evaluated interactions between recombinant PrP monomer and TUDCA and the effect of buffer conditions on the structure and stability of recombinant PrP in the presence of TUDCA.
For SPR studies, PrP(90-231) was immobilized on an NTA-sensor chip. As a control, we used Congo red at concentrations ranging from 4 to 250 μM and measured a dissociation equilibrium constant (KD) of 23.3 ± 3.8 μM (Fig. 2A). In contrast, TUDCA interacted very weakly with PrP(90-231), as reflected by the much lower response and the inability to reach a saturation response even at millimolar concentrations (Fig. 2B). Based on our results, any KD for TUDCA must be >200 μM. Similar results were obtained when the full-length version of PrP(23-231) was used (data not shown).
FIG 2.

Interaction of TUDCA with PrP(90-231) as measured by SPR and CD analyses. (A and B) Sensograms of His-tagged PrP(90-231), captured on an NTA-sensor chip, interacting with serial dilutions of Congo red (A) or TUDCA (B). (C) Ellipticity of monomeric PrP(90-231) in the absence (dashed line) or presence (filled line) of 1 mM TUDCA determined under the RT-QuIC and Gdn-IF (inset) buffer conditions. Ten spectra were recorded at 25°C and averaged. (D) Thermal denaturation of PrP(90-231) in the absence (triangles) or presence (squares) of 1 mM TUDCA was monitored by measuring ellipticity at 222 nm.
Using far-UV CD under the buffer conditions for RT-QuIC and for Gdn-IF, we assessed whether TUDCA could induce changes in the secondary structure of PrP(90-231). As expected, PrP(90-231) showed a predominant α-helical signal with minima at 208 and 222 nm (Fig. 2C). The addition of 1 mM TUDCA induced a small loss of signal intensity, but overall the spectrum remained unaltered (Fig. 2C), indicating no significant structural change.
Lastly, because TUDCA has been reported to act as a chaperone and stabilize protein structure (3), we used CD to compare the thermal denaturation profile of PrP(90-231) in the presence and absence of 1 mM TUDCA by monitoring the 222-nm intensity as a function of temperature. In the absence of TUDCA, the melting temperature (Tm) for PrP(90-231) was 66°C. The presence of 1 mM TUDCA did not substantially affect the thermal stability of PrP, only producing a 2°C decrease in Tm (Fig. 2D). Therefore, the ability of TUDCA to reduce seeding efficiency in the RT-QuIC reactions was not explained by an interaction with the monomer.
Ability of bile acids to clear PrPSc in chronically infected cell culture.
If TUDCA's ability to reduce infectious prion seed efficiency is not dependent on an interaction with monomer, then we should be able to substitute cellular PrP (PrPC) as the substrate and still see an effect. For this experiment, we turned to prion-infected cell cultures, in which cells express membrane-associated PrPC substrate under physiological conditions and replicate true infectious prions (unlike the aggregation assays, which contain denaturant, are shaken, and lack membrane-associated PrPC substrate).
We evaluated the ability of TUDCA and its precursor, UDCA, to inhibit the propagation of PrPSc in RML-infected mouse neuroblastoma cells (ScN2a) and measured the levels of PK-resistant PrP (PrPres), which correlates with PrPSc levels. After performing toxicity profiles of TUDCA and UDCA in uninfected N2a cells (data not shown), we determined that, while TUDCA was nontoxic at 1 mM, UDCA caused ∼60% cell death at this concentration. We therefore chose to treat cultures with 100 μM. Both bile acids decreased the levels of PrPres by 40% after the second passage; the amount of PrPres then remained constant for the following passages (Fig. 3). Neither compound completely cleared PrPres after six passages. There was also no discernible effect on PrPC distribution with TUDCA or UDCA treatment (Fig. 4).
FIG 3.

TUDCA and UDCA treatment of N2a cells chronically infected with RML (ScN2a cells). (A) Representative immunoblots of PrPres in ScN2a cells through six passages (3 to 4 days each) in the presence of 100 μM TUDCA (top panel) or 100 μM UDCA (bottom panel). Cells were harvested and treated with 20 μg of PK/ml for 1 h at 37°C. (B) Mean ± the SE densitometries of PrPres from two experiments (*, P < 0.05 [statistically significant difference from control]). TUDCA, diamonds; UDCA, triangles. Signal was normalized to untreated control cells (P0).
FIG 4.

Effect of bile acids on PrPC distribution and cell morphology. N2a cells were exposed to 100 μM UDCA or 500 μM TUDCA for 24 h and then fixed, permeabilized, and labeled with SAF83 for PrPC (green) and DAPI (blue). Images were obtained at ×20 using a Zeiss LSM. Scale bars, 40 μm.
Effect of bile acids on the efficiency of PrPSc infection in SSCA.
Given the ability for TUDCA and UDCA to reduce PrPres propagation cells without a direct interaction with monomeric PrP, we sought to determine whether the bile acids might interfere instead with PrPSc, altering its ability to infect cells or cause PrP conversion. To assess this possibility, we performed infections using infected brain homogenate preincubated with TUDCA or UDCA. We could not do this experiment with ScN2a cells, since they are chronically infected, so we instead use the standard scrapie cell assay (SSCA) based on acute infection and subsequent passage of L929 fibroblast cultures. The SSCA is a powerful tool for quantifying the amount of infectivity in a sample (28, 29).
