

ABSTRACT
Worldwide G-glycoprotein phylogeny of human respiratory syncytial virus (hRSV) group A sequences revealed diversification in major clades and genotypes over more than 50 years of recorded history. Multiple genotypes cocirculated during prolonged periods of time, but recent dominance of the GA2 genotype was noticed in several studies, and it is highlighted here with sequences from viruses circulating recently in Spain and Panama. Reactivity of group A viruses with monoclonal antibodies (MAbs) that recognize strain-variable epitopes of the G glycoprotein failed to correlate genotype diversification with antibody reactivity. Additionally, no clear correlation was found between changes in strain-variable epitopes and predicted sites of positive selection, despite both traits being associated with the C-terminal third of the G glycoprotein. Hence, our data do not lend support to the proposed antibody-driven selection of variants as a major determinant of hRSV evolution. Other alternative mechanisms are considered to account for the high degree of hRSV G-protein variability.
IMPORTANCE An unusual characteristic of the G glycoprotein of human respiratory syncytial virus (hRSV) is the accumulation of nonsynonymous (N) changes at higher rates than synonymous (S) changes, reaching dN/dS values at certain sites predictive of positive selection. Since these sites cluster preferentially in the C-terminal third of the G protein, like certain epitopes recognized by murine antibodies, it was proposed that immune (antibody) selection might be driving the apparent positive selection, analogous to the antigenic drift observed in the influenza virus hemagglutinin (HA). However, careful antigenic and genetic comparison of the G glycoprotein does not provide evidence of antigenic drift in the G molecule, in agreement with recently published data which did not indicate antigenic drift in the G protein with human sera. Alternative explanations to the immune-driven selection hypothesis are offered to account for the high level of G-protein genetic diversity highlighted in this study.
INTRODUCTION
Human respiratory syncytial virus (hRSV) is recognized as the major cause of severe acute lower respiratory tract infections (ALRI) in infants and young children worldwide (1). hRSV causes annual epidemics, and reinfections are common throughout life, although they are usually less severe than the primary infections. hRSV is also an important cause of morbidity and mortality in the elderly and in adults with cardiopulmonary disease or with an impaired immune system (2).
hRSV is an enveloped, nonsegmented, negative-sense RNA virus, classified in the genus Pneumovirus within the Paramyxoviridae family (for a recent review, see reference 3). The hRSV genome encodes 11 proteins, two of them being the major surface glycoproteins of the virus envelope. These are (i) the attachment (G) protein, which mediates binding of the virus to the cell surface (4), and (ii) the fusion (F) protein, which promotes fusion of the virus and cell membrane, allowing cell entry of the viral genome (5).
The G protein is a type II glycoprotein synthesized as a 32-kDa polypeptide precursor of 297 to 310 amino acids (aa), depending on the strain, and modified posttranslationally by the addition of several N-linked oligosaccharides and multiple O-linked sugar chains (6). The G-protein ectodomain (from residue 67 to the C terminus) has a central conserved region (aa 163 to 189) that includes four Cys residues (residues 173, 176, 182, and 186), and it is essentially devoid of potential glycosylation sites. This conserved region is flanked by two highly variable mucin-like segments, very rich in Ser and Thr, that are potential sites of O glycosylation. The extensive glycosylation of the G protein shapes its reactivity with both murine monoclonal antibodies (MAbs) (7) and human convalescent-phase sera (8).
hRSV isolates were originally classified into two antigenic groups (A and B) based on reactivity with hyperimmune serum and later with G-specific MAbs (9, 10). Antigenic groups A and B were found to correlate with genetically distinct viral groups. Studies of hRSV evolution have focused mainly on the G glycoprotein, since G is the most divergent gene product among hRSV isolates. Recent full-genome sequence analysis has confirmed that G is most informative for studies of hRSV evolution (11).
Three types of epitopes recognized by murine MAbs have been identified in the G molecule: (i) conserved epitopes, which are present in all virus isolates; (ii) group-specific epitopes, which are shared by all viruses of the same antigenic group; and (iii) strain-specific or -variable epitopes, which are shared by a subset of viruses of the same antigenic group (12). Whereas the conserved and group-specific epitopes were mapped in the central conserved region of the G-protein ectodomain, the strain-variable epitopes clustered mainly in the C-terminal third of the G protein.
One of the main evolutionary hallmarks of hRSV G protein is that whereas nucleotide changes spread uniformly along the gene, nonsynonymous (N) changes accumulate at higher rates than synonymous (S) changes in the two variable regions, reaching dN/dS values at certain sites predictive of positive selection (12–14). The fact that these sites cluster preferentially in the C-terminal third of the G-protein primary structure, like the strain-variable epitopes, was taken as tentative evidence of immune (antibody)-driven positive selection, which was proposed as an important determinant of hRSV evolution (13, 15). This type of immune selection postulated for hRSV G protein would then be similar to the well-established antigenic drift described for the influenza virus hemagglutinin (HA) (16). In this case, new influenza virus strains are positively selected with changes in residues of the HA head which are part of epitopes recognized by neutralizing Abs. The new strains can thus reinfect the same human population despite the presence of preexisting antibodies against strains of previous epidemics.
General patterns of virus evolution are better discerned when viruses are sampled from different places over long time periods. Hence, we decided to reassess the genetic evolution of the antigenic group A of hRSV, since sequence information is most abundant for this group of viruses and since two sets of MAbs isolated in our laboratory (17, 18) could be used to compare genetic and antigenic changes in hRSV G protein. The results obtained indicate that group A viruses have diversified during their recorded history in branches (or clades) of different evolutionary significance and temporal dominance. However, epitopes recognized by strain-variable MAbs remain unchanged for long time periods, not showing signs of antigenic drift despite extensive sequence variation of the G glycoprotein.