We first demonstrated an antiprion effect of 100 μM TUDCA or UDCA in the SSCA after infection with 0.1% (wt/vol) RML-infected brain homogenate (Fig. 5). Cultures were treated either starting immediately after infection (D0) or 5 days later at the first passage (P1). Both bile acids reduced PrPres levels and were more effective when given immediately after infection. TUDCA reduced PrPres levels to 57% ± 9% and 73% ± 11% (mean ± the SE) that of the control when treatment started at D0 or P1, respectively. For UDCA, the levels were 38% ± 4% and 62% ± 6%, respectively. We then preincubated 100 μM TUDCA or UDCA in 1% RML-infected brain homogenate at 4°C for 24 h and then used this homogenate to infect the cells (Fig. 5, Pre-Rx only). Because the homogenate is diluted in the process of infection, the final concentration of bile acid in the medium was only 15 μM during the first passage in these preincubated samples. Despite the lower final concentration of bile acid in the media, preincubated samples produced PrPres levels comparable to cells treated with 100 μM bile acid at D0. Further supplementing the culture medium to reach 100 μM bile acid after preincubation did not lead to further inhibition (Fig. 5, Pre + Post-Rx), indicating that maximal inhibition was achieved from contact between the inoculum and bile acid.
FIG 5.
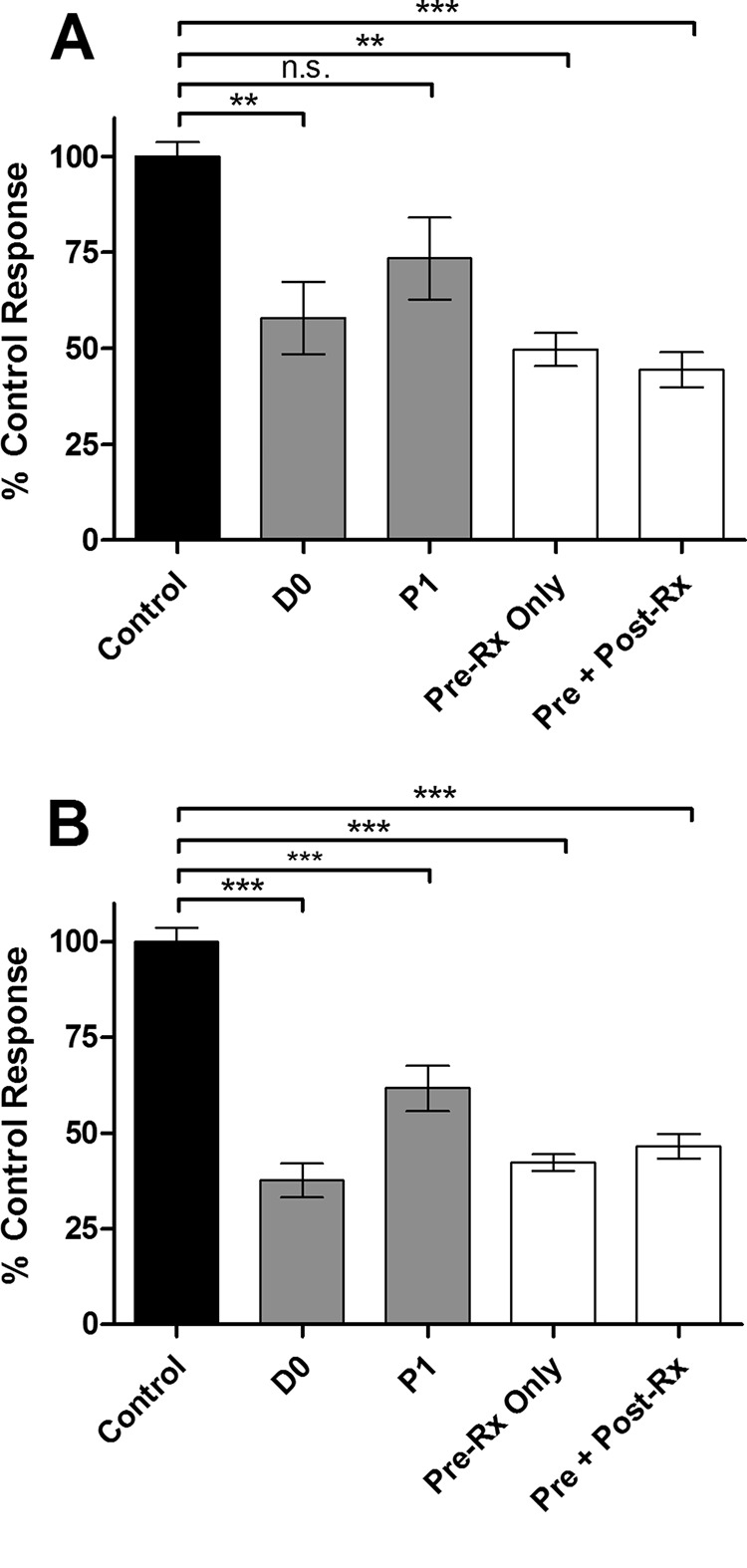
TUDCA and UDCA treatment of L929 cells acutely infected with RML (scrapie cell assay). Levels of PrPres after three passages (5 days each) in a scrapie cell assay in the presence of 100 μM TUDCA (A) or UDCA (B). The cells were treated immediately after RML infection (D0), starting after the first passage (P1), or were exposed to infected brain homogenate preincubated with bile acid (Pre-Rx), followed by no further addition (Pre-Rx only) or the addition of 100 μM TUDCA at passage 1 (Pre + Post-Rx). The readout was normalized to untreated infected control wells. The data represent mean ± the SEM. n = 8 for D0 and P1; n = 4 for Pre-Rx only and Pre + Post-Rx. ***, P < 0.001; **, P < 0.01 (statistically significant difference from the control). n.s., P > 0.05.
Effect of bile acids on infected cerebellar slice culture.
Given the partial efficacy of the bile acids in two cell culture models of prion infection, we examined their effect on the replication of PrPSc in cerebellar slice culture. The prion organotypic slice culture assay (POSCA) allows prion replication ex vivo under conditions that closely resemble intracerebral infection (17, 18). In addition, it has been demonstrated that this assay predicts in vivo efficacy of drug treatment more accurately than cell-based assays (30).
We first confirmed that 100 μM TUDCA or UDCA was not toxic to POSCA (data not shown). We then infected cerebellar slices with 1% RML-infected brain homogenate and treated with TUDCA starting at day 0. As expected, in untreated samples, PrPres was detected by day 17 postinfection, and the levels of PrPres continued to increase for the following 10 days (Fig. 6A). In treated samples, PrPres was still first detectable by day 17 postinfection, but the PrPres levels remained stable from day 21 onward, with the total amount of PrPres reduced by ca. 50% (Fig. 6B). This reduction was not because of a reduction of PrPC, since the total amount of PrP remained relatively constant over 28 days, and it was the same in both treated and untreated cultures (Fig. 6A). This effect was dose dependent, since treatment with a lower dose of TUDCA (10 μM), starting at day 7, did not affect PrPSc levels (data not shown).
FIG 6.

TUDCA treatment of RML-infected cerebellar slice cultures. (A) Immunoblot for PrPres, total PrP, BiP, and p-eIF2α of RML-infected cerebellar slice cultures left untreated or treated with 100 μM TUDCA starting immediately after infection. Samples were harvested at days 14, 17, 24, and 27 postinfection. (B) Quantification of PrPres and p-eIF2α signals from three independent samples harvested at 27 dpi. The data represent means ± the SEM. *, P < 0.05 (statistically significant difference from the control).
Neuroprotective effect of bile acids on cerebellar slice culture.
Another advantage of POSCA is that it not only propagates PrPres but also closely reproduces prion pathogenesis (18, 30). To evaluate whether the antiprion propagation effect observed for the bile acids was accompanied by a neuroprotective effect, we assessed neuronal viability and overall levels of cell death in the cerebellar slice cultures over the course of the infection. When the slices where exposed to noninfectious brain homogenate (NBH), the slices maintained a stable density of neurons, including Purkinje and cerebellar granule cells, for 49 dpi (Fig. 7). TUDCA and UDCA treatment did not induce neuronal loss or affect overall viability relative to controls, as measured by TUNEL staining. As expected, infection with 10 μg of RML-infected brain homogenate/ml induced the loss of granule neurons and Purkinje cells, beginning at day 42 and progressing through day 49 postinfection.
FIG 7.

Bile acid-induced neuroprotection ex vivo. Cerebellar slices were stained with antibodies to Calbindin (green) and NeuN (white), in addition to TUNEL (red) and DAPI (blue) counterstains. n = 3 (representative images are shown). RML-infected slices were treated with 500 μM TUDCA or 100 μM starting at day 14 (D14) or 21 (D21) postinfection. Slices were collected at days 35 (D35), 42 (D42), and 49 (D49), stained, and analyzed by confocal microscopy. Prion infection elicited a significant loss of granule cell neurons (NeuN+) and Purkinje cells (Calbindin+). Uninfected controls were inoculated with normal brain homogenate (NBH) and were either left untreated or treated with TUDCA or UDCA starting at day 14 or 21.
For our treatment applications, we chose to maximize tolerable dosages and also test delayed treatments, because effective prion therapies need to have efficacy if started after symptom onset. Based on toxicity profiles (data not shown), we used 500 μM TUDCA and 100 μM UDCA and tested treatments starting at day 14 (D14), which is ∼3 days before PrPres is first detectable, or at day 21 (D21), when the levels of PrPres are clearly detectable. Treatment with either bile acid starting at day 14 had a neuroprotective effect in infected cultures, maintaining the levels of granule and Purkinje cells for 49 days. Less neuroprotection was observed when treatment was started at day 21 postinfection. Comparing the two compounds, a qualitatively greater neuroprotective effect was seen with UDCA than with TUDCA when treatment started at day 21.