MATERIALS AND METHODS
Clinical samples and virus isolation.
Samples from Hospital Gregorio Marañón (HGM) (Madrid, Spain) were kindly provided by the HGM BioBank. Informed consent was obtained from the patients' parents or guardians. Samples, diluted in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum, were used to infect HEp-2 cell monolayers growing in 24-well plates, as described previously (15). When cytopathic effect was evident, cells were scrapped off into the medium and the suspension was stored at −80°C. Samples from Instituto Conmemorativo Gorgas (Panamá, Panama) were collected as part of surveillance activities for influenza virus and other respiratory viruses, as approved by the Institute Ethical Committee.
RNA extraction, DNA amplification, and sequencing of the G-protein gene.
Sequencing was done from total RNA automatically extracted from the frozen clinical specimens using QIAamp MinElute virus spin kit and the QIAcube (Qiagen), following the manufacturer's instructions. The full-length G-protein gene was amplified by SuperScript III one-Step reverse transcription-PCR (RT-PCR) (Invitrogen) and primers OG1-21 (5′-GGGGCAAATGCAACCATGTCC-3′; nucleotides [nt] 1 to 21 of the G gene; positive sense) and F164 (5′-GTTATGACACTGGTATACCAACC-3′; nt 141 to 164 of the F gene, negative sense). PCR products were subjected to forward and reverse cycle sequencing with the BigDye Terminator 3.1 kit (Applied Biosystems) and the above-described primers.
Sequence data and BLAST search.
The sequences reported here were aligned with ClustalX 1.81 (19) and manually edited with BioEdit version 7.0.9.1 (20). Other sequences were retrieved from GenBank. Since many sequences were not full length, a total of 2,167 sequences which spanned nt 312 to the end of the G-protein gene (i.e., most of the protein ectodomain) were selected and included in the study. These sequences were aligned with the online version of MAFFT v7 software (21). Duplicate sequences were identified with the ElimDupes tool (http://hcv.lanl.gov/content/sequence/ELIMDUPES/elimdupes.html) (22) and deleted. The remaining 1,485 unique sequences used in this study and the corresponding GenBank accession numbers are listed in Table S1 in the supplemental material.
Phylogenetic analysis by Bayesian MCMC and maximum-likelihood methods.
Phylogenetic analysis by the Markov chain Monte Carlo (MCMC) method was performed with the BEAST v1.7.4 package (http://beast.bio.ed.ac.uk) (23, 24), using the GTR + invariant + Gamma model selected as the best-fitting nucleotide substitution model for hRSV G-protein sequences by using hierarchical likelihood ratio testing, implemented in the ModelTest software version 3.06 (25) The data set was analyzed using the Bayesian skyline model, assuming a relaxed (uncorrelated log normal) molecular clock. MCMC chains were run to achieve convergence, which was confirmed with Tracer v1.6.0 (http://beast.bio.ed.ac.uk/Tracer). Statistical uncertainty in parameter estimates is given by the 95% highest-probability density (HPD) values. The data obtained in the MCMC analysis were also used to infer a maximum clade credibility (MCC) tree with Tree-Annotator v1.4.7 and FigTree v1.4.2 (http://tree.bio.ed.ac.uk/software/figtree/). MEGA software version 6 was used for the maximum-likelihood phylogenetic analysis (26).
Fluorescent labeling of infected cells with MAbs.
HEp-2 cells growing in 96-well microtiter plates were infected at a multiplicity of infection (MOI) of ∼ 0.5 PFU/cell with viruses representative of the hRSV A genotypes. Twenty-four hours later, cells were washed with phosphate-buffered saline (PBS) containing 0.05% Tween 20, fixed with 80% acetone, and incubated with anti-G MAbs, followed by fluorescein-linked antibody (GE Healthcare). Cell-associated fluorescence was measured with a Tecan Infinite 200 Pro (Tecan Group Ltd.). In addition, cells were examined with a UV-illuminated Nikon Eclipse TS100 microscope.
Nucleotide sequence accession numbers.
The sequences reported here were deposited in the GenBank database under accession numbers KF300969 and KF300971 to KF301019 for sequences from Panama and KP792352 to KP792376 for sequences from Madrid.
RESULTS
Evolution and dominance of group A genotypes over time.
Group A viruses were classified by Peret et al. into five genotypes (27), to which nine new genotypes have been added over time by different authors without uniform criteria. It was thus considered necessary to reevaluate the current classification of group A genotypes. Hence, 1,485 unique sequences (from nucleotide 312 to the end) of the G gene were withdrawn from GenBank, together with genotype information, when available. The aligned sequences were used to assemble the maximum clade credibility (MCC) tree shown in Fig. 1A. Eleven of the 14 different genotypes previously described (GA1, GA2, GA3, GA4, GA5, GA6, GA7, NA1, NA2, NA4, and ON1) were identified in the tree. The NA3, SAA1, and SAA2 genotypes were not included, since only partial C-terminal sequences are available in the databases. The genetic P distances between individual genotypes, as well as within each genotype, were calculated (Table 1). The highest intragenotypic P distance (0.049) was found in the GA1 genotype which includes some of the oldest hRSV strains (Long and A2). This P value was thus taken as the minimal threshold for sorting viruses into different genotypes. Using this criterion, the NA1, NA2, NA4, and ON1 genotypes were reclassified into the GA2 genotype, leaving only seven well-recognized genotypes (GA1 to GA7) within group A of hRSV (Table 2).
FIG 1.