UDCA treatment reduces astrogliosis in male Gfap-luc mice.
Based on the marked astrogliosis observed in prion diseases, Tamguney et al. demonstrated that prion infection could be measured by bioluminescence of prion-inoculated mice that express luciferase under the Gfap gene promoter (21). We used this mouse model here to test the efficacy of orally administered UDCA against prion infection. We infected the mice with 1% RML brain homogenate and scanned the animals approximately weekly starting at day 49 postinfection. As previously reported (21), infected mice started to show bioluminescence at ∼60 dpi, reaching a maximum between 80 and 90 dpi. Taking advantage of the water solubility and ability of UDCA to cross BBB, we supplied the mice with water containing 0.03% (wt/vol) UDCA. Starting UDCA treatment at day 7 or 35 postinfection significantly reduced bioluminescence in the male mice (Fig. 8A and B), indicating possible protection of UDCA against the inflammatory reaction induced by prion infection. Starting treatment at day 35 postinfection also significantly reduces bioluminescence in Gfap-luc male mice brain (Fig. 8B). Surprisingly, astrogliosis inhibition was gender dependent, being only observed in male mice and not in females (Fig. 8B).
FIG 8.

Bioluminescence and survival curves of RML-infected Tg(Gfap-luc) and C57BL/6 mice treated with UDCA. Tg(Gfap-luc) mice were intracerebrally inoculated with 1% RML-infected brain homogenate and treated with 0.03% (wt/vol) UDCA, whereas C57BL/6 mice were intracerebrally inoculated with 0.1% RML-infected brain homogenate and treated with 0.05% (wt/vol) UDCA. (A) Representative scans of one infected untreated male mouse (top panels) and one infected treated male mouse (bottom panel). Treatment started at 7 dpi. Scans were taken at 49, 68, 71, 78, 85, 91, and 98 dpi. (B) Bioluminescence quantification from the brains of male (left) and female (right) infected Tg(Gfap-luc) mice untreated (circles) and treated starting at 7 dpi (diamonds) or 35 dpi (triangles). The data show means ± the SEM (n = 6 for control and n = 4 for mice treated at day 35 and females treated at day 7; n = 3 for males treated at day 7; **, P < 0.01). (C) Kaplan-Meier plots for survival time of male (left) and female (right) infected C57BL/6 mice untreated (dashed curve) and treated starting at day 35 (solid curve). Log-rank (Mantel-Cox) and Gehan-Breslow-Wilcoxon tests were applied for statistical analysis obtaining P values of 0.021 and 0.039, respectively (n = 6 for control [3 males, 3 females]; n = 5 for treated males; n = 5 for treated females; *, P < 0.05).
UDCA treatment increases the survival time in male C57BL/6 mice.
Based on the promising reduction in bioluminescence in UDCA-treated Gfap-luc mice, we decided to adjust the conditions to maximize the UDCA effects. In this case, C57BL/6 mice were infected with 0.1% (wt/vol) RML (instead of 1% [wt/vol] used for Gfap-luc mice). We increased the concentration of UDCA from 0.03 to 0.05%, the maximal concentration at which this bile acid remains soluble. Since UDCA treatment in Gfap-luc mice reduced bioluminescence whether started at day 7 or 35 postinfection, we started treatment at day 35 for this experiment. The mice tolerated the daily treatment without signs of distress or weight loss. The mean survival was 169 ± 1 dpi (mean ± the SE) for untreated animals, with no difference between male and female mice. Treatment with UDCA extended the mean survival of male mice to 180 ± 1 dpi without effect on the female mice, whose mean survival was 166 ± 3 dpi (Fig. 8C).
Levels of PSD95 and p-eIF2α after TUDCA treatment.
Although it is possible that the reduction in PrPres levels alone accounts for the neuroprotective effects seen in POSCA and the change in survival curves, TUDCA, and UDCA have been reported to have a number of downstream effects on the pathogenesis in other diseases (2). Using POSCA, we have previously observed early changes in postsynaptic density protein 95 (PSD95) induced by prion infection, occurring around the time of first PrPres detection (18). We therefore chose to examine whether this early marker is affected by TUDCA. When we examined PSD95 levels in infected slices treated with or without 100 μM TUDCA, we found that TUDCA reduced the usual increase in PSD95 seen at day 24 (Fig. 9A), although this effect was not statistically significant (P = 0.051).
FIG 9.

Effect of TUDCA on PSD95 levels of infected cerebellar slices. (A and B) Western blots and densitometric analysis of PSD95 expression levels in POSCA untreated and treated with 100 μM TUDCA starting at day 0 (A) or 10 μM TUDCA starting at day 7 (B) postinfection. Samples were harvested at days 14, 17, 21, 24, and 27. The data represent means ± the SEM (n = 3 for untreated and n = 2 for treated samples).
To test whether any effect could simply be secondary to the reduced PrPres levels in treated slices, we studied the levels of PSD95 in samples treated with 10 μM TUDCA at day 7 postinfection, a treatment condition that we previously found had no effect on PrPres accumulation. A similar trend to reduce PSD95 levels was observed in these samples (Fig. 9B), again not reaching statistical significance (P = 0.126).
Because eIF2α phosphorylation has been implicated in prion disease pathogenesis (9), and TUDCA has been reported to decrease p-eIF2α (31, 32), we also examined p-eIF2α in our slices. Interestingly, TUDCA induced a significant increase in p-eIF2α by day 27 (Fig. 6A). In addition, a lower-molecular-weight band of p-eIF2α (the cleaved form [33]) became more prominent in treated samples at day 21. When misfolded proteins accumulate in the endoplasmic reticulum (ER), binding immunoglobulin protein (BiP) expression increases, and this is used as a marker of ER stress. Elevation of BiP is one trigger for the phosphorylation of eIF2α. However, no significant changes in BiP were observed in our model (Fig. 6A).
DISCUSSION
The bile acids TUDCA and UDCA have protective effects in several protein folding neurodegenerative disease models, including AD, HD, and PD (10–12). In the present study, we tested the efficacy of these compounds in prion disease, both in terms of their effect on prion protein aggregation and their ability to prevent neuronal loss induced by prion infection.
We demonstrated that TUDCA has antiaggregation effects on recombinant prion protein (recPrP) in vitro without any direct interaction with monomeric recPrP, as determined by our aggregation kinetics, CD, and SPR studies. This inhibitory effect was interesting in that TUDCA interfered with the seeding ability of three strains of infectious prions (seen as a prolongation of the lag phase in RT-QuIC), but not of recPrP fibrils. This effect of TUDCA on PrPSc seeds was confirmed by preincubation experiments using the scrapie cell assay. Interestingly, the level of PrPres produced by the cells was reduced to the same degree whether the cells were exposed to RML-infected brain homogenate preincubated with bile acid or whether they were treated with 100 μM bile acid starting immediately after infection. Treating cells with 100 μM bile acid after exposure to preincubated brain homogenate did not reduce levels any further. Thus, preincubation was as effective a method of inhibition as was immediate treatment, implying an interaction between TUDCA and the PrPSc seeds was occurring, as seen in our RT-QuIC experiments.
In addition to interfering with PrPSc seeding efficiency, TUDCA reduced by half the total amount of ThT-positive aggregates produced in kinetic studies and the amount of PrPres produced in chronically infected cell culture and POSCA. Given that TUDCA does not bind monomeric PrP, explanations for this reduction include either a sequestration or a breakdown of substrate (other than monomer) that would normally be incorporated into the fibrils. Given the maximal reduction in infection of the scrapie cell assay by preincubation of TUDCA with the brain homogenate, it is most likely that this substrate is the seed itself. This is why, in the RT-QuIC reactions, the addition of TUDCA does not delay the lag phase beyond that of the normal brain homogenate (spontaneous seed formation) control; TUDCA does not affect the formation of the seed itself. Whether TUDCA actually sequesters the seed or enhances its breakdown (or its clearance in cells) is not clear, but the balance between production and clearance is definitely altered, because we see lower levels of PrPres maintained over serial passage in chronically infected N2a cells treated with TUDCA or UDCA, and we see steady-state levels maintained in POSCA over time.
Because neuropathology does not necessarily correlate with the levels of PrPres, and because bile acids have proposed mechanisms of action beyond effects on protein aggregation, we looked for evidence of bile acid neuroprotection in POSCA. Encouragingly, for slices treated starting at day 14 (∼3 days before PrPres is detectable), neuronal loss was reduced. This effect was more pronounced for UDCA than TUDCA. When treatment started later, at day 21 (when the levels of PrPres are clearly detectable), the effect was not seen for TUDCA but was still apparent for UDCA. This change in efficacy with delayed treatment was also apparent in our scrapie cell assay, where cells treated starting at day 5 (first passage) produced more PrPres than those treated on day 0.
In our animal studies, mice received daily dosages of 0.006 to 0.01% (wt/wt) UDCA. By comparison, a recent study showing efficacy of TUDCA in human ALS patients used 1 g twice a day, which translates to 0.003% (wt/wt) daily. TUDCA has been shown to reduce both astrocytosis (10) and glial activation (34). The reduction in bioluminescence seen in UDCA-treated Gfap-luc mice was consistent with these other studies. What was striking was the gender predominance of the effect. This benefit to males was also seen in the prolonged incubation periods of male C57BL/6 mice. There are no prior studies that have observed this gender bias, but we speculate that the most likely reason is a gender difference in the metabolism of the drug itself. The absorption of UDCA from the intestine is incomplete but when it is absorbed, it is extracted by the liver, conjugated with glycerine and taurine, secreted in bile, and then undergoes enterohepatic circulation with the endogenous bile acids. The enterohepatic circulation is efficient, which results in relatively low serum concentrations (35). The more hydrophilic TUDCA is better absorbed than UDCA, and, although it can be partially deconjugated and reconjugated with glycine, it has reduced biotransformation to hydrophobic metabolites which leads to higher bioavailability (36). Therefore, we have now begun studies of TUDCA at higher dosages in mice to determine whether we can overcome this gender bias.