Phylogeny of hRSV group A viruses and genotype temporal dominance. (A) Maximum clade credibility (MCC) tree from Bayesian analysis of 1,485 unique nucleotide sequences of the G-protein gene ectodomain of hRSV group A retrieved from GenBank. Clades are colored according the genotype classification shown in Table 1. (B) Frequencies of the different genotypes in 5-year periods from 1956 to 2013. The number of sequences (n) included in each period is indicated below the charts.
TABLE 1.
Genetic distances between and within the genotypes described in the literature for 2,167 hRSV group A sequences of the G-protein ectodomain
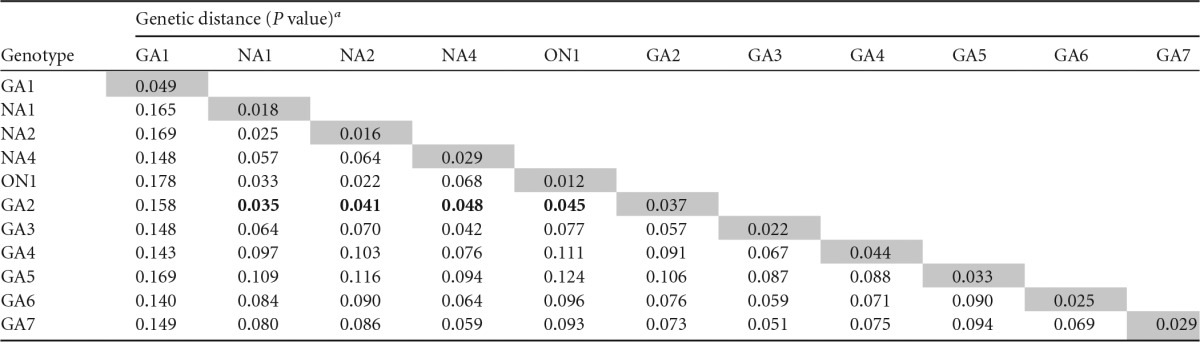
Pairwise distances were calculated between individual genotypes, as well as within each genotype, using MEGA software version 6. BioEdit version 7.0.9.1 was used for amino acid analysis (20). Boldface indicates P values between genotypes GA2 and NA1, NA2, NA4, and ON1 that are below the intragenotypic distance for GA1, which was taken as the minimal threshold for clustering sequences in different genotypes. Therefore, the NA1, NA2, NA4, and ON1 genotypes were regrouped within the GA2 genotype in Table 2.
TABLE 2.
Genetic distances (P values) within and between the genotypes in which group A sequences of the G-protein ectodomain of hRSV were reclassified in this study
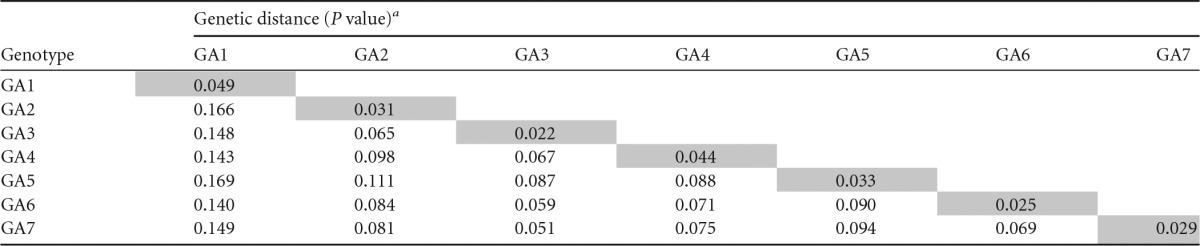
Pairwise distances were calculated as for Table 1. Note that values for GA2 slightly differ from those in Table 1 as result of genotype reclassification.
The most recent common ancestor (MRCA) of all group A sequences dated back to the 1940s, when a major split into two branches occurred (Fig. 1A). One of the branches included viruses of the GA1 genotype. The other branch split in the early 1970s into two new branches. One of them gave rise to the GA4 and GA5 genotypes. Whereas only few GA4 viruses circulated for a short time period, the GA5 genotype has dominated that branch and has survived until today. The other new branch diversified with time in the group A genotypes, GA2, GA3, GA6, and GA7. It is worth emphasizing that the major branching events seen in Fig. 1A occurred only occasionally, while diversification into genotypes occurred more frequently.
It is also evident from Fig. 1A that cocirculation of genotypes occurred throughout most of the known history of group A viruses, except on two occasions. One was before mid-1970s, and the other is today. This is best visualized in Fig. 1B, where the numbers of recorded sequences from different genotypes are grouped and color coded in 5-year intervals. GA1 prevailed before 1980, although the low number of samples from this time period prevents a definitive conclusion about genotype dominance. During the 1980s and 1990s, viruses from almost all genotypes were circulating, with fluctuating dominance. This situation was highlighted in numerous publications from those dates (see, for instance references 27 and 28); however, after 2000 and particularly after 2005, the proportion of viruses belonging to the GA2 genotype steadily increased, to become almost exclusive after 2010. Since the data in Fig. 1B have been extracted from multiple studies using different set of primers and slightly different methods (see Table S1 in the supplemental material), it is unlikely that sampling or geographical bias may account for the observed shift in GA2 dominance. However, very low-level circulation of viruses belonging to other genotypes cannot be excluded, as recently exemplified for group B viruses (29).
Current situation.
Recent publications have drawn attention to the shift from “multiple genotype circulation to prolonged circulation of predominant genotypes,” as seen in Belgium between 1996 and 2011 (30). In addition, a novel GA2 variant with a 72-nt duplication, named ON1 and first detected in Ontario (Canada) in December 2010 (31), has spread rapidly worldwide (32–34), exacerbating the current dominance of the GA2 genotype.
Access to very recent hRSV samples from Madrid (Spain) and Panama allowed us to assess the present genotype dominance in these two places, which are geographically distant and with different climates. Ectodomain G sequences of 49 samples from Panama and 25 samples from Madrid were determined and used to build the phylogenetic tree shown in Fig. 2 with representatives of the main group A genotypes. The Madrid samples were from the epidemics in 2007 to 2008, 2012 to 2013, and 2013 to 2014. The Panama sequences were from 2010, 2011, and 2012. Among the Madrid samples, the GA5 genotype represented a minority of sequences from 2007 to 2008; in contrast, most samples from this epidemic together with all sequences from the last two epidemics were clustered in the GA2 genotype. Madrid viruses from the last two epidemics contained the 72-nt duplication characteristic of the ON1 variant. Sequences from Panama were all clustered within GA2, most of them within ON1. Clearly, sequences from both places were interlocked in the tree, strengthening the idea of temporal rather than local clustering of hRSV strains and the present dominance of the GA2 genotype.
FIG 2.

Phylogenetic tree of hRSV group A sequences from recent epidemics in Madrid and Panama. The maximum-likelihood phylogenetic tree was constructed on the basis of nucleotide sequences of the G-protein ectodomain obtained from Madrid (diamonds) and Panama (circles) samples. Virus nomenclature follows the general consensus, with the last two digits referring to year of isolation. The bar represents 0.02 nucleotide substitution per site, and the tree is unrooted. Numbers at the internal nodes represent the bootstrap probabilities (1,000 replicates). Only bootstrap values of >70 are shown. The number of sequences identical to those shown in the figure is indicated in parentheses at right of the sample name. Asterisks denote viruses included in the analysis shown in Fig. 3 and 4.
Reactivity with strain-specific anti-G monoclonal antibodies.
Some of the strain-variable MAbs isolated in our laboratory have been used previously to assess the antigenic relatedness of hRSV strains collected through relatively short time periods (15, 35, 36). Hence, a set of hRSV group A viruses, covering the entire recorded history of this antigenic group and representing all genotypes shown in Table 2 except GA4 and GA6 (for which viruses were not available), was used to reevaluate reactivity with the strain-variable MAbs.
The results shown in Fig. 3 show two apparent antigenic subgroups according to reactivity with the MAb panel. One subgroup included the viruses representative of the GA1 genotype, which reacted efficiently with MAbs 25G, 78G, and 68G raised against the Long strain but lacked reactivity with the strain-variable MAbs raised against the Mon/3/88 virus. The exception was MAb 63G, which, as originally reported, showed cross-reactivity with an extended set of viruses (but not all) without any obvious trend (17). The other subgroup included viruses from genotype GA2, including its ON1 variant and genotypes GA3, GA7, and GA5. All these viruses lacked reactivity with the MAbs raised against the Long strain (except the noted 63G) but reacted with most MAbs raised against Mon/3/88, with some exceptions discussed below. The results shown in Fig. 3 were in good agreement with the fluorescence patterns of infected cells stained with the MAbs (Fig. 4).
FIG 3.

Reactivity of group A viruses with MAbs. Sequences of the viral strains used in this experiment were used to build the phylogenetic tree shown on the left, as for Fig. 2. Each virus was used to infect HEp-2 cell cultures, which were stained at 24 h after infection with the indicated MAbs and anti-mouse fluorescein-linked antibody (GE Healthcare). Two panels of strain-variable MAbs were used: one panel included MAbs 63G, 25G, 78G, and 68G, obtained from mice inoculated with the Long strain of hRSV (17), and the other included MAbs 021/12G, 021/10G, 021/9G, 021/8G, 021/7G, and 021/16G, obtained from mice inoculated with Mon/3/88 virus (18). MAb 021/1G, which recognizes a conserved epitope of hRSV G protein, was included as control. Numbers shown within the boxes are the fluorescence values after normalization, so that fluorescence of Long with each Long-specific MAb was normalized to 100% and similarly for the Mon/3/88 virus with the MAbs specific for this virus. Squares with fluorescence values of >50% have a black background, those with values between 25% and 50% have a gray background, and those with values of <25% have a white background. The results are representative of five independent determinations.
FIG 4.

UV light photographs of cultures used for the quantitative analysis in Fig. 3.
It is worth stressing that the patterns of MAb reactivity shown in Fig. 3 and 4 did not show a clear association with viral genotypes, except as noted with GA1. For instance, MAb 021/16G did not react with Mon/4/90 but reacted efficiently with Mad/4/90 from the same genotype (GA3). Exactly the opposite was true for MAb 021/7G with the same two viruses. Antibody reactivity also could not be associated with time of virus isolation. Remarkably, the epitopes of MAbs 25G, 78G, and 68G raised against the Long strain of 1956 were preserved in viruses isolated almost 40 years later in Montevideo (Fig. 3 and 4). Similarly, the epitopes recognized by MAbs raised against Mon/3/88 were preserved in most viruses isolated 24 years later in Madrid and that contained the ON1 72-nt duplication.
Figure 5A shows the alignment of sequences of the G-protein C-terminal third from viruses included in the antigenic analysis shown in Fig. 3. Residues that were changed in previously described mutants that are resistant to certain MAbs (15, 18) are indicated. When the sequence changes shown in Fig. 5A are compared with the MAb reactivities shown in Fig. 3, three main conclusions can be reached.
FIG 5.

Sequence alignment of the C-terminal thirds of G-protein sequences. (A) Alignment of partial (C-terminal) G-protein sequences of the viruses used for Fig. 3. Numbering is shown above the Long sequence, which is used as a reference for the next two viruses. The entire sequence of Mon/3/88 is also shown as a reference for the rest of viruses. Only the amino acid changes are indicated. A lack of change is denoted by a dot. Asterisks indicate stop codons. Note the 72-nt duplicated sequence in four viruses, which forces the gaps denoted by hyphens in the other sequences. Residues that showed changes in mutants selected with the indicated MAbs are indicated by arrows at the top. (B) Sites of positive selection predicted in the indicated studies (11, 13, 30, 31, 33, 49–53) are denoted by small circles below the corresponding amino acid.