There is a wealth of data from studies looking at TUDCA and UDCA in other models of disease, with many mechanisms of action proposed (3–8, 31, 32). It is beyond the scope of this study to present a definitive and unifying mechanism for these bile acids, since it would appear that effects can depend to some extent on cell type and disease process. Nevertheless, several results from our study are worth discussing in light of previous findings.
In POSCA, we have previously identified a transient increase in postsynaptic density 95 protein (PSD95) after prion infection (18). This transient early increase has also been found in prion-infected Tg37 mouse models, occurring 1 week before PrPres detection by Western blotting (9), and in the related protein folding neurodegenerative disease, Alzheimer's disease (AD) (37). PSD95 interacts with N-methyl-d-aspartate (NMDA) receptors to modulate their activity and this modulation is deregulated during the excitotoxicity associated with neurodegenerative diseases. Given this, the modulation of PSD95 levels has been proposed as a therapeutic strategy to treat NMDA receptor-mediated excitotoxicity (37). We found that TUDCA may reduce this increase in PSD95, independent of its effect on PrPres, although the change in POSCA is subtle, not reaching statistical significance, so it will need to be confirmed in animal studies.
Related to this, and perhaps of more interest, is TUDCA's effect on the phosphorylation of eIF2α in POSCA. There is a wealth of data supporting TUDCA's ability to reduce ER stress in models of bowel inflammation, liver disease, and pulmonary fibrosis (22, 31, 32, 38). Some of these effects have been observed as a reduction in levels of phosphorylated eIF2α. In our study, by contrast, we have found a consistent and statistically significant increase in p-eIF2α in the early stages of POSCA infection. eIF2α regulates protein translation and, in its phosphorylated form, it prevents the translation of a number of proteins, one of which is PSD-95 (9). The modest increase in p-eIF2α that we observed in the early stages of POSCA infection did in fact correlate well in time with the lack of increase in PSD-95. This reduction of translation is thought to reduce protein burden in a cell. The accumulation of misfolded protein in the ER is one cause of ER stress that can lead to phosphorylation of eIF2α. However, the chaperone BiP is usually upregulated under such circumstances, an upregulation that is upstream of eIF2α phosphorylation. In POSCA, BiP levels did not change, suggesting that the increasing p-eIF2α levels were induced by a different pathway, one induced by TUDCA. Thus, an early increase in p-eIF2α may actually be a protective and adaptive response, while sustained phosphorylation may be detrimental late in prion disease (9). Further studies of TUDCA's effect on p-eIF2α levels in early and late prion disease in vivo will help confirm this effect.
To conclude, we present here evidence that TUDCA and UDCA can interfere with the seeding efficiency of prions and provide neuroprotection in prion-infected brain slice cultures, with pilot studies indicating some efficacy in vivo. The upstream effect that we describe in the present study complements the diverse array of downstream neuroprotective mechanisms of these bile acids and increases the potential for efficacy in human prion disease. Further studies in prion-infected animals will be required to fully elucidate all other mechanisms of action in prion disease, but the fact that these agents may act at different levels of the pathogenic cascade, coupled with the advantages of being orally bioavailable, permeable to the blood-brain barrier, nontoxic and FDA-approved for human use, make these natural compounds promising alternatives for the treatment of prion diseases.
ACKNOWLEDGMENTS
This study was supported by the University Hospital Foundation (grant RES0021090) and the Alberta Prion Research Institute (grant RES0020661).
REFERENCES
- 1.Sim VL. 2012. Prion disease: chemotherapeutic strategies. Infect Disord Drug Targets 12:144–160. doi: 10.2174/187152612800100161. [DOI] [PubMed] [Google Scholar]
- 2.Cortez L, Sim V. 2014. The therapeutic potential of chemical chaperones in protein folding diseases. Prion 8:197–202. doi: 10.4161/pri.28938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Geier A, Wagner M, Dietrich CG, Trauner M. 2007. Principles of hepatic organic anion transporter regulation during cholestasis, inflammation, and liver regeneration. Biochim Biophys Acta 1773:283–308. doi: 10.1016/j.bbamcr.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 4.Rodrigues CM, Fan G, Wong PY, Kren BT, Steer CJ. 1998. Ursodeoxycholic acid may inhibit deoxycholic acid-induced apoptosis by modulating mitochondrial transmembrane potential and reactive oxygen species production. Mol Med 4:165–178. doi: 10.1007/s0089480040165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rodrigues CM, Fan G, Ma X, Kren BT, Steer CJ. 1998. A novel role for ursodeoxycholic acid in inhibiting apoptosis by modulating mitochondrial membrane perturbation. J Clin Invest 101:2790–2799. doi: 10.1172/JCI1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rodrigues CM, Sola S, Sharpe JC, Moura JJ, Steer CJ. 2003. Tauroursodeoxycholic acid prevents Bax-induced membrane perturbation and cytochrome C release in isolated mitochondria. Biochemistry 42:3070–3080. doi: 10.1021/bi026979d. [DOI] [PubMed] [Google Scholar]