(i) Loss of reactivity with some MAbs coincided with certain sequence changes. For instance, MAb 63G did not react with viruses Mon/9/91, Mad/GM2_14/12, and Mon/4/90, which have the changes P206Q, K205E, and F208P, respectively, within the stretch of amino acids where epitope 63G has been mapped (37). Note, however, that the changes F208I/L in the same region did not alter reactivity with MAb 63G. Similarly, the total or partial loss of reactivity of MAbs 021/16G and 021/9G with Mon/4/90 coincided with the R244S change.
(ii) In other cases, however, residues that changed in escape mutants were totally conserved in natural isolates. For instance, residue 234, which changed in certain mutants resistant to MAb 68G, was unaltered in natural viruses. Similarly, residues 237 and 239, which changed in mutants selected with MAb 021/8G, or residue 284, which changed in mutants resistant to MAb 78G, were conserved in all sequences shown in Fig. 5A, irrespective of their MAb reactivity pattern.
(iii) The MAbs used for Fig. 3 have been reported to react in Western blotting with the G proteins of viruses used in their selection (7, 38). Each epitope should thus encompass several contiguous amino acids of the G-protein primary structure. It was therefore surprising to find the relatively high level of epitope conservation shown in Fig. 3 notwithstanding the extensive sequence variation of the G protein in that region (Fig. 5A). Once more, although sporadic changes were observed in individual viruses, no clear accumulation of antigenic changes with time or genetic distance was discernible.
DISCUSSION
Two enthralling findings stand out from this study: (i) the diversification of group A viruses in major branches after relatively long periods of time followed by periodic dominance of certain genotypes and (ii) the level of epitope conservation in the G glycoprotein despite the high level of sequence variation.
It is clear from Fig. 1 that diversification in major branches differs from genotype divergence not only in the magnitude of the genetic distances involved but additionally in the frequency of their respective splitting events. Hence, it is plausible that the two types of diversification have different causes, hitherto unknown. Genotype GA1, which originated from the main branching event shown in Fig. 1 is now apparently extinct. It may be that GA1 viruses exhausted the repertoire of functional amino acids that could be changed in the G glycoprotein. It is also obvious from Fig. 1B that genotype dominance has alternated in different time periods. While several genotypes cocirculated most of the time with alternating dominance, the GA2 genotype has become almost exclusive since 2005, as reported in several studies (30, 33) and observed with recent viruses from Madrid and Panama (Fig. 2). Although studied in less detail, similar shifts in genotype dominance have been reported for group B viruses (39).
What, then, are the selective forces driving branching, genotype divergence, temporal dominance, and intragenotypic evolution of hRSV? By analogy with other viruses, such as influenza A virus, antibody-driven positive selection has been proposed as a major determinant of hRSV evolution (13) to enable reinfections of the same population, an epidemiological hallmark of hRSV (40). Positive selection is supported by the high rate of dN/dS substitutions and by predictions of positively selected changes at certain sites of the G glycoprotein. Figure 5B shows sites of positive selection in the C-terminal third of the G glycoprotein predicted in different studies. The accumulation of those sites (but not all) in the same region of the G protein where strain-variable epitopes are clustered has been a major argument for the antibody-driven positive selection hypothesis of hRSV evolution.
Often, however, changes in sites of positive selection do not correlate with changes in MAb reactivity. For instance, one of the most recurrently predicted sites of positive selection is residue 237, where MAb 021/8G selected an escape mutant with the change N237Y (18). Paradoxically, however, amino acid 237 is conserved in all sequences shown in Fig. 5A, including those of viruses of the GA1 genotype which are not recognized by MAb 021/8G. In other sites of positive selection, such as residue 274, changes found in certain viruses (L to P, I, or T) do not correlate with the MAb reactivity shown in Fig. 3. Hence, no definitive association between altered antibody reactivity and sites of predicted positively selected changes could be shown.
Additionally, when Fig. 3 and 5 are globally examined, no obvious correspondence between genotype genetic relatedness and MAb reactivity pattern is observed. These results are generally in agreement with previous reports that detected sporadic antigenic changes in viruses circulating in Argentina and Chile (35, 41) or in Germany (36) with the MAbs used in this study but without any distinctive trend.
It may be argued that murine MAbs may not represent the repertoire of human antibodies raised after hRSV infection. Without excluding this possibility, it is worth mentioning that linear epitopes recognized by antibodies present in human sera have been detected in peptides (42) or protein segments from the G glycoprotein C-terminal third (8).
It should be also stressed that most murine MAbs raised against the G glycoprotein are weak neutralizers (17, 43). Furthermore, most of the neutralizing activity found in human immunoglobulins is directed against highly conserved epitopes of the F glycoprotein (44, 45). In other words, the immune pressure afforded by anti-G antibodies is expected to be only marginal, if any. Hence, it may be that hRSV reinfections are determined by short-lived (or weak) antibody responses rather than selection of antigenic variants. Indeed, recent studies have provided evidence that reinfections in children are caused almost as frequently by heterologous viruses as by viruses of the homologous antigenic group (46, 47). In addition, although partial group-specific neutralizing responses were noted in very young children after hRSV infections, neutralization was reported to be equally effective against contemporary or historical viruses, suggesting no significant antigenic drift (48). Indeed, similar results have been obtained in our laboratory with a limited set of infant sera, in which neither neutralizing nor G-protein binding antibodies were strain dependent within the same antigenic group as the infecting virus (A. Trento et al., unpublished data).
In summary, this study highlights a complex pattern of group A hRSV diversification, with major branching and temporal genotype dominance over time that could not be directly related to antigenic changes. Although some weak antibody selection cannot be excluded, our results do not support the idea that the high level of sequence variation in the G glycoprotein is the result of an antigenic drift similar to that of influenza A virus HA (16). It is thus likely that other factors contribute to the accumulation of sequence changes in hRSV G protein. The high plasticity of this protein to incorporate drastic sequence changes without apparent alterations in virus fitness should be stressed (12). Is it possible, then, that the seemingly positive selection of changes in RSV G protein is the result of a high mutation rate (as generally in RNA viruses) together with selective constrains other than immune selection? For instance, is it possible that bottleneck effects occurring during virus transmission together with a very malleable molecule may result in an apparent positive selection in hRSV G protein? It may be that hRSV has found an “entropically” favorable solution for the G protein so that its unusual amino acid sequence and its added malleability maintain functionality together with an apparent positive selection. Further studies with well-selected hRSV strains should help to discern among this possibility and others lying behind the intriguing paradoxes of hRSV evolution.
Supplementary Material
ACKNOWLEDGMENTS
We thank Juan Ortín, Isidoro Martínez, and Jason McLellan for critical readings of the manuscript and very useful comments and members of the Genomic Core Facility at the Instituto de Salud Carlos III for technical help. We particularly acknowledge the patients in this study for their participation and the HGM BioBank integrated in RETICS, National Network Biobanks, and collaborating centers for the generous gifts of clinical samples used in this work.
This work was supported by grant SAF2012-31217 from Plan Nacional I+D+I (J.A.M., Ministerio de Economía y Competitividad, Spain) and grant CAP10-001, Senacyt-Panamá. The HGM BioBank, integrated in National Network Biobanks, is supported by Instituto de Salud Carlos III, Spanish Science and Innovation Ministry (grant number PT13/0010/0028) and Fundación para la Investigación y Prevención del SIDA (FIPSE), Spain.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.00467-15.
REFERENCES
- 1.Nair H, Nokes DJ, Gessner BD, Dherani M, Madhi SA, Singleton RJ, O'Brien KL, Roca A, Wright PF, Bruce N, Chandran A, Theodoratou E, Sutanto A, Sedyaningsih ER, Ngama M, Munywoki PK, Kartasasmita C, Simoes EA, Rudan I, Weber MW, Campbell H. 2010. Global burden of acute lower respiratory infections due to respiratory syncytial virus in young children: a systematic review and meta-analysis. Lancet 375:1545–1555. doi: 10.1016/S0140-6736(10)60206-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Falsey AR, Hennessey PA, Formica MA, Cox C, Walsh EE. 2005. Respiratory syncytial virus infection in elderly and high-risk adults. N Engl J Med 352:1749–1759. doi: 10.1056/NEJMoa043951. [DOI] [PubMed] [Google Scholar]
- 3.Collins PL, Melero JA. 2011. Progress in understanding and controlling respiratory syncytial virus: still crazy after all these years. Virus Res 162:80–99. doi: 10.1016/j.virusres.2011.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Levine S, Klaiber-Franco R, Paradiso PR. 1987. Demonstration that glycoprotein G is the attachment protein of respiratory syncytial virus. J Gen Virol 68:2521–2524. doi: 10.1099/0022-1317-68-9-2521. [DOI] [PubMed] [Google Scholar]
- 5.Walsh EE, Hruska J. 1983. Monoclonal antibodies to respiratory syncytial virus proteins: identification of the fusion protein. J Virol 47:171–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gruber C, Levine S. 1985. Respiratory syncytial virus polypeptides. V. The kinetics of glycoprotein synthesis. J Gen Virol 66:1241–1247. [DOI] [PubMed] [Google Scholar]
- 7.Garcia-Beato R, Martinez I, Franci C, Real FX, Garcia-Barreno B, Melero JA. 1996. Host cell effect upon glycosylation and antigenicity of human respiratory syncytial virus G glycoprotein. Virology 221:301–309. doi: 10.1006/viro.1996.0379. [DOI] [PubMed] [Google Scholar]
- 8.Palomo C, Cane PA, Melero JA. 2000. Evaluation of the antibody specificities of human convalescent-phase sera against the attachment (G) protein of human respiratory syncytial virus: influence of strain variation and carbohydrate side chains. J Med Virol 60:468–474. doi:. [DOI] [PubMed] [Google Scholar]
- 9.Anderson LJ, Hierholzer JC, Tsou C, Hendry RM, Fernie BF, Stone Y, McIntosh K. 1985. Antigenic characterization of respiratory syncytial virus strains with monoclonal antibodies. J Infect Dis 151:623–633. [DOI] [PubMed] [Google Scholar]
- 10.Mufson MA, Orvell C, Rafnar B, Norrby E. 1985. Two distinct subtypes of human respiratory syncytial virus. J Gen Virol 66:2111–2124. doi: 10.1099/0022-1317-66-10-2111. [DOI] [PubMed] [Google Scholar]
- 11.Tan L, Lemey P, Houspie L, Viveen C, Jansen NJ, van Loon AM, Wiertz E, van Bleek GM, Martin DP, Coenjaerts FE. 2012. Genetic variability among complete human respiratory syncytial virus subgroup A genomes: bridging molecular evolutionary dynamics and epidemiology. PLoS One 7:e51439. doi: 10.1371/journal.pone.0051439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Melero JA, Garcia-Barreno B, Martinez I, Pringle CR, Cane PA. 1997. Antigenic structure, evolution and immunobiology of human respiratory syncytial virus attachment (G) protein. J Gen Virol 78:2411–2418. [DOI] [PubMed] [Google Scholar]