- 7.Azzaroli F, Mehal W, Soroka CJ, Wang L, Lee J, Crispe IN, Boyer JL. 2002. Ursodeoxycholic acid diminishes Fas-ligand-induced apoptosis in mouse hepatocytes. Hepatology 36:49–54. doi: 10.1053/jhep.2002.34511. [DOI] [PubMed] [Google Scholar]
- 8.Hylemon PB, Zhou H, Pandak WM, Ren S, Gil G, Dent P. 2009. Bile acids as regulatory molecules. J Lipid Res 50:1509–1520. doi: 10.1194/jlr.R900007-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moreno JA, Radford H, Peretti D, Steinert JR, Verity N, Martin MG, Halliday M, Morgan J, Dinsdale D, Ortori CA, Barrett DA, Tsaytler P, Bertolotti A, Willis AE, Bushell M, Mallucci GR. 2012. Sustained translational repression by eIF2α-P mediates prion neurodegeneration. Nature 485:507–511. doi: 10.1038/nature11058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nunes AF, Amaral JD, Lo AC, Fonseca MB, Viana RJ, Callaerts-Vegh Z, D'Hooge R, Rodrigues CM. 2012. TUDCA, a bile acid, attenuates amyloid precursor protein processing and amyloid-beta deposition in APP/PS1 mice. Mol Neurobiol 45:440–454. doi: 10.1007/s12035-012-8256-y. [DOI] [PubMed] [Google Scholar]
- 11.Keene CD, Rodrigues CM, Eich T, Chhabra MS, Steer CJ, Low WC. 2002. Tauroursodeoxycholic acid, a bile acid, is neuroprotective in a transgenic animal model of Huntington's disease. Proc Natl Acad Sci U S A 99:10671–10676. doi: 10.1073/pnas.162362299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Castro-Caldas M, Carvalho AN, Rodrigues E, Henderson CJ, Wolf CR, Rodrigues CM, Gama MJ. 2012. Tauroursodeoxycholic acid prevents MPTP-induced dopaminergic cell death in a mouse model of Parkinson's disease. Mol Neurobiol 46:475–486. doi: 10.1007/s12035-012-8295-4. [DOI] [PubMed] [Google Scholar]
- 13.Elia AE, Lalli S, Monsurro MR, Sagnelli A, Taiello AC, Reggiori B, La Bella V, Tedeschi G, Albanese A. 2015. Tauroursodeoxycholic acid in the treatment of patients with amyotrophic lateral sclerosis. Eur J Neurol doi: 10.1111/ene.12664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cortez LM, Kumar J, Renault L, Young HS, Sim VL. 2013. Mouse prion protein polymorphism Phe-108/Val-189 affects the kinetics of fibril formation and the response to seeding: evidence for a two-step nucleation polymerization mechanism. J Biol Chem 288:4772–4781. doi: 10.1074/jbc.M112.414581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Butler DA, Scott MR, Bockman JM, Borchelt DR, Taraboulos A, Hsiao KK, Kingsbury DT, Prusiner SB. 1988. Scrapie-infected murine neuroblastoma cells produce protease-resistant prion proteins. J Virol 62:1558–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mahal SP, Baker CA, Demczyk CA, Smith EW, Julius C, Weissmann C. 2007. Prion strain discrimination in cell culture: the cell panel assay. Proc Natl Acad Sci U S A 104:20908–20913. doi: 10.1073/pnas.0710054104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Falsig J, Julius C, Margalith I, Schwarz P, Heppner FL, Aguzzi A. 2008. A versatile prion replication assay in organotypic brain slices. Nat Neurosci 11:109–117. doi: 10.1038/nn2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Campeau JL, Wu G, Bell JR, Rasmussen J, Sim VL. 2013. Early increase and late decrease of Purkinje cell dendritic spine density in prion-infected organotypic mouse cerebellar cultures. PLoS One 8:e81776. doi: 10.1371/journal.pone.0081776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fischer M, Rulicke T, Raeber A, Sailer A, Moser M, Oesch B, Brandner S, Aguzzi A, Weissmann C. 1996. Prion protein (PrP) with amino-proximal deletions restoring susceptibility of PrP knockout mice to scrapie. EMBO J 15:1255–1264. [PMC free article] [PubMed] [Google Scholar]
- 20.Zhu L, Ramboz S, Hewitt D, Boring L, Grass DS, Purchio AF. 2004. Non-invasive imaging of GFAP expression after neuronal damage in mice. Neurosci Lett 367:210–212. doi: 10.1016/j.neulet.2004.06.020. [DOI] [PubMed] [Google Scholar]
- 21.Tamguney G, Francis KP, Giles K, Lemus A, DeArmond SJ, Prusiner SB. 2009. Measuring prions by bioluminescence imaging. Proc Natl Acad Sci U S A 106:15002–15006. doi: 10.1073/pnas.0907339106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Berger E, Haller D. 2011. Structure-function analysis of the tertiary bile acid TUDCA for the resolution of endoplasmic reticulum stress in intestinal epithelial cells. Biochem Biophys Res Commun 409:610–615. doi: 10.1016/j.bbrc.2011.05.043. [DOI] [PubMed] [Google Scholar]