- 13.Woelk CH, Holmes EC. 2001. Variable immune-driven natural selection in the attachment (G) glycoprotein of respiratory syncytial virus (RSV). J Mol Evol 52:182–192. [DOI] [PubMed] [Google Scholar]
- 14.Tan L, Coenjaerts FE, Houspie L, Viveen MC, van Bleek GM, Wiertz EJ, Martin DP, Lemey P. 2013. The comparative genomics of human respiratory syncytial virus subgroups A and B: genetic variability and molecular evolutionary dynamics. J Virol 87:8213–8226. doi: 10.1128/JVI.03278-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garcia O, Martin M, Dopazo J, Arbiza J, Frabasile S, Russi J, Hortal M, Perez-Brena P, Martinez I, Garcia-Barreno B. 1994. Evolutionary pattern of human respiratory syncytial virus (subgroup A): cocirculating lineages and correlation of genetic and antigenic changes in the G glycoprotein. J Virol 68:5448–5459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Knossow M, Skehel JJ. 2006. Variation and infectivity neutralization in influenza. Immunology 119:1–7. doi: 10.1111/j.1365-2567.2006.02421.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garcia-Barreno B, Palomo C, Penas C, Delgado T, Perez-Brena P, Melero JA. 1989. Marked differences in the antigenic structure of human respiratory syncytial virus F and G glycoproteins. J Virol 63:925–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martinez I, Dopazo J, Melero JA. 1997. Antigenic structure of the human respiratory syncytial virus G glycoprotein and relevance of hypermutation events for the generation of antigenic variants. J Gen Virol 78:2419–2429. [DOI] [PubMed] [Google Scholar]
- 19.Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. 1997. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hall TA. 1999. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 93/98/NT41. Nucleic Acids Symp Ser 41:95–98. [Google Scholar]
- 21.Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuiken C, Yusim K, Boykin L, Richardson R. 2005. The Los Alamos hepatitis C sequence database. Bioinformatics 21:379–384. doi: 10.1093/bioinformatics/bth485. [DOI] [PubMed] [Google Scholar]
- 23.Drummond AJ, Rambaut A, Shapiro B, Pybus OG. 2005. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol Biol Evol 22:1185–1192. doi: 10.1093/molbev/msi103. [DOI] [PubMed] [Google Scholar]
- 24.Drummond AJ, Rambaut A. 2007. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol 7:214. doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Posada D, Crandall KA. 1998. MODELTEST: testing the model of DNA substitution. Bioinformatics 14:817–818. doi: 10.1093/bioinformatics/14.9.817. [DOI] [PubMed] [Google Scholar]
- 26.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. 2013. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol 30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peret TC, Hall CB, Schnabel KC, Golub JA, Anderson LJ. 1998. Circulation patterns of genetically distinct group A and B strains of human respiratory syncytial virus in a community. J Gen Virol 79:2221–2229. [DOI] [PubMed] [Google Scholar]
- 28.Cane PA, Matthews DA, Pringle CR. 1994. Analysis of respiratory syncytial virus strain variation in successive epidemics in one city. J Clin Microbiol 32:1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Agoti CN, Gitahi CW, Medley GF, Cane PA, Nokes DJ. 2013. Identification of group B respiratory syncytial viruses that lack the 60-nucleotide duplication after six consecutive epidemics of total BA dominance at coastal Kenya. Influenza Other Respir Viruses 7:1008–1012. doi: 10.1111/irv.12131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Houspie L, Lemey P, Keyaerts E, Reijmen E, Vergote V, Vankeerberghen A, Vaeyens F, De BH, Van RM. 2013. Circulation of HRSV in Belgium: from multiple genotype circulation to prolonged circulation of predominant genotypes. PLoS One 8:e60416. doi: 10.1371/journal.pone.0060416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eshaghi A, Duvvuri VR, Lai R, Nadarajah JT, Li A, Patel SN, Low DE, Gubbay JB. 2012. Genetic variability of human respiratory syncytial virus A strains circulating in Ontario: a novel genotype with a 72 nucleotide G gene duplication. PLoS One 7:e32807. doi: 10.1371/journal.pone.0032807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Prifert C, Streng A, Krempl CD, Liese J, Weissbrich B. 2013. Novel respiratory syncytial virus a genotype, Germany, 2011-2012. Emerg Infect Dis 19:1029–1030. doi: 10.3201/eid1906.121582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martinelli M, Frati ER, Zappa A, Ebranati E, Bianchi S, Pariani E, Amendola A, Zehender G, Tanzi E. 2014. Phylogeny and population dynamics of respiratory syncytial virus (Rsv) A and B. Virus Res 189:293–302. doi: 10.1016/j.virusres.2014.06.006. [DOI] [PubMed] [Google Scholar]
- 34.Agoti CN, Otieno JR, Gitahi CW, Cane PA, Nokes DJ. 2014. Rapid spread and diversification of respiratory syncytial virus genotype ON1, Kenya. Emerg Infect Dis 20:950–959. doi: 10.3201/eid2006.131438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Galiano MC, Luchsinger V, Videla CM, De Souza L, Puch SS, Palomo C, Ricarte C, Ebekian B, Avendano L, Carballal G. 2005. Intragroup antigenic diversity of human respiratory syncytial virus (group A) isolated in Argentina and Chile. J Med Virol 77:311–316. doi: 10.1002/jmv.20456. [DOI] [PubMed] [Google Scholar]