- 23.Song S, Liang JJ, Mulhern ML, Madson CJ, Shinohara T. 2011. Cholesterol-derived bile acids enhance the chaperone activity of alpha-crystallins. Cell Stress Chaperones 16:475–480. doi: 10.1007/s12192-011-0259-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Makarava N, Kovacs GG, Savtchenko R, Alexeeva I, Budka H, Rohwer RG, Baskakov IV. 2011. Genesis of mammalian prions: from noninfectious amyloid fibrils to a transmissible prion disease. PLoS Pathog 7:e1002419. doi: 10.1371/journal.ppat.1002419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sano K, Atarashi R, Ishibashi D, Nakagaki T, Satoh K, Nishida N. 2014. Conformational properties of prion strains can be transmitted to recombinant prion protein fibrils in real-time quaking-induced conversion. J Virol 88:11791–11801. doi: 10.1128/JVI.00585-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ferreira NC, Marques IA, Conceicao WA, Macedo B, Machado CS, Mascarello A, Chiaradia-Delatorre LD, Yunes RA, Nunes RJ, Hughson AG, Raymond LD, Pascutti PG, Caughey B, Cordeiro Y. 2014. Anti-prion activity of a panel of aromatic chemical compounds: in vitro and in silico approaches. PLoS One 9:e84531. doi: 10.1371/journal.pone.0084531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oelschlegel AM, Weissmann C. 2013. Acquisition of drug resistance and dependence by prions. PLoS Pathog 9:e1003158. doi: 10.1371/journal.ppat.1003158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Klohn PC, Stoltze L, Flechsig E, Enari M, Weissmann C. 2003. A quantitative, highly sensitive cell-based infectivity assay for mouse scrapie prions. Proc Natl Acad Sci U S A 100:11666–11671. doi: 10.1073/pnas.1834432100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mahal SP, Demczyk CA, Smith EW Jr, Klohn PC, Weissmann C. 2008. Assaying prions in cell culture: the standard scrapie cell assay (SSCA) and the scrapie cell assay in end point format (SCEPA). Methods Mol Biol 459:49–68. doi: 10.1007/978-1-59745-234-2_4. [DOI] [PubMed] [Google Scholar]
- 30.Falsig J, Sonati T, Herrmann US, Saban D, Li B, Arroyo K, Ballmer B, Liberski PP, Aguzzi A. 2012. Prion pathogenesis is faithfully reproduced in cerebellar organotypic slice cultures. PLoS Pathog 8:e1002985. doi: 10.1371/journal.ppat.1002985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ben Mosbah I, Alfany-Fernandez I, Martel C, Zaouali MA, Bintanel-Morcillo M, Rimola A, Rodes J, Brenner C, Rosello-Catafau J, Peralta C. 2010. Endoplasmic reticulum stress inhibition protects steatotic and non-steatotic livers in partial hepatectomy under ischemia-reperfusion. Cell Death Dis 1:e52. doi: 10.1038/cddis.2010.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, Gorgun CZ, Hotamisligil GS. 2006. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 313:1137–1140. doi: 10.1126/science.1128294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marissen WE, Guo Y, Thomas AA, Matts RL, Lloyd RE. 2000. Identification of caspase 3-mediated cleavage and functional alteration of eukaryotic initiation factor 2α in apoptosis. J Biol Chem 275:9314–9323. doi: 10.1074/jbc.275.13.9314. [DOI] [PubMed] [Google Scholar]
- 34.Yanguas-Casas N, Barreda-Manso MA, Nieto-Sampedro M, Romero-Ramirez L. 2014. Tauroursodeoxycholic acid reduces glial cell activation in an animal model of acute neuroinflammation. J Neuroinflamm 11:50. doi: 10.1186/1742-2094-11-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Crosignani A, Setchell KD, Invernizzi P, Larghi A, Rodrigues CM, Podda M. 1996. Clinical pharmacokinetics of therapeutic bile acids. Clin Pharmacokinetics 30:333–358. doi: 10.2165/00003088-199630050-00002. [DOI] [PubMed] [Google Scholar]
- 36.Invernizzi P, Setchell KD, Crosignani A, Battezzati PM, Larghi A, O'Connell NC, Podda M. 1999. Differences in the metabolism and disposition of ursodeoxycholic acid and of its taurine-conjugated species in patients with primary biliary cirrhosis. Hepatology 29:320–327. doi: 10.1002/hep.510290220. [DOI] [PubMed] [Google Scholar]
- 37.Leuba G, Savioz A, Vernay A, Carnal B, Kraftsik R, Tardif E, Riederer I, Riederer BM. 2008. Differential changes in synaptic proteins in the Alzheimer frontal cortex with marked increase in PSD-95 postsynaptic protein. J Alzheimer's Dis 15:139–151. [DOI] [PubMed] [Google Scholar]
- 38.Omura T, Asari M, Yamamoto J, Oka K, Hoshina C, Maseda C, Awaya T, Tasaki Y, Shiono H, Yonezawa A, Masuda S, Matsubara K, Shimizu K. 2013. Sodium tauroursodeoxycholate prevents paraquat-induced cell death by suppressing endoplasmic reticulum stress responses in human lung epithelial A549 cells. Biochem Biophys Res Commun 432:689–694. doi: 10.1016/j.bbrc.2013.01.131. [DOI] [PubMed] [Google Scholar]