- 36.Adams O, Werzmirzowsky J, Hengel H. 2013. Genetic analysis and antigenic characterization of human respiratory syncytial virus group A viruses isolated in Germany 1996-2008. Virus Genes 47:210–218. doi: 10.1007/s11262-013-0936-9. [DOI] [PubMed] [Google Scholar]
- 37.Garcia-Barreno B, Delgado T, Akerlind-Stopner B, Norrby E, Melero JA. 1992. Location of the epitope recognized by monoclonal antibody 63G on the primary structure of human respiratory syncytial virus G glycoprotein and the ability of synthetic peptides containing this epitope to induce neutralizing antibodies. J Gen Virol 73:2625–2630. doi: 10.1099/0022-1317-73-10-2625. [DOI] [PubMed] [Google Scholar]
- 38.Palomo C, Garcia-Barreno B, Penas C, Melero JA. 1991. The G protein of human respiratory syncytial virus: significance of carbohydrate side-chains and the C-terminal end to its antigenicity. J Gen Virol 72:669–675. doi: 10.1099/0022-1317-72-3-669. [DOI] [PubMed] [Google Scholar]
- 39.Trento A, Casas I, Calderon A, Garcia-Garcia ML, Calvo C, Perez-Brena P, Melero JA. 2010. Ten years of global evolution of the human respiratory syncytial virus BA genotype with a 60-nucleotide duplication in the G protein gene. J Virol 84:7500–7512. doi: 10.1128/JVI.00345-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Henderson FW, Collier AM, Clyde WA Jr, Denny FW. 1979. Respiratory-syncytial-virus infections, reinfections and immunity. A prospective, longitudinal study in young children. N Engl J Med 300:530–534. [DOI] [PubMed] [Google Scholar]
- 41.Galiano MC, Palomo C, Videla CM, Arbiza J, Melero JA, Carballal G. 2005. Genetic and antigenic variability of human respiratory syncytial virus (groups a and b) isolated over seven consecutive seasons in Argentina (1995 to 2001). J Clin Microbiol 43:2266–2273. doi: 10.1128/JCM.43.5.2266-2273.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cane PA. 1997. Analysis of linear epitopes recognised by the primary human antibody response to a variable region of the attachment (G) protein of respiratory syncytial virus. J Med Virol 51:297–304. doi:. [DOI] [PubMed] [Google Scholar]
- 43.Anderson LJ, Bingham P, Hierholzer JC. 1988. Neutralization of respiratory syncytial virus by individual and mixtures of F and G protein monoclonal antibodies. J Virol 62:4232–4238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Magro M, Mas V, Chappell K, Vazquez M, Cano O, Luque D, Terron MC, Melero JA, Palomo C. 2012. Neutralizing antibodies against the preactive form of respiratory syncytial virus fusion protein offer unique possibilities for clinical intervention. Proc Natl Acad Sci U S A 109:3089–3094. doi: 10.1073/pnas.1115941109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McLellan JS, Chen M, Leung S, Graepel KW, Du X, Yang Y, Zhou T, Baxa U, Yasuda E, Beaumont T, Kumar A, Modjarrad K, Zheng Z, Zhao M, Xia N, Kwong PD, Graham BS. 2013. Structure of RSV fusion glycoprotein trimer bound to a prefusion-specific neutralizing antibody. Science 340:1113–1117. doi: 10.1126/science.1234914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yui I, Fujino M, Sawada A, Nakayama T. 2014. Novel clinical features of recurrent human respiratory syncytial virus infections. J Med Virol 86:1629–1638. doi: 10.1002/jmv.23809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Agoti CN, Mwihuri AG, Sande CJ, Onyango CO, Medley GF, Cane PA, Nokes DJ. 2012. Genetic relatedness of infecting and reinfecting respiratory syncytial virus strains identified in a birth cohort from rural kenya. J Infect Dis 206:1532–1541. doi: 10.1093/infdis/jis570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sande CJ, Mutunga MN, Medley GF, Cane PA, Nokes DJ. 2013. Group- and genotype-specific neutralizing antibody responses against respiratory syncytial virus in infants and young children with severe pneumonia. J Infect Dis 207:489–492. doi: 10.1093/infdis/jis700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zlateva KT, Lemey P, Vandamme AM, Van RM. 2004. Molecular evolution and circulation patterns of human respiratory syncytial virus subgroup a: positively selected sites in the attachment G glycoprotein. J Virol 78:4675–4683. doi: 10.1128/JVI.78.9.4675-4683.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gaunt ER, Jansen RR, Poovorawan Y, Templeton KE, Toms GL, Simmonds P. 2011. Molecular epidemiology and evolution of human respiratory syncytial virus and human metapneumovirus. PLoS One 6:e17427. doi: 10.1371/journal.pone.0017427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pretorius MA, van NS, Tempia S, Moyes J, Cohen C, Madhi SA, Venter M. 2013. Replacement and positive evolution of subtype A and B respiratory syncytial virus G-protein genotypes from 1997-2012 in South Africa. J Infect Dis 208(Suppl 3):S227–S237. doi: 10.1093/infdis/jit477. [DOI] [PubMed] [Google Scholar]
- 52.Kim YJ, Kim DW, Lee WJ, Yun MR, Lee HY, Lee HS, Jung HD, Kim K. 2014. Rapid replacement of human respiratory syncytial virus A with the ON1 genotype having 72 nucleotide duplication in G gene. Infect Genet Evol 26:103–112. doi: 10.1016/j.meegid.2014.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Botosso VF, Zanotto PM, Ueda M, Arruda E, Gilio AE, Vieira SE, Stewien KE, Peret TC, Jamal LF, Pardini MI, Pinho JR, Massad E, Sant'anna OA, Holmes EC, Durigon EL. 2009. Positive selection results in frequent reversible amino acid replacements in the G protein gene of human respiratory syncytial virus. PLoS Pathog 5:e1000254. doi: 10.1371/journal.ppat.1000254. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